

Abstract
The direct synthetic organic use of electricity is currently experiencing a renaissance. More synthetically oriented laboratories working in this area are exploiting both novel and more traditional concepts, paving the way to broader applications of this niche technology. As only electrons serve as reagents, the generation of reagent waste is efficiently avoided. Moreover, stoichiometric reagents can be regenerated and allow a transformation to be conducted in an electrocatalytic fashion. However, the application of electroorganic transformations is more than minimizing the waste footprint, it rather gives rise to inherently safe processes, reduces the number of steps of many syntheses, allows for milder reaction conditions, provides alternative means to access desired structural entities, and creates intellectual property (IP) space. When the electricity originates from renewable resources, this surplus might be directly employed as a terminal oxidizing or reducing agent, providing an ultra‐sustainable and therefore highly attractive technique. This Review surveys recent developments in electrochemical synthesis that will influence the future of this area.
Keywords: electrochemistry, oxidation, reduction, sustainable chemistry, synthetic methods
1. Introduction
For the first time since the industrial revolution, excessive energy consumption and energy sources are major topics of political and social discourse.1 Current innovations are rated not only in terms of their benefit and utility, but also with regard to their ecological footprint. The development of green industrial processes is becoming more and more important and requires unobjectionable energy sources. Particularly over the past decade, energy from renewable resources has been increasingly exploited all over the world to reduce pollution, carbon dioxide emission, and waste generation.2 While these energy sources help to render industrial processes “greener” with regard to energy consumption, it is worthwhile to adapt this principle also in chemical synthesis. Electroorganic synthesis is the synthetic organic chemistry discipline that enables the direct use of electricity to generate valuable compounds. Hence, it is possible to transfer green aspects of sustainable energy sources to the whole production process.
Electrochemistry is mostly taught within physical chemistry and therefore focuses on the creation and interpretation of electric signals. This way of thinking is due to the importance of electroanalytical methods in battery research, for example, or in electron transfer in general. However, these electric signals do not provide much information on synthetic utility. Therefore, electrogenerated compounds must be isolated. Consequently, the synthetic potential of electrochemistry lay dormant, and this area was mostly considered as a niche technology. Several laboratory‐scale synthetic applications have been known for a long time, but mostly faded into oblivion in the second half of the 20th century. Nevertheless, a comprehensive guide on the technical aspects of laboratory‐scale preparative electrolysis can be found in the book “Organic Electrochemistry”.3
Over the past decade, organic electrochemistry has experienced a renaissance in the field of preparative organic methods. Conventional chemical oxidizing or reducing agents are being replaced by electric current as an inexpensive, renewable, and inherently safe reagent.4 Limitations on the redox potential, associated with every chemical reagent, can be circumvented as the reactivity can be tuned by changing the applied potential. The latter point is important as it enables electrochemical reactions to readily use extraordinary reaction pathways, enabling the shortening of conventional multi‐step reaction sequences. The use of electric current avoids the generation of reagent waste and increases atom economy as only the addition or removal of electrons is required for reactivity. By combining all of these advantages with long‐lasting and non‐sacrificial electrode materials, this method represents real “green chemistry”, and is an important component in the solution of current and future challenges.5 The development of green processes is particularly interesting for large‐scale technical applications. In this Review, recent developments in this field are discussed, showcasing the exciting potential of “electrified” organic synthesis.
The electron transfer at the electrode–electrolyte interface is mostly considered to be the decisive factor for electroconversion. For very small molecules such as CO, methane, formaldehyde, methanol, or ethylene, this is generally true, and electrocatalysts can be key for synthetic conversion.6, 7 Owing to the stability and lifetime of many organic radicals, there is a second regime that needs to be controlled to achieve the desired transformation.8
In addition to particular factors influencing the electron transfer, such as the electrode material and the overpotential, it is, with regard to chemical follow‐up processes, crucial not to underestimate typical effects of organic synthesis. The supporting electrolyte, its concentration, as well as the solvation of charged intermediates and the ionic strength can have a significant effect on selectivity and efficiency (Figure 1). Whereas it is necessary to control both regimes, this can also be used as a means to control the outcome of a reaction. The application of low current densities in many electrosynthetic processes indicates that the conversion was investigated under optimal conditions on a small laboratory scale over acceptable time frames. Nevertheless, large‐scale applications aim for high current throughput, which can be achieved, for example, by using porous electrode materials9 or stabilizing electrolytes.10
Figure 1.
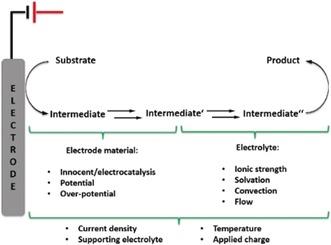
Processes and parameters of electrosynthesis.
Recently, Baran and co‐workers published a general overview on electroorganic developments since 2000.11 It was our aim to write a Review with a clear focus on synthetic developments that are of great importance in contemporary synthesis:
electrochemical fluorination
electrochemical C−N functionalization of arenes
Kolbe electrolysis
electrochemical arene couplings
electrochemical construction of heterocycles
electrochemistry in the synthesis of natural products, related compounds, and late‐stage functionalization
In a second Review,173 other conceptual aspects of organic electrosynthesis will be discussed. This thematic structure completes the puzzle of different overviews published over the last few months, and surveys the most important achievements of the last decades.
2. Electrochemical Fluorination
Only very few fluorinated organic compounds occur naturally.12 Owing to their interesting chemical, biological, and physical properties as well as their applications, for instance, in materials science or medicinal chemistry, the introduction of fluorine atoms into organic substrates is highly desired.13 Methods for the selective chemical fluorination of organic substrates suffer from the hazardous character of most fluorinating reagents, as well as their significant costs.14 Electrochemical perfluorination of organic substrates in anhydrous HF at Ni anodes, known as the commercially used Simons process, was already developed in 1949,15 and later enhanced by using molten KF⋅2 HF and carbon anodes.16 The first report on selective partial fluorination dates back to 1970, and this process was further developed mainly by Fuchigami and co‐workers. Most of the advances accomplished in this area have recently been summarized.14, 17 Thus, only the key features of selective electrochemical fluorination are discussed here.
Ionic liquid fluoride salts with the general formula Et3N⋅n HF or R4NF⋅n HF (n=3–5; R=Me, Et or nPr)18 are used as the fluoride donor either in aprotic solvents or as the neat ionic liquid. Olah's reagent (70 % HF/pyridine) can only be used in a few processes owing to the nucleophilicity of pyridine (Section 3). In the presence of water, the nucleophilicity of fluoride is drastically decreased. Thus the use of NBu4F⋅3 H2O is not viable. The anodic stability of alkylamine/HF ionic liquids increases with the HF content. Therefore, the substrate scope could be broadened by using the neat ionic liquid containing 4 or 5 equiv of HF.19 Furthermore, the fluoride salt can tune the selectivity of the reaction (e.g., α‐fluorination vs. fluorodesulfurization) as a result of different amounts of free amine base present in the electrolyte. The selectivity can also be tuned by changing the solvent system and thus stabilizing or destabilizing the initially formed radical cations (Scheme 1).20, 21
Scheme 1.
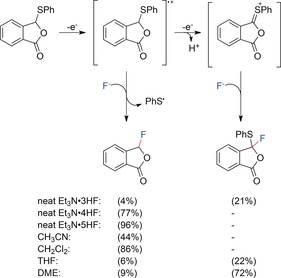
Solvent effects in the anodic fluorination of 3‐phenylthiophthalide. Yields determined by 19F NMR analysis.20
Initially, electrochemical fluorination was performed in aprotic solvents. However, significant anode passivation and, in the case of CH3CN, concurring acetamidation were observed. Furthermore, the electrochemical window was limited by the oxidation of the solvent. To overcome these drawbacks, either mediated systems and/or neat ionic liquids were used for electrochemical fluorination. Under such conditions, the scope was expanded, and the current efficiency was increased. Neat ionic liquids are more viscous than common organic solvents, resulting in slower mass transport. Mass transport and current efficiency can be improved by ultrasonication of the electrolyte.22 The challenge of the low atom economy could partially be overcome by recycling and reusing the ionic liquids. The electrochemical window of the electrolyte was further extended by using a combination of Et3N⋅n HF (n=4–5) and imidazolium‐based ionic liquids. The addition of ether‐containing additives (dimethoxyethane (DME), ethylene glycol, or poly(ethylene glycol) (PEG)) to coordinate the counterions led to an increase in the fluoride nucleophilicity and thus in the reaction efficiency (Scheme 2).23, 24
Scheme 2.
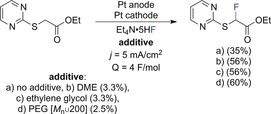
Influence of ether‐containing additives on the electrochemical fluorination of α‐(2‐pyrimidylthio)acetate.23
However, even in neat ionic liquids, anode passivation was not completely suppressed. Therefore, various systems have been developed for indirect electrochemical fluorination, for example, by using triarylamines, halides, or hypervalent difluoroiodoarenes as mediators. This led to significant improvements in terms of reaction efficiency and selectivity. Particular interest has been paid to hypervalent difluoroiodoarenes, which are anodically generated in situ from the corresponding iodoarenes. Thus only catalytic amounts of the iodoarene were required. Interestingly, anisole‐derived iodoarenes performed best (Scheme 3).25, 26
Scheme 3.
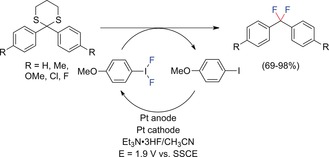
Indirect anodic gem‐difluorination of dithioketals with para‐methoxyiodobenzene difluoride.25 SSCE=saturated sodium calomel electrode.
Task‐specific iodoarenes, carrying, for example, an imidazolium moiety,27 and polymer‐supported iodoarenes28 have also been developed. These are stable during the electrochemical reaction and product extraction, and can be reused in subsequent runs. These developments culminated in a highly selective method for the electrochemical fluorination of (hetero)aromatic compounds under mild reaction conditions.14,17 However, although the used alkylamine/HF salts are less hazardous than most fluorinating agents used in chemical fluorination methods, they are still toxic and highly corrosive and therefore need to be handled with care, especially with higher HF contents. In addition, they are expensive and difficult to obtain commercially. A less expensive and less toxic alternative is the use of inorganic fluoride salts, such as alkali‐metal fluorides (MFs), which are stable and therefore easier to handle, as the fluoride source and supporting electrolyte. The challenges associated with the poor solubility and therefore low nucleophilicity of MFs in organic solvents was overcome by the addition of poly(ethylene glycol) (PEG) to coordinate the cation. Thus the anodic fluorination of triphenylmethane, for example, as well as fluorodesulfurization were successfully achieved by using a PEG/MF (M=K or Cs) system (Scheme 4).29
Scheme 4.
Anodic fluorination with alkali‐metal fluorides.29
3. Electrochemical C−N Functionalization of Arenes
Primary aromatic amines are important structural motifs in organic chemistry. They are used for the synthesis of pharmaceuticals,30 natural products,31 dyes,32 polymers,33 and functional materials.34 Therefore, an easy access to aniline derivatives is highly desired. Electrochemical methods for the C−N functionalization of aromatic compounds comprise pyridination35 and acetamidation,36 which date back to 1957 and 1966, respectively. Another strategy is the indirect cathodic amination of unsaturated and aromatic compounds with the TiIII/TiIV hydroxylamine system, which was extensively studied by Lisitsyn and co‐workers.37 Direct anodic amination with ammonia or primary alkylamines is not viable owing to the low oxidation potentials of alkylamines (1.5‐1.6 V vs. the saturated calomel electrode (SCE))38 and aniline derivatives (1.0 V vs. Ag/AgCl/saturated KCl).39 Thus selective oxidation of aromatic substrates in the presence of alkylamines or the resulting anilines is not possible or leads to overoxidation of the aromatic substrate. Electrochemical C−N functionalization has been extensively reviewed.40 Therefore, we will only focus on recent developments in this area.
3.1. Electrochemical C−H Amination
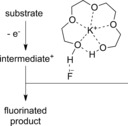
In 2013, Yoshida and co‐workers presented a highly efficient method for the electrochemical C−H amination of activated arenes.41, 42, 43 Upon anodic oxidation at a carbon felt anode and nucleophilic trapping of the generated radical cations with pyridine, pyridinium intermediates were obtained. Overoxidation was prevented by the strongly electron‐withdrawing effect of the positive charge. Subsequent aminolysis with piperidine released the free anilines (Scheme 5). The functional group tolerance was remarkable (e.g., iodine substituents). However, only electron‐rich substrates (e.g., anisoles, π‐extended aromatic compounds) were successfully converted.
Scheme 5.
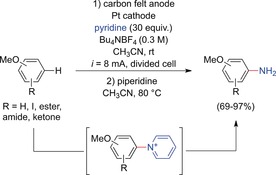
Electrochemical C−H amination of activated arenes.41, 42, 43
By employing boron‐doped diamond (BDD) anodes, Waldvogel and co‐workers were able to overcome the limitation to electron‐rich substrates,44 and the method was successfully expanded to less activated 1,3‐dialkylarenes (Scheme 6). Other anode materials including Pt showed strong fouling behavior as electropolymerization and carbonization is promoted at such positive potentials.
Scheme 6.
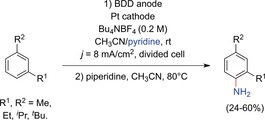
Electrochemical C−H amination of less activated alkylated arenes at BDD anodes.44
Electrochemical amination exclusively resulted in the formation of 2,4‐dialkylanilines. No functionalization of the benzylic position was observed. Even p‐xylene, mesitylene, and tetrahydronaphthalene were successfully aminated.
By using BDD anodes, Waldvogel and co‐workers were also the first to observe the direct twofold amination of naphthalene via a dipyridinium derivative (Scheme 7).45 This method provides an innovative access to the technically important bulk chemical 1,5‐diaminonaphthalene. Aromatic diamines are valuable precursors for the synthesis of polyurethanes, indispensable in our daily life.46 In particular, the highly regioselective introduction of two amino moieties is of interest as it is a major challenge of conventional multistep processes.47 The twofold amination method was successfully applied to diphenylmethane, triphenylmethane, biphenyl, and phenanthrene. However, in all cases, the formation of regioisomers was observed.
Scheme 7.
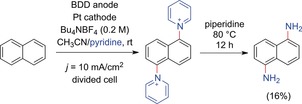
Twofold electrochemical C−H amination of naphthalene.45
Electrochemical amination of benzoxazoles by oxidative cross‐dehydrogenative couplings with secondary amines was reported by Zeng, Little, and co‐workers in 2014 (Scheme 8).48 Ring opening of the benzoxazole with an amine gave an ortho‐hydroxyamidine adduct, which underwent intramolecular cyclization into a benzoxazoline. Oxidation to the 2‐aminobenzoxazole was achieved by reaction with I+, electrochemically generated from tetrabutylammonium iodide. Thus the overall process is a mediated electrochemical dehydrogenative oxidation. The reaction tolerated methyl, chloro, and nitro substituents at the benzene moiety but nitro substituents led to significantly reduced yields. A variety of cyclic and acyclic secondary amines were successfully employed, whereas primary amines gave complex product mixtures.
Scheme 8.
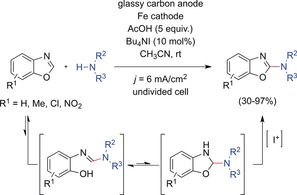
Electrochemical amination of benzoxazoles through oxidative dehydrogenative couplings.48
The challenge of coupling functionalized primary alkylamines with arene substrates was solved by the Yoshida group with a heterocyclization approach (Scheme 9).49 The direct use of unprotected or protected primary amines was not possible owing to unselective oxidation, low nucleophilicity, or overoxidation. In contrast, heterocyclic compounds obtained by the reaction of functionalized amines with nitriles exhibit higher oxidation potentials than the aromatic starting materials, enabling selective oxidation. Furthermore, heterocycles are sufficiently nucleophilic towards the anodically generated radical cations. In addition, overoxidation was avoided as the positively charged intermediates are rather electron‐poor and electrostatically repelled. Subsequent ring opening of these cationic intermediates was achieved under non‐oxidative conditions by reaction with sodium bicarbonate or ethylenediamine (EDA). The obtained products feature common structural motifs of biologically and pharmaceutically active compounds.50 This method was applicable to primary alkylamines with a hydroxy or amino group in the β‐ or γ‐position, providing five‐ and six‐membered heterocyclic compounds. The reaction scope included aromatic products generated from polycyclic arenes (naphthalene, phenanthrene, 9,9‐dimethylfluorene) as well as functionalized anisole derivatives containing iodo, ester, amide, nitrile, or tert‐butyl moieties.
Scheme 9.
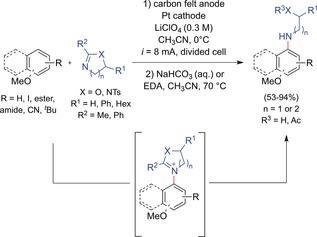
Anodic coupling of functionalized primary amines with aromatic compounds according to the heterocyclization approach.49
3.2. Electrochemical Arylation of N‐Heterocycles
In 2014, Yoshida and co‐workers reported the direct C−N coupling of N‐protected imidazoles with electron‐rich aromatic or benzylic substrates (Scheme 10).42,43, 51 To avoid overoxidation, N‐protected imidazoles, in particular N‐mesylimidazole, were used, yielding positively charged imidazolium intermediates after nucleophilic attack at the anodically generated cation. Treatment with piperidine yielded N‐aryl or N‐benzyl imidazoles, which are of significant relevance as biologically active compounds. The overall yields ranged from moderate to excellent (R=p‐OTs: 36 %, R=p‐Et: 99 %). Interestingly, a methyl group para to an electron‐releasing group promotes exclusive C−H functionalization at this benzylic position.
Scheme 10.
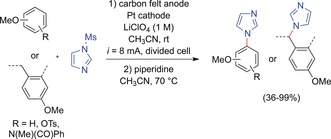
Anodic coupling of N‐protected imidazoles with activated aromatic compounds.42,43,51 Ts=para‐toluenesulfonyl.
3.3. Synthesis of N‐Heterocyclic Compounds by Electrochemical C−N Functionalization
In contrast to previous Sections, we will herein discuss the construction of new N‐heterocyclic moieties by C−N bond formation (compare Section 6). A promising process for the electrochemical synthesis of 2‐aminobenzoxazoles and 2‐aminobenzothiazoles from (thio)phenols was also established by the Yoshida group (Scheme 11).42,43, 52 The intramolecular C−N coupling process circumvented the issue of regioselectivity, and the conversion of less activated substrates became viable. After installation of a 2‐pyrimidyl moiety at the hydroxy or thiol function, intramolecular anodic C−N functionalization yielded benzoxazolopyrimidinium and benzothiazolopyrimidinium derivatives. Ring opening with piperidine generated a broad variety of 2‐aminobenzoxazoles or the corresponding benzothiazoles. The functional group tolerance was remarkable, and even substrates with strongly electron‐withdrawing substituents were successfully converted.
Scheme 11.
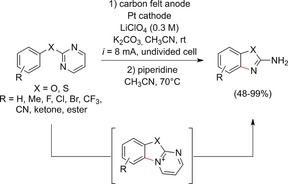
Intramolecular anodic C−N functionalization yielding 2‐aminobenzoxazoles or ‐thiazoles.42,43,52
An electrochemical route to 1,4‐benzoxazin‐3‐ones, important structural motifs in natural products and in pharmaceutically active compounds, by anodic C−H amination of phenoxyacetate derivatives was established by the groups of Yoshida and Waldvogel (Scheme 12).53, 54 Electrochemical oxidation of phenoxyacetate substrates afforded the corresponding pyridinium derivatives. Treatment with piperidine released the primary amine, which immediately attacked the ester moiety, yielding the desired benzoxazinone. To enable ring closure, the amination needs to occur ortho to the carboxymethoxy substituent. Thus the ortho position must be accessible and activated.
Scheme 12.
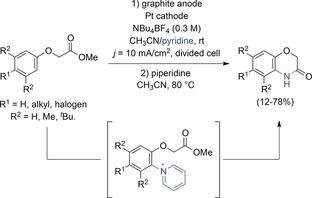
Synthesis of 1,4‐benzoxazin‐3‐ones by electrochemical C−H amination.53
This method was applicable to a broad range of phenoxyacetates and tolerated halogen substituents, which may serve as valuable leaving groups for subsequent functionalization. BDD anodes provided an appropriate electrochemical window for the conversion of fluorinated substrates. Despite the cationic nature of the intermediates, tert‐butyl groups were also tolerated.
3.4. Electrochemical Benzylic C−H Amination via Benzylaminosulfonium Ions
Yoshida and co‐workers elaborated an efficient procedure for the electrochemical benzylic C−H amination of different toluene derivatives (Scheme 13).55, 56 Recently, an extension of the cation‐pool method to highly unstable benzylic cations was reported.55 The electrochemically generated cations were stabilized as benzylaminosulfonium ions by reaction with N‐tosyl diphenylsulfilimine. Again, overoxidation was avoided because of the positive charge of the intermediate. Upon electrolysis, different aromatic nucleophiles were added to obtain the corresponding benzylic/aromatic cross‐coupling products. However, to achieve benzylic C−H amination, N−S bond cleavage without cleavage of the C−N bond was envisioned.56 In fact, the Yoshida group successfully converted the N‐tosyl benzylaminosulfonium ion into the corresponding N‐tosyl benzylamine by treatment with Bu4NI. This procedure was applicable to various toluene derivatives, tolerating halogen, ketone, ester, ether, and nitro moieties, to provide the corresponding benzylamines in good yields (up to 90 %). For substrates with two benzylic C−H bonds, the monoaminated product was selectively obtained, and secondary benzylic C−H bonds could also be aminated.
Scheme 13.
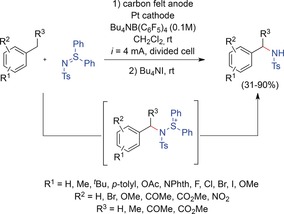
Electrochemical benzylic C−H amination of toluene derivatives.56 NPhth=phthalimide.
3.5. Metal‐Catalyzed Electrochemical C−N Bond Formation
Recently, Baran and co‐workers reported an electrochemically enabled nickel‐catalyzed cross‐coupling between aryl halides and alkyl amines that proceeded under mild reaction conditions to give the desired products in yields of up to 87 % (Scheme 14).57 Starting from NiII, a low‐valent Ni catalyst is formed in situ at the cathode to enable oxidative addition of the aryl halide. Concurrently, the anodic reaction is used for the intermediate formation of a nickel species in a high oxidation state to facilitate reductive elimination of the cross‐coupling products. Aside from secondary and primary amines, amides and alcohols were used as nucleophiles. Owing to the higher reactivity of the Ni catalyst compared to a Pd or Cu catalyst, less reactive electrophiles (e.g., aryl chlorides) could be employed. Heteroaryl halides as well as substrates with various electron‐withdrawing groups were also tolerated. As no excess of a strong base such as an alkoxide is required, the scope is unusually broad and the process practical.
Scheme 14.
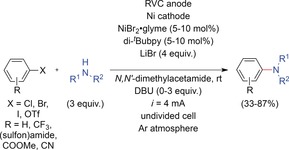
Electrochemically enabled nickel‐catalyzed amination of aryl halides.57 DBU=1,8‐diazabicyclo[5.4.0]undec‐7‐ene, di‐tBubpy=4,4′‐di‐tert‐butyl‐2,2′‐dipyridine, RVC= reticulated vitreous carbon.
A manganese‐catalyzed electrochemical route to 1,2‐diazides proceeding through oxidation of an azide or a MnII–azide complex and addition of the resulting azidyl radicals to alkenes was established by Lin and co‐workers.58 This process has recently been reviewed by the Baran group.11
4. Kolbe Electrolysis
The Kolbe electrolysis is one of the oldest electroorganic reactions. First performed by Faraday in 183459 and further studied by Kolbe in 1849,60 it is a general method for the electrochemical conversion of ubiquitous carboxylic acids. Radical and cation intermediates determine the outcome of the reaction. The Kolbe electrolysis is generally applicable to a broad range of substrates. However, this method requires very high current densities, leading to limited functional group tolerance. Moreover, the intermediate generation of radicals or cations may result in side reactions. The radical generation in the Kolbe electrolysis relies on an oxidative decarboxylation. Radicals can undergo dimerization, cross‐coupling, or cyclization, depending on the substrate structure and available reaction partners. If a second oxidation to the corresponding cation occurs, this “non‐Kolbe” pathway includes fragmentation, rearrangement, elimination, or nucleophile addition (Scheme 15). In case of water addition, this process is called Hofer–Moest reaction.61 If the electron transfer to the carboxylic acid occurs from a tethered moiety with a lower oxidation potential, such as electron‐rich arenes, the reaction is termed pseudo‐Kolbe reaction.62
Scheme 15.
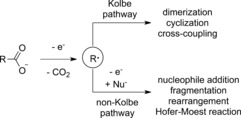
General steps of Kolbe electrolysis. Nu=nucleophile.
The outcome of a Kolbe electrolysis depends mainly on the reaction conditions. Current density, substrate concentration, and pH value are major factors.63 The formation of a carboxylate layer on the anode seems to be crucial for the reactions, while solvent gets desorbed. High current densities (>250 mA cm−2) are recommended to favor this layer formation, and a suitable pH value ensures carboxylate formation. As the Kolbe method was developed a long time ago, most work was done in the last century. Nevertheless, there are some examples of synthetically relevant applications developed after 2000. Schäfer and co‐workers summarized the work done until 1990 in a clear review article.64 With regards to the conversion of fatty acids as renewables, a more recent article exists.65 In general, present‐day research on Kolbe electrolysis focuses on the generation of large‐scale products such as biofuels66 or adiponitrile.67 In the following, we will highlight some contributions that represent synthetically relevant developments of the Kolbe electrolysis.
A cyclization process described by Markó and co‐workers is a promising example of a Kolbe electrolysis. Five‐ and six‐membered rings were formed in a radical cascade reaction in yields of up to 90 % (Scheme 16).68 This reaction combined a two‐fold Kolbe electrolysis of both the substrate and acetic acid with an intramolecular radical cyclization reaction. This cyclization was particularly successful when the nucleophilic carbon radical added to the double bond of an α,β‐unsaturated carbonyl compound.
Scheme 16.
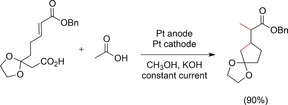
Radical cascade reaction induced by a Kolbe electrolysis.68 Bn=benzyl.
Becker and co‐workers developed a method to access 1,2‐disilylethanes in promising yields, whereby the concurrence of Kolbe dimerization and non‐Kolbe products was studied. The current efficiency was relatively high, and current densities of 250 mA cm−2 were applied (Scheme 17).69
Scheme 17.
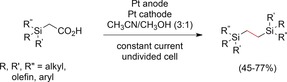
Kolbe dimerization of α‐silylacetic acids.69
Seebach and co‐workers developed a non‐Kolbe electrolysis with protected 4‐hydroxyproline derivatives to obtain the corresponding methoxy‐substituted products in high yields for follow‐up reactions (Scheme 18).70
Scheme 18.
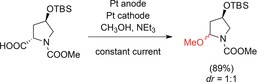
Non‐Kolbe conversion of an enantiomerically pure protected 4‐hydroxyproline.70 TBS=tert‐butyldimethylsilyl.
Markó and co‐workers developed a process to access ketones by twofold oxidative decarboxylation of disubstituted malonic acids.71 The addition of NH3 was crucial for the success of the reaction as it stabilized the corresponding carboxylate. Typical for Kolbe reactions, high current densities of up to 90 mA cm−2 were used. However, the current efficiency was low (Scheme 19).
Scheme 19.
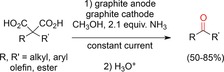
Non‐Kolbe electrolysis for the generation of ketones from disubstituted malonic acids.71
The important roles of additives, bases, and their interactions with the carboxylic acid substrates were further studied by Tajima and co‐workers. A solid‐supported piperidine base was used for a non‐Kolbe methoxylation,72 a Kolbe dimerization,73 as well as a mixed dimerization74 without the need for any further electrolytes. Schäfer and co‐workers recently developed a Kolbe homo‐coupling and cross‐coupling of menthol derivatives with different butyric acids.75 This method has only restricted applicability, but nevertheless, radical‐based diastereoselective additions are mechanistically interesting.
There are further examples of exotic synthetic applications and detailed studies of the Kolbe electrolysis.76 However, this method has some major drawbacks related to the harsh reaction conditions and the limited functional group tolerance. This process is mainly interesting with regard to the large‐scale synthesis of small molecules as high current densities are favorable for industrial processes.
5. Electrochemical Arene Couplings
The chemical modification of aromatic substrates is of general interest for modern organic chemistry. In particular, biaryls are structural motifs of importance in a variety of different disciplines, such as in materials and pharmaceutical science, and are especially common in natural products and potent ligand systems for several applications.77 Biaryls can be synthesized by transition‐metal catalysis in combination with prefunctionalized arenes or by using chemical redox agents to activate C−H or C−X bonds. All of these methods produce a significant amount of reagent waste or suffer from unselective bond formation. Different methods have been developed to combine common coupling reactions with electrochemical synthesis or to carry out metal‐ and reagent‐free electrochemical coupling reactions. This section is divided into several subsections. The first two subsections will deal with cathodic and anodic electrochemical conversions of prefunctionalized aromatic compounds into biaryls, whereas the latter two subsections focus on more modern and atom‐economic direct electrochemical coupling reactions by C−H activation.
5.1. Cathodic Couplings of Prefunctionalized Arenes
The prefunctionalization of both substrates is the most reliable means to ensure regioselective formation of the desired C−C bond. Methods for cathodic coupling reactions of prefunctionalized arenes focus on the transformation of aryl halides. Typically, either sacrificial materials, such as Mg electrodes, are directly used for the desired coupling reaction of aryl halides, or transition‐metal catalysts (e.g., Pd‐ or Ni‐based) can be electrochemically regenerated and combined with prefunctionalized arenes. The main disadvantage of these coupling reactions is the poor atom economy. Prefunctionalization of both coupling partners has to be carried out by conventional methods, and both activating groups as well as the transition‐metal catalyst and the sacrificed electrode material result in reagent waste. Furthermore, many methods are limited to homo‐coupling, in the sense that one arene undergoes dimerization, accompanied by loss of the activating group (Scheme 20).
Scheme 20.
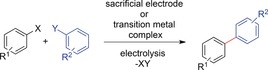
General reaction scheme for electrochemical coupling reactions of prefunctionalized arenes. For most reactions, R1=R2 and X=Y.
An interesting protocol dealing with the cathodic coupling of aromatic halides by utilization of Mg electrodes was reported by Kim and co‐workers.78 Electrolysis was carried out at a high current density of 41 mA cm−2 in THF with LiClO4 as the supporting electrolyte. The authors reported minimization of Mg electrode consumption to 5 wt % when cathode and anode were interchanged every 30 s. However, this statement was not precise as this value referred to 95 % of the mass of the Mg electrode being recovered and was not normalized to the amount of consumed starting material. Electrolysis of bromobenzene gave biphenyl in a good yield of 62 %, and mono‐ and dibenzyl bromides could also be coupled with this method. Nevertheless, the scope was limited.
The use of transition‐metal catalysts, in particular Pd systems, in combination with electrochemistry for the reductive coupling of aryl halides has been intensively studied. The cathodic action leads to umpolung of the Pd intermediate, which generates a catalytically active species that can be used for C−C bond couplings between aryl halides (Scheme 21).
Scheme 21.
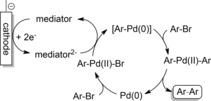
Catalytic cycle for the reductive coupling of aryl halides by electrochemical regeneration of active Pd complexes.
Early studies in this field were carried out by Torii and co‐workers.79 Electrolysis was carried out under constant current conditions (2.5 mA cm−2) in an H‐type divided cell, with a Pb cathode and a Pt anode in DMF with Et4NOTs as the supporting electrolyte. The use of Pd catalysts and their direct electroreductive regeneration enabled the formation of a variety of symmetric biaryls from aryl, naphthyl, and pyridyl halides in good yields (16–99 %). Arene cross‐couplings have only been achieved with equimolar amounts of a Pd catalyst in a two‐step reaction sequence. Further studies on Pd catalysts in combination with electrochemistry have been carried out by Tanaka and co‐workers. The electrolysis of aryl halides (Scheme 22) was carried out in the presence of a Pd catalyst and either N,N′‐dialkyl 4,4‐bipyridinium‐80 or N‐alkyl 4‐alkoxycarbonylpyridinium based‐mediators81 at Pt cathodes in undivided cells. The homo‐coupling of a variety of aryl bromides with electron‐withdrawing substituents proceeded in good yields, whereas the presence of electron‐donating substituents resulted in lower yields. To achieve electrolysis in undivided cells, sacrificial anodes such as Mg rods or Zn plates were required to prevent electrochemical oxidation of the mediator or halides prior to reactivation of the Pd catalyst. The synthesized biaryl derivatives are depicted in Scheme 22.
Scheme 22.
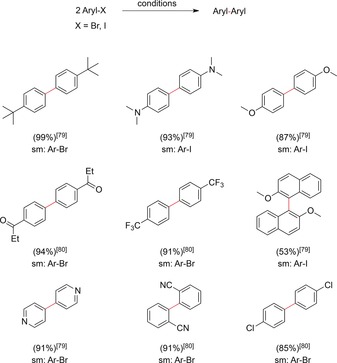
A selection of biaryls synthesized by electroreductive coupling reactions.79,80 sm=starting material.
Aside from the electrochemical regeneration of soluble Pd catalysts, Rothenberg and co‐workers reported on the use of a solid palladium electrode for Ullmann‐type couplings of aryl iodides and aryl bromides in 2006 (Scheme 23).82 The electrolysis was carried out in an ionic liquid as the electrolyte. Constant current conditions of 10 mA were applied on a solution of the aryl halide and a Pd anode in combination with a Pt cathode. First, the in situ formation of palladium nanoparticles was observed. These nanoparticles were responsible for the catalytic Ullmann‐type homo‐coupling and the selective formation of biaryls in good yields. The authors reported the overall loss of Pd being equivalent to 0.1 mol % relative to the used amount of aryl halide.
Scheme 23.
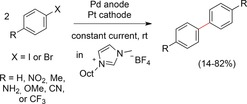
Electroreductive homo‐coupling with palladium nanoparticles electrochemically generated from solid palladium electrodes.82
A cost‐efficient alternative to the use of Pd catalysts is based on the combination of Ni catalysts and aryl halides for electroreductive coupling reactions. The concept behind this transformation is the electrochemical regeneration of low‐valent Ni complexes. These are well‐known to enable C−C bond formation when applied in equimolar amounts in conventional syntheses. Numerous investigations on these aryl–aryl coupling reactions have been carried out. Early reports by Jennings83 were followed by mechanistic investigations by Périchon and co‐workers,84 and the applied catalyst was further improved by Fox and co‐workers.85
Here, the reactions were carried out under argon atmosphere in a divided cell. The use of a carbon cloth cathode offered a large surface area while the applied sacrificial lithium anodes seem to be untypical. The use of DMSO as a polar solvent with a high donor number was crucial for effective coupling reactions. The authors surmised that this effect was due to the increased stability of the generated Ni0 catalysts, as well as the large separation of the electrochemical potentials for reduction of the aryl halide and the propagation of the catalytic cycle in DMSO, which prevents competing dehalogenation. Instrument‐based simplifications for the application of Ni catalysts were initiated by Troupel and co‐workers.86 This setup has been further optimized by Léonel, Gosmini, and Périchon in several publications, which extended the scope of this reaction to the cross‐coupling of N‐heterocyclic halides, for example, pyridine,87 pyridazine,88 pyrimidine,89, 90 and pyrazine derivatives,89 with aryl halides, as well as the cross‐coupling of heterocyclic halides.87 The general setup with nickel 2,2′‐bipyridine complexes as catalysts, a nickel foam cathode, and a sacrificial anode (Mg, Zn, Fe, or Fe/Ni) remained unchanged. For the selective formation of cross‐coupling products, the combination of different halogen substituents on the coupling partners or reactivity differences with the catalyst due to the distinct electronic structures of both aryl halides are necessary. A collection of the synthesized heterobiaryls are shown in Scheme 24.
Scheme 24.
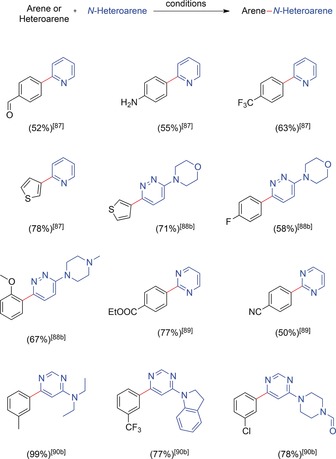
Heterobiaryls synthesized by reductive electrochemical cross‐couplings of aryl halides mediated by nickel complexes.87, 88, 89, 90
In addition, Gosmini and co‐workers reported cobalt catalysts for the cross‐coupling of aryl halides with heteroaryl halides91 or with aryl halides92 under otherwise similar conditions.
5.2. Anodic Couplings of Prefunctionalized Arenes
Anodic couplings of prefunctionalized arenes can be performed with electropositive substituents, such as aryl boronic acids, aryl boronates, and aryl trifluoroborates, in combination with Pd catalysts. Similar to the reductive protocols, the main disadvantages are the poor atom economy and the sophisticated preparation of starting materials by conventional methods. Electrooxidative homo‐couplings to biaryls with Pd catalysts have been investigated by the groups of Amatore, Tanaka, and others.93, 94 The key step in all proposed mechanisms is the anodic regeneration of active PdII species from Pd0, generated in situ by the coupling reaction. As depicted in Scheme 25, the use of organic mediators such as benzoquinone or TEMPO was essential for the reported reactions to proceed.
Scheme 25.
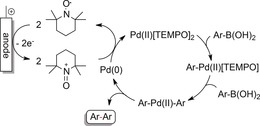
Oxidative coupling of aryl boronic acids by electrooxidative regeneration of PdII species using TEMPO as a mediator.94
The proposed mechanism is closely related to those of the reductive homo‐coupling reactions described before. The reactions are typically carried out in the anodic compartment of a divided cell. Constant current conditions, with a current density of 3.3 mA cm−2 at the Pt electrode, were reported with TEMPO as a mediator.94 Electrochemical oxidation with para‐benzoquinone as the mediator was investigated at constant potentials of +0.75 V using a carbon cloth anode and a saturated calomel reference electrode.93 In contrast to the reductive pathway, aryl boronic acids gave the desired biaryls in good yields with electron‐withdrawing as well as electron‐releasing substituents. A collection of different coupling products are depicted in Scheme 26. Whereas the electrochemical reactions had to be carried out under inert atmosphere (argon) and in divided cells, one positive feature was the use of water or water‐dominated organic solvent mixtures.
Scheme 26.
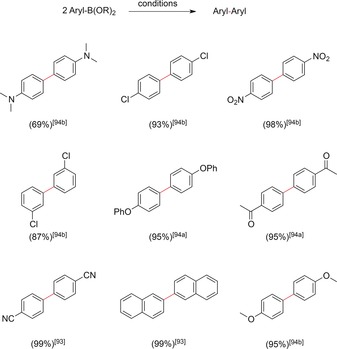
A selection of biaryls synthesized by electrooxidative couplings of aryl boronic acids.93,94
5.3. Reductive Couplings by C−H Activation
In general, the application of electrochemistry enables the development of sustainable processes. To prevent the formation of chemical waste in coupling reactions, C−H bond activation is favorable. Only a few attempts have been made towards the cathodic coupling of arenes by C−H activation. Reductive electrochemical couplings focus on the C−H activation of one coupling partner, with an aryl halide as the other coupling partner. Procedures following this concept include early reports by Thiébault and co‐workers, who described cross‐couplings of phenols with arenes95 as well as heteroarenes.96 The electrolysis was carried out in liquid ammonia under constant‐current conditions. The reactions took place at a Pt grid cathode in combination with a sacrificial Mg anode. To prevent the formation of regioisomers, the authors focused on the cross‐coupling of 2,4‐ or 2,6‐di‐tert‐butylphenols with various arenes. The application of various mediators and basic additives was also studied.
Such electroreductive coupling reactions were recently investigated by Atobe and co‐workers,97 who achieved the C−C cross‐coupling of an aryl halide with a second arene. The mechanistic rationale is shown in Scheme 27, and was described to involve a single electron transfer to the aryl halide and subsequent dehalogenation to give an aryl radical. This radical is attacked by the coupling partner to form the C−C bond. Rearomatization is achieved by base‐assisted proton abstraction and electron transfer from the radical anion to the starting material.
Scheme 27.
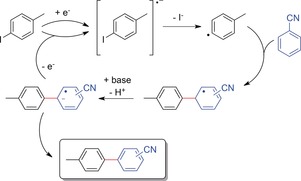
Mechanism for the electroreductive coupling of aryl halides with arenes, shown for the reaction of 4‐iodotoluene with benzonitrile.97
These reactions were carried out in a divided cell in DMF under constant‐current conditions (15–30 mA cm−2) using Pt electrodes. The presence of base was indispensable. Unfortunately, most yields were determined by NMR analysis with an internal standard (11–49 %), and only one product was isolated, namely that of the model reaction of 4‐iodotoluene and benzonitrile, in rather low yield (21 %, with regioisomers o/p=62:38).
5.4. Anodic Couplings by C−H Activation
Electrochemical coupling reactions by twofold C−H activation are ideal candidates for the development of sustainable processes as no leaving groups are required. Typically, twofold C−H activation for biaryl formation was performed by anodic treatment of starting materials. Several conversions of this type were already investigated in the early‐ to mid‐20th century.98 Nevertheless, these investigations mostly comprised uncontrolled electrolysis reactions of arenes and in particular phenols, leading to unpredictable product mixtures. Therefore, this Section will focus on a selection of reports, summarizing strategies to achieve selective C−H activation reactions of arenes. General problems for twofold anodic C−H activation include the formation of diverse product mixtures because of unselective oxidation processes, the decomposition of oxidized intermediates, and high tendencies towards side reactions. Furthermore, the formed biaryls can have lower oxidation potentials than the starting materials, making them prone to overoxidation.99 Several strategies have been investigated to overcome these problems.
The application of Pd catalysts for electrooxidative coupling reactions by C−H activation was investigated by Kakiuchi and co‐workers.100 When aryl pyridines were used in combination with a Pd catalyst and iodine as a redox mediator, the aryl pyridines were homo‐coupled in the ortho position of the arene moiety owing to precoordination of the mediator (Scheme 28).
Scheme 28.
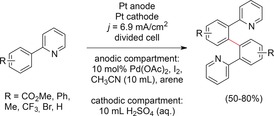
General reaction scheme for the anodic coupling of aryl pyridines by C−H activation.100
The activation of remote C−H bonds by pyridines in the ortho‐position of an attached aryl group is well‐known for conventional Pd coupling reactions.101 Regeneration of the catalyst, typically carried out with oxidizers, was realized in the anodic compartment of a divided cell at a Pt electrode. This method was limited to aryl pyridines, which significantly lowers its viability.
Whereas prefunctionalization was not necessary, transition‐metal catalysts (Pd‐based) were still required. The direct conversion of the starting material at the anode is favorable to design green processes. Typically, the mechanistic rationale for a direct anodic coupling of arenes comprises several steps (Scheme 29). First, one arene is oxidized, forming a radical cation. This reactive intermediate is attacked by a nucleophilic coupling partner to form the desired C−C bond. Rearomatization of the biaryl occurs in a second oxidation step, either at the anode or in solution.
Scheme 29.
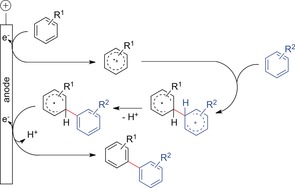
Mechanistic rationale for anodic aryl–aryl coupling reactions.
A well‐investigated example is the anodic trimerization of 1,2‐dialkoxybenzenes.102 Waldvogel and co‐workers reported an effective procedure for the anodic synthesis of triphenyleneketals by trimerization of catechols (Scheme 30).103 The authors optimized the electrochemical protocols, which usually only gave low yields. Key for successful electrolysis in an undivided cell was the application of propylene carbonate in combination with Me4NBF4 or Bu4NBF4 as the supporting electrolyte. As the trimerization product is poorly soluble in this mixture, overoxidation was circumvented. The products were obtained in yields of up to 80 %.
Scheme 30.
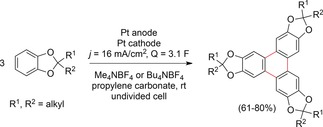
Anodic couplings of catechols to triphenyleneketals.103
An elegant approach to guarantee regioselective bond formation and inhibit overoxidation involves template‐directed reactions. Waldvogel and co‐workers reported a protocol for template‐directed anodic phenol coupling reactions (Scheme 31).104 Here, NaB(OAr)4 was prepared by a two‐step synthesis. This salt served as starting material and supporting electrolyte in the subsequent anodic coupling reaction. The electrolysis was carried out in an undivided cell equipped with Pt electrodes. A current density of 12.5 mA cm−2 was applied, and the polarity was reversed every 60 s to avoid electrode coating. The authors found that a ligand interchange between the formed borates was crucial for achieving conversions greater than 50 %. This approach can be used for the conversion of large amounts of substrate (20–3000 g).
Scheme 31.
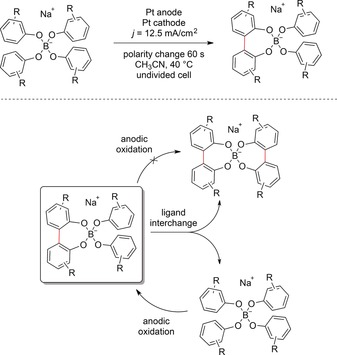
Template‐directed anodic phenol coupling sequence.104
The direct oxidative C−H/C−H cross‐coupling of two different arenes is challenging as one coupling partner has to be selectively oxidized. Otherwise, statistical formation of homo‐ and cross‐coupling products will take place upon non‐selective oxidation. This would lead to moderate yields of the desired cross‐coupling products in the best case. Additionally, overoxidation of the obtained biaryls might lower the overall yield and result in the formation of oligomeric side products. An extremely elegant way to achieve the formation of non‐symmetric biaryls by electrochemical oxidation is the “radical cation pool” method, which was established by Yoshida and co‐workers (Scheme 32).105 The idea behind this method is based on the separation of the oxidation and coupling events in time and space to prevent homo‐coupling and overoxidation. This concept will be surveyed in detail in a follow‐up Review on electrochemical methods.173 It should be noted that unactivated arenes can be coupled in good yields and with high selectivity with this method. However, the reactions have to be carried out at low temperatures and on a very small scale (0.1 mmol). Therefore, this method is less attractive for large‐scale synthesis.
Scheme 32.
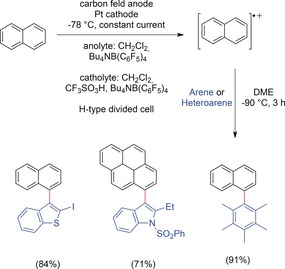
The radical cation pool method for C−H/C−H cross‐couplings of arenes.105
Atobe and co‐workers developed another approach to separate the electrochemical oxidation from the coupling event to obtain cross‐coupling products by twofold arene C−H activation.106 The use of a microflow reactor with two inlets enabled the formation of a liquid–liquid parallel laminar flow phase and enabled the selective formation of the desired cross‐coupling products (Scheme 33). This unique method will also be discussed in detail in our Review on electrochemical methods.173
Scheme 33.
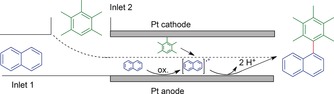
Schematic illustration of the liquid–liquid parallel laminar flow in an electrochemical microflow reactor for arene–arene cross‐couplings. The dashed line represents the liquid–liquid border of the laminar flow.106
A different approach was investigated by Waldvogel and co‐workers for the selective electrochemical homo‐ and cross‐coupling of phenols, arenes, and aniline derivatives. The distinct solvation properties of the starting materials and reactive intermediates and the unique solvent effects of 1,1,1,3,3,3‐hexafluoropropan‐2‐ol (HFIP) led to high yields and selectivities in the anodic synthesis of coupling products in undivided cells.107 Solvation by HFIP not only stabilized the reactive intermediates, but also decoupled the nucleophilicity from the oxidation potential owing to the diverse solvation properties of the coupling partners, making cross‐couplings possible. The yield and selectivity could be increased in many cases with additives, such as water or methanol, that influence the solvation. Electrochemical cross‐couplings of phenols with arenes108 or other phenols109, 110 showcase the broad applicability of this method. Additionally, comparison of the electrochemical cross‐couplings of phenols with the same products synthesized by oxidative couplings using chemical oxidizers demonstrated that this electrochemical cross‐coupling can easily compete with conventional methods.109 Recent studies have highlighted its robustness,111 and extended the applicability of this process to the electrochemical synthesis of partially protected non‐symmetric biphenols,112 protected bianiline derivatives113 (Scheme 34, top), and m‐terphenyl‐2,2′′‐diols by twofold C−C cross‐coupling114 (Scheme 34, bottom). The cross‐coupling of phenols with heterocycles was recently described.115 All products can be synthesized on multigram scale. The use of high current densities was established in first experiments, and has to be tested for further substrate combinations.111
Scheme 34.
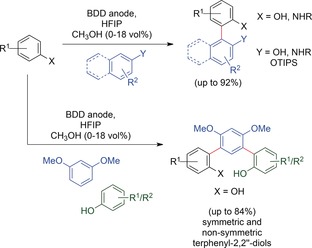
Various electrochemical cross‐couplings enabled by C−H activation developed by Waldvogel and co‐workers.107, 108, 109, 110, 111, 112, 113, 114
6. Electrochemical Synthesis of Heterocycles
Heterocycles are omnipresent structural motifs in organic chemistry. Most natural products and pharmaceutically active compounds contain heterocycles. In classic organic chemistry, methods for the synthesis of heterocycles are often based on condensation reactions. Elevated temperatures and the use of acid or base catalysts are a major attribute of these processes. Over the last decades, innovative methods such as transition‐metal‐catalyzed C−H activations and oxidations with hypervalent iodine have facilitated the synthesis of several valuable structures.116
Electrochemical routes for the construction of heterocycles have always been an important area of research. Direct or mediated electrochemical oxidation or reduction can initiate an intramolecular or intermolecular cyclization by formation of a radical or radical ion. This umpolung step normally proceeds at the functional group with the easiest accessible redox potential. Ring closure to generate the heterocycle proceeds by C−C, C−Het, or Het−Het bond formation. The progress made in this field has previously been summarized in the 1970s, 1980s, and 1990s by Lund and Tabacovic.117 More recently, Francke118 and Zeng et al.119 discussed some of these studies. The aim of this Section is to focus on progress made in recent years. It mainly deals with the reactivity of nitrogen‐centered radicals, which has turned into a topic of great significance.
Moeller and co‐workers recently reported several ways to trap radical cations that had been anodically generated from electron‐rich olefins by intramolecular cyclization. Different tethered nucleophiles served as the trapping moiety, including alcohols, amines, and sulfonamides. By variation of the nucleophile and the ring size, five‐, six‐,120 and seven‐membered121 rings containing oxygen and nitrogen heteroatoms were accessible. In general, oxidation could also occur at the nucleophile followed by intramolecular electron transfer or addition of the olefin to the generated radical (Scheme 35).
Scheme 35.
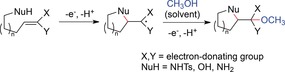
General cyclization of olefins with trapping nucleophiles.
After cyclization, a second oxidation occurred, and the intermediate was trapped with either methanol or another tethered nucleophile. The enormous potential of such conversions was greatly expanded by variation of the reaction conditions. The replacement of methanol as the second trapping group with another tethered nucleophile enabled the sequential formation of two heterocycles in a one‐pot process. The example in Scheme 36 shows only one of several possibilities in detail. The use of 2,6‐lutidine instead of lithium methoxide led to fast trapping by the sulfonamide and subsequent attack of the alcohol, while the yields were still high.
Scheme 36.
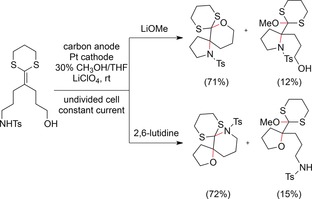
Exemplary competitive cyclizations with different bases.120b
Moeller and co‐workers expanded the synthetic scope to amides and reported the first direct electrochemical generation of amidyl radicals.122 The use of amides and anilides enabled the direct synthesis of lactams as valuable core structures (Scheme 37). Mechanistically, the olefin served as the trapping group as the oxidation potential of the deprotonated amide anion was shown to be lower in cyclic voltammetry studies.
Scheme 37.
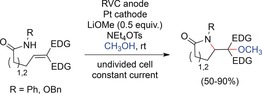
Anodic cyclization of amides to lactams.122 EDG=electron‐donating group.
These aminomethoxylation methods were limited to electron‐rich olefins, and methanol was mainly used to trap the cyclized intermediate. More recently, Xu and co‐workers developed various reaction protocols and demonstrated that this method could be used to access a broader scope of amino‐functionalized products.
With 2,2,6,6‐tetramethylpiperidine‐N‐oxyl (TEMPO) as the radical trapping reagent, non‐activated alkenes were converted.123 After the first oxidation step and cyclization, the carbon‐centered radical did not undergo a second oxidation, but reacted with TEMPO instead (Scheme 38, top). Here, the TEMPO group served as a mediator and a trapping agent and provided the possibility for subsequent functionalization. Acyclic alkenes were also converted, and spiro compounds were accessed with excellent diastereoselectivity. A cyclopropane derivative was used as a radical clock substrate to confirm the radical nature of the reaction mechanism. Xu and co‐workers also developed an elegant modification of their method that is based on the addition of an excess of 1,4‐cyclohexadiene instead of TEMPO (Scheme 38, bottom).124 The use of 1,4‐cyclohexadiene as a hydrogen atom donor leads to a redox‐neutral process with potential applications in natural product synthesis. Ferrocene was required as a mediator for the oxidation to avoid electrode passivation. With a 5:1 mixture of THF and methanol, the oxidation potential of the carbamate was lowered, and efficient electron transfer from ferrocene was possible. Suitable substrates also underwent redox‐neutral radical cascade reactions and, in the absence of 1,4‐cyclohexadiene, oxidative cascade reactions. Tricyclic products and indolines are thus accessible.
Scheme 38.
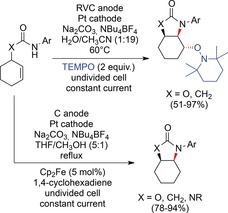
Aminooxygenation of non‐activated alkenes and redox‐neutral amination of substituted olefins.123,124 Cp=cyclopentadienide.
If no radical trapping groups or hydrogen donors are present, the β‐hydrogen atom can eliminate upon cyclization.125 With this modified method, vinyl‐substituted heterocycles were generated (Scheme 39). This moiety is usually accessible by transition‐metal catalysis, and the amination of tri‐ and tetrasubstituted olefins is difficult.126 The diastereoselectivity of the reaction was high, and the scope broad.
Scheme 39.
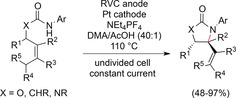
Electrochemical amination of substituted olefins.125 DMA=N,N‐dimethylacetamide.
Based on the hydroamination work, Xu and co‐workers applied the method for indoline synthesis124 to the formation of indoles. The authors employed the same conditions and urea substrates, whereby the urea moiety served as a connecting bridge between a tethered arene and an alkyne. These substrates were directly converted into substituted indoles. A useful feature of this procedure is the fact that 4‐, 5‐, and 6‐azaindoles are also accessible in high yields (Scheme 40).127
Scheme 40.
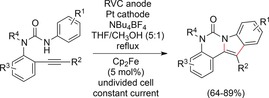
Electrochemical formation of fused indoles.127
Recently, Xu and co‐workers reported a method to access 3‐fluorooxindoles. At 0 °C with cyclopentadienyllithium as the base, this process enabled the oxidation of the acidic α‐position in N‐aryl amides. The base was essential to lower the oxidation potential of the α‐carbon atom by deprotonation and enabled the electron transfer from the mediator. This C−C coupling reaction is an elegant means to access 3‐fluorooxindoles (Scheme 41), which can be functionalized in subsequent reactions.128
Scheme 41.
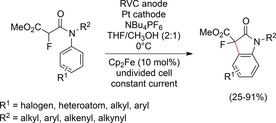
Electrochemical formation of 3‐fluorooxindoles.128
The studies presented thus far focused on the reactivity of amidyl radicals towards olefins and alkynes. Waldvogel and co‐workers recently developed a method for the intramolecular dimerization of two anilides via an amidyl radical. N−N bonds were electrochemically formed without the need for highly toxic hydrazines as precursors.129 Easily accessible malonic acid dianilides were directly converted into pharmaceutically relevant pyrazolidine‐3,5‐diones (Scheme 42).
Scheme 42.
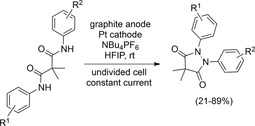
Electrochemical synthesis of pyrazolidine‐3,5‐diones.129
Interestingly, the reactions only worked well in HFIP. Other solvents failed or gave only traces of the desired product. The prominent role of HFIP as a radical‐stabilizing solvent as well as the aromatic substitution pattern were key factors for a successful reaction.130 The authors also reported another interesting feature of this transformation. If N−N bond formation is too slow, it competes with the formation of a C−O bond, affording benzoxazoles.131 This led to the development of a procedure for the reagent‐free synthesis of benzoxazoles starting from monoanilides in very good yields (Scheme 43). Comparison of two commonly used simple electrochemical set‐ups revealed the optimization potential in terms of the final outcome of the reaction.
Scheme 43.
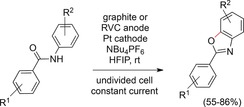
Direct electrochemical formation of benzoxazoles from anilides.131
Waldvogel and co‐workers showed by cyclic voltammetry studies that the mechanism involves a diradical for N−N bond formation and a cation for the benzoxazole synthesis.132 Electrochemical methods for the simple construction of benzoxazoles have also been developed by other groups. Zheng and co‐workers reported a 2,3‐dichloro‐5,6‐dicyano‐p‐hydroquinone (DDH) mediated process to convert a Schiff base with a tethered 2‐aminoarene into the corresponding benzoxazole.133 A large amount of mediator (30 mol %) was necessary under the optimized reaction conditions, but several aromatic functional groups were tolerated by this method. This approach constitutes an alternative to the use of a biphasic electrolyte, which is mandatory when sodium iodide serves as the mediator.134 As reported above, Yoshida and co‐workers also developed an elegant way to construct this important heterocyclic structure by using their amination procedure in an intramolecular fashion (see Section 3.3).52
Xu et al. reported an elegant synthesis of benzothiazoles from thioanilides. In an undivided cell under constant‐current conditions, many different functional groups and substitution patterns were tolerated (Scheme 44).135
Scheme 44.
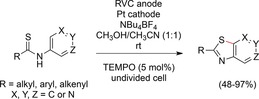
TEMPO‐mediated synthesis of benzothiazoles.135
The authors again confirmed that this was a valuable alternative to classic synthetic routes, and that this method can be used to generate natural products or drugs more efficiently. A similar substrate underwent the same cyclization reaction.136 When amidine functional groups were oxidized at the anode, the generated amidinyl radicals underwent cyclization with the attached aromatic ring system (Scheme 45). This reaction exhibited high chemoselectivity, and several functional groups were tolerated.
Scheme 45.
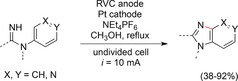
Electrochemical generation of benzimidazoles and pyridoimidazoles.136
Huang and co‐workers developed another method to synthesize simpler benzimidazoles, benzoxazoles, and benzothiazoles.137 In a CoIII‐mediated reaction, alcohols were fused with aromatic ortho‐amino‐functionalized arenes, as shown in Scheme 46. The reaction is based on the oxidation of the alcohol to an aldehyde by the cobalt(III) mediator. The aldehyde subsequently undergoes a condensation reaction with the amine, oxygen, or sulfur moiety. The final product is formed by oxidation in air.
Scheme 46.
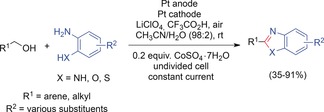
Cobalt(III)‐mediated synthesis of benzimidazoles and benzothiazoles.137
Wang and co‐workers developed a procedure towards isatins by combining the formation of C−O bonds with the synthesis of heterocycles (Scheme 47).138 Isatins are an important motif in natural products and pharmaceutically active compounds. Iodine served as a mediator for the initial oxidation of the methyl group while pure oxygen was used as the oxygen source. Follow‐up oxidations could be initiated by oxygen, the electrode, or the iodine mediator. The use of pure oxygen might impact on the scalability because of safety considerations.
Scheme 47.
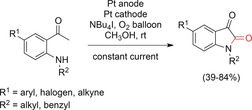
Iodine‐mediated synthesis of isatins.138
There are rare examples for the electrochemical synthesis of three‐membered heterocycles. However, these are very valuable synthetic intermediates because of their inherent ring strain.139 Yudin and co‐workers synthesized aziridines from N‐aminophthalimides under potentiostatic control with a silver wire as a pseudo‐reference electrode (Scheme 48).140
Scheme 48.
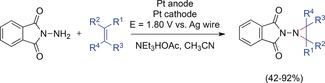
Potentiostatic aziridine synthesis with N‐aminophthalamide.140
The electrolysis had to be conducted in a divided cell as olefin reduction occurred as a side reaction when an undivided cell was used. This approach was further developed into an interesting method for the amination of sulfoxides in two subsequent electrolysis processes in divided cells, with a potentiostatic anodic oxidation followed by constant‐current cathodic reduction.141 Direct epoxidation at the electrode required a catalyst that mediates the generation of the oxygen species as olefins are difficult to oxidize or reduce electrochemically (Scheme 49). Further work was done on electrochemical epoxidations. Page, Marken, and co‐workers used electrochemically generated peroxodicarbonate and peroxodisulfate for the epoxidation of an iminium catalyst.142
Scheme 49.
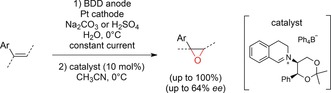
The enantiomeric excess was generally higher when persulfates were employed. Styrene‐type alkenes were successfully converted, and boron‐doped diamond was crucial for the generation of the peroxo species. High current densities were applied, and the electrolysis step was performed prior to catalyst addition.
Another mediated anodic epoxidation protocol was developed by Rossen and co‐workers.143 The synthesis of the HIV protease inhibitor indinavir included an epoxidation step, which was realized by constant‐current electrolysis with sodium bromide as the supporting electrolyte (Scheme 50). Anodically generated HOBr led to the formation of a bromohydrin, which cyclized to the epoxide. The use of sodium bromide as a mediator has advantages over peracids in terms of scale‐up and safety aspects. This work showcased the potential of electroorganic synthesis for a broad range of applications.
Scheme 50.

Hypobromide‐mediated epoxidation for the synthesis of indinavir.143
Another indirect epoxidation method for olefins is based on the use of reduced oxygen as the epoxidation reagent. Oxygen reduction can lead to the formation of hydrogen peroxide in acidic media, which subsequently reacts with an epoxidation catalyst. Electrochemical reduction of molecular oxygen is mainly studied in the field of fuel cells.144 This approach demonstrated the interesting implementation of fundamental transformations in simple electrochemical reactions, such as oxygen reduction, but the reported yields and conversions were quite low. The complex set‐ups and procedures exacerbate optimization and applicability.145
7. Natural Products, Related Compounds, and Late‐Stage Functionalization
Electrochemical synthesis comprises a broad variety of possible transformations, such as C−C coupling reactions, functional group transformations, and reactions of heteroatom moieties. Nevertheless, synthetic applications of electrochemistry are mostly associated with transformations of inorganic compounds or the synthesis of small organic molecules. Only a few attempts have been made towards the synthesis of complex organic structures, such as natural products. This may also be due to the reluctance of synthetic chemists to use electrochemical methods on rather complex starting materials. Nevertheless, it has been shown that electrochemical methods provide a green and sustainable alternative means to access even complex natural products. Additionally, electrochemical transformations can lead to superior results in situations where conventional methods fail. A collection of investigated electrochemical transformations applied in natural product synthesis and late‐stage functionalization will be surveyed.
7.1. Electrochemical Dehydrodimerization
Dehydrogenative couplings or dehydrodimerization reactions are of particular interest as only protons are liberated, which can be discharged at the cathode. The generated hydrogen can potentially be directly employed for a coupled synthesis.146 One remarkable example is the N−N heteroatom coupling of carbazoles at a carbon anode, investigated by Baran and co‐workers as a pivotal step of the first total synthesis of dixiamycin B (Scheme 51).147
Scheme 51.
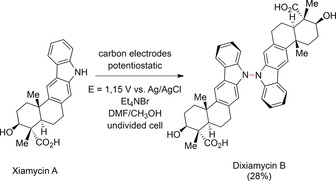
N−N dehydrodimerization reaction of xiamycin A to dixiamycin B.147
The authors demonstrated that this reaction can be generally applied to substituted carbazoles and β‐carbolines with yields of up to 66 %. Here, the electrochemical approach was superior to classic organic oxidation reactions. In this protocol, the supporting electrolyte (Et4NBr) acted as a mediator, resulting in a mild oxidative reaction. Baran and co‐workers also observed unusual selectivity in that only a single atropisomer of the desired coupling product was formed.
7.2. Synthesis by Electrochemically Initiated Cyclization
Many synthetic targets involve cyclic moieties; consequently, electrochemical cyclization steps play an important role. A large variety of different natural products were synthesized by including an intramolecular anodic olefin coupling reaction as a key step. Important work has been done by Moeller and co‐workers for the synthesis of (−)‐alliacol A,148 (+)‐nemorensic acid,149 (−)‐crobarbatic acid,150 and the arteannuin ring skeleton.151 Trauner et al. synthesized a precursor towards guanacastepene152 with this method, whereas conventional transformations, such as Rh‐catalyzed cyclopropanation/rearrangement or diazotization reactions, did not succeed in delivering the desired compound. Wright et al. reported on the synthesis of the hamigeran ring skeleton.153
These reactions were mostly carried out with a reticulated vitreous carbon (RVC) anode in the presence of methanol and 2,6‐lutidine as a proton scavenger (Schemes 52 and 53), and under constant‐current conditions in an undivided cell. On the other hand, owing to the large, but not well defined, surface of the porous glassy carbon anode, the reaction conditions cannot be easily transferred to other setups. A general reaction mechanism and a collection of interesting natural products are shown in Scheme 52.
Scheme 52.
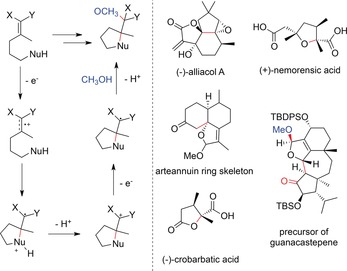
Postulated mechanism for anodic cyclizations of olefins. The exact mechanism, order of steps, and rates of each step may vary for each substrate and different reaction conditions (X, Y=electron‐donating groups, e.g., OMe, SMe/S‐Alkyl). Synthesized natural product skeletons are shown on the right.148, 149, 150, 151, 152 TBDPS=tert‐butyldiphenylsilyl, TBS=tert‐butyldimethylsilyl.
Scheme 53.
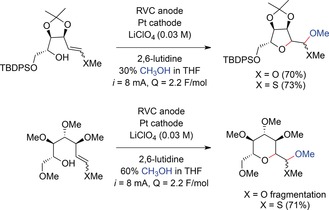
Synthesis of C‐glycosides by anodic olefin couplings.154
The proposed mechanism of the intramolecular coupling reaction involves a single electron oxidation at the HOMO of a functional group attached to an electron‐rich double bond, forming the initial radical cation. Subsequent attack of an electron‐rich nucleophile leads to the formation of mostly five‐ to seven‐membered rings. After proton abstraction, an additional oxidation step results in the formation of a cation, which will be quenched by MeOH as a solvent.
Trends regarding the ring sizes generated by electrochemical cyclization reactions were studied by Moeller and co‐workers for the synthesis of C‐glycosides by anodic olefin coupling reactions.154 Cyclization to a furanose system was achieved by the intramolecular coupling of a hydroxy group to either a methoxy enol ether or a vinyl sulfide in yields of up to 73 %. For the pyranose systems, the use of vinyl sulfide substrates was favorable. This means that trapping of the radical cation intermediates was accelerated by the formation of less polarized radical cations. With this modification, the desired pyranose glycosides were isolated in yields of up to 71 % (Scheme 53). With vinyl sulfides, mixtures were obtained because of partial substitution of the methyl sulfide with methoxy groups.
The anodic formation of spiro compounds was used as a key step in the enantioselective synthesis of heliannuol E by Nishiyama and co‐workers.155 Under constant‐current conditions, oxidation at a carbon beaker anode was carried out to form a spirodienone derivative (Scheme 54). Nishiyama et al. also applied the electrochemical synthesis of spiro acetals to the synthesis of ossamycin, following a similar approach.156
Scheme 54.
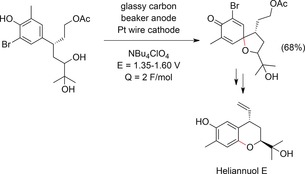
Anodic synthesis of a spiro compound for the synthesis of heliannuol E.155
Chromans and spiro chromans such as euglobal skeletons play an important role in a variety of polycyclic aromatic natural products. Electrochemical syntheses of these moieties were achieved by Chiba and co‐workers by intermolecular cycloaddition reactions of olefins and in situ generated ortho‐quinone methides.157, 158 The reaction can be carried out in a two‐phase reaction medium consisting of MeNO2 and LiClO4 as the electrolyte for the oxidation reaction at a glassy carbon anode and hexane, in order to dissolve the reaction products and prevent oxidative decomposition at the anode (Scheme 55).157 LiClO4 was found to be crucial for significant conversion in the cycloaddition reaction as both ions have a promotional effect on Diels–Alder reactions. In the second approach, an electrode coated with PTFE fiber was used to increase the selectivity of the electrochemical cycloaddition reaction.158
Scheme 55.
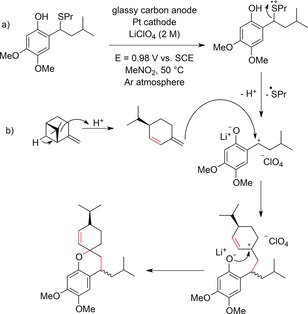
Electrochemical synthesis of chromans and spiro chromans by intermolecular cycloadditions of terpenes and in situ generated ortho‐quinone methides.157
Anodic phenol–olefin couplings have been extensively studied by the groups of Yamamura and others. For example, Yamamura and co‐workers synthesized a highly oxygenated isocedrene. The anodic intramolecular coupling of a phenol to an olefin was used as a key step to construct the bicyclic ring skeleton. Unfortunately, constant‐current conditions in a defined potential range were applied, and no current densities were given (Scheme 56).159
Scheme 56.
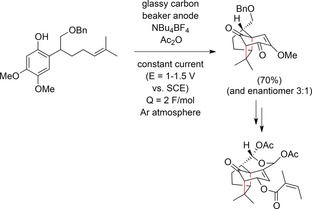
Electrochemical synthesis of isocedrene by an anodic phenol–olefin coupling.159
Intermolecular anodic phenol–olefin couplings were also investigated by Einaga, Nishiyama, and co‐workers.160 Electrolysis of isoeugenol in MeOH at a constant potential provided the neolignan (±)‐licarin A by coupling and cyclization reactions. After extensive optimization, the authors found that the combination of a batch cell with BDD electrodes is superior for this electrochemical synthesis. This can be rationalized by the proposed mechanism, wherein methoxyl radicals play an important role. The authors demonstrated by ESR studies that the amount of methoxy radicals was higher at the BDD than at other anodes. The desired natural product was obtained in yields of up to 40 % along with two side products (Scheme 57).
Scheme 57.
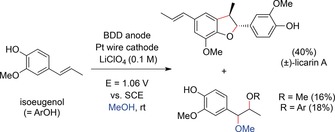
Electrochemical synthesis of licarin A by an anodic phenol–olefin coupling.160
Another interesting example is the synthesis of o‐methylthalibrine investigated by Nishiyama and co‐workers.161 This synthesis involves the electrochemical formation of diaryl ethers by substitution of a halogen with a phenolic substrate and subsequent reduction of the formed quinone acetal intermediate at a Zn cathode. In general, anodic treatment of phenol derivatives is a powerful approach to synthesize various natural products or natural product skeletons. This topic has been surveyed for example by Yamamura and Nishiyama.159, 162
A list of potentially useful anodic cyclization reactions for the construction of alkaloids has been complied by Schäfer.163 One remarkable example, namely the anodic intramolecular macrocyclization of a phenol and an indole moiety, was reported by Harran and co‐workers.164 The multistep synthesis of a diazonamide‐based drug (DZ‐2384; Scheme 58) involves macrocyclization by intramolecular C−O and C−C bond formation. The selectivity of the oxidative cyclization was increased and the reagent waste was minimized by development of an electrochemical protocol. Both factors were of significant importance considering that the reaction was carried out on multigram scale (up to 60 g of starting material). Constant‐potential electrolysis in DMF/H2O with Et4NBF4 as the supporting electrolyte produced the macrocycle in 43 % yield (based on recovered starting material).
Scheme 58.
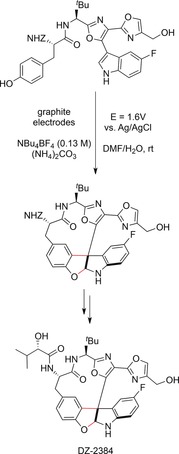
Electrochemical synthesis of the precursor for DZ‐2384 on multigram scale by anodic macrocyclization.164
The anodic treatment of amines has been used as part of a multistep procedure towards the total synthesis of (+)‐N‐methylanisomycin.165 Lithiation of an amine and subsequent oxidation of the obtained lithium amide resulted in an intramolecular cyclization reaction. Electrolysis was carried out under constant‐current conditions at a Pt anode in a divided cell (Scheme 59). The observed stereoselectivity was rationalized by adsorption of the radical species via its less hindered face at the electrode surface.
Scheme 59.
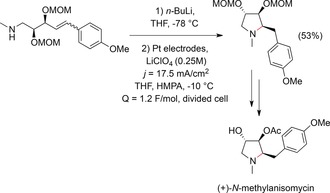
Synthesis of (+)‐N‐methylanisomycin by anodic cyclization of a δ‐alkenylamine.165 HMPA=hexamethylphosphoric acid triamide, MOM=methoxymethyl.
The late‐stage functionalization of natural products and their precursors is an effective means to synthesize various derivatives, for example, to investigate the effect of different substituents. In addition, they may represent metabolites, which can then be easily evaluated in terms of their biological action or toxicity. Aubé and co‐workers developed a procedure for the anodic late‐stage functionalization of polycyclic lactams. Electrolysis in an undivided cell with MeOH as the solvent and Et4NOTs or LiClO4 as the supporting electrolyte at carbon electrodes resulted in the methoxylation of cyclic and noncyclic amides. The authors demonstrated that these methoxy amides can be used for various diversification reactions (see Scheme 60).166
Scheme 60.
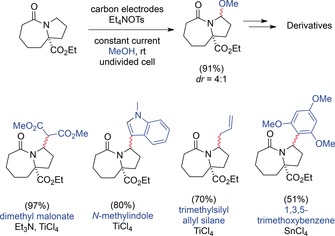
Late‐stage functionalization of cyclic and noncyclic amides shown for one bicyclic lactam.166
7.3. Kolbe and Minisci Reactions
Kolbe electrolysis is a well‐studied transformation, best known for the coupling of two aliphatic carboxylic acid moieties, and has been discussed in detail in Section 4. Its application in natural product synthesis was studied by Schäfer and co‐workers, for example. The electrochemical synthesis of insect pheromones or precursors thereof is one remarkable example.167
Another decarboxylative Kolbe‐type transformation applied in natural product synthesis was studied by Mori and co‐workers. The total synthesis of the bicyclic β‐lactam (+)‐PS‐5 features a decarboxylative Hofer–Moest‐type/non‐Kolbe reaction as a key step.168
7.4. C−H Oxidation
Baran and co‐workers recently developed a method for the electrochemical synthesis of enones from the corresponding olefins. This process proceeds through oxidation in the α‐position of the double bond, using Cl4NHPI (N‐hydroxytetrachlorophthalimide) as the mediator, and subsequent peroxide formation/elimination to afford the final α,β‐unsaturated ketone.169 This work is a perfect example of the further development of a known method.170 More than ten different natural products, as well as several steroids and terpene derivatives, were easily obtained (Scheme 61). Furthermore, the yields of the electrochemical steps in the synthesis of nootkatone, isolongifolenone, verbenone, and carvone were comparable or even superior to those described in the literature for using Cr‐, Rh‐, or Mn‐based reagents for the same transformation.
Scheme 61.
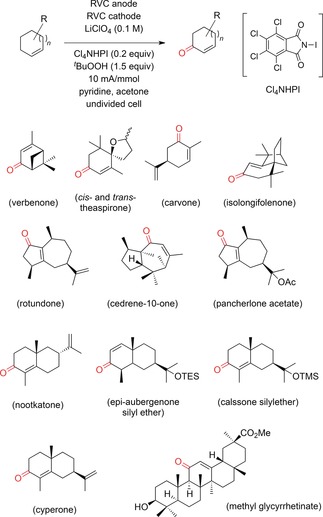
Electrochemical synthesis of enones by allylic C−H oxidation for the synthesis of several natural compounds.169
The authors further exploited the potential of this transformation by functionalizing non‐activated C−H bonds with a simple redox mediator such as quinuclidine.171 Methylene and methine moieties were electrochemically transformed into ketones or tertiary alcohols. This enabled the synthesis of (+)‐2‐oxo‐yahazunone on a large scale and in six steps starting from sclareolide (Scheme 62), in addition to the divergent synthesis of various meroterpenoids.
Scheme 62.
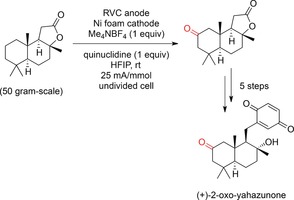
Total synthesis of (+)‐2‐oxo‐yahazunone by C−H activation.171
Both approaches by Baran et al. lead the way to the development of versatile, broadly applicable chemical transformations. However, mediators were necessary, and efforts should be taken to achieve direct electrochemical oxidation at the electrode.172
8. Future Perspectives
Although electroorganic synthesis has been studied for more than 150 years, significant progress was made over the last few decades. More research groups are now focusing on this topic as it combines various advantages of social and political importance with efficient synthetic applications. It can be expected that different groups will approach synthetic problems and challenges from different directions. Electrified organic synthesis will provide several synthetic tools in the near future that will be second to none in terms of sustainability and very useful. In combination with renewable electricity sources, the electrosynthesis of value‐added chemicals will be a game changer for the chemical industries.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Anton Wiebe obtained his Diploma in chemistry from the Johannes Gutenberg University Mainz in 2014, working in the group of Prof. Dr. S. R. Waldvogel for his diploma thesis. During his studies, he went to the University of Otago, New Zealand (Prof. Dr. S. Brooker) for a research stay. He is currently a PhD student under the supervision of Prof. Dr. S. R. Waldvogel and a member of the Max Planck Graduate Center, studying electrochemical oxidative coupling reactions of aromatic molecules.

Biographical Information
Tile Gieshoff obtained his Diploma in chemistry from the Johannes Gutenberg University Mainz in 2014 after conducting research towards his diploma thesis at Sanofi‐Aventis Deutschland GmbH. He is currently a PhD student under the supervision of Prof. Dr. S. R. Waldvogel and a member of the MAINZ graduate school, working on the electroorganic synthesis of heterocycles. In 2016, he was a visiting researcher at Washington University in St. Louis (Prof. Dr. K. Moeller).

Biographical Information
Sabine Möhle obtained her B.Sc. in chemistry from the University of Regensburg in 2013. After a research stay at the University of California, Santa Barbara (Prof. Dr. R. D. Little) in 2014, she finished her M.Sc. in chemistry in the group of Prof. Dr. O. Reiser at the University of Regensburg in 2014. She is currently a PhD student under the supervision of Prof. Dr. S. R. Waldvogel, working on the electrochemical amination of arenes. Her PhD studies are supported by a Kekulé Fellowship of the Funds of the Chemical Industry.

Biographical Information
Eduardo Rodrigo obtained his B.Sc. in chemistry in 2009 and his M.Sc. in organic chemistry in 2011 from the University Autónoma of Madrid. In 2014, he was a visitor at the University of Jyväskylä, Finland (Prof. Dr. P. M. Pihko). He finished his PhD studies in 2016 at the University Autónoma of Madrid in the field of organocatalysis under the supervision of Dr. M. B. Cid. Currently, he is a postdoctoral researcher at the Johannes Gutenberg University Mainz in the group of Prof. Dr. S. R. Waldvogel, working in the field of organic electrosynthesis.

Biographical Information
Michael Zirbes obtained his B.Sc. in chemistry in 2014 from the Johannes Gutenberg University Mainz. After working there as an undergraduate research assistant in 2015, he obtained his M.Sc. in organic chemistry in 2017 from the same university. Currently, he is a PhD student under the supervision of Prof. Dr. S. R. Waldvogel, investigating the electrochemical conversion of renewable raw materials.

Biographical Information
Siegfried R. Waldvogel studied chemistry in Konstanz and received his PhD in 1996 from the University of Bochum/Max Planck Institute for Coal Research under the supervision of Prof. Dr. M. T. Reetz. After postdoctoral research at the Scripps Research Institute in La Jolla, California (Prof. Dr. J. Rebek, Jr.), he began his habilitation in 1998 at the University of Münster. In 2004, he moved to the University of Bonn as a Professor of Organic Chemistry. In 2010, he became Full Professor at the Johannes Gutenberg University Mainz. His main research interests are organic electrochemistry, oxidative coupling reactions with Mo V reagents, and supramolecular sensing.

Acknowledgements
The authors thank the DFG (Wa1276/14‐1 and Wa 1276/17‐1) for financial support. Support of the Advanced Lab of Electrochemistry and Electrosynthesis—ELYSION (Carl Zeiss Stiftung) is gratefully acknowledged. We appreciate financial support by the Center for INnovative and Emerging MAterials (CINEMA). A.W. acknowledges the Max Planck Graduate Center for financial support. S.M. thanks the Fonds der Chemischen Industrie (FCI) for a Kekulé fellowship. T.G. is a recipient of a DFG fellowship by the Excellence Initiative at the Graduate School Materials Science in Mainz (GSC 266).
A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes, S. R. Waldvogel, Angew. Chem. Int. Ed. 2018, 57, 5594.
Contributor Information
Anton Wiebe, http://www.chemie.uni-mainz.de/OC/AK-Waldvogel/.
Prof. Dr. Siegfried R. Waldvogel, Email: waldvogel@uni-mainz.de.
References
- 1.J. G. J. Olivier, G. Janssens-Maenhout, M. Muntean, J. A. H. W. Peters, Trends in Global CO2 Emissions: 2016 Report, The Hague, 2016.
- 2.Renewables 2017: Global Status Report, REN21, Paris, 2017.
- 3. Jörissen J., Speiser B., Preparative Electrolysis on the Laboratory Scale, 5th ed. (Eds.: O. Hammerich, B. Speiser), CRC Press, Boca Raton, 2016, pp. 263–330. [Google Scholar]
- 4. Horn E. J., Rosen B. R., Baran P. S., ACS Cent. Sci. 2016, 2, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Frontana-Uribe B. A., Little R. D., Ibanez J. G., Palma A., Vasquez-Medrano R., Green Chem. 2010, 12, 2099–2119; [Google Scholar]
- 5b. Schäfer H. J., C. R. Chim. 2011, 14, 745–765. [Google Scholar]
- 6.Z. W. She, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov, T. F. Jaramillo, Science 2017, 355, https://doi.org/10.1126/science.aad4998. [DOI] [PubMed]
- 7.
- 7a. Turner J. A., Science 2004, 305, 972–974; [DOI] [PubMed] [Google Scholar]
- 7b. Chu S., Majumdar A., Nature 2012, 488, 294–303; [DOI] [PubMed] [Google Scholar]
- 7c. Lewis N. S., Nocera D. G., Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gütz C., Klöckner B., Waldvogel S. R., Org. Process Res. Dev. 2016, 20, 26–32. [Google Scholar]
- 9. Schmitt D., Regenbrecht C., Hartmer M., Stecker F., Waldvogel S. R., Beilstein J. Org. Chem. 2015, 11, 473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kulisch J., Nieger M., Stecker F., Fischer A., Waldvogel S. R., Angew. Chem. Int. Ed. 2011, 50, 5564–5567; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 5678–5682. [Google Scholar]
- 11. Yan M., Kawamata Y., Baran P. S., Chem. Rev. 2017, 117, 13230–13319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kirsch P., Modern Fluoroorganic Chemistry. Synthesis, Reactivity, Applications, Wiley-VCH, Weinheim, 2004. [Google Scholar]
- 13.
- 13a. Purser S., Moore P. R., Swallow S., Gouverneur V., Chem. Soc. Rev. 2008, 37, 320–330; [DOI] [PubMed] [Google Scholar]
- 13b. Bégué J.-P., Bonnet-Delpon D., Bioorganic and Medicinal Chemistry of Fluorine, Wiley, Hoboken, 2008; [Google Scholar]
- 13c. Hiyama T., Organofluorine Compounds. Chemistry and Applications (Ed.: H. Yamamoto), Springer, Berlin, Heidelberg, 2000. [Google Scholar]
- 14. Fuchigami T., Inagi S., Chem. Commun. 2011, 47, 10211–10223. [DOI] [PubMed] [Google Scholar]
- 15. Simons J. H., J. Electrochem. Soc. 1949, 95, 47–67. [Google Scholar]
- 16. Lund H., Organic Electrochemistry. An Introduction and Guide, 3rd ed. (Ed.: H. Lund), Dekker, New York, 1991. [Google Scholar]
- 17. Fuchigami T., Inagi S., Fluorination in Organic Electrochemistry, 5th ed. (Eds.: O. Hammerich, B. Speiser), CRC Press, Boca Raton, 2016, pp. 807–826. [Google Scholar]
- 18. Momota K., Morita M., Matsuda Y., Electrochim. Acta 1993, 38, 1123–1130. [Google Scholar]
- 19. Chen S.-Q., Hatakeyama T., Fukuhara T., Hara S., Yoneda N., Electrochim. Acta 1997, 42, 1951–1960. [Google Scholar]
- 20.
- 20a. Hasegawa M., Ishii H., Fuchigami T., Green Chem. 2003, 5, 512–515; [Google Scholar]
- 20b. Hasegawa M., Fuchigami T., Electrochim. Acta 2004, 49, 3367–3372. [Google Scholar]
- 21. Ishii H., Yamada N., Fuchigami T., Chem. Commun. 2000, 1617–1618. [Google Scholar]
- 22. Sunaga T., Atobe M., Inagi S., Fuchigami T., Chem. Commun. 2009, 956–958. [DOI] [PubMed] [Google Scholar]
- 23. Inagi S., Sawamura T., Fuchigami T., Electrochem. Commun. 2008, 10, 1158–1160. [Google Scholar]
- 24. Sawamura T., Inagi S., Fuchigami T., J. Electrochem. Soc. 2009, 156, E26–E28. [Google Scholar]
- 25. Fuchigami T., Fujita T., J. Org. Chem. 1994, 59, 7190–7192. [Google Scholar]
- 26. Fujita T., Fuchigami T., Tetrahedron Lett. 1996, 37, 4725–4728. [Google Scholar]
- 27. Sawamura T., Kuribayashi S., Inagi S., Fuchigami T., Org. Lett. 2010, 12, 644–646. [DOI] [PubMed] [Google Scholar]
- 28. Sawamura T., Kuribayashi S., Inagi S., Fuchigami T., Adv. Synth. Catal. 2010, 352, 2757–2760. [Google Scholar]
- 29. Sawamura T., Takahashi K., Inagi S., Fuchigami T., Angew. Chem. Int. Ed. 2012, 51, 4413–4416; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 4489–4492. [Google Scholar]
- 30.
- 30a. Hili R., Yudin A. K., Nat. Chem. Biol. 2006, 2, 284–287; [DOI] [PubMed] [Google Scholar]
- 30b. Czarnik A. W., Acc. Chem. Res. 1996, 29, 112–113; [DOI] [PubMed] [Google Scholar]
- 30c. Bikker J. A., Brooijmans N., Wissner A., Mansour T. S., J. Med. Chem. 2009, 52, 1493–1509. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Bowie A. L., Hughes C. C., Trauner D., Org. Lett. 2005, 7, 5207–5209; [DOI] [PubMed] [Google Scholar]
- 31b. Fukuyama T., Xu L., Goto S., J. Am. Chem. Soc. 1992, 114, 383–385. [Google Scholar]
- 32.
- 32a. Sousa M. M., Melo M. J., Parola A. J., Morris P. J. T., Rzepa H. S., Seixas de Melo J. S., Chem. Eur. J. 2008, 14, 8507–8513; [DOI] [PubMed] [Google Scholar]
- 32b. Zollinger H., Color Chemistry. Syntheses, Properties, and Applications of Organic Dyes and Pigments , 3rd ed., Wiley-VCH, Weinheim, 2004. [Google Scholar]
- 33.
- 33a. MacDiarmid A. G., Epstein A. J., Faraday Discuss. Chem. Soc. 1989, 88, 317–332; [Google Scholar]
- 33b. MacDiarmid A. G., Synth. Met. 1997, 84, 27–34; [Google Scholar]
- 33c. Weissermel K., Arpe H.-J., Industrial Organic Chemistry , 3rd ed., Wiley-VCH, Weinheim, 2007. [Google Scholar]
- 34. Yella A., Lee H.-W., Tsao H. N., Yi C., Chandiran A. K., Nazeeruddin M. K., Diau E. W.-G., Yeh C.-Y., Zakeeruddin S. M., Grätzel M., Science 2011, 334, 629–634. [DOI] [PubMed] [Google Scholar]
- 35.
- 35a. Shang D. T., Blount H. N., J. Electroanal. Chem. 1974, 54, 305–311; [Google Scholar]
- 35b. Shine H. J., Ristagno C. V., J. Org. Chem. 1971, 36, 4050–4055; [Google Scholar]
- 35c. Lund H., Tegnér C., Takman B., Acta Chem. Scand. 1957, 11, 1323–1330; [Google Scholar]
- 35d. Reitstöen B., Parker V. D., Lillerud K.-P., Högfeldt E., Spielbüchler P., Pedersen J. B., Krogsgaard-Larsen P., Acta Chem. Scand. 1992, 46, 464–468. [Google Scholar]
- 36.
- 36a. Eberson L., Olofsson B., Haug A., Painter T., Rydberg J., Craig J. C., Acta Chem. Scand. 1969, 23, 2355–2366; [Google Scholar]
- 36b. Bewick A., Edwards G. J., Mellor J. M., Electrochim. Acta 1976, 21, 1101–1104; [Google Scholar]
- 36c. Hammerich O., Parker V. D., J. Chem. Soc. Chem. Commun. 1974, 245–246; [Google Scholar]
- 36d. Eberson L., Nyberg K., Tetrahedron Lett. 1966, 7, 2389–2393. [Google Scholar]
- 37.
- 37a. Lisitsyn Y. A., Sukhov A. V., Russ. J. Electrochem. 2013, 49, 91–95; [Google Scholar]
- 37b. Lisitsyn Y. A., Sukhov A. V., Russ. J. Electrochem. 2011, 47, 1180–1185; [Google Scholar]
- 37c. Lisitsyn Y. A., Sukhov A. V., Russ. J. Gen. Chem. 2013, 83, 1457–1458; [Google Scholar]
- 37d. Lisitsyn Y. A., Sukhov A. V., Russ. J. Gen. Chem. 2017, 87, 16–21; [Google Scholar]
- 37e. Lisitsyn Y. A., Sukhov A. V., Russ. J. Org. Chem. 2015, 51, 439–440; [Google Scholar]
- 37f. Lisitsyn Y. A., Kargin Y. M., Russ. J. Electrochem. 2004, 40, 864–867; [Google Scholar]
- 37g. Lisitsyn Y. A., Grigor′eva L. V., Russ. J. Electrochem. 2009, 45, 132–138; [Google Scholar]
- 37h. Lisitsyn Y. A., Busygina N. V., Zyavkina Y. I., Shtyrlin V. G., Russ. J. Electrochem. 2010, 46, 512–523; [Google Scholar]
- 37i. Lisitsyn Y. A., Grigor′eva L. V., Russ. J. Phys. Chem. 2009, 83, 509–510; [Google Scholar]
- 37j. Lisitsyn Y. A., Kargin Y. M., Russ. J. Electrochem. 2004, 40, 977–980; [Google Scholar]
- 37k. Lisitsyn Y. A., Kargin Y. M., Russ. J. Electrochem. 2000, 36, 89–99. [Google Scholar]
- 38. Bourdelande J. L., Gallardo I., Guirado G., J. Am. Chem. Soc. 2007, 129, 2817–2821. [DOI] [PubMed] [Google Scholar]
- 39.
- 39a. Jaworski J. S., Aromatic Nitrogen-Containing Compounds in Organic Electrochemistry, 5th ed. (Eds.: O. Hammerich, B. Speiser), CRC Press, Taylor & Francis Group, Boca Raton, 2016, pp. 1121–1148; [Google Scholar]
- 39b. Mu S., Kan J., Electrochim. Acta 1996, 41, 1593–1599. [Google Scholar]
- 40.“Oxidative Substitution and Addition Reactions”: Hammerich O., Utley J. H. P. in Organic Electrochemistry, 5th ed. (Eds.: O. Hammerich, B. Speiser), CRC Press, Taylor & Francis Group, Boca Raton, 2016, pp. 775–806. [Google Scholar]
- 41. Morofuji T., Shimizu A., Yoshida J.-i., J. Am. Chem. Soc. 2013, 135, 5000–5003. [DOI] [PubMed] [Google Scholar]
- 42. Yoshida J.-i., Shimizu A., Ashikari Y., Morofuji T., Hayashi R., Nokami T., Nagaki A., Bull. Chem. Soc. Jpn. 2015, 88, 763–775. [Google Scholar]
- 43. Waldvogel S. R., Möhle S., Angew. Chem. Int. Ed. 2015, 54, 6398–6399; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6496–6497. [Google Scholar]
- 44. Herold S., Möhle S., Zirbes M., Richter F., Nefzger H., Waldvogel S. R., Eur. J. Org. Chem. 2016, 1274–1278. [Google Scholar]
- 45. Möhle S., Herold S., Richter F., Nefzger H., Waldvogel S. R., ChemElectroChem 2017, 4, 2196–2210. [Google Scholar]
- 46.
- 46a. Engels H.-W., Pirkl H.-G., Albers R., Albach R. W., Krause J., Hoffmann A., Casselmann H., Dormish J., Angew. Chem. Int. Ed. 2013, 52, 9422–9441; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 9596–9616. [Google Scholar]
- 47.
- 47a. Booth G. in Ullmann's Encyclopedia of Industrial Chemistry, Vol. A 17, 5th ed. (Ed.: W. Gerhartz), Wiley-VCH, Weinheim, 1991, pp. 9–57; [Google Scholar]
- 47b. Behre H., Heinz U., Hammerschmidt E., DE 3308883 (A1), 1984;
- 47c. Lindner O., Pelster H., Steffan G., Stüwe A., EP 0012260 (A1), 1980;
- 47d. Alles H.-U., Schulz N., Wunderlich H., Jakob R., EP 0000155 (A1), 1979;
- 47e. Behre H., Jakob L., Heinz U., Mayer D., Busse R., DE 3840618 (C1), 1990;
- 47f. Laue J., Steffens C., Krause J., Wershofen S., Kilian W., Seekamp M., Ruhland M., US 2015246873 (A1), 2015.
- 48. Gao W.-J., Li W.-C., Zeng C.-C., Tian H.-Y., Hu L.-M., Little R. D., J. Org. Chem. 2014, 79, 9613–9618. [DOI] [PubMed] [Google Scholar]
- 49. Morofuji T., Shimizu A., Yoshida J.-i., J. Am. Chem. Soc. 2015, 137, 9816–9819. [DOI] [PubMed] [Google Scholar]
- 50.
- 50a. Amado J. A., Pesquera C., Gonzalez E. M., Otero M., Freijanes J., Alvarez A., Postgrad. Med. J. 1990, 66, 221–223; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50b. Shimura K., Kodama E., Sakagami Y., Matsuzaki Y., Watanabe W., Yamataka K., Watanabe Y., Ohata Y., Doi S., Sato M., Kano M., Ikeda S., Matsuoka M., J. Virol. 2008, 82, 764–774; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50c. Warot D., Berlin I., Patat A., Durrieu G., Zieleniuk I., Puech A. J., J. Clin. Pharmacol. 1996, 36, 942–950. [DOI] [PubMed] [Google Scholar]
- 51. Morofuji T., Shimizu A., Yoshida J.-i., J. Am. Chem. Soc. 2014, 136, 4496–4499. [DOI] [PubMed] [Google Scholar]
- 52. Morofuji T., Shimizu A., Yoshida J.-i., Chem. Eur. J. 2015, 21, 3211–3214. [DOI] [PubMed] [Google Scholar]
- 53. Wesenberg L., Herold S., Shimizu S., Yoshida Y.-i., Waldvogel S. R., Chem. Eur. J. 2017, 23, 12096–12099. [DOI] [PubMed] [Google Scholar]
- 54. Brahma K., Das B., Chowdhury C., Tetrahedron 2014, 70, 5863–5871. [Google Scholar]
- 55. Hayashi R., Shimizu A., Yoshida J.-i., J. Am. Chem. Soc. 2016, 138, 8400–8403. [DOI] [PubMed] [Google Scholar]
- 56. Hayashi R., Shimizu A., Song Y., Ashikari Y., Nokami T., Yoshida J.-i., Chem. Eur. J. 2017, 23, 61–64. [DOI] [PubMed] [Google Scholar]
- 57. Li C., Kawamata Y., Nakamura H., Vantourout J. C., Liu Z., Hou Q., Bao D., Starr J. T., Chen J., Yan M., Baran P. S., Angew. Chem. Int. Ed. 2017, 56, 13088–13093; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13268–13273. [Google Scholar]
- 58. Fu N., Sauer G. S., Saha A., Loo A., Lin S., Science 2017, 357, 575–579. [DOI] [PubMed] [Google Scholar]
- 59. Faraday M., Ann. Phys. Chem. 1834, 109, 433–451. [Google Scholar]
- 60. Kolbe H., Ann. Chem. Pharm. 1849, 69, 257–294. [Google Scholar]
- 61.
- 61a. Hofer H., Moest M., Justus Liebigs Ann. Chem. 1902, 323, 284–323; [Google Scholar]
- 61b. Stapley J. A., Bemiller J. N., Carbohydr. Res. 2007, 342, 610–613. [DOI] [PubMed] [Google Scholar]
- 62.
- 62a. Coleman J. P., Lines R., Utley J. H. P., Weedon B. C. L., J. Chem. Soc. Perkin Trans. 2 1974, 1064; [Google Scholar]
- 62b. Galicia M., González-Fuentes M. A., Valencia D. P., González F. J., J. Electroanal. Chem. 2012, 672, 28–33. [Google Scholar]
- 63. Stang C., Harnisch F., ChemSusChem 2016, 9, 50–60. [DOI] [PubMed] [Google Scholar]
- 64. Schäfer H. J., Top. Curr. Chem. 1990, 152, 91–151. [Google Scholar]
- 65. Schäfer H. J., Eur. J. Lipid Sci. Technol. 2012, 114, 2–9. [Google Scholar]
- 66. dos Santos T. R., Harnisch F., Nilges P., Schröder U., ChemSusChem 2015, 8, 886–893. [DOI] [PubMed] [Google Scholar]
- 67. Dai J.-J., Huang Y.-B., Fang C., Guo Q.-X., Fu Y., ChemSusChem 2012, 5, 617–620. [DOI] [PubMed] [Google Scholar]
- 68. Lebreux F., Buzzo F., Markó I. E., Synlett 2008, 2815–2820. [Google Scholar]
- 69. Shtelman A. V., Becker J. Y., J. Org. Chem. 2011, 76, 4710–4714. [DOI] [PubMed] [Google Scholar]
- 70. Renaud P., Seebach D., Helv. Chim. Acta 1986, 69, 1704–1710. [Google Scholar]
- 71. Ma X., Luo X., Dochain S., Mathot C., Markó I. E., Org. Lett. 2015, 17, 4690–4693. [DOI] [PubMed] [Google Scholar]
- 72. Tajima T., Kurihara H., Fuchigami T., J. Am. Chem. Soc. 2007, 129, 6680–6681. [DOI] [PubMed] [Google Scholar]
- 73. Kurihara H., Fuchigami T., Tajima T., J. Org. Chem. 2008, 73, 6888–6890. [DOI] [PubMed] [Google Scholar]
- 74. Kurihara H., Tajima T., Fuchigami T., Electrochemistry 2006, 74, 615–617. [Google Scholar]
- 75. Letzel M. C., Schäfer H. J., Fröhlich R., Beilstein J. Org. Chem. 2017, 13, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.
- 76a. Koutsaftis D., Marinis D., Karantonis A., Electrochim. Acta 2012, 59, 376–381; [Google Scholar]
- 76b. Lateef S., Reddy S., Mohan K., Reddy S., Reddy J., Tetrahedron Lett. 2007, 48, 77–80; [Google Scholar]
- 76c. Hernández-Muñoz L. S., Galano A., Astudillo-Sánchez P. D., Abu-Omar M. M., González F. J., Electrochim. Acta 2014, 136, 542–549. [Google Scholar]
- 77.
- 77a. Franke R., Selent D., Börner A., Chem. Rev. 2012, 112, 5675–5732; [DOI] [PubMed] [Google Scholar]
- 77b. Okamoto K., Zhang J., Housekeeper J. B., Marder S. R., Luscombe C. K., Macromolecules 2013, 46, 8059–8078; [Google Scholar]
- 77c. von Nussbaum F., Brands M., Hinzen B., Weigand S., Habich D., Angew. Chem. Int. Ed. 2006, 45, 5072–5129; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 5194–5254. [Google Scholar]
- 78. Kweon D., Jang Y., Kim H., Bull. Korean Chem. Soc. 2003, 24, 1049–1050. [Google Scholar]
- 79. Torii S., Tanaka H., Morisaki K., Tetrahedron Lett. 1985, 26, 1655–1658. [Google Scholar]
- 80. Kuroboshi M., Shiba T., Tanaka H., Tetrahedron Lett. 2013, 54, 3666–3668. [Google Scholar]
- 81. Tanaka H., Kuroboshi M., Katoaka R., Suzuki R., Electrochemistry 2013, 81, 356–358. [Google Scholar]
- 82. Pachón L. D., Elsevier C. J., Rothenberg G., Adv. Synth. Catal. 2006, 348, 1705–1710. [Google Scholar]
- 83. Jennings P. W., Pillsbury D. G., Hall J. L., Brice V. T., J. Org. Chem. 1976, 41, 719–722. [Google Scholar]
- 84. Troupel M., Rollin Y., Sibille S., Périchon J., Fauvarque J.-F., J. Organomet. Chem. 1980, 202, 435–446. [Google Scholar]
- 85. Fox M. A., Chandler D. A., Lee C., J. Org. Chem. 1991, 56, 3246–3255. [Google Scholar]
- 86. Courtois V., Barhdadi R., Troupel M., Périchon J., Tetrahedron 1997, 53, 11569–11576. [Google Scholar]
- 87. Gosmini C., Lasry S., Nédélec J.-Y., Périchon J., Tetrahedron 1998, 54, 1289–1298. [Google Scholar]
- 88.
- 88a. Sengmany S., Leonel E., Polissaint F., Nédélec J.-Y., Pipelier M., Thobie-Gautier C., Dubreuil D., J. Org. Chem. 2007, 72, 5631–5636; [DOI] [PubMed] [Google Scholar]
- 88b. Sengmany S., Vitu-Thiebaud A., Le Gall E., Condon S., Leonel E., Thobie-Gautier C., Pipelier M., Lebreton J., Dubreuil D., J. Org. Chem. 2013, 78, 370–379. [DOI] [PubMed] [Google Scholar]
- 89. Gosmini C., Nédélec J. Y., Périchon J., Tetrahedron Lett. 2000, 41, 201–203. [Google Scholar]
- 90.
- 90a. Sengmany S., Vasseur S., Lajnef A., Le Gall E., Léonel E., Eur. J. Org. Chem. 2016, 4865–4871; [Google Scholar]
- 90b. Sengmany S., Le Gall E., Léonel E., Molecules 2011, 16, 5550–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Le Gall E., Gosmini C., Nédélec J.-Y., Périchon J., Tetrahedron Lett. 2001, 42, 267–269. [Google Scholar]
- 92. Gomes P., Fillon H., Gosmini C., Labbé E., Périchon J., Tetrahedron 2002, 58, 8417–8424. [Google Scholar]
- 93. Amatore C., Cammoun C., Jutand A., Eur. J. Org. Chem. 2008, 4567–4570. [Google Scholar]
- 94.
- 94a. Mitsudo K., Shiraga T., Tanaka H., Tetrahedron Lett. 2008, 49, 6593–6595; [Google Scholar]
- 94b. Mitsudo K., Shiraga T., Kagen D., Shi D., Becker J. Y., Tanaka H., Tetrahedron 2009, 65, 8384–8388. [Google Scholar]
- 95. Alam N., Amatore C., Combellas C., Thiébault A., Verpeaux J. N., Tetrahedron Lett. 1987, 28, 6171–6174. [Google Scholar]
- 96. Boy P., Combellas C., Suba C., Thiébault A., J. Org. Chem. 1994, 59, 4482–4489. [Google Scholar]
- 97. Qu Y., Tateno H., Matsumura Y., Kashiwagi T., Atobe M., Molecules 2017, 22, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.
- 98a. Nyberg K., Lindberg B., Pilotti Å., Lindberg A. A., Lamvik A., Sunde E., Sørensen N. A., Acta Chem. Scand. 1970, 24, 1609–1617; [Google Scholar]
- 98b. Nyberg K., Larsen P. K., Lemmich J., Tørset O., Lagerlund I., Ehrenberg L., Acta Chem. Scand. 1971, 25, 534–542; [Google Scholar]
- 98c. Nyberg K., Kimmel E. C., Holm B., Widmark G., Koskikallio J., Kachi S., Acta Chem. Scand. 1971, 25, 2983–2988; [Google Scholar]
- 98d. Nyberg K., Ekström B., Sjöberg B., Husebye S., Klæboe P., Swahn C.-G., Acta Chem. Scand. 1973, 27, 503–509. [Google Scholar]
- 99. Schubert M., Franzmann P., Wunsche von Leupoldt A., Koszinowski K., Heinze K., Waldvogel S. R., Angew. Chem. Int. Ed. 2016, 55, 1156–1159; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1168–1172. [Google Scholar]
- 100. Saito F., Aiso H., Kochi T., Kakiuchi F., Organometallics 2014, 33, 6704–6707. [Google Scholar]
- 101. Gensch T., Hopkinson M. N., Glorius F., Wencel-Delord J., Chem. Soc. Rev. 2016, 45, 2900–2936. [DOI] [PubMed] [Google Scholar]
- 102. Bechgaard K., Parker V. D., J. Am. Chem. Soc. 1972, 94, 4749–4750. [Google Scholar]
- 103.
- 103a. Regenbrecht C., Waldvogel S. R., Beilstein J. Org. Chem. 2012, 8, 1721–1724; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103b. Schopohl M. C., Faust A., Mirk D., Fröhlich R., Kataeva O., Waldvogel S. R., Eur. J. Org. Chem. 2005, 2987—2999; [Google Scholar]
- 103c. Waldvogel S. R., Fröhlich R., Schalley C. A., Angew. Chem. Int. Ed. 2000, 39, 2472—2475; [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 2580–2583. [Google Scholar]
- 104.
- 104a. Malkowsky I. M., Rommel C. E., Fröhlich R., Griesbach U., Pütter H., Waldvogel S. R., Chem. Eur. J. 2006, 12, 7482–7488; [DOI] [PubMed] [Google Scholar]
- 104b. Malkowsky I. M., Fröhlich R., Griesbach U., Pütter H., Waldvogel S. R., Eur. J. Inorg. Chem. 2006, 1690–1697. [DOI] [PubMed] [Google Scholar]
- 105. Morofuji T., Shimizu A., Yoshida J.-i., Angew. Chem. Int. Ed. 2012, 51, 7259–7262; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7371–7374. [Google Scholar]
- 106. Arai T., Tateno H., Nakabayashi K., Kashiwagi T., Atobe M., Chem. Commun. 2015, 51, 4891–4894. [DOI] [PubMed] [Google Scholar]
- 107. Elsler B., Wiebe A., Schollmeyer D., Dyballa K. M., Franke R., Waldvogel S. R., Chem. Eur. J. 2015, 21, 12321–12325. [DOI] [PubMed] [Google Scholar]
- 108.
- 108a. Kirste A., Elsler B., Schnakenburg G., Waldvogel S. R., J. Am. Chem. Soc. 2012, 134, 3571–3576; [DOI] [PubMed] [Google Scholar]
- 108b. Kirste A., Schnakenburg G., Stecker F., Fischer A., Waldvogel S. R., Angew. Chem. Int. Ed. 2010, 49, 971–975; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 983—987. [Google Scholar]
- 109. Riehl B., Dyballa K., Franke R., Waldvogel S., Synthesis 2016, 49, 252–259. [Google Scholar]
- 110. Elsler B., Schollmeyer D., Dyballa K. M., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2014, 53, 5210–5213; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5311–5314. [Google Scholar]
- 111. Wiebe A., Riehl B., Lips S., Franke R., Waldvogel S. R., Sci. Adv. 2017, 3, eaao3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wiebe A., Schollmeyer D., Dyballa K. M., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2016, 55, 11801–11805; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11979–11983. [Google Scholar]
- 113. Schulz L., Enders M., Elsler B., Schollmeyer D., Dyballa K. M., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2017, 56, 4877–4881; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4955–4959. [Google Scholar]
- 114. Lips S., Wiebe A., Elsler B., Schollmeyer D., Dyballa K. M., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2016, 55, 10872–10876; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11031–11035. [Google Scholar]
- 115. Wiebe A., Lips S., Schollmeyer D., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2017, 56, 14727–14731; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14920–14925. [Google Scholar]
- 116. Alvarez-Builla J., Vaquero J. J., Barluenga J., Modern Heterocyclic Chemistry, Wiley-VCH, Weinheim, 2011. [Google Scholar]
- 117.
- 117a. Lund H., Adv. Heterocycl. Chem. 1970, 12, 213–316; [Google Scholar]
- 117b. Lund H., Tabaković I., Adv. Heterocycl. Chem. 1984, 36, 235–341; [Google Scholar]
- 117c. Tabaković I., Top. Curr. Chem. 1997, 185, 87–139. [Google Scholar]
- 118. Francke R., Beilstein J. Org. Chem. 2014, 10, 2858–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Y. Jiang, K. Xu, C. Zeng, Chem. Rev 2017, https://doi.org/10.1021/acs.chemrev.7b00271. [DOI] [PubMed]
- 120.
- 120a. Xu H.-C., Moeller K. D., Angew. Chem. Int. Ed. 2010, 49, 8004–8007; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 8176–8179; [Google Scholar]
- 120b. Campbell J. M., Xu H.-C., Moeller K. D., J. Am. Chem. Soc. 2012, 134, 18338–18344; [DOI] [PubMed] [Google Scholar]
- 120c. Redden A., Perkins R. J., Moeller K. D., Angew. Chem. Int. Ed. 2013, 52, 12865–12868; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 13103–13106. [Google Scholar]
- 121. Moeller K. D., Tetrahedron 2000, 56, 9527–9554. [Google Scholar]
- 122. Xu H.-C., Campbell J. M., Moeller K. D., J. Org. Chem. 2014, 79, 379–391. [DOI] [PubMed] [Google Scholar]
- 123. Xu F., Zhu L., Zhu S., Yan X., Xu H.-C., Chem. Eur. J. 2014, 20, 12740–12744. [DOI] [PubMed] [Google Scholar]
- 124. Zhu L., Xiong P., Mao Z.-Y., Wang Y.-H., Yan X., Lu X., Xu H.-C., Angew. Chem. Int. Ed. 2016, 55, 2226–2229; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2266–2269. [Google Scholar]
- 125. Xiong P., Xu H.-H., Xu H.-C., J. Am. Chem. Soc. 2017, 139, 2956–2959. [DOI] [PubMed] [Google Scholar]
- 126. Weinstein A. B., Schuman D. P., Tan Z. X., Stahl S. S., Angew. Chem. Int. Ed. 2013, 52, 11867–11870; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12083–12086. [Google Scholar]
- 127. Hou Z.-W., Mao Z.-Y., Zhao H.-B., Melcamu Y. Y., Lu X., Song J., Xu H.-C., Angew. Chem. Int. Ed. 2016, 55, 9168–9172; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9314–9318. [Google Scholar]
- 128. Wu Z.-J., Xu H.-C., Angew. Chem. Int. Ed. 2017, 56, 4734–4738; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4812–4816. [Google Scholar]
- 129. Gieshoff T., Schollmeyer D., Waldvogel S. R., Angew. Chem. Int. Ed. 2016, 55, 9437–9440; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9587–9590. [Google Scholar]
- 130.
- 130a. Eberson L., Hartshorn M. P., Persson O., J. Chem. Soc. Perkin Trans. 2 1995, 1735; [Google Scholar]
- 130b. Colomer I., Chamberlain A. E. R., Haughey M. B., Donohoe T. J., Nat. Rev. Chem. 2017, 1, 0088. [Google Scholar]
- 131.
- 131a. Gieshoff T., Kehl A., Schollmeyer D., Moeller K. D., Waldvogel S. R., Chem. Commun. 2017, 53, 2974–2977; [DOI] [PubMed] [Google Scholar]
- 131b. Kehl A., Gieshoff T., Schollmeyer D., Waldvogel S. R., Chem. Eur. J. 2018, 24, 590—593.. [DOI] [PubMed] [Google Scholar]
- 132. Gieshoff T., Kehl A., Schollmeyer D., Moeller K. D., Waldvogel S. R., J. Am. Chem. Soc. 2017, 139, 12317–12324. [DOI] [PubMed] [Google Scholar]
- 133. Kang L.-S., Xiao H.-l., Zeng C.-C., Hu L.-M., Little R. D., J. Electroanal. Chem. 2016, 767, 13–17. [Google Scholar]
- 134. Li W.-C., Zeng C.-C., Hu L.-M., Tian H.-Y., Little R. D., Adv. Synth. Catal. 2013, 355, 2884–2890. [Google Scholar]
- 135. Qian X.-Y., Li S.-Q., Song J., Xu H.-C., ACS Catal. 2017, 7, 2730–2734. [Google Scholar]
- 136. Zhao H.-B., Hou Z.-W., Liu Z.-J., Zhou Z.-F., Song J., Xu H.-C., Angew. Chem. Int. Ed. 2017, 56, 587–590; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 602–605. [Google Scholar]
- 137. Lai Y.-L., Ye J.-S., Huang J.-M., Chem. Eur. J. 2016, 22, 5425–5429. [DOI] [PubMed] [Google Scholar]
- 138. Qian P., Su J.-H., Wang Y., Bi M., Zha Z., Wang Z., J. Org. Chem. 2017, 82, 6434–6440. [DOI] [PubMed] [Google Scholar]
- 139. Aziridines and Epoxides in Organic Synthesis (Ed.: A. K. Yudin), Wiley-VCH, Weinheim, 2006. [Google Scholar]
- 140.
- 140a. Siu T., Yudin A. K., J. Am. Chem. Soc. 2002, 124, 530–531; [DOI] [PubMed] [Google Scholar]
- 140b. Siu T., Picard C. J., Yudin A. K., J. Org. Chem. 2005, 70, 932–937. [DOI] [PubMed] [Google Scholar]
- 141. Siu T., Yudin A. K., Org. Lett. 2002, 4, 1839–1842. [DOI] [PubMed] [Google Scholar]
- 142. Page P. C. B., Marken F., Williamson C., Chan Y., Buckley B. R., Bethell D., Adv. Synth. Catal. 2008, 350, 1149–1154. [Google Scholar]
- 143.
- 143a. Rossen K., Volante R. P., Reider P. J., Tetrahedron Lett. 1997, 38, 777–778; [Google Scholar]
- 143b. LeBlond C. R., Rossen K., Gortsema F. P., Zavialov I. A., Cianciosi S. J., Andrews A. T., Sun Y., Tetrahedron Lett. 2001, 42, 8603–8606. [Google Scholar]
- 144. Debe M. K., Nature 2012, 486, 43–51. [DOI] [PubMed] [Google Scholar]
- 145.
- 145a. Espinal L., Suib S. L., Rusling J. F., J. Am. Chem. Soc. 2004, 126, 7676–7682; [DOI] [PubMed] [Google Scholar]
- 145b. Nishihara H., Pressprich K., Murray R. W., Collman J. P., Inorg. Chem. 1990, 29, 1000–1006. [Google Scholar]
- 146. Llorente M. J., Nguyen B. H., Kubiak C. P., Moeller K. D., J. Am. Chem. Soc. 2016, 138, 15110–15113. [DOI] [PubMed] [Google Scholar]
- 147. Rosen B. R., Werner E. W., O'Brien A. G., Baran P. S., J. Am. Chem. Soc. 2014, 136, 5571–5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Mihelcic J., Moeller K. D., J. Am. Chem. Soc. 2004, 126, 9106–9111. [DOI] [PubMed] [Google Scholar]
- 149. Liu B., Moeller K. D., Tetrahedron Lett. 2001, 42, 7163–7165. [Google Scholar]
- 150. Xu H.-C., Brandt J. D., Moeller K. D., Tetrahedron Lett. 2008, 49, 3868–3871. [Google Scholar]
- 151. Wu H., Moeller K. D., Org. Lett. 2007, 9, 4599–4602. [DOI] [PubMed] [Google Scholar]
- 152. Hughes C. C., Miller A. K., Trauner D., Org. Lett. 2005, 7, 3425–3428. [DOI] [PubMed] [Google Scholar]
- 153. Sperry J. B., Wright D. L., Tetrahedron Lett. 2005, 46, 411–414. [Google Scholar]
- 154. Xu G., Moeller K. D., Org. Lett. 2010, 12, 2590–2593. [DOI] [PubMed] [Google Scholar]
- 155. Doi F., Ogamino T., Sugai T., Nishiyama S., Tetrahedron Lett. 2003, 44, 4877–4880. [Google Scholar]
- 156. Honjo E., Kutsumura N., Ishikawa Y., Nishiyama S., Tetrahedron 2008, 64, 9495–9506. [Google Scholar]
- 157. Chiba K., Arakawa T., Tada M., J. Chem. Soc. Perkin Trans. 1 1998, 2939–2942. [Google Scholar]
- 158. Chiba K., Sonoyama J., Tada M., J. Chem. Soc. Perkin Trans. 1 1996, 1435–1443. [Google Scholar]
- 159. Takakura H., Yamamura S., Tetrahedron Lett. 1999, 40, 299–302. [Google Scholar]
- 160. Sumi T., Saitoh T., Natsui K., Yamamoto T., Atobe M., Einaga Y., Nishiyama S., Angew. Chem. Int. Ed. 2012, 51, 5443–5446; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5539–5542. [Google Scholar]
- 161. Naito Y., Tanabe T., Kawabata Y., Ishikawa Y., Nishiyama S., Tetrahedron Lett. 2010, 51, 4776–4778. [Google Scholar]
- 162.
- 162a. Yamamura S., Nishiyama S., Synlett 2002, 0533–0543; [Google Scholar]
- 162b. Yamamura S., Shizuri Y., Shigemori H., Okuno Y., Ohkubo M., Tetrahedron 1991, 47, 635–644. [Google Scholar]
- 163. Schäfer H. J., Oxidative Coupling in Organic Electrochemistry, 5th ed. (Eds.: O. Hammerich, B. Speiser), CRC Press, Taylor & Francis Group, Boca Raton, 2016, pp. 705–773. [Google Scholar]
- 164. Ding H., DeRoy P. L., Perreault C., Larivée A., Siddiqui A., Caldwell C. G., Harran S., Harran P. G., Angew. Chem. Int. Ed. 2015, 54, 4818–4822; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4900–4904. [Google Scholar]
- 165. Tokuda M., Fujita H., Miyamoto T., Suginome H., Tetrahedron 1993, 49, 2413–2426. [Google Scholar]
- 166. Frankowski K. J., Liu R., Milligan G. L., Moeller K. D., Aubé J., Angew. Chem. Int. Ed. 2015, 54, 10555–10558; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10701–10704. [Google Scholar]
- 167.
- 167a. Klünenberg H., Schäfer H. J., Angew. Chem. Int. Ed. Engl. 1978, 17, 47–48; [Google Scholar]; Angew. Chem. 1978, 90, 48–49; [Google Scholar]
- 167b. Knolle J., Schäfer H. J., Angew. Chem. Int. Ed. Engl. 1975, 14, 758; [Google Scholar]; Angew. Chem. 1975, 87, 777; [Google Scholar]
- 167c. Seidel W., Knolle J., Schäfer H. J., Chem. Ber. 1977, 110, 3544–3552; [Google Scholar]
- 167d. Seidel W., Schäfer H. J., Chem. Ber. 1980, 113, 451–456. [Google Scholar]
- 168. Mori M., Kagechika K., Tohjima K., Shibasaki M., Tetrahedron Lett. 1988, 29, 1409–1412. [Google Scholar]
- 169. Horn E. J., Rosen B. R., Chen Y., Tang J., Chen K., Eastgate M. D., Baran P. S., Nature 2016, 533, 77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.
- 170a. Masui M., Hara S., Ueshima T., Kawaguchi T., Ozaki S., Chem. Pharm. Bull. 1983, 31, 4209–4211; [Google Scholar]
- 170b. Masui M., Hosomi K., Tsuchida K., Ozaki S., Chem. Pharm. Bull. 1985, 33, 4798–4802; [Google Scholar]
- 170c. Masui M., Ueshima T., Ozaki S., J. Chem. Soc. Chem. Commun. 1983, 479. [Google Scholar]
- 171. Kawamata Y., Yan M., Liu Z., Bao D.-H., Chen J., Starr J. T., Baran P. S., J. Am. Chem. Soc. 2017, 139, 7448–7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Waldvogel S. R., Selt M., Angew. Chem. Int. Ed. 2016, 55, 12578–12580; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12766–12768. [Google Scholar]
- 173.S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe, S. R. Waldvogel, Angew. Chem. Int Ed 2018, 57, DOI: https://doi.org/10.1002/anie.201712732; Angew. Chem 2018, 130, DOI: https://doi.org/10.1002/ange.201712732.