

Abstract
Aim
To compare alirocumab, a proprotein convertase subtilisin‐kexin type 9 inhibitor, with usual care (UC) in individuals with type 2 diabetes (T2DM) and mixed dyslipidaemia not optimally managed by maximally tolerated statins in the ODYSSEY DM‐DYSLIPIDEMIA trial (NCT02642159).
Materials and Methods
The UC options (no additional lipid‐lowering therapy; fenofibrate; ezetimibe; omega‐3 fatty acid; nicotinic acid) were selected prior to stratified randomization to open‐label alirocumab 75 mg every 2 weeks (with increase to 150 mg every 2 weeks at week 12 if week 8 non‐HDL cholesterol concentration was ≥2.59 mmol/L [100 mg/dL]) or UC for 24 weeks. The primary efficacy endpoint was percentage change in non‐HDL cholesterol from baseline to week 24.
Results
The randomized population comprised 413 individuals (intention‐to‐treat population, n = 409; safety population, n = 412). At week 24, the mean non‐HDL cholesterol reductions were superior with alirocumab (−32.5% difference vs UC, 97.5% confidence interval −38.1 to −27.0; P < .0001). Overall, 63.6% of alirocumab‐treated individuals were maintained on 75 mg every 2 weeks. Alirocumab also reduced LDL cholesterol (−43.0%), apolipoprotein B (−32.3%), total cholesterol (−24.6%) and LDL particle number (−37.8%) at week 24 vs UC (all P < .0001). Consistent with the overall trial comparison, alirocumab reduced non‐HDL cholesterol to a greater degree within each UC stratum at week 24. The incidence of treatment‐emergent adverse events was 68.4% (alirocumab) and 66.4% (UC). No clinically meaningful effect on glycated haemoglobin, or change in number of glucose‐lowering agents, was seen.
Conclusions
In individuals with T2DM and mixed dyslipidaemia on maximally tolerated statin, alirocumab showed superiority to UC in non‐HDL cholesterol reduction and was generally well tolerated.
Keywords: mixed dyslipidaemia, non‐HDL cholesterol, PCSK9, type 2 diabetes
1. INTRODUCTION
Atherosclerotic cardiovascular disease (ASCVD) is the leading cause of morbidity and mortality among individuals with type 2 diabetes mellitus (T2DM).1 The reasons are probably multifactorial, but a relevant contributory factor may be the greater prevalence of mixed dyslipidaemia, which is characterized by elevated triglyceride (TG) levels and thus elevated TG‐rich lipoprotein (TRL) and TRL cholesterol levels, as well as low levels of HDL cholesterol.2 Mixed dyslipidaemia in T2DM might not be detected with measurement of LDL cholesterol levels, as these may remain within a normal range.3 Non‐HDL cholesterol (calculated by subtracting HDL cholesterol from total cholesterol), accounts for the sum of all atherogenic lipoproteins (LDL cholesterol, intermediate‐density lipoprotein, very‐low‐density lipoprotein [VLDL], VLDL remnants, chylomicron remnants, and lipoprotein a [Lp(a)]) and has been suggested to be a better indicator of cardiovascular (CV) risk than LDL cholesterol among individuals with elevated TG levels, including individuals with dyslipidaemia.3, 4, 5 Populations with mixed dyslipidaemia also have qualitative changes in LDL particles, with a higher number of smaller, more dense LDL particles; these are believed to be more atherogenic than larger, more buoyant particles.2
Lipid‐lowering therapy (LLT) with statins increases the clearance of atherogenic lipoproteins and thus reduces plasma cholesterol levels, principally through reductions in LDL cholesterol.6 This results in a significantly lower risk of ASCVD with the proportional benefit related to the absolute reduction in LDL cholesterol.7 Other therapeutic approaches that further increase clearance of atherogenic lipoproteins include ezetimibe8 and the inhibitors of proprotein convertase subtilisin‐kexin type 9 (PCSK9), alirocumab9 and evolocumab.10 Adding ezetimibe or evolocumab to statin significantly reduces CV events (the CV outcomes study with alirocumab [NCT01663402] is ongoing)11, 12; however, no previous study has prospectively evaluated PCSK9 inhibition in individuals with diabetes and mixed dyslipidaemia or compared different therapeutic options among individuals with elevated TG levels despite maximally tolerated statin therapy, an important consideration given the “real‐world” clinical uncertainty around potential therapeutic agents which principally reduce either the synthesis of TRL particles (fibrates), lipolysis of TGs (omega‐3 fatty acids), clearance of atherogenic lipoproteins (ezetimibe), or a combination of these mechanisms (nicotinic acid).
The ODYSSEY DM‐DYSLIPIDEMIA trial was designed to address these clinical uncertainties and assessed the efficacy and safety of alirocumab vs usual lipid‐lowering care (UC), stratified by an investigator's predefined option for add‐on therapy (fenofibrate, omega‐3 fatty acids, ezetimibe, nicotinic acid or no additional LLT) to maximally tolerated statins among individuals with T2DM at high ASCVD risk who had mixed dyslipidaemia and in whom non‐HDL cholesterol was not adequately controlled (≥2.59 mmol/L [≥100 mg/dL]). The primary endpoint (not used in previous randomized studies) was the difference in the percentage change from baseline in non‐HDL cholesterol between alirocumab and UC (overall; ie, all options). A prespecified analysis was used to compare the superiority of alirocumab vs fenofibrate (recommended in guidelines for treating individuals with elevated TGs4, 5).
2. MATERIALS AND METHODS
2.1. Study design
ODYSSEY DM‐DYSLIPIDEMIA (NCT02642159) was a phase IIIb/IV, randomized, open‐label, parallel group, multicentre trial. The trial was conducted at 110 sites in 14 countries; screening started in March 2016 and recruitment was completed in September 2016. The study design and methods have been published previously.13 Brief methods are summarized below and further details are provided in Appendix S1.
The trial was conducted in accordance with the ethical principles laid down by the 18th World Medical Assembly (Helsinki, 1964) and all applicable amendments laid down by the World Medical Assemblies, and the International Conference on Harmonisation guidelines. The trial protocol was approved by the relevant institutional review boards or independent ethics committees, and all participating individuals provided written informed consent.
2.2. Trial participants
The trial included individuals (aged ≥18 years) with T2DM and mixed dyslipidaemia whose non‐HDL cholesterol was not adequately controlled despite stable maximally tolerated statin dose for ≥4 weeks prior to screening visit, without other LLTs, and who had either a documented history of ASCVD or at least 1 additional CV risk factor. Study participants had to have a glycated haemoglobin (HbA1c) of <9% (74.9 mmol/mol); changes to antihyperglycaemic medications were to be limited and made only in circumstances of clinical need for the duration of the study.
Mixed dyslipidaemia was defined as non‐HDL cholesterol ≥2.59 mmol/L (≥100 mg/dL) and TGs ≥1.70 mmol/L (150 mg/dL; but <5.65 mmol/L [500 mg/dL]) at the screening visit. The maximally tolerated dose of statin was based on the judgment of the investigator. Individuals with documented statin intolerance (as judged by the investigator) and therefore not receiving statin therapy could also be enrolled. ASCVD was defined as coronary heart disease, peripheral arterial disease or ischaemic stroke. Full inclusion and exclusion criteria have been reported previously.13
2.3. Study procedures
After a screening period of up to 3 weeks, investigators selected before randomization the most appropriate choice from a range of 5 therapeutic options based on their usual clinical practice, namely, not to add any LLT, or to add 1 of the following: ezetimibe, fenofibrate, omega‐3 fatty acid formulation or nicotinic acid. Participants were randomized in a 2:1 ratio to receive, on top of maximally tolerated statin (or no statin if intolerant), either open‐label alirocumab or UC for 24 weeks. Randomization was stratified by the UC option selected by the investigator prior to randomization.
Alirocumab was initiated at a dose of 75 mg every 2 weeks, with blinded dose increase to 150 mg every 2 weeks at week 12 if week 8 non‐HDL cholesterol was ≥2.59 mmol/L (≥100 mg/dL), henceforth referred to as “alirocumab 75/150 mg every 2 weeks.”
2.4. Endpoints and assessments
The primary efficacy endpoint was the percentage change in non‐HDL cholesterol from baseline to week 24, analysed using an intention‐to‐treat approach. Further details on secondary endpoints and laboratory and safety assessments are given in Appendix S1.
2.5. Statistical analysis
The primary efficacy endpoint was analysed using a mixed‐effect model with a repeated measures approach to account for missing data. Further information on analysis methods is presented in Appendix S1.
3. RESULTS
3.1. Participating individuals
Eligible individuals were allocated to UC options by the investigator prior to randomization, and were subsequently randomized within each stratum to either alirocumab or UC option in a 2:1 ratio (Figure 1). A total of 413 individuals were randomized to alirocumab (n = 276) or UC (n = 137). The median daily doses of UC treatments are given in Table S1.
Figure 1.
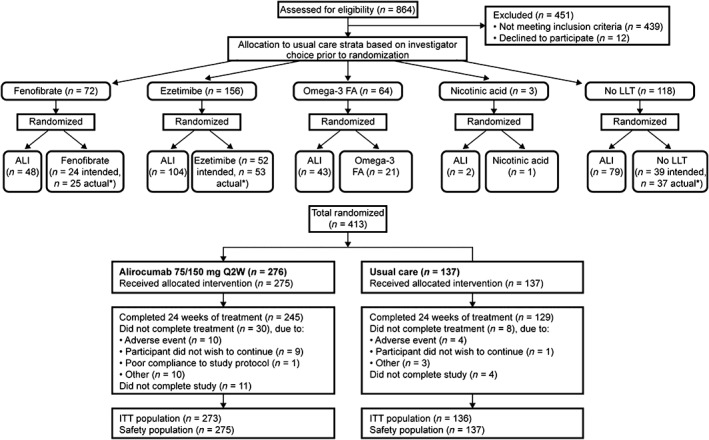
Disposition of individuals for the DM‐DYSLIPIDEMIA study. Abbreviations: ALI, alirocumab; FA, fatty acids; ITT, intention‐to‐treat; LLT, lipid‐lowering therapy; Q2W, every 2 weeks. aThirty‐nine individuals were intended for “no LLT” prior to randomization; however, 37 actually received “no LLT.” One individual intended for “no LLT” received fenofibrate and another received ezetimibe. In total, 4 individuals were not included in the ITT analysis (no non‐HDL cholesterol value available within 1 of the analysis windows up to week 24) and 1 individual was not included in the safety analysis (individual did not wish to continue prior to treatment)
Baseline characteristics and lipid variables were generally similar regardless of treatment allocation (Tables 1 and S2). At baseline, 84.0% of individuals in the alirocumab group and 76.6% in the UC group were receiving statin therapy (of these, 46.3% [alirocumab] and 36.2% [UC] were receiving high‐intensity statin). Treatment groups included 34.4% and 34.3% of individuals with a history of ASCVD and 65.6% and 65.7% without ASCVD but with additional CV risk factors in the alirocumab and UC groups, respectively (Table 1).
Table 1.
Baseline characteristics (randomized population)
| Alirocumab 75/150 mg every 2 weeksn = 276 | UC n = 137 | |
|---|---|---|
| Age, years | 62.8 (9.3) | 64.1 (8.8) |
| Men | 147 (53.3) | 69 (50.4) |
| Race | ||
| White | 247 (89.5) | 123 (89.8) |
| Black | 16 (5.8) | 6 (4.4) |
| Ethnicity: Hispanic/Latin | 35 (12.7) | 14 (10.2) |
| Body mass index, kg/m2 | 32.7 (5.4) | 33.2 (4.9) |
| HbA1c, % [mmol/mol] | 7.1 (0.8) [53.5 (9.1)] | 7.1 (0.9) [54.5 (9.4)] |
| HbA1c | ||
| <7% (<53.0 mmol/mol) | 134 (48.6) | 57 (41.6) |
| ≥7% to <8% (≥53.0 mmol/mol to <63.9 mmol/mol) | 100 (36.2) | 56 (40.9) |
| ≥8% to <9 (≥63.9 mmol/mol to <74.9 mmol/mol) | 42 (15.2) | 24 (17.5) |
| FPG | ||
| mmol/L | 8.04 (2.11) | 8.22 (2.19) |
| mg/dL | 144.9 (38.1) | 148.1 (39.4) |
| Median (Q1:Q3) duration of DM, years | 10.7 (5.5:17.5) | 11.5 (6.2:18.7) |
| Hypertensiona | 241 (87.3) | 123 (89.8) |
| Current cigarette smoker | 38 (13.8) | 23 (16.8) |
| CKDb | 41 (14.9) | 23 (16.8) |
| Individuals with ASCVD | 95 (34.4) | 47 (34.3) |
| Individuals without ASCVD but with additional CV risk factor | 181 (65.6) | 90 (65.7) |
| Any statinc | 231 (84.0) | 105 (76.6) |
| High‐intensity statind | 107 (46.3) | 38 (36.2) |
| Moderate‐intensity statind | 103 (44.6) | 64 (61.0) |
| Low‐intensity statind | 21 (9.1) | 3 (2.9) |
| Any LLT other than statinse before randomization | 1 (0.4) | 2 (1.5) |
| Fenofibrates | 1 (0.4) | 1 (0.7) |
| Cholesterol absorption inhibitor | 0 | 1 (0.7) |
| Nutraceuticals impacting lipids/other | 0 | 1 (0.7) |
| No LLTc (no statin or other LLT) | 44 (16.0) | 32 (23.4) |
| Statin‐intolerant | 43 (15.6) | 31 (22.6) |
| Concomitant antihyperglycemic drugsc | ||
| Biguanides | 211 (76.7) | 106 (77.4) |
| Insulin | 102 (37.1) | 56 (40.9) |
| Sulphonylureas | 67 (24.4) | 31 (22.6) |
| SGLT2 inhibitors | 39 (14.2) | 18 (13.1) |
| DPP‐4 inhibitors | 36 (13.1) | 20 (14.6) |
| GLP‐1RA | 32 (11.6) | 21 (15.3) |
| Thiazolidinediones | 9 (3.3) | 5 (3.6) |
| α‐glucosidase inhibitors | 2 (0.7) | 2 (1.5) |
| Other blood glucose‐lowering drugs | 9 (3.3) | 2 (1.5) |
| Baseline lipidsf | ||
| Non‐HDL cholesterol | ||
| mmol/L | 4.02 (1.20) | 4.18 (1.26) |
| mg/dL | 155.1 (46.2) | 161.5 (48.8) |
| LDL cholesterol, measuredg | ||
| mmol/L | 2.86 (1.04) | 3.04 (1.13) |
| mg/dL | 110.4 (40.3) | 117.3 (43.5) |
| ApoB, mg/dL [g/L] | 101.9 (25.8) [1.0 (0.3)] | 106.1 (28.7) [1.1 (0.3)] |
| Total cholesterol | ||
| mmol/L | 5.06 (1.19) | 5.25 (1.32) |
| mg/dL | 195.4 (46.0) | 202.5 (51.1) |
| Median (Q1:Q3) Lp(a), mg/dL | 16.0 (5.0:54.0) | 15.0 (5.0:40.0) |
| Median (Q1:Q3) TGs | ||
| mmol/L | 2.43 (1.91:3.22) | 2.40 (1.90:3.12) |
| mg/dL | 214.5 (169.0:285.0) | 212.0 (168.0:276.0) |
| HDL cholesterol | ||
| mmol/L | 1.04 (0.25) | 1.06 (0.30) |
| mg/dL | 40.3 (9.8) | 41.1 (11.6) |
| LDL particle number, nmol/L | 1404.1 (456.1) | 1483.8 (482.8) |
| ApoA1, mg/dL | 138.6 (21.2) | 139.4 (22.9) |
| LDL particle size, nm | 20.3 (0.6) | 20.3 (0.6) |
Abbreviations: Alirocumab 75/150 mg Q2W, alirocumab 75 mg Q2W with possible dose increase to 150 mg Q2W at Week 12; Apo, apolipoprotein; ASCVD, atherosclerotic cardiovascular disease; CKD, chronic kidney disease; CV, cardiovascular; DM, diabetes mellitus; DPP‐4, dipeptidyl peptidase 4; FPG, fasting plasma glucose; eGFR, estimated glomerular filtration rate; GLP‐1RA, glucagon‐like peptide 1 receptor agonist; HbA1c, glycated haemoglobin; LLT, lipid‐lowering therapy; Lp(a), lipoprotein a; Q1/Q3, first/third quartile; SGLT2, sodium‐glucose co‐transporter‐2; TG, triglyceride; UC, usual care.
Data are mean (SD) or n (%), unless otherwise indicated.
Established based on use of antihypertensive medication.
Defined as eGFR 15–60 mL/min/1.73 m2.
Data presented for safety population (275 alirocumab; 137 UC).
High‐intensity statin: atorvastatin 40 to 80 mg, rosuvastatin 20 to 40 mg or simvastatin 80 mg. Moderate‐intensity statin: atorvastatin 10 to 20 mg, fluvastatin 40 mg, fluvastatin extended release 80 mg, lovastatin 40 mg, pitavastatin 2 to 4 mg, pravastatin 10 to 20 mg, rosuvastatin 5 to 10 mg or simvastatin 20 to 40 mg. Low‐intensity statin: fluvastatin 20 to 40 mg, lovastatin 20 mg, pitavastatin 1 mg, pravastatin 10 to 20 mg or simvastatin 10 mg.
In combination with statins or not.
Order based on hierarchical order, except for LDL particle size.
β‐quantification.
3.2. Lipid variables
The least‐squares mean (SE) percentage change from baseline to week 24 in non‐HDL cholesterol was −37.3 (3.0)% with alirocumab and −4.7 (3.3)% with UC (−32.5% difference vs UC, 97.5% confidence interval [CI] –38.1 to −27.0; P < .0001 [Figure 2A]). Alirocumab also significantly lowered levels of measured LDL cholesterol, apolipoprotein (Apo)B, total cholesterol and Lp(a) vs UC (all P < .0001; Figure 2A). Non‐HDL cholesterol and measured LDL cholesterol reductions were observed from the first measured time point at week 8 and maintained through the 24‐week treatment period (Figure S1). TG levels were decreased in both arms at week 24, with no significant difference between alirocumab (−13.0%) vs UC (−8.8%; Figure 2A). As a result of the hierarchical testing procedure used, P values from subsequent testing of secondary endpoints are nominal only. At week 24, alirocumab treatment resulted in improvements from baseline (nominal P value <.025 vs UC) in HDL cholesterol, and LDL particle number and size (Figure 2A). Results for fenofibrate stratum and other individual UC strata were similar to the overall analysis (Figure 2B‐E); as a result of small patient numbers, data were not analysed for the nicotinic acid stratum. Similar results to those at week 24 were seen at week 12, when all individuals in the alirocumab arm were receiving the 75‐mg dose (Figure S2). In the alirocumab group, the dose of 75 mg every 2 weeks was maintained in 63.6% of individuals after week 12.
Figure 2.
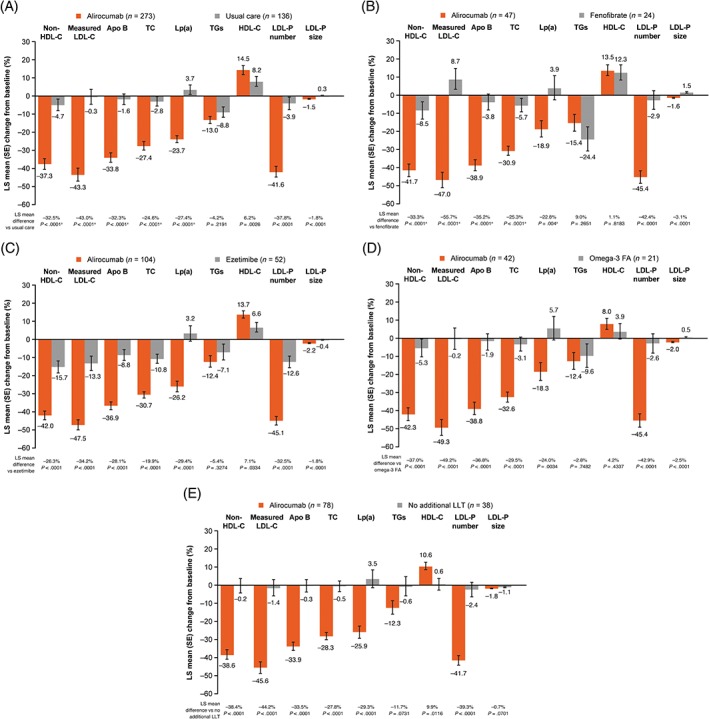
Primary and selected key secondary efficacy endpoints at week 24 for A, the overall study population; B, fenofibrate; C, ezetimibe; D, omega‐3 fatty acids and E, no lipid‐lowering therapy (ITT analysis). Abbreviations: Apo, apolipoprotein; FA, fatty acids; HDL‐C, HDL cholesterol; ITT, intenttion‐to‐treat; LDL‐C, LDL cholesterol; LDL‐P, LDL particle; LLT, lipid‐lowering therapy; Lp(a), lipoprotein (a); LS, least‐squares; TC, total cholesterol; TG, triglyceride. aStatistically significant according to the fixed hierarchical approach used to ensure a strong control of the overall type I error rate at the 0.025 level. ITT analysis includes individuals according to planned treatment (see footnote to Figure 1)
At week 24, more than two‐thirds of alirocumab‐treated individuals achieved levels of non‐HDL cholesterol <2.59 mmol/L (<100 mg/dL), measured LDL cholesterol <1.81 mmol/L (<70 mg/dL) and ApoB <80 mg/dL (<0.8 g/L; Figure 3). In addition, a greater proportion of individuals in the alirocumab group vs UC achieved a reduction in LDL cholesterol from baseline of ≥50% (55.2% vs 3.8%).
Figure 3.
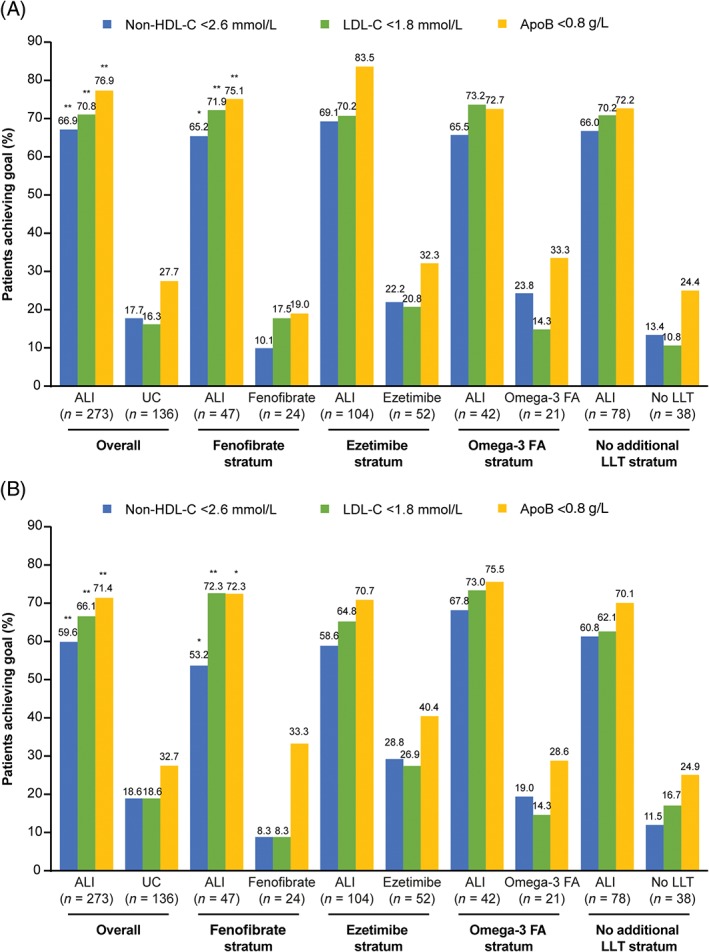
Proportion of individuals achieving predefined lipid goals at A, week 24 and B, week 12 (intention‐to‐treat analysis; as‐planned study cohorts). Abbreviations: ALI, alirocumab; Apo, apolipoprotein; FA, fatty acids; LDL‐C, LDL cholesterol; LLT, lipid‐lowering therapy; non‐HDL‐C, non‐HDL cholesterol; UC, usual care. *P < .05; **P < .0001 vs control. Non‐HDL cholesterol: 2.6 mmol/L = 100 mg/dL; LDL cholesterol: 1.8 mmol/L = 70 mg/dL; ApoB: 0.8 g/L = 80 mg/dL
Results were consistent across the various subgroups analysed (Figure S3).
3.3. Free and total PCSK9
In the alirocumab group, free PCSK9 levels changed by −43.3% and −60.6% at week 12 and week 24, respectively (UC: +18.2% and +11.8%, respectively; Figure 4). Total PCSK9 levels changed by +357.6% at week 12 and +413.3% at week 24 in the alirocumab group (UC: +13.9% and +10.8%, respectively; Figure 4). Corresponding data within individual UC strata are also shown in Figure 4.
Figure 4.
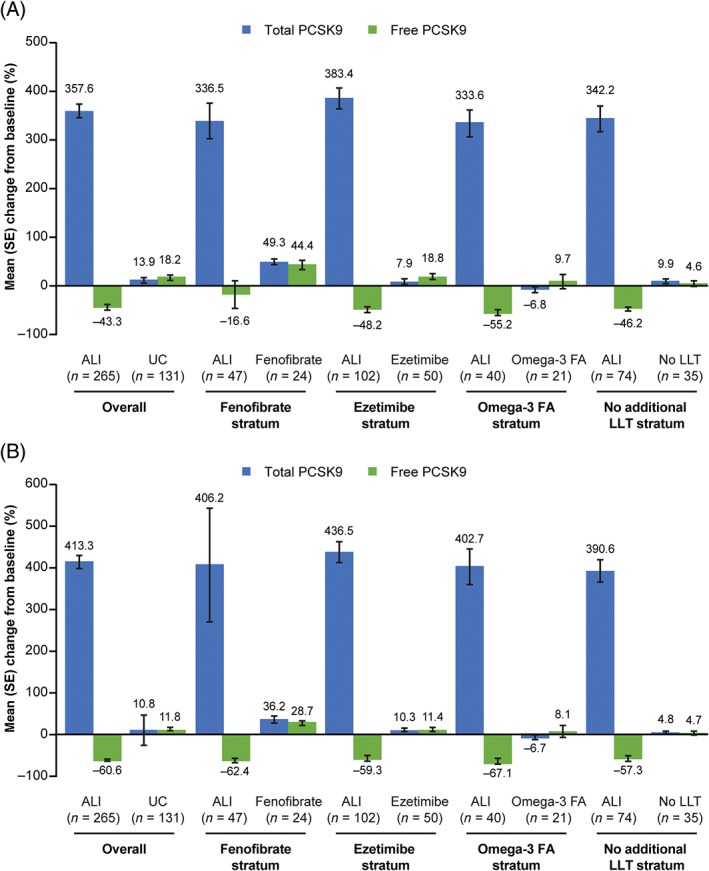
Percent change from baseline to A, week 12 and B, week 24 in levels of free and total PCSK9 in individuals receiving alirocumab vs usual care (PCSK9 analysis). Abbreviations: ALI, alirocumab; FA, fatty acids; LLT, lipid‐lowering therapy; LS, least squares; PCSK9, proprotein convertase subtilisin‐kexin type 9; UC, usual care
3.4. Diabetes‐related endpoints
The mean HbA1c and fasting plasma glucose (FPG) levels observed during the study are shown in Figure S4. The mean (SE) absolute change from baseline in HbA1c at week 24 was +2.58 (0.41) mmol/mol in the alirocumab group and +2.10 (0.57) mmol/mol in the UC group (P = .48); corresponding values in % for HbA1c were +0.24 (0.04)% (alirocumab group) and +0.19 (0.05)% (UC group). The mean (SE) absolute change in FPG from baseline at week 24 was +0.32 (0.13) and −0.01 (0.17) mmol/L in the alirocumab and UC groups, respectively (P = .12 vs UC); corresponding values for FPG were +5.70 (2.25) and −0.10 (3.07) mg/dL for alirocumab and UC, respectively. The median total number of glucose‐lowering treatments received remained stable over time, with no change between baseline and week 24 (Table S3).
3.5. Safety
The percentage of individuals who experienced any treatment‐emergent adverse events (TEAEs), treatment‐emergent serious adverse events and TEAEs leading to discontinuation was similar in the alirocumab and UC groups (Table S4).
The TEAEs occurring in ≥2% of individuals were reported at generally similar frequencies in the alirocumab and UC groups, with some TEAEs occurring at higher frequency in the alirocumab vs UC group and vice versa; urinary tract infection (alirocumab: 5.8%; UC: 3.6%) and diarrhoea (alirocumab: 5.1%; UC: 6.6%) were the most common TEAEs (Table S5).
In total, 3.0% (n = 8) of individuals receiving alirocumab and 0.8% (n = 1) of those receiving UC had low‐titre persistent anti‐drug antibodies. At week 12, 0.7% (n = 2) of individuals in the alirocumab group demonstrated positive neutralizing anti‐drug antibodies; none were observed at week 24. In the UC group, no neutralizing anti‐drug antibodies were observed.
4. DISCUSSION
ODYSSEY DM‐DYSLIPIDEMIA is the first dedicated study of a PCSK9 inhibitor to evaluate efficacy and safety vs UC among individuals with T2DM and mixed dyslipidaemia and elevated non‐HDL cholesterol levels despite maximally tolerated statin therapy, and the first randomized trial to use non‐HDL cholesterol as the primary endpoint. The pragmatic design of this study with the choice of UC (in addition to maximally tolerated statin therapy) based on the investigator's predefined option allows, for the first time, a direct comparison with multiple therapeutic alternatives to alirocumab and their effects on non‐HDL cholesterol as well as a range of lipoprotein markers believed to be causally related to ASCVD.
Alirocumab demonstrated superiority from baseline to week 24 in reducing non‐HDL cholesterol (by 32.5%), ApoB (32.3%), Lp(a) (27.4%), total cholesterol (24.6%) and measured LDL cholesterol (43.0%) vs UC. Alirocumab was not superior to UC in reducing TGs and, because of the hierarchical nature of testing, the significant increase in HDL cholesterol levels (+6.2%) and significant reductions in LDL particle number (−37.8%) and LDL particle size (−1.8%) relative to UC should be considered nominal. Moreover, in the present study, 66.9% of alirocumab‐treated individuals achieved non‐HDL cholesterol levels <2.59 mmol/L (100 mg/dL; 17.7% with UC) and 70.8% achieved LDL cholesterol levels <1.81 mmol/L (70 mg/dL) vs 16.3% with UC.
Alirocumab was also superior to fenofibrate in reducing non‐HDL cholesterol (33.3% vs fenofibrate), ApoB (35.2%), Lp(a) (22.8%), total cholesterol (25.3%) and measured LDL cholesterol (55.7%). At week 24, the percentage change from baseline in the fenofibrate group was equivalent to the alirocumab group in lowering TG and raising HDL cholesterol when added to maximally tolerated statin therapy. However, participants in the present study had moderately elevated TG levels (median baseline TGs of ~2.4 mmol/L [~210 mg/dL]), and conclusions cannot be extrapolated to those with more severely elevated TG levels (>5 mmol/L), who were excluded from the study. Nominally greater reductions from baseline to week 24 in LDL particle number (42.4%) and LDL particle size (3.1%) favoured alirocumab over fenofibrate. Furthermore, at week 24, 65.2% of patients achieved non‐HDL cholesterol levels <2.59 mmol/L (<100 mg/dL) and 71.9% LDL cholesterol levels <1.81 mmol/L (<70 mg/dL) with alirocumab vs 10.1% and 17.5%, respectively, with fenofibrate. Directionally concordant results were observed favouring alirocumab vs no LLT, omega‐3 fatty acids or ezetimibe where sample size allowed such comparisons.
The present study also offers novel insights into the potential mechanisms behind changes in different lipid variables when other LLTs are added to statins and the potential impact of these differential changes on the putative likelihood of future ASCVD. In the overall UC group, mean total and free PCSK9 concentrations changed by +10.8% and +11.8%, respectively, at week 24 compared with baseline; larger increases were seen in the fenofibrate group and also (to a lesser extent) in the ezetimibe group, in line with previous observations that treatment with these LLTs increases PCSK9 levels.14, 15 Omega‐3 fatty acids appeared to have a negligible effect on PCSK9 levels. The increases in PCSK9 levels in the overall UC group were associated with only modest reductions from baseline to week 24 in ApoB (−1.6%) and LDL particle number (−3.9%), and increase in LDL particle size (+0.3%) and therefore modest reductions in non‐HDL cholesterol (−4.7%) and measured LDL cholesterol levels (−0.3%), despite an 8.8% reduction in TG levels and an 8.2% increase in HDL cholesterol levels. In contrast, at week 24 total PCSK9 levels increased among alirocumab‐treated individuals by +413.3% and free PCSK9 decreased by −60.6%, reflecting that most circulating PCSK9 was bound to alirocumab. This, in turn, reduced ApoB and non‐HDL cholesterol levels and LDL particle number by approximately one‐third and measured LDL cholesterol by approximately two‐fifths, compared with the more modest reductions in the UC group, and resulted in a greater proportion (66.9%) of individuals achieving non‐HDL cholesterol <2.59 mmol/L (<100 mg/dL) with alirocumab vs the addition of UC to statins (17.7%). Taken together, these results demonstrate the significant impact on clearance of atherogenic particles by targeting PCSK9 for inhibition with alirocumab compared with the different modes of actions of the UC therapies. Conversely, extracellular PCSK9 inhibition with alirocumab does not appear to substantially affect TG metabolism, although reductions in TG levels with alirocumab were similar to those observed with fenofibrate.
Based on post hoc data from the ACCORD trial or from meta‐analyses of fibrate trials,16, 17 many clinicians add fibrates to statins in individuals with high TGs or high TGs/low HDL cholesterol. However, in the present prespecified analyses we demonstrate that the addition of fenofibrate to statins results in little or no reduction in atherogenic lipoproteins and hence a trivial reduction in their cholesterol cargo, despite favourable but clinically modest changes in TG and HDL cholesterol. These data underscore the importance of therapeutic approaches that increase the clearance of atherogenic lipoproteins rather than other currently available therapies (apart from statins) that target either synthesis or lipolysis of TG, but which have little impact on atherogenic cholesterol levels, as demonstrated by modest improvements in non‐HDL cholesterol goal attainment.
Whilst the present data cannot be used to assess whether favourable changes in atherogenic particle clearance will translate into better clinical outcomes in patients with T2DM and atherogenic dyslipidaemia, it should be noted that UC failed to achieve non‐HDL cholesterol levels <2.59 mmol/L (<100 mg/dL; recommended as a treatment target for individuals with T2DM in guidelines4, 5) for the majority (82.3%). In contrast, at week 24, only 33.1% of the alirocumab group failed to achieve non‐HDL cholesterol levels <2.59 mmol/L (<100 mg/dL). The findings were consistent with previous subgroup or post hoc analyses which have reported the effect of PCSK9 inhibitors according to mixed dyslipidaemia or diabetes status.18, 19, 20, 21, 22, 23, 24
This study was not designed to assess CV outcomes. Post hoc analyses of alirocumab ODYSSEY trials have suggested that event reduction continues to very‐low levels of LDL cholesterol (~25–50 mg/dL)25; however, this requires confirmation in the forthcoming ODYSSEY OUTCOMES study, which includes a prespecified subgroup analysis in individuals with diabetes mellitus (DM). CV outcomes data are available for other PCSK9 monoclonal antibodies. In the FOURIER study, in individuals with CV disease (CVD) with and without T2DM, evolocumab reduced LDL cholesterol by 56 mg/dL (~1.4 mmol/L) from baseline with a 20% reduction in major CV events (CV death, myocardial infarction or stroke).26 Similar results were observed in individuals with DM and stable ASCVD.11 Among individuals with higher CV risk (46.1–47.8% with DM) a benefit in reducing major CV events (non‐fatal myocardial infarction, non‐fatal stroke, hospitalization for angina requiring revascularization or CV death) was shown with bococizumab (hazard ratio 0.79; 95% CI 0.65‐0.97; P = .02).27
Non‐HDL cholesterol was chosen as the primary endpoint in this study after reports that it represents a better risk marker than LDL cholesterol when TG levels are elevated4; however, we acknowledge that there is no direct strong evidence from randomized trials that additional changes in non‐HDL cholesterol, on top of LDL cholesterol reductions, contribute to further CVD reduction. Moreover, it is difficult to separate reductions in non‐HDL cholesterol, LDL cholesterol, ApoB and LDL particle number, which are highly correlated. There are data from meta‐analyses suggesting that greater reductions in non‐HDL cholesterol and ApoB are related to further reductions in CVD,28, 29 analogous to well‐established data for LDL cholesterol reduction.7, 12, 26 The importance of reducing atherogenic particle number has gained further credence with the large genetic analyses by Ference et al30 demonstrating that, with add‐on therapy, which affects both the quality and content of atherogenic particles, any CV benefit is more accurately predicted by particle number (as depicted by changes in ApoB) rather than by LDL cholesterol. This is supported by findings from the recent REVEAL trial with the cholesteryl ester transfer protein inhibitor anacetrapib, in which the observed clinical risk reduction was considerably less than that anticipated by the observed reductions in LDL cholesterol.31 A meta‐regression analysis of statin and non‐statin therapies by Robinson et al28 suggested that every 10 mg/dL (0.1 g/L) reduction in ApoB would result in a ~6% proportional reduction in CVD risk. Applying those data to the present population with a starting ApoB level of ~100 mg/dL (1 g/L), alirocumab, fenofibrate, omega‐3 fatty acids and ezetimibe would be expected to achieve an absolute reduction of 33.8 mg/dL (0.3 g/L), 3.8 mg/dL (0.04 g/L), 1.9 mg/dL (0.02 g/L) and 8.8 mg/dL (0.1 g/L), respectively. Extrapolating from the meta‐regression, this would be expected to translate into 20.3%, 2.3%, 1.1% and 5.3% reductions in the risk of CVD. Notably, the estimated 20.3% risk reduction is consistent with the observed results from the FOURIER study,11 and the estimated 5.3% risk reduction is roughly consistent with the results of the ezetimibe IMPROVE‐IT study.12 Our data also suggest that, unless there is some benefit of TG‐lowering per se on CVD, as yet unidentified and independent of the modest reductions in ApoB and LDL particle number, the results of the ongoing outcomes trials for fibrates and fish oils (PROMINENT: NCT03071692; STRENGTH: NCT02104817; REDUCE‐IT: NCT01492361) are unlikely to show significant CVD risk reduction. Furthermore, based on these data, there is no rationale for the routine use of fenofibrate as add‐on to statin therapy if the goal of adding it is to reduce non‐HDL cholesterol or ApoB as a means to reduce CV risk.
Statin use has been associated with an increase in risk of T2DM, and Mendelian randomization studies have reported an association between PCSK9 loss‐of‐function mutations and risk of diabetes30, 32, 33; however, we did not see any clinically relevant effect of alirocumab on change in glycaemic variables or in use of antihyperglycaemic agents in the present study, supporting previous pooled analyses and sub‐analyses,22, 34, 35 and the more recent analysis from the FOURIER study,11, 26 which indicated no meaningful effect of PCSK9 inhibitors on either HbA1c or FPG levels or on rates of new‐onset diabetes. However, larger study populations and longer‐term studies are required to further validate the long‐term effects of PCSK9 inhibition, as use of these therapies is likely to be lifelong.
In this study, alirocumab was generally well tolerated, with comparable rates of TEAEs between alirocumab and usual care. No local injection‐site reactions (defined as those deemed to be allergic and requiring medical consultation) were reported in this study in either treatment arm.
The rate of persistent anti‐drug antibodies observed in the present study was similar to the overall rate seen in a pooled analysis of 10 ODYSSEY studies, which demonstrated substantial LDL cholesterol reductions that were maintained over the course of studies, regardless of anti‐drug antibody status.36
Limitations of the present study include its relatively short duration and the number of individuals enrolled, which did not allow analysis of rare adverse events. The awareness of treatment might have introduced bias by study participants and investigators.37 Safety reporting could have been influenced as study participants and investigators would have known what treatment they were receiving. Similarly, treatment adherence to diet and other medication may have been influenced by the participants' knowledge about treatment. Simultaneous addition of UC therapies was not included in the protocol, although it is acknowledged that this may be recommended in real‐life practice.
In conclusion, among individuals with T2DM and mixed dyslipidaemia whose total atherogenic cholesterol burden was inadequately controlled despite maximally tolerated statin therapy, increasing the clearance of atherogenic lipoproteins with a PCSK9 inhibitor more effectively reduced total atherogenic cholesterol levels compared with the usual lipid‐lowering therapeutic approaches currently used.
Supporting information
Appendix S1. Supplementary Appendix.
Table S1. UC dose details (randomized population).
Table S2. Baseline characteristics in the alirocumab vs intent‐to‐prescribe fenofibrate, ezetimibe, omega‐3 fatty acids and no LLT study cohorts (randomized population).
Table S3. Number of glucose‐lowering treatments over time (ITT analysis).
Table S4. TEAEs, adverse events of special interest and safety laboratory values (potentially clinically significant abnormality values; safety analysis).
Table S5. Treatment‐emergent adverse events by preferred term occurring in ≥2% of individuals in either group (safety analysis).
Figure S1. Percentage change from baseline in levels of A, non‐HDL‐C and B, LDL‐C (determined by beta‐quantification) over time in individuals receiving alirocumab 75/150 mg Q2W vs usual care (ITT analysis)
Figure S2. Primary and selected key secondary efficacy endpoints at Week 12 for A, the overall study population; B, fenofibrate; C, ezetimibe; D, omega‐3 fatty acids and E, no LLT (ITT analysis; as‐planned study cohorts).
Figure S3. Percent change from baseline in non‐HDL‐C at Week 24: Subgroup analysis according to baseline A, demographic characteristics; B, statin therapy and C, lipids (ITT analysis).
Figure S4. Mean A, A1C and B, FPG levels over time in individuals receiving alirocumab 75/150 mg Q2W versus usual care (ITT analysis).
ACKNOWLEDGMENTS
The authors would like to thank the participants, their families, and all investigators involved in this study. The following people from the study sponsors reviewed and provided editorial comments on the manuscript: Lisa Aurand, Ameen Ghannam, Corinne Hanotin and Michael Howard (Sanofi), and Carol Hudson, Robert Pordy and Robert Sanchez (Regeneron Pharmaceuticals, Inc.). The sponsor was involved in the study design, collection, analysis and interpretation of data, as well as data checking of information provided in the manuscript. The authors had unrestricted access to study data, were responsible for all content and editorial decisions, and received no honoraria related to the development of this publication.
Conflict of interest
K.K.R. has received: personal fees (data safety monitoring board) from AbbVie, Inc.; consultant fees/honoraria from Aegerion, Algorithm, Amgen, AstraZeneca, Boehringer Ingelheim, Cerenis, Eli Lilly and Company, Ionis Pharmaceuticals, Kowa, Medicines Company, MSD, Novartis, Pfizer, Inc., Regeneron Pharmaceuticals, Inc., Resverlogix, Sanofi and Takeda; and research grants from Kowa, Pfizer, Inc., and Regeneron Pharmaceuticals, Inc. L.A.L. has received: personal fees from Esperion; grants and personal fees from Amgen, AstraZeneca, Eli Lilly and Company, Merck, Regeneron Pharmaceuticals, Inc. and Sanofi; and grants from Kowa and the Medicine Company. D.M.‐W. has received speaker's bureau and consultant/advisory board fees from Amgen, AstraZeneca, Boehringer Ingelheim, MSD (Merck), Novartis, Novo Nordisk and Sanofi. B.C. has received: research funding and personal fees from Sanofi and Regeneron Pharmaceuticals, Inc. during the conduct of the study; research funding from Pfizer, Inc.; and honoraria from AstraZeneca, Pierre Fabre, Janssen, Eli Lilly and Company, MSD Merck & Co., Novo Nordisk, Sanofi and Takeda. H.M.C. has received grants, personal fees and non‐financial support from Sanofi and Regeneron Pharmaceuticals, Inc. during the conduct of the study; grants, personal fees and non‐financial support from Eli Lilly and Company; grants and other support from Roche Pharmaceuticals; grants from Pfizer, Inc., Boehringer Ingelheim, and AstraZeneca LP; and other support from Bayer. R.R.H. has received research funding from AstaMed, Eli Lilly and Company, Hitachi, Lexicon, Novo Nordisk and Viacyte and is a consultant for and/or advisory panel member of Alere, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol‐Myers Squibb, Elcelyx, Gilead, Intarcia, Ionis, Janssen/Johnson & Johnson, Merck and Sanofi‐Aventis. F.J.T. has received speaker's bureau and consultant/advisory board fees from AstraZeneca, Amgen, Boehringer Ingelheim, Bristol‐Myers Squibb, Eli Lilly and Company, GlaxoSmithKline, Janssen Pharmaceuticals, Merck Sharpe & Dohme, Novartis Pharmaceuticals Co., Novo Nordisk, Sanofi and Regeneron Pharmaceuticals, Inc. C.D., M.B.‐B. and A.L. are employees of and shareholders in Sanofi. R.S. is an employee of and shareholder in Regeneron Pharmaceuticals, Inc. S.D.P. has received research funding from AstraZeneca, Boheringer Ingelheim, Novartis Pharmaceuticals Co. and Merck Sharpe & Dohme; and is a consultant for or has received honoraria from AstraZeneca, Boehringer Ingelheim, Eli Lilly and Company, GlaxoSmithKline, Janssen Pharmaceuticals, Laboratoires Servier, Merck Sharpe & Dohme, Novartis Pharmaceuticals Co., Novo Nordisk, Sanofi, Servier and Takeda Pharmaceuticals.
Author contributions
K.K.R., L.A.L., D.M.‐W., B.C., H.M.C., R.R.H., F.J.T., M.B.‐B., C.D., A.L., R.S. and S.D.P. contributed to the study design or concept and the interpretation of the data, and critically reviewed and edited the manuscript. In addition, S.D.P. and F.J.T. were investigators who contributed to the data acquisition. All authors approved the final version.
Ray KK, Leiter LA, Müller‐Wieland D, et al. Alirocumab vs usual lipid‐lowering care as add‐on to statin therapy in individuals with type 2 diabetes and mixed dyslipidaemia: The ODYSSEY DM‐DYSLIPIDEMIA randomized trial. Diabetes Obes Metab. 2018;20:1479–1489. https://doi.org/10.1111/dom.13257
Funding information Sanofi and Regeneron Pharmaceuticals, Inc.
REFERENCES
- 1. American Diabetes Association . 9. Cardiovascular disease and risk management. Diabetes Care. 2017;40:S75‐S87. [DOI] [PubMed] [Google Scholar]
- 2. Vergès B. Pathophysiology of diabetic dyslipidaemia: where are we? Diabetologia. 2015;58:886‐899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ryden L, Grant PJ, Anker SD, et al. ESC guidelines on diabetes, pre‐diabetes, and cardiovascular diseases developed in collaboration with the EASD: the task force on diabetes, pre‐diabetes, and cardiovascular diseases of the European Society of Cardiology (ESC) and developed in collaboration with the European Association for the Study of Diabetes (EASD). Eur Heart J. 2013;34:3035‐3087. [DOI] [PubMed] [Google Scholar]
- 4. Bays HE, Jones PH, Orringer CE, Brown WV, Jacobson TA. National lipid association annual summary of clinical lipidology 2016. J Clin Lipidol. 2016;10:S1‐43. [DOI] [PubMed] [Google Scholar]
- 5. Catapano AL, Graham I, De Backer G, et al. 2016 ESC/EAS guidelines for the management of dyslipidaemias. Eur Heart J. 2016;37:2999‐3058. [DOI] [PubMed] [Google Scholar]
- 6. Ginsberg HN. REVIEW: efficacy and mechanisms of action of statins in the treatment of diabetic dyslipidemia. J Clin Endocrinol Metab. 2006;91:383‐392. [DOI] [PubMed] [Google Scholar]
- 7. Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta‐analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670‐1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tremblay AJ, Lamarche B, Cohn JS, Hogue JC, Couture P. Effect of ezetimibe on the in vivo kinetics of apoB‐48 and apoB‐100 in men with primary hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2006;26:1101‐1106. [DOI] [PubMed] [Google Scholar]
- 9. Reyes‐Soffer G, Pavlyha M, Ngai C, et al. Effects of PCSK9 inhibition with alirocumab on lipoprotein metabolism in healthy humans. Circulation. 2017;135:352‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Watts GF, Chan DC, Dent R, et al. Factorial effects of evolocumab and atorvastatin on lipoprotein metabolism. Circulation. 2017;135:338‐351. [DOI] [PubMed] [Google Scholar]
- 11. Sabatine MS, Leiter LA, Wiviott SD, et al. Cardiovascular safety and efficacy of the PCSK9 inhibitor evolocumab in patients with and without diabetes and the effect of evolocumab on glycaemia and risk of new‐onset diabetes: a prespecified analysis of the FOURIER randomised controlled trial. Lancet Diabetes Endocrinol. 2017;5:941‐950. [DOI] [PubMed] [Google Scholar]
- 12. Cannon CP, Blazing MA, Giugliano RP, et al. IMPROVE‐IT Investigators; Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387‐2397. [DOI] [PubMed] [Google Scholar]
- 13. Müller‐Wieland D, Leiter LA, Cariou B, et al. Design and rationale of the ODYSSEY DM‐DYSLIPIDEMIA trial: lipid‐lowering efficacy and safety of alirocumab in individuals with type 2 diabetes and mixed dyslipidaemia at high cardiovascular risk. Cardiovasc Diabetol. 2017;16:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rey J, Poitiers F, Paehler T, et al. Relationship between low‐density lipoprotein cholesterol, free proprotein convertase subtilisin/kexin type 9, and alirocumab levels after different lipid‐lowering strategies. J Am Heart Assoc. 2016;5:e003323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Costet P, Hoffmann MM, Cariou B, Guyomarc'h Delasalle B, Konrad T, Winkler K. Plasma PCSK9 is increased by fenofibrate and atorvastatin in a non‐additive fashion in diabetic patients. Atherosclerosis. 2010;212:246‐251. [DOI] [PubMed] [Google Scholar]
- 16. ACCORD Study Group , Ginsberg HN, Elam MB, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563‐1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sacks FM, Carey VJ, Fruchart JC. Combination lipid therapy in type 2 diabetes. N Engl J Med. 2010;363:692‐694. author reply 694–695. [DOI] [PubMed] [Google Scholar]
- 18. Robinson JG, Farnier M, Krempf M, et al. ODYSSEY LONG TERM Investigators; Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489‐1499. [DOI] [PubMed] [Google Scholar]
- 19. Taskinen MR, Del Prato S, Bujas‐Bobanovic M, Louie MJ, Lorenzato C, Colhoun HM. Alirocumab in individuals with diabetes and mixed dyslipidemia: pooled analyses of five phase 3 trials: presented at the International Diabetes Federation World Congress 2015, Abstract 0272‐PD.2015. http://conference.idf.org/IDF2015/CM.NET.WebUI/CM.NET.WEBUI.SCPR2/SCPRfunctiondetail.aspx?confID=05000000-0000-0000-0000-000000000003&sesID=05000000-0000-0000-0000-000000001704&absID=07000000-0000-0000-0000-000000011066. Accessed July 27, 2017.
- 20. Ginsberg HN, Farnier M, Robinson JG, et al. Efficacy and safety of alirocumab: pooled analyses of 1048 individuals with diabetes mellitus from five placebo‐controlled phase 3 studies of at least 52 weeks duration. Circulation. 2015;132:A17070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kastelein JJ, Ginsberg HN, Langslet G, et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J. 2015;36:2996‐3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leiter LA, Zamorano JL, Bujas‐Bobanovic M, et al. Lipid‐lowering efficacy and safety of alirocumab in patients with or without diabetes: a sub‐analysis of ODYSSEY COMBO II. Diabetes Obes Metab. 2017;19:989‐996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sattar N, Preiss D, Robinson JG, et al. Lipid‐lowering efficacy of the PCSK9 inhibitor evolocumab (AMG 145) in patients with type 2 diabetes: a meta‐analysis of individual patient data. Lancet Diabetes Endocrinol. 2016;4:403‐410. [DOI] [PubMed] [Google Scholar]
- 24. Rosenson RS, Jacobson TA, Preiss D, et al. Efficacy and safety of the PCSK9 inhibitor evolocumab in patients with mixed hyperlipidemia. Cardiovasc Drugs Ther. 2016;30:305‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ray KK, Ginsberg HN, Davidson MH, et al. Reductions in atherogenic lipids and major cardiovascular events: a pooled analysis of 10 ODYSSEY trials comparing alirocumab with control. Circulation. 2016;134:1931‐1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713‐1722. [DOI] [PubMed] [Google Scholar]
- 27. Ridker PM, Revkin J, Amarenco P, et al. SPIRE Cardiovascular Outcome Investigators; Cardiovascular efficacy and safety of bococizumab in high‐risk patients. N Engl J Med. 2017;376:1527‐1539. [DOI] [PubMed] [Google Scholar]
- 28. Robinson JG, Wang S, Jacobson TA. Meta‐analysis of comparison of effectiveness of lowering apolipoprotein B versus low‐density lipoprotein cholesterol and nonhigh‐density lipoprotein cholesterol for cardiovascular risk reduction in randomized trials. Am J Cardiol. 2012;110:1468‐1476. [DOI] [PubMed] [Google Scholar]
- 29. Robinson JG, Wang S, Smith BJ, Jacobson TA. Meta‐analysis of the relationship between non‐high‐density lipoprotein cholesterol reduction and coronary heart disease risk. J Am Coll Cardiol. 2009;53:316‐322. [DOI] [PubMed] [Google Scholar]
- 30. Ference BA, Robinson JG, Brook RD, et al. Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. N Engl J Med. 2016;375:2144‐2153. [DOI] [PubMed] [Google Scholar]
- 31. Bowman L, Hopewell JC, Chen F, et al. Effects of anacetrapib in patients with atherosclerotic vascular disease. N Engl J Med. 2017;377:1217‐1227. [DOI] [PubMed] [Google Scholar]
- 32. Lotta LA, Sharp SJ, Burgess S, et al. Association between low‐density lipoprotein cholesterol‐lowering genetic variants and risk of type 2 diabetes: a meta‐analysis. JAMA. 2016;316:1383‐1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schmidt AF, Swerdlow DI, Holmes MV, et al. LifeLines Cohort study group; UCLEB consortium; PCSK9 genetic variants and risk of type 2 diabetes: a mendelian randomisation study. Lancet Diabetes Endocrinol. 2017;5:97‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blom DJ, Koren MJ, Roth E, et al. Evaluation of the efficacy, safety and glycaemic effects of evolocumab (AMG 145) in hypercholesterolaemic patients stratified by glycaemic status and metabolic syndrome. Diabetes Obes Metab. 2017;19:98‐107. [DOI] [PubMed] [Google Scholar]
- 35. Colhoun HM, Ginsberg HN, Robinson JG, et al. No effect of PCSK9 inhibitor alirocumab on the incidence of diabetes in a pooled analysis from 10 ODYSSEY phase 3 studies. Eur Heart J. 2016;37:2981‐2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roth EM, Goldberg AC, Catapano AL, et al. Antidrug antibodies in patients treated with alirocumab. N Engl J Med. 2017;376:1589‐1590. [DOI] [PubMed] [Google Scholar]
- 37. Chan AW, Tetzlaff JM, Gotzsche PC, et al. SPIRIT 2013 explanation and elaboration: guidance for protocols of clinical trials. BMJ. 2013;346:e7586. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supplementary Appendix.
Table S1. UC dose details (randomized population).
Table S2. Baseline characteristics in the alirocumab vs intent‐to‐prescribe fenofibrate, ezetimibe, omega‐3 fatty acids and no LLT study cohorts (randomized population).
Table S3. Number of glucose‐lowering treatments over time (ITT analysis).
Table S4. TEAEs, adverse events of special interest and safety laboratory values (potentially clinically significant abnormality values; safety analysis).
Table S5. Treatment‐emergent adverse events by preferred term occurring in ≥2% of individuals in either group (safety analysis).
Figure S1. Percentage change from baseline in levels of A, non‐HDL‐C and B, LDL‐C (determined by beta‐quantification) over time in individuals receiving alirocumab 75/150 mg Q2W vs usual care (ITT analysis)
Figure S2. Primary and selected key secondary efficacy endpoints at Week 12 for A, the overall study population; B, fenofibrate; C, ezetimibe; D, omega‐3 fatty acids and E, no LLT (ITT analysis; as‐planned study cohorts).
Figure S3. Percent change from baseline in non‐HDL‐C at Week 24: Subgroup analysis according to baseline A, demographic characteristics; B, statin therapy and C, lipids (ITT analysis).
Figure S4. Mean A, A1C and B, FPG levels over time in individuals receiving alirocumab 75/150 mg Q2W versus usual care (ITT analysis).