

Key Points
Single-cell transcriptional landscape of 44 802 hematopoietic stem/progenitor cells defines entry points to 8 different blood lineages.
Comparison with 13 815 c-Kit mutant cells identifies pleiotropic changes in cell type abundance and underlying molecular profiles.
Abstract
Hematopoietic stem and progenitor cells (HSPCs) maintain the adult blood system, and their dysregulation causes a multitude of diseases. However, the differentiation journeys toward specific hematopoietic lineages remain ill defined, and system-wide disease interpretation remains challenging. Here, we have profiled 44 802 mouse bone marrow HSPCs using single-cell RNA sequencing to provide a comprehensive transcriptional landscape with entry points to 8 different blood lineages (lymphoid, megakaryocyte, erythroid, neutrophil, monocyte, eosinophil, mast cell, and basophil progenitors). We identified a common basophil/mast cell bone marrow progenitor and characterized its molecular profile at the single-cell level. Transcriptional profiling of 13 815 HSPCs from the c-Kit mutant (W41/W41) mouse model revealed the absence of a distinct mast cell lineage entry point, together with global shifts in cell type abundance. Proliferative defects were accompanied by reduced Myc expression. Potential compensatory processes included upregulation of the integrated stress response pathway and downregulation of proapoptotic gene expression in erythroid progenitors, thus providing a template of how large-scale single-cell transcriptomic studies can bridge between molecular phenotypes and quantitative population changes.
Visual Abstract
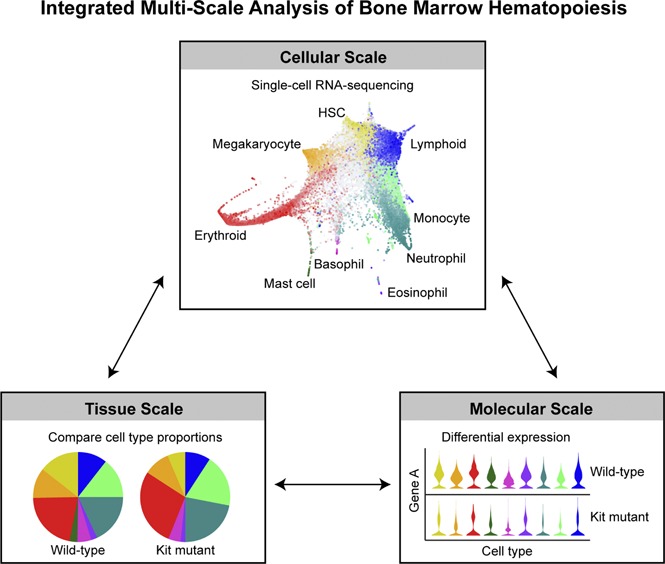
Introduction
The generation of mature blood lineages from hematopoietic stem cells (HSCs) has long served as a paradigm for the wider field of stem cell biology.1 Through a combination of advanced purification protocols and functional validation assays, high-purity HSC and progenitor populations have been defined. Classically, these populations have been considered as discrete steps within an ordered and hierarchical branching process. However, introduction of additional surface marker combinations indicated that there are likely multiple routes that can lead to functionally equivalent myeloid progenitors.2 Moreover, single-cell profiling in the mouse,3 single-cell functional assays in the human system,4 and transposon tracking during unperturbed hematopoiesis5 all emphasized that many single cells within traditionally defined multipotent populations may already be fated toward just one lineage.
Single-cell expression profiling has provided new insights into hematopoietic regulatory networks,6,7 cellular states associated with cell fate decision making,8,9 the cellular heterogeneity of stem and progenitor populations,3,10 and the recognition that transcriptional changes associated with early lineage diversification may be of a continuous nature.11 When performed at a large enough scale, single-cell expression snapshots can be used to reconstruct entire transcriptional landscapes of a given differentiation process, with multiple computational methods now in place to recover complex branching within such differentiation landscapes.12,13 However, since HSCs as well as some of the progenitor populations are exceedingly rare, systematic application of such approaches to the hematopoietic system will be most powerful once tens of thousands of single-cell transcriptomes have been processed.
Here, we report the generation and analysis of over 40 000 single-cell transcriptomes covering the hematopoietic stem/progenitor (HSPC) compartment from mouse bone marrow. A transcriptional landscape representation distinguishes entry points for 8 distinct mature lineages, with all transcriptomic data readily accessible through a user-friendly web interface. We resolve the early diversification between mast cell and basophil progenitors, and we go on to identify and validate a common progenitor cell for these 2 lineages within mouse bone marrow. We demonstrate that bipotent basophil-mast cell progenitor activity is present in the bone marrow of c-Kit mutant W41/W41 mice, despite severe mast cell deficiencies in the periphery. Single-cell expression profiling of over 13 000 HSPCs from W41/W41 bone marrow reveals a number of quantitative shifts and underlying gene expression changes in the stem cell, myeloid, and erythroid compartments, as well as the absence of cells with a transcriptome characteristic for the entry point into mast cell differentiation, thus highlighting the broad relevance of our new reference dataset for the unbiased interpretation of mutant phenotypes.
Materials and methods
For detailed materials and methods, see supplemental Methods (available on the Blood Web site).
Cell isolation
The femora, tibiae, and ilia of C57BL/6 or W41/W41 mice were crushed, the bone marrow cells were released and the red blood cells were lysed with ammonium chloride solution (STEMCELL Technologies, Vancouver, Canada). Antibodies used for isolation are listed in supplemental Methods. Cells were sorted using an Influx cell sorter (BD Biosciences, San Jose, CA). For Smart-Seq2, cells were sorted into lysis buffer and processed as described previously.10,14 For 10x Chromium (10x Genomics, Pleasanton, CA) experiments, cells were sorted and processed according to manufacturer’s protocol. The data were deposited in National Center for Biotechnology Information’s Gene Expression Omnibus (accession numbers GSE106973 and GSE107727, respectively). For in vitro culture assays of hematopoietic progenitors, cells were sorted twice and cultured. The single-cell precision mode was used for all clonogenic assays to minimize the probability of plating doublets.
In vitro culture of primary hematopoietic progenitor cells
Cells were cultured in Iscove modified Dulbecco medium (Sigma-Aldrich, St Louis, MO) with 20% heat-inactivated fetal calf serum (Sigma-Aldrich), 100 U/mL penicillin (Sigma-Aldrich), 0.1 mg/mL streptomycin (Sigma-Aldrich), 2 mM l-glutamine (Sigma-Aldrich), and 0.1 μM 2-mercaptoethanol (Thermo Fisher Scientific, Waltham, MA). The medium was supplemented with 20 ng/mL interleukin-3 (IL-3), 50 ng/mL IL-5, 50 ng/mL IL-9, 10 ng/mL granulocyte-macrophage colony-stimulating factor, and 20 ng/mL stem cell factor (SCF) to analyze myeloid potential (all were recombinant mouse cytokines from Peprotech, Rocky Hill, NJ).15 Erythroid potential was analyzed by culturing cells in the presence of 2 U/mL human erythropoietin (Janssen-Cilag, High Wycombe, UK) with 20 ng/mL mouse IL-3 and 20 ng/mL mouse SCF. Both the myeloid and the erythroid conditions support basophil and mast cell differentiation.
Single cells were expanded in 72-well Terasaki plates (Greiner Bio-One, Kremsmünster, Austria), stained with fluorophore-conjugated antibodies in the wells, diluted in buffer containing 4′,6-diamidino-2-phenylindole (BD Biosciences), and analyzed with flow cytometry without washing the cells. At least 5 events in a gate were required to score single colonies positive for a cell population. Typically, wells containing at least 20 cells were analyzed.
In vitro culture of HSCs
E-SLAM HSCs (CD45+EPCR+CD48−CD150+) were isolated as described previously.16 Single E-SLAM HSCs were sorted into round bottom 96-well plates. Cells were sorted and cultured in StemSpan (STEMCELL Technologies) containing fetal calf serum, 300 ng/mL SCF (Bio-techne, Minneapolis, MN), and 20 ng/mL IL-11 (Bio-techne). Daily cell counts were performed to assess cell divisional kinetics.17
Processing of droplet-based single-cell RNA sequencing (scRNA-seq) data
Data were processed using the Scanpy Python module.18 After quality control, 44 802 wild-type (WT) cells and 13 815 W41/W41 cells were retained. Data were visualized using force-directed graphs, similar to previously described.19
Clustering analysis on WT and W41/W41 Lin−c-Kit+ (LK) cells
WT LK cells were clustered using Louvain clustering (Scanpy igraph method) with 15 nearest neighbors. To assign W41/W41 LK cells to clusters, these data were projected into the principal-component analysis space of the WT data, and nearest neighbors calculated between the 2 data sets based on Euclidean distance in the top 50 components. W41/W41 cells were assigned to the same cluster that the majority of their 15 nearest WT neighbors belonged to.
Results
Single-cell profiling reveals entry points to 8 blood lineages in the bone marrow stem/progenitor compartment
Recent technologies have allowed the capture and RNA sequencing of ever-increasing numbers of single cells.20 To obtain a comprehensive view of hematopoiesis in mouse bone marrow, we sorted cells in 2 broad gates: LK, capturing HSPCs, and Lin−Sca-1+c-Kit+ (LSK), a subset of the LK gate enriched for HSCs and more immature progenitors (Figure 1A). Sorting LSK cells separately ensured that we profiled sufficient numbers of the less abundant cell types. Droplet-based scRNA-seq was performed, resulting in 44 802 individual transcriptional profiles, with a median of over 2500 genes detected per cell (Figure 1B).
Figure 1.

Transcriptional profiling of 44 802 single hematopoietic stem and progenitor cells reveals a differentiation landscape with 8 lineage entry points. (A) Diagram indicating relative maturity of cells captured in LSK and LK sorting gates. (B) Sorting gate used to isolate LSK and LK cells based on c-Kit and Sca-1 surface expression for droplet-based scRNA-seq. Table indicates numbers of cells that passed quality control and median number of genes detected in scRNA-seq samples. (C) Force-directed graph embedding of scRNA-seq data on LSK and LK cells. Individual cells are connected to their 7 nearest neighbors based on similarities in their transcriptional profiles, and the resulting nearest-neighbor graph is used to calculate 2-dimensional coordinates. Top panel shows LSK cells highlighted in red, with LK cells in gray in the background. Similarly, the bottom panel highlights LK cells in purple, with LSK cells in gray behind. (D) Expression of marker genes plotted on the force-directed graph embedding. Gene expression is plotted on a log(normalized count + 1) scale, with gray equal to no counts and dark red representing the maximum value detected. (E) Expression of marker genes plotted on the force-directed graph embedding using a log(normalized count + 1) scale, with gray equal to no counts and dark red representing the maximum value detected. Insets in the bottom left of each panel display magnified branch of interest. Panels display genes relating to (i) eosinophil, (ii) mast cell and basophil, (iii) mast cell, and (iv) basophil lineages, respectively. Bone marrow cells were harvested simultaneously and pooled from 6 mice.
Recent work has demonstrated the power of force-directed graph layouts for their ability to separate cells from different lineages in 2-dimensional embeddings.19,21 The force-directed graph embedding on the 44 802 HSPC profiles showed a central region of earlier stem and progenitor cells mainly from the LSK gate, with the majority of more differentiated LK cells residing on several branches (Figure 1C). To locate the stem cells within our data, we calculated a score for each cell representing the combined expression of a set of genes previously shown to be enriched in functional HSCs,10 which highlighted a region populated by mainly LSK cells (supplemental Figure 1A). Plotting the expression of marker genes on the graph allowed us to define entry points for differentiation toward mature cell types (Figure 1D; supplemental Figure 1B). Expression of Procr, Fgd5, and Hoxb5 marked the location of HSCs22-24; Dntt, Flt3, and Rag2 indicated lymphoid progenitors25; and Pf4, Itga2b, and Gp1bb26 highlighted megakaryocyte progenitors. The largest branch showed high expression of erythroid genes such as Klf1, Epor, and Gata1,27 and neutrophil and monocyte progenitors were marked by expression of genes such as Elane, Gfi1, and Cebpe or Irf8, Csf1r, and Ly86, respectively.9 Expression of Prg2 and Prg3 indicated a small population of eosinophil progenitors (Figure 1Ei).9,28
The graph layout also showed additional branches with strong expression of genes such as Ms4a2 and Cpa3 (Figure 1Eii).29 Closer inspection revealed a separation of cells appearing to enter mast cell (Gzmb and Cma1 expression)29,30 and basophil lineages (Prss34 and Mcpt8 expression)29,31 (Figure 1Eiii-iv). The existence of these progenitor populations was confirmed by visualizing the data using diffusion maps32 and t-distributed stochastic neighbor embedding33 (supplemental Figure 2). We also found that force-directed graphs calculated on LSK and LK cells separately showed a more homogeneous structure for the LSK cells, with less prominent entry points to the different blood lineages, whereas the LK embedding recapitulated the structure calculated on the LSK and LK cells together (supplemental Figure 3A-B). Pairwise distances between the LSK cells were smaller than distances between LK cells, supporting the difference in heterogeneity (supplemental Figure 3C). To allow exploration of gene expression by the wider community, we created freely accessible Web sites (http://gottgens-lab.stemcells.cam.ac.uk/adultHSPC10X/; http://app.stemcells.cam.ac.uk/adultHSPC10X/).
Single-cell cultures reveal a population of bipotent basophil/mast cell progenitors in the bone marrow
Visualizations of the scRNA-seq data revealed entry points to both mast cell and basophil lineages with these branches positioned next to each other in the graph layout, yet the early steps of basophil and mast cell lineage diversification remain poorly defined. Bipotent basophil/mast cell progenitors (BMCPs) have been described in the spleen as Lin−c-Kit+ integrin β7hi CD16/32hi cells,34 but this study failed to detect integrin β7hi progenitors in the bone marrow. Here, we analyzed in excess of 2.5 million mouse bone marrow cells, which allowed us to identify a population of Lin−Sca-1−c-Kit+ integrin β7hi CD16/32hi bone marrow cells that fell outside the classic common myeloid progenitor, granulocyte-monocyte progenitor (GMP), and megakaryocyte/erythroid progenitor (MEP) gates (Figure 2A; supplemental Figure 4A). These cells had a blast-like morphology and sometimes contained a small number of scattered granules (Figure 2B). We next sorted Lin−Sca-1−c-Kit+ integrin β7hi CD16/32hi cells in bulk and cultured them in a myeloid cytokine medium that is capable of supporting the growth of eosinophils, neutrophils, mast cells, and basophils (see “Materials and methods”). The sorted cells readily formed mast cells and basophils but no (or very few) eosinophils and neutrophils, as assessed by flow cytometry and confirmed with cytochemical staining (Figure 2B-C; supplemental Figure 4B-C). We therefore designated this population as bone marrow BMCPs. GMPs and Lin−Sca-1−c-Kit+ progenitors falling outside the newly defined BMCP gate, here referred to as mixed progenitors (MPs), predominantly gave rise to neutrophils (Figure 2B-C; supplemental Figure 4B-C). Monocytes/macrophages could not be identified with our flow cytometry panel but were seen to be present using cytochemical staining (supplemental Figure 4D).
Figure 2.

Lin−Sca-1−c-Kit+integrin β7hiCD16/32hibone marrow cells constitute bipotent mast cell/basophil progenitors. Bone marrow cells from WT mice were analyzed by flow cytometry and bipotent BMCPs, MPs, and GMPs were sorted and cultured in myeloid promoting-conditions or in erythroid-promoting conditions. (A) The gating strategy of primary BMCPs, MPs, and GMPs and the experimental setup are shown. The frequency of BMCPs is indicated as percentage of Lin−Sca-1−c-Kit+ cells. (B) Sorted cells were cultured in myeloid-promoting conditions for 5 days and stained with May-Grünwald Giemsa. Photo width, 33 μm. Images were captured using the Axio Imager.Z2, Axiocam 506, and Zen software (Zeiss). (C) BMCPs, MPs, and GMPs were cultured in bulk with myeloid-promoting cytokines and analyzed by flow cytometry. Supplemental Figure 4B shows the gating strategy. Day 7 cells were analyzed without the CD49b antibody. (D) Colonies derived from single BMCPs, MPs, and GMPs cultured in myeloid-promoting conditions were analyzed with flow cytometry. Supplemental Figure 4E shows the gating strategy. (E-F) BMCPs and MEPs were sorted and cultured in erythroid-promoting conditions. The cultured cells were analyzed by flow cytometry. Bulk cultures (E) and single-cell cultures (F) are shown. Supplemental Figure 5B shows the gating strategy. Panels C and D show data pooled from 2 experiments per time point from 4 independent sorts. Panels E and F show data pooled from 2 independent experiments. The number of single-cell colonies in which the cell types were determined is indicated above each bar. Supplemental Figures 4F-H and 5C-D show the colony sizes. Bulk indicates 50 to 100 sorted cells. Bone marrow cells were pooled from 2 to 4 mice per experiment. Ba, basophil; Bl, blast; Eo, eosinophil; Ery, erythroid; MC, mast cell; N, neutrophil.
We next sorted individual cells from the newly defined bone marrow BMCP gate to initiate clonal cultures for subsequent analysis by flow cytometry. Single bone marrow BMCP cells differentiated into mast cells, basophils, and mixed mast cell/basophil colonies in 4:4:1 ratio (day 5) (Figure 2D-E). By contrast, single MPs and single GMPs predominantly formed neutrophil colonies (Figure 2D). When cultured in conditions promoting erythroid differentiation, BMCPs formed mast cells and basophils, with no detectable erythroid output (Figure 2E-F). In contrast, MEPs cultured in the same conditions readily formed erythroid cells, but not mast cells or basophils (Figure 2E-F; supplemental Figure 5A-B). Although we cannot exclude the possibility that a small minority of bipotent colonies may be the result of a sorted doublet, all our experimental protocols and procedures were designed to ensure that wells contained single cells (supplemental Figure 5E). Taken together, therefore, Lin−Sca-1−c-Kit+ integrin β7hi CD16/32hi bone marrow cells constitute a mixture of unipotent and bipotent progenitors with mast cell– and basophil-forming capacity.
BMCPs exhibit a distinct transcriptional profile that shows evidence of priming toward the mast cell and basophil lineages
To interrogate the molecular profile of the newly defined bone marrow BMCP population, we performed high-coverage scRNA-seq analysis in parallel to our single-cell culture assays (Figures 2 and 3A). GMPs were processed in parallel as an outgroup for both the culturing assays and molecular profiling. We visualized the transcriptomic data using diffusion maps,35 a dimensionality reduction technique previously applied to single-cell profiles,32 which clearly separated the BMCPs and the GMPs (Figure 3B). Unsupervised clustering separated the BMCP population into 2 clusters (Figure 3C). Genes highly expressed in BMCPs included genes found in mature mast cells and/or basophils, such as the α and β subunits of the immunoglobulin E receptor, Ms4a2 and Fcer1a (supplemental Figure 6).29 By contrast, genes specifically expressed in the GMPs included characteristic neutrophil markers such as Elane, Prtn3, and Mpo (supplemental Figure 6).
Figure 3.

scRNA-seq of bone marrow basophil/mast cell progenitors reveals transcriptional profile distinct from GMPs. (A) Schematic showing process of isolating BMCP and GMP cells for parallel single-cell sequencing and culture experiments (Figure 2). Table indicates numbers of cells that passed quality and median number of genes detected in scRNA-seq samples. (B) Diffusion map calculated on 4267 highly variable genes from scRNA-seq data colored by cell type. (C) Diffusion map colored by clusters assigned by hierarchical clustering. (D) Heatmap displaying Z-score transformed expression of 15 most significantly differentially expressed genes specific to each cluster. Genes in top rows are upregulated in cluster 1 vs cluster 2, genes in middle rows are upregulated in cluster 2 when compared with cells in clusters 1 and 3, and genes in bottom rows are upregulated in cluster 3 cells when compared with cells in clusters 1 and 2. The colored bars at the top of the heatmap indicate cluster and cell type identity of cells in the columns. (E) Expression of groups of genes from heatmap in panel D in the droplet-based scRNA-seq data. A geometric mean score of counts was calculated for each cell across the genes in a group. The color of cells indicates the value of this score, with gray being the lowest value and red the highest value. Insets in left and center panels show magnifications of regions in black boxes. DC, diffusion component.
The BMCP cells within cluster 1 displayed higher expression of genes associated with mature mast cells and basophils (Cma1, Mcpt8, and Ndst2) (Figure 3D), suggesting that these cells were more differentiated than the cluster 2 BMCPs. Of interest, retrospective analysis of the indexed sorting data revealed that cluster 1 BMCPs had higher expression of several surface markers including FcεRI and a higher side scatter, consistent with a more differentiated status (supplemental Figure 7). Visualization of the most differentially expressed genes between the clusters (Figure 3D) and in the droplet scRNA-seq landscape (Figure 3E) demonstrated that the cluster 1 gene set was most similar to the tips of the mast cell and basophil branches, whereas the cluster 2 gene set highlighted less differentiated cells. The cluster 3 gene set, which represents genes highly expressed in GMPs, mapped to the neutrophil branch. Taken together, this analysis provides the molecular profiles of the functionally defined bone marrow BMCP progenitor cells and places them into the context of the broader bone marrow HSPC transcriptional landscape.
W41/W41 mice with mutant c-Kit retain bipotent BMCPs but lack a distinct mast cell trajectory in bone marrow
The importance of c-Kit for both the maintenance of LT-HSCs and the maturation of mast cells in vivo, prompted us to investigate the W41/W41 mice, which have a V831M mutation in the Kit gene, causing impaired c-Kit kinase activity.36 To interrogate the mast cell phenotype and additional alterations of the HSPC compartment in the presence of defective Kit signaling at a global level, we therefore performed molecular profiling on LK cells from age- and sex-matched W41/W41 mice. To compare cells from different lineages, we firstly clustered the WT LK compartment and then proceeded to map W41/W41 LK cells to their closest corresponding cluster (Figure 4A-B; supplemental Figure 8A-B). Comparison of WT and W41/W41 mice showed dramatic alterations in the structure of the transcriptional landscape. Most notably, the distinct mast cell branch is absent in W41/W41 mice, in agreement with the severe mast cell deficiency observed in these mice (Figure 4C).37 The LK data from the W41/W41 mice are also included on our interactive website to simultaneously plot expression of any chosen gene in both the WT and W41/W41 landscapes.
Figure 4.

The transcriptional landscape is altered in W41/W41mice and lacks cells entering a mast cell differentiation program. (A) Cluster identity of WT LK cells. Cells were clustered into 13 clusters using Louvain clustering on the k-nearest-neighbor graph connected scRNA-seq profiles. Cluster identifiers are ordered by size, with cluster 1 containing the most cells and cluster 13 the least. (B) W41/W41 LK cells from 2 animals were sequenced and then assigned to WT clusters based on the cluster identities of their nearest WT neighbors. The color in the plot indicates which WT cluster the W41/W41 cell was mapped to. (C) The 15 nearest neighbors of W41/W41 LK cells from cluster 9 were identified in the WT LK data. Nearest neighbor score represents the number of times a WT is one of those nearest neighbors. (D) Sorting strategy used to isolate BMCPs from bone marrow of W41/W41 mice. The frequency of BMCPs is indicated as percentage of Lin−Sca-1−c-Kit+ cells. (E) BMCPs, MPs, and GMPs were cultured in 86 to 100 cell bulk pools for 5 days with myeloid-promoting cytokines and analyzed by flow cytometry. (F) Colonies derived from single BMCPs and MPs cultured in myeloid-promoting conditions were analyzed with flow cytometry. The number of single-cell colonies in which the cell types were determined is indicated above each bar. Supplemental Figure 8C shows the colony sizes. Single GMP colonies were not analyzed. The gating strategy is shown in supplemental Figure 4B,E. The data are pooled from 2 independent experiments. Pooled bone marrow cells from 2 or 3 mice were used in each experiment.
Although the molecular cluster of mast cell progenitors was absent from the HSPC landscape in W41/W41 mice, the Lin−Sca-1−c-Kit+ integrin β7hi CD16/32hi BMCP cells were present in the bone marrow of these mice (Figure 4D) and, when sorted in bulk, gave rise to mast cells and basophils in vitro (Figure 4E). Subsequent single-cell analysis yielded pure mast cell and basophil colonies as well as mixed basophil/mast cell colonies, similar to WT mice (Figures 4F and 2D), consistent with the finding that IL-3 is sufficient for mast cell generation in vitro38 and a previous report that showed that mast cell cultures can be obtained from W41/W41 bone marrow despite the mast cell deficiency of these animals.36
Global effects of impaired c-Kit signaling across the hematopoietic landscape
W41/W41 mice are mast cell deficient and exhibit mild macrocytic anemia.37,39,40 To further investigate the effects of impaired c-Kit signaling across the entire HSPC transcriptional landscape, comparison of the proportions of LK cells within each cluster was performed (Figure 5A). This analysis demonstrated that cluster 5, corresponding to the most mature erythroid progenitors, and cluster 10, containing neutrophil progenitors, showed large increases in relative size, whereas other clusters, including cluster 9 containing mast cell and basophil progenitors, relatively decreased in size. Flow cytometry analysis of LK cells using a recently described antibody panel41 that enables the isolation of progenitors at different stages of erythroid commitment confirmed the relative increase in vivo in the most mature erythroid progenitors in the W41/W41 mice (supplemental Figure 9A-B).
Figure 5.

Local and global differences in signaling programs of c-Kit mutant mice are revealed by single-cell transcriptional profiling and functional assays. (A) Log2 fold-change of the percentage of cells in a cluster in the W41/W41 WT data divided by the percentage of cells mapped to that cluster in the WT data. Left-hand and right-hand bars indicate fold-changes for samples from 2 separate mice. Fold changes are only displayed for WT clusters with >100 cells. (B) Division kinetics of single WT and W41/W41 HSCs. (C) Colony size of single WT and W41/W41 HSCs at day 10. The results in panel B are derived from 5 WT and 5 W41/W41 mice, in which at total of at least 500 single HSCs were analyzed per genotype. The results in panel C are derived from 6 WT and 7 W41/W41 mice. A total of 598 WT and 629 W41/W41 HSCs were analyzed in panel C. The frequency of HSCs that formed colonies was 94% in WT and 95% in W41/W41 mice. The means and standard error of the mean of the mice are indicated. Unpaired 2-tailed Student t tests for each time point; *P < .05, **P < .01. Clone size estimates: VS, very small (<50 cells); S, small (50-5000 cells); M, medium (5000-10 000 cells); L, large (10 000-50 000); and XL, extra large (≥50 000 cells). (D-G) Violin plots showing the distribution of selected genes in WT and corresponding W41/W41 clusters, as measured by scRNA-seq. Distributions are shown for clusters containing >100 WT cells. Colors correspond to those in Figure 4. Data from WT and W41/W41 mice were normalized independently.
Impaired c-Kit signaling is known to affect the long-term repopulation capacity of HSCs.40,42,43 Furthermore, the colony-forming unit granulocyte/erythroid/macrophage/megakaryocyte frequencies, but not colony-forming unit granulocyte/monocyte frequencies, were reduced in W41/W41 mice (supplemental Figure 9Ci),44 consistent with a reduced frequency of early HSPCs (Figure 5A). To study the impact of the reduced kinase activity on the long-term HSCs (LT-HSCs), E-SLAM HSCs were isolated and placed in in vitro culture. W41/W41 HSCs were slower to divide than WT HSCs (Figure 5B). This reduction in proliferation was even more apparent after 10 days in culture (Figure 5C). Although the frequency of small colonies produced from the WT and W41/W41 HSCs was comparable, the frequency of very small colonies was significantly increased and large colonies of 5000 cells or more were dramatically reduced or absent in the W41/ W41 cultures. E-SLAMs from W41/W41 mice gave rise to monocytes, neutrophils, and megakaryocytes, similar to WT E-SLAMs (supplemental Figure 9Cii). This first analysis of division time and colony output of ESLAM W41/W41 LT-HSCs therefore clearly showed reduced proliferation at the upper tiers of the hematopoietic tree, implicating SCF signaling both in exit from quiescence and in cell cycle transit time in LT-HSCs and immature progenitors. To investigate whether cell cycle differences could be seen in the molecular profiles captured by scRNA-seq, we scored cells by combined expression of G2/M marker genes.45 This revealed an uneven distribution of the expression of G2/M markers across the transcriptional landscape (supplemental Figure 9D-E). The G2M signature was particularly low in the intermediate erythroid population of cluster 3 (supplemental Figure 9D-G), which was seen to be much depleted in the W41/W41 mice (Figure 5A), in contrast to the erythroid clusters with higher G2M scores, which were found to be enriched.
To determine specific genes showing different behavior in W41/W41 when compared with WT cells at corresponding stages of hematopoiesis, we performed differential expression between corresponding clusters. Myc was found to be within the most significantly downregulated genes for each of the W41/W41 clusters 1 to 11 (Figure 5D; supplemental Table 1), consistent with reduced c-Kit signaling. Computation of overlaps between genes upregulated in W41/W41 clusters and gene sets from the Molecular Signatures Database Hallmark Gene Set Collection45 revealed unfolded protein response to be among the significant gene sets, suggesting a possible induction of a stress response program (HALLMARK_UNFOLDED_PROTEIN_RESPONSE; false discovery rate = 5.74 × 10−5). Genes in this set showing significant upregulation across multiple clusters included Atf4 (Figure 5E), Psat1, Mthfd2, and Imp3 (supplemental Figure 9H).
While previous reports have shown that the W41/W41 HSPCs are defective when subjected to both in vivo and in vitro assays,42-44,46 the mutant mice have relatively normal steady-state hematopoiesis apart from mild anemia. No differences in the distribution of expression of cytokine receptors such as Epor (Figure 5F) could be seen, suggesting that in more lineage-restricted cells, the dependency upon c-Kit signaling is diminished and the lineage-specific receptors are able to compensate and maintain an almost normal lineage output.47,48 To identify possible additional molecular processes compensating for impaired Kit activity, differential gene expression analysis was performed on the most mature erythroid progenitor clusters. Gene set enrichment analysis of the genes most strongly downregulated in the later erythroid clusters (clusters 2 and 5) showed significant overlap with apoptosis-related genes (HALLMARK_APOPTOSIS; false discovery rate = 1.19 × 10−2). Among these were Casp3 (Figure 5G), which was specifically downregulated in the W41/ W41 later erythroid clusters (clusters 2 and 5), and Casp6, Pdcd4, and Bid (supplemental Figure 9I). This analysis therefore highlights molecular alterations that underpin the phenotypic changes across the hematopoietic system and sheds light on how the perturbed system compensates for the decline of an essential signaling pathway.
Discussion
Here, we report comprehensive single-cell expression profiling of the mouse HSPC compartment that allowed us to reconstruct a transcriptional landscape leading toward entry points into 8 distinct mature lineages. Through a combination of single-cell in vitro assays and molecular profiling, we identified and characterized a common mast cell/basophil progenitor in mouse bone marrow, thus opening up future research into the diversification of these 2 key lineages of the innate immune system. More broadly, we also show how our new comprehensive single-cell transcriptome landscape represents a highly informative template for the identification and subsequent interpretation of population alterations and molecular signatures of mutant phenotypes.
Because the whole transcriptome can be exploited in an entirely data-driven approach, single-cell genomics is widely expected to transform our ability to define cell types and cellular states. However, performing such analysis on the bone marrow with its complex mixture of both abundant and very rare cell types requires sampling of large numbers of cells. LT-HSCs for example are <1:1000 Lin−c-Kit+ cells, which is why we complemented our 22 000 Lin−c-Kit+ single-cell transcriptomes with another 23 000 LSK single-cell transcriptomes, containing at least 1% LT-HSCs. Our analysis now demonstrates that progenitors representing the entry points for mature lineages can also be exceedingly rare, since the bone marrow BMCP identified in this paper represents only 1:23 000 bone marrow cells, within the same order of magnitude as some previously defined rare multipotent progenitor cells.49 One of the major current goals in the field is to use single-cell transcriptome data for the reconstruction of differentiation trajectories by ordering single-cell transcriptomes along putative differentiation journeys.12,13,50 Our identification of the bone marrow BMCP progenitor as a rare cell type highlights the notion that large cell numbers will be required to define both the starting as well as ending points of hematopoietic differentiation trajectories within the stem/progenitor compartment.
Building on a data-driven approach to define the cellular composition of the bone marrow HSPC compartment, we also demonstrate the potential power of such unbiased analysis to define the phenotypic consequences of mutations in major regulatory pathways, such as the defect in c-Kit signaling present in the W41 mouse model. Historically, flow cytometry would have been used to assess potential alterations in population structure within the HSPC compartment. By contrast, comparative large-scale single-cell transcriptome profiling has several advantages. These include (1) analysis does not depend on a set of markers that need to be chosen a priori but instead is based on the transcriptome; (2) the whole compartment is analyzed at once, including subpopulations that may not be distinguishable by surface markers; and (3) both quantitative and qualitative alterations can be not only readily observed but also linked immediately to the underlying transcriptional differences and potential biological mechanisms.
The W41 mutation is a missense mutation in the kinase domain of the c-Kit gene36 and represents a partial loss of function. The viability of the W41/W41 mutant animals provides an attractive opportunity to investigate the pleiotropic effects across the entire HSPC compartment. Our functional analysis identifies defects right at the top of the hematopoietic hierarchy, while later progenitors maintain functionality. Since the absolute number of c-Kit+ cells is reduced in the W41 model,44 the dynamic shifts in relative abundance across the stem/progenitor compartment could be viewed as an adaptive system-wide response to maintain near-normal hematopoietic function despite the major defects at the HSC and immature progenitor levels.
Importantly, high-throughput single-cell transcriptomics provides tissue-scale information that is rooted in molecular-level gene expression data. For the interpretation of pleiotropic mutant phenotypes such as the W41/W41, this provides direct access to both the underlying molecular defects as well as the molecular nature of system-wide coping mechanisms. Our observation of reduced Myc expression is consistent with both the delay in G0 exit as well as the reduced proliferation of early progenitors. Possible coping mechanisms include the system-wide induction of the transcription factor, Atf4, which is involved in the integrated stress response51 as well as the erythroid-specific reduction of caspase-3 expression, consistent with an attempt to restore homeostasis to the compartment by promoting a “prosurvival” molecular signature.52 Mutations in c-Kit constitute driver mutations within hematopoietic malignancies, such as acute myeloid leukemia and mastocytosis.53-55 These mutations are known to confer cytokine independence and resistance to apoptosis,56 implicating a role in cell survival. The molecular, cellular, and tissue-scale insights gained here on a loss-of-function model may also shed light on the role of c-Kit in malignancies. More broadly, we demonstrate how unbiased investigation at the molecular, cellular, and tissue-scale levels has the potential to provide a system-level understanding that is rooted in molecular information, thus opening up the possibility to design new strategies aiming to “reset” diseased tissues.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Reiner Schulte, Chiara Cossetti, and Gabriela Grondys-Kotarba at the Cambridge Institute for Medical Research Flow Cytometry Core for their help with cell sorting and Caroline Oedekoven, Miriam Belmonte, Dean Pask, and Tina Hamilton for technical assistance. We would also like to thank Allon Klein for helpful discussions.
Work in the authors’ laboratory is supported by grants from Wellcome; Bloodwise; Cancer Research UK; Leukemia Lymphoma Society; National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (DK106766); and core support grants by Wellcome to the Cambridge Institute for Medical Research and Wellcome–Medical Research Council Cambridge Stem Cell Institute. D.G.K. is supported by a Bloodwise Bennett Fellowship (15008) and a European Research Council starting grant (StG-2016). F.K.H. and S.N. are recipients of a Medical Research Council Studentship. B.P.-S. is funded by the Wellcome Trust 4 Year programme in Stem Cell Biology and Medicine and the University of Cambridge. M.S. is the recipient of a Biotechnology and Biological Sciences Research Council Industrial CASE Studentship. J.S.D. is supported by a grant from the Swedish Research Council.
Footnotes
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession numbers GSE106973 and GSE107727).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.S.D., M.S., W.W.Y.L., S.N., and N.K.W. performed experiments; F.K.H. analyzed single-cell sequencing data; B.P.-S. created the interactive website; R.H. and E.D. mapped sequencing data; C.W. and S.W. provided methods for doublet removal and visualization of droplet-based scRNA-seq data; D.G.K., N.K.W., and B.G. designed and supervised the study; and J.S.D., F.K.H., N.K.W., and B.G. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Nicola K. Wilson, Department of Haematology, University of Cambridge, Cambridge Institute for Medical Research and Wellcome–Medical Research Council Cambridge Stem Cell Institute, Hills Rd, Cambridge CB2 0XY, United Kingdom; e-mail: nkw22@cam.ac.uk; and Berthold Göttgens, Department of Haematology, University of Cambridge, Cambridge Institute for Medical Research and Wellcome–Medical Research Council Cambridge Stem Cell Institute, Hills Rd, Cambridge CB2 0XY, United Kingdom; e-mail: bg200@cam.ac.uk.
References
- 1.Ema H, Morita Y, Suda T. Heterogeneity and hierarchy of hematopoietic stem cells. Exp Hematol. 2014;42(2):74-82.e2. [DOI] [PubMed] [Google Scholar]
- 2.Adolfsson J, Månsson R, Buza-Vidas N, et al. Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell. 2005;121(2):295-306. [DOI] [PubMed] [Google Scholar]
- 3.Paul F, Arkin Y, Giladi A, et al. Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell. 2015;163(7):1663-1677. [DOI] [PubMed] [Google Scholar]
- 4.Notta F, Zandi S, Takayama N, et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science. 2016;351(6269):aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun J, Ramos A, Chapman B, et al. Clonal dynamics of native haematopoiesis. Nature. 2014;514(7522):322-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moignard V, Macaulay IC, Swiers G, et al. Characterization of transcriptional networks in blood stem and progenitor cells using high-throughput single-cell gene expression analysis. Nat Cell Biol. 2013;15(4):363-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamey FK, Nestorowa S, Kinston SJ, Kent DG, Wilson NK, Göttgens B. Reconstructing blood stem cell regulatory network models from single-cell molecular profiles. Proc Natl Acad Sci USA. 2017;114(23):5822-5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pina C, Fugazza C, Tipping AJ, et al. Inferring rules of lineage commitment in haematopoiesis. Nat Cell Biol. 2012;14(3):287-294. [DOI] [PubMed] [Google Scholar]
- 9.Olsson A, Venkatasubramanian M, Chaudhri VK, et al. Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature. 2016;537(7622):698-702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilson NK, Kent DG, Buettner F, et al. Combined single-cell functional and gene expression analysis resolves heterogeneity within stem cell populations. Cell Stem Cell. 2015;16(6):712-724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Velten L, Haas SF, Raffel S, et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol. 2017;19(4):271-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qiu X, Mao Q, Tang Y, et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods. 2017;14(10):979-982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haghverdi L, Büttner M, Wolf FA, Buettner F, Theis FJ. Diffusion pseudotime robustly reconstructs lineage branching. Nat Methods. 2016;13(10):845-848. [DOI] [PubMed] [Google Scholar]
- 14.Picelli S, Faridani OR, Björklund ÅK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9(1):171-181. [DOI] [PubMed] [Google Scholar]
- 15.Drissen R, Buza-Vidas N, Woll P, et al. Distinct myeloid progenitor-differentiation pathways identified through single-cell RNA sequencing. Nat Immunol. 2016;17(6):666-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kent DG, Copley MR, Benz C, et al. Prospective isolation and molecular characterization of hematopoietic stem cells with durable self-renewal potential. Blood. 2009;113(25):6342-6350. [DOI] [PubMed] [Google Scholar]
- 17.Kent DG, Li J, Tanna H, et al. Self-renewal of single mouse hematopoietic stem cells is reduced by JAK2V617F without compromising progenitor cell expansion. PLoS Biol. 2013;11(6):e1001576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weinreb C, Wolock S, Klein A. SPRING: a kinetic interface for visualizing high dimensional single-cell expression data. Bioinformatics. 2017;34(7):1246-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng GXY, Terry JM, Belgrader P, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spitzer MH, Gherardini PF, Fragiadakis GK, et al. An interactive reference framework for modeling a dynamic immune system. Science. 2015;349(6244):1259425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balazs AB, Fabian AJ, Esmon CT, Mulligan RC. Endothelial protein C receptor (CD201) explicitly identifies hematopoietic stem cells in murine bone marrow. Blood. 2006;107(6):2317-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen JY, Miyanishi M, Wang SK, et al. Hoxb5 marks long-term haematopoietic stem cells and reveals a homogenous perivascular niche. Nature. 2016;530(7589):223-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gazit R, Mandal PK, Ebina W, et al. Fgd5 identifies hematopoietic stem cells in the murine bone marrow. J Exp Med. 2014;211(7):1315-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rothenberg EV. Transcriptional control of early T and B cell developmental choices. Annu Rev Immunol. 2014;32(1):283-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rowley JW, Oler AJ, Tolley ND, et al. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood. 2011;118(14):e101-e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dzierzak E, Philipsen S. Erythropoiesis: development and differentiation. Cold Spring Harb Perspect Med. 2013;3(4):a011601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Graaf CA, Choi J, Baldwin TM, et al. Haemopedia: an expression atlas of murine hematopoietic cells. Stem Cell Reports. 2016;7(3):571-582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dwyer DF, Barrett NA, Austen KF, Consortium TIGP; Immunological Genome Project Consortium. Expression profiling of constitutive mast cells reveals a unique identity within the immune system. Nat Immunol. 2016;17(7):878-887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pardo J, Wallich R, Ebnet K, et al. Granzyme B is expressed in mouse mast cells in vivo and in vitro and causes delayed cell death independent of perforin. Cell Death Differ. 2007;14(10):1768-1779. [DOI] [PubMed] [Google Scholar]
- 31.Ugajin T, Kojima T, Mukai K, et al. Basophils preferentially express mouse Mast Cell Protease 11 among the mast cell tryptase family in contrast to mast cells. J Leukoc Biol. 2009;86(6):1417-1425. [DOI] [PubMed] [Google Scholar]
- 32.Haghverdi L, Buettner F, Theis FJ. Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics. 2015;31(18):2989-2998. [DOI] [PubMed] [Google Scholar]
- 33.Van Der Maaten L, Hinton G. Visualizing data using t-SNE. J Mach Learn Res. 2008;9:2579-2605. [Google Scholar]
- 34.Arinobu Y, Iwasaki H, Gurish MF, et al. Developmental checkpoints of the basophil/mast cell lineages in adult murine hematopoiesis. Proc Natl Acad Sci USA. 2005;102(50):18105-18110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coifman RR, Lafon S, Lee AB, et al. Geometric diffusions as a tool for harmonic analysis and structure definition of data: diffusion maps. Proc Natl Acad Sci USA. 2005;102(21):7426-7431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nocka K, Tan JC, Chiu E, et al. Molecular bases of dominant negative and loss of function mutations at the murine c-kit/white spotting locus: W37, Wv, W41 and W. EMBO J. 1990;9(6):1805-1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ingram DA, Yang FC, Travers JB, et al. Genetic and biochemical evidence that haploinsufficiency of the Nf1 tumor suppressor gene modulates melanocyte and mast cell fates in vivo. J Exp Med. 2000;191(1):181-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eklund KK, Ghildyal N, Austen KF, Friend DS, Schiller V, Stevens RL. Mouse bone marrow-derived mast cells (mBMMC) obtained in vitro from mice that are mast cell-deficient in vivo express the same panel of granule proteases as mBMMC and serosal mast cells from their normal littermates. J Exp Med. 1994;180(1):67-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geissler EN, McFarland EC, Russell ES. Analysis of pleiotropism at the dominant white-spotting (W) locus of the house mouse: a description of ten new W alleles. Genetics. 1981;97(2):337-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geissler EN, Russell ES. Analysis of the hematopoietic effects of new dominant spotting (W) mutations of the mouse. I. Influence upon hematopoietic stem cells. Exp Hematol. 1983;11(6):452-460. [PubMed] [Google Scholar]
- 41.Tusi BK, Wolock SL, Weinreb C, et al. Population snapshots predict early haematopoietic and erythroid hierarchies. Nature. 2018;555(7694):54-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharma Y, Astle CM, Harrison DE. Heterozygous kit mutants with little or no apparent anemia exhibit large defects in overall hematopoietic stem cell function. Exp Hematol. 2007;35(2):214-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller CL, Rebel VI, Lemieux ME, Helgason CD, Lansdorp PM, Eaves CJ. Studies of W mutant mice provide evidence for alternate mechanisms capable of activating hematopoietic stem cells. Exp Hematol. 1996;24(2):185-194. [PubMed] [Google Scholar]
- 44.Lacombe J, Krosl G, Tremblay M, et al. Genetic interaction between Kit and Scl. Blood. 2013;122(7):1150-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller CL, Rebel VI, Helgason CD, Lansdorp PM, Eaves CJ. Impaired steel factor responsiveness differentially affects the detection and long-term maintenance of fetal liver hematopoietic stem cells in vivo. Blood. 1997;89(4):1214-1223. [PubMed] [Google Scholar]
- 47.Wu H, Klingmüller U, Acurio A, Hsiao JG, Lodish HF. Functional interaction of erythropoietin and stem cell factor receptors is essential for erythroid colony formation. Proc Natl Acad Sci USA. 1997;94(5):1806-1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu H, Klingmüller U, Besmer P, Lodish HF. Interaction of the erythropoietin and stem-cell-factor receptors. Nature. 1995;377(6546):242-246. [DOI] [PubMed] [Google Scholar]
- 49.Cabezas-Wallscheid N, Klimmeck D, Hansson J, et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell. 2014;15(4):507-522. [DOI] [PubMed] [Google Scholar]
- 50.Setty M, Tadmor MD, Reich-Zeliger S, et al. Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat. Biotechnol. 2016;34(6):637-645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Reports. 2016;17(10):1374-1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.B’chir W, Maurin A-C, Carraro V, et al. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41(16):7683-7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beghini A, Peterlongo P, Ripamonti CB, et al. C-kit mutations in core binding factor leukemias. Blood. 2000;95(2):726-727. [PubMed] [Google Scholar]
- 54.Nagata H, Worobec AS, Oh CK, et al. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci USA. 1995;92(23):10560-10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Longley BJJ Jr, Metcalfe DD, Tharp M, et al. Activating and dominant inactivating c-KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proc Natl Acad Sci USA. 1999;96(4):1609-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ning ZQ, Li J, Arceci RJ. Signal transducer and activator of transcription 3 activation is required for Asp(816) mutant c-Kit-mediated cytokine-independent survival and proliferation in human leukemia cells. Blood. 2001;97(11):3559-3567. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.