

Abstract
The neuronal ceroid lipofuscinoses are a class of inherited neurodegenerative diseases characterized by the accumulation of autofluorescent storage material. The most common neuronal ceroid lipofuscinosis has juvenile onset with rapid onset blindness and progressive degeneration of cognitive processes. The juvenile form is caused by mutations in the CLN3 gene, which encodes the protein CLN3. While mouse models of Cln3 deficiency show mild disease phenotypes, it is apparent from patient tissue- and cell-based studies that its loss impacts many cellular processes. Using Cln3 deficient mice, we previously described defects in mouse brain endothelial cells and blood-brain barrier (BBB) permeability. Here we expand on this to other components of the BBB and show that Cln3 deficient mice have increased astrocyte endfeet area. Interestingly, this phenotype is corrected by treatment with a commonly used GAP junction inhibitor, carbenoxolone (CBX). In addition to its action on GAP junctions, CBX has also been proposed to alter lipid microdomains. In this work, we show that CBX modifies lipid microdomains and corrects membrane fluidity alterations in Cln3 deficient endothelial cells, which in turn improves defects in endocytosis, caveolin-1 distribution at the plasma membrane, and Cdc42 activity. In further work using the NIH Library of Integrated Network-based Cellular Signatures (LINCS), we discovered other small molecules whose impact was similar to CBX in that they improved Cln3-deficient cell phenotypes. Moreover, Cln3 deficient mice treated orally with CBX exhibited recovery of impaired BBB responses and reduced auto-fluorescence. CBX and the compounds identified by LINCS, many of which have been used in humans or approved for other indications, may find therapeutic benefit in children suffering from CLN3 deficiency through mechanisms independent of their original intended use.
Keywords: Neuronal ceroid lipofuscinoses, JNCL, CLN3, Cav-1, Cdc42, Blood-brain barrier, Carbenoxolone, Membrane fluidity
1. Introduction
The neuronal ceroid lipofuscinoses (NCLs) are progressive neuro-degenerative diseases characterized by the accumulation of auto-fluorescent material in multiple cell types. The most common NCL has a juvenile onset and is commonly caused by mutations in CLN3 (referred to as CLN3 deficiency) (Williams and Mole, 2012). CLN3 deficiency generally manifests with early visual deficits followed several years later by seizures with mental and physical decline (Phillips et al., 2005). To date, only palliative care is available for this invariably fatal disease.
The CLN3 gene encodes the protein CLN3 (also known as battenin), which is a 438 amino acid hydrophobic, multi-pass transmembrane protein of unresolved function (Cotman and Staropoli, 2012; Phillips et al., 2005). Work in model systems over the last two decades suggest that CLN3 impacts multiple cellular functions including lysosomal pH (Golabek et al., 2000; Holopainen et al., 2001; Pearce et al., 1999; Pearce and Sherman, 1998), vesicular trafficking (Cao et al., 2006; Codlin and Mole, 2009; Fossale et al., 2004; Kama et al., 2011; Metcalf et al., 2008), palmitoyl desaturase activity (Narayan et al., 2006), endocytosis (Codlin et al., 2008; Fossale et al., 2004; Luiro et al., 2001; Luiro et al., 2004; Schultz et al., 2014; Tecedor et al., 2013; Vidal-Donet et al., 2013), and membrane microdomain formation or stability (Tecedor et al., 2013).
Using a Cln3 reporter mouse we previously showed that Cln3 is highly expressed in brain vasculature endothelial cells (Eliason et al., 2007). Brain endothelial cells are a component of the blood-brain barrier (BBB) and are vital for the maintenance of neuronal health and CNS function. In some neurodegenerative diseases, such as Alzheimer’s disease, endothelial cell dysfunction (Sagare et al., 2013) at the BBB is a critical element in disease pathogenesis. Intriguingly, CLN3 disease patients and mouse models display autoantibodies against CNS antigens in peripheral blood. Concurrently, high levels of storage material accumulate in endothelial cells (Lamb et al., 2006; Lim et al., 2007). Additionally, Cln3-null mice have abnormal BBB responses to hypotonic stress in vivo (Tecedor et al., 2013). Whether other components of the BBB, for example astrocytes whose endfeet are a constituent of the BBB, are affected in CLN3 deficiency is not known.
Here, we report that Cln3-null mice have increased astrocyte endfeet area in the BBB. Increased astrocyte endfeet size could reflect defects in astrocyte communication, which occurs at GAP junctions. Indeed prior work in slice culture models have shown excessive astrocytic GAP junction connectivity (Burkovetskaya et al., 2014). Interestingly, we found that oral treatment of Cln3−/− mice with the GAP junction inhibitor Carbenoxolone (CBX) significantly corrects astrocyte endfeet area. CBX exerts its impact on GAP junction/hemichannel activity upon intercalation into the plasma membrane, which likely alters membrane microfluidity (Davidson and Baumgarten, 1988; Goldberg et al., 1996; Tovar et al., 2009). Interestingly, we previously described altered plasma membrane fluidity in endothelial cells derived from Cln3−/− mice (Tecedor et al., 2013).
We next tested the hypothesis that CBX improves Cln3 phenotypes in vitro and in vivo by improving membrane fluidity defects. Our results show that, in vitro, CBX corrects previously reported lipid microdomain changes, caveolar and fluid phase endocytosis defects, and other downstream cellular pathologies reported in Cln3−/− mice cells including altered Cdc42-GTP levels. In vivo, CBX corrected altered BBB responses in Cln3 deficient mice in addition to improving astrocyte endfeet phenotypes. Also, we took advantage of the NIH Library of Integrated Network-based Cellular Signatures (LINCS) database to identify other compounds that behave similarly to CBX in model systems. Overall our data present evidence supporting further testing of 7 compounds for potential treatment for CLN3 deficiency.
2. Materials and methods
2.1. Mice
All animal experiments were approved by the University of Iowa animal care and use committee and conducted in accordance with institutional and federal guidelines. Here we used WT, Cln3lacZ/+ (Cln3−/+), and Cln3lacZ/lacZ (Cln3−/−) mice (Eliason et al., 2007) backcrossed to the C57BL/6J background. Mixes of male and female mice were used for experiments.
2.2. MBEC cell lines
Due to the low yield of primary MBECs, we used previously described immortalized MBEC from Cln3−/− mice (Tecedor et al., 2013). Cln3 expression was stably restored in Cln3−/− MBEC using a lentivirus vector as described previously (Tecedor et al., 2013), creating the Cln3R, sister cell line. Cln3R and Cln3−/− MBECs behave similarly to primary wildtype and Cln3−/− MBECs (Schultz et al., 2014; Tecedor et al., 2013). Immortalized MBECs are positive for Von Willebrand factor and ZO-1 (Tecedor et al., 2013).
2.3. FRAP
2.3.1. Membrane fluidity
Cells were incubated with Alexa 488-CTB (Invitrogen) in the dark for 30 min on ice. Cells were briefly rinsed with ice-cold media and placed on ice in the dark until use. Imaging and photobleaching were conducted on a Zeiss LSM510 microscope (Carl Zeiss) equipped with an Ar 488 nm laser. 60 iterations at 100% of 488 nm excitation was used to photobleach a small portion (31 px diameter) of the membrane. Adjacent non photobleached membrane areas of the same size were used as a control for sequential bleaching as previously described (Tecedor et al., 2013).
2.3.2. GAP junction connectivity
MBECs were incubated with 5 μM calcein-AM (Molecular probes) for 30 min at 37 °C in the dark, then rinsed briefly with cold cell culture media. Whole cells were photobleached as described above in the Membrane fluidity section. Post-bleached images were captured every 5 s. Neighboring cells were used as controls for sequential bleaching due to the imaging process. FRAP was quantified as described previously (Kenworthy, 2007). Results from three independent experiments were analyzed together (n = 76, 42, 52, 52 for Cln3−/−, Cln3R, Cln3−/−+CBX, Cln3R+CBX respectively). Two-way ANOVA resulted with significant interaction. Data was analyzed for each time point by one-way ANOVA followed by Tukey’s post-hoc analysis.
2.4. Cholesterol quantification
Detergent-free carbonate extraction and discontinuous sucrose density centrifugation was used to enrich plasma membrane and separate lipid microdomain fractions as before (Tecedor et al., 2013). Cholesterol was quantified by amplex red assay (Life Technologies). Briefly, cell fractions were diluted in reaction buffer and incubated with the amplex red reagent per the manufactures instructions. Cholesterol intensity was quantified for each membrane fraction with a monochrome microplate reader Safire2 (Tecan Group Ltd. Mannedorf, Switzerland). Results were normalized to total amount of quantified cholesterol per the manufacturer’s instructions.
2.5. Cdc42-GTP analysis
The Cdc42 G-Lisa® from Cytoskeleton Inc. was utilized for measurement of Cdc42-GTP levels. Briefly, 0.8 mg/ml of snap frozen lysate was incubated in the ELISA plate and the manufactures instructions were followed for detection and analysis as previously described (Schultz et al., 2014). Data were normalized to the protein standard and significance was tested by one-way ANOVA with Tukey post-hoc analysis.
2.6. Fluid-phase endocytosis
Subconfluent cells were incubated with 0.5 mg/ml Alexa 488 10,000 MW dextran (Invitrogen) for 20 min. Then cells were briefly washed 3× with 37°C PBS to remove unbound dextran and immediately fixed with 4% PFA at 37 °C. Membrane impermeable 200 mM Red-40 was added to quench extra cellular dextran signal allowing quantification of only internalized dextran (Schultz et al., 2014). Cells were immediately imaged on an Olympus IX81 microscope and fluorescent intensity was calculated by ImageJ. Significance was determined by a one-way ANOVA with Tukey post-hoc (Fig. 5C) analysis or one-way ANOVA with Holm-Sidak’s multiple comparisons post-hoc (Fig. 6B) comparing each treatment with the Cln3−/− MBEC + vehicle group.
Fig. 5.
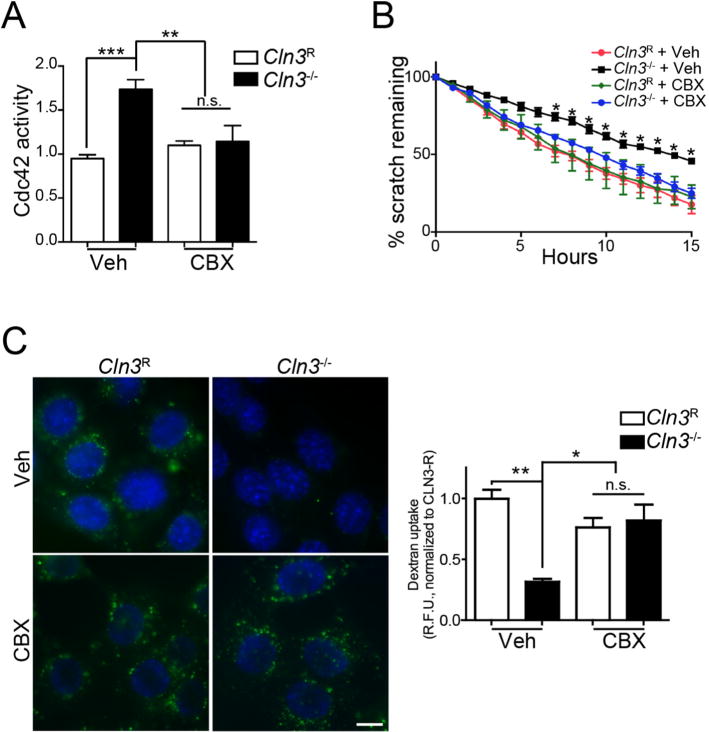
Correction of Cdc42-dependent defects in Cln3−/− MBECs with CBX. A, MBECs were treated for 2 h with CBX (50 μM) or vehicle and Cdc42-GTP levels were measured. Data are mean ± SEM of 5 independent experiments, one-way ANOVA with Tukey’s multiple comparison correction. B, MBECs were grown to confluence, a scratch wound made, and CBX (3.5 μM) or vehicle added to cells. Cell migration was imaged overnight and quantified by T-Scratch software. Data are mean ± s.e.m. from 4 independent experiments evaluated by one-way ANOVA with Tukey’s post-hoc correction. C, MBECs were treated with CBX (25 μM) or vehicle and endocytic uptake assessed by A488-Dextran uptake (green). Hoechst 33342 was used to label nuclei (blue). Non-internalized extracellular dextran was quenched by Red-40 and epifluorescent images were quantified by ImageJ. Data are mean ± s.e.m. from ~70 cells from 3 independent experiments, and evaluated one-way ANOVA with Tukey’s post-hoc. For all panels *, p < 0.05, **, p < 0.01, ***, p < 0.001, n.s. = not significant. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 6.
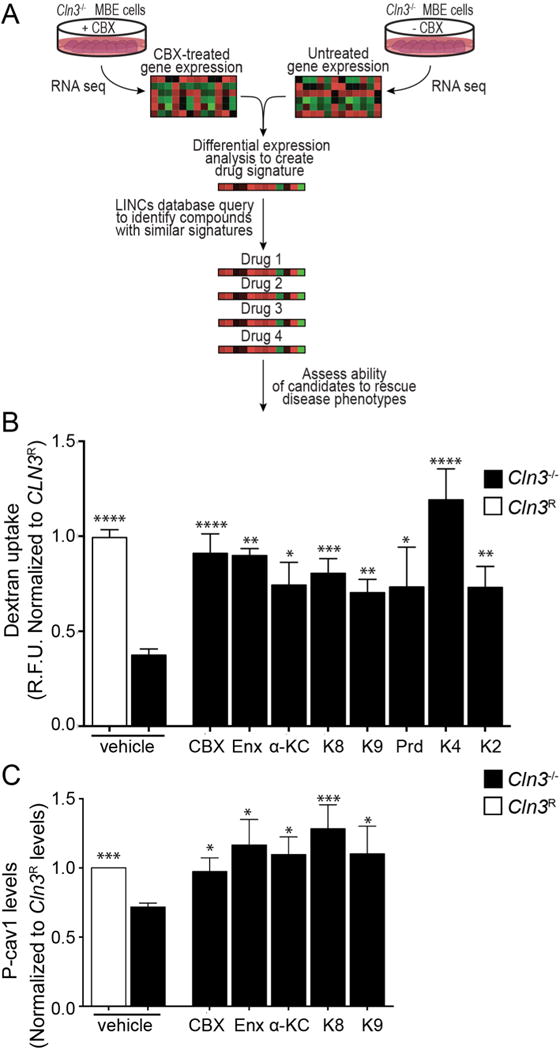
Multidrug screening. A, LINCs strategy to identify compounds with signatures similar to CBX. B, Fluid-phase endocytosis was analyzed by Alexa 488-dextran uptake (as in Fig. 5C). Data are mean ± s.e.m. from 4 to 8 independent experiments for each small molecule, one-way ANOVA with Holm-Sidak’s multiple comparisons post-hoc for analyzing differences of vehicle treated Cln3R MBECs with respect to all other groups. C, Phosphorylated Cav-1(P-cav1) levels in Cln3R and Cln3−/− MBECs after small molecule treatment. Data represent mean ± s.e.m. from six independent experiments, non-parametric Mann-Whitney test with Bonferroni‘s multiple comparison correction for vehicle treated Cln3−/− MBECs with respect to all other groups. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Carbenoxolone (CBX), Enoxolone (Enx), Prednisolone (Prd), α-ketocholesterol (α-KC), BRD-K95814727 (K8), BRD-K30446755 (K4), BRD-K33258928 (K2), BRD-K95985487 (K9).
2.7. Scratch assay
MBECs were grown to confluence and a pipette tip was used to create a wound. Cells were briefly washed with PBS and cell culture media was added for the remainder of the experiment. Cells were imaged every hour on an automated live cell Olympus IX81 microscope overnight. T-Scratch software (Geback et al., 2009) was used to quantify wound closure. Significance was assessed by 2-way ANOVA with Bonferroni post-hoc test (Schultz et al., 2014).
2.8. Immunofluorescence
Sub-confluent MBECs were briefly washed and fixed in 4% PFA, permeabilized with 0.01% Trition-X-100, washed three times with PBS, and blocked for 1 h with 10% goat serum. Cells were incubated overnight with Cav-1 antibody (1:200) Abcam, washed three times, and incubated with goat anti-rabbit Alexa 488 secondary for 45 min in the dark. After mounting with Vectashield, cells were imaged on a Zeiss 710 confocal microscope.
2.9. Protein analysis
Three confluent 160-mm-diameter dishes of MBECs were harvested and plasma membranes were isolated as previously described (Yao et al., 2009). Protein concentrations were determined by Bio-Rad Dc-Protein assay (Bio-Rad). Samples were mixed with Bio-Rad XT sample buffer and reducing agent, run on Criterion™ XT 4–12% Bis-Tris Gels (Bio-Rad), and transferred onto PVDF membranes. Immunodetection was performed with (1:100) rabbit anti-connexin 43 antibody (Invitrogen), (1:500) mouse anti phospho-caveolin1 (pY14) (BD Transduction Laboratories), (1:5000) rabbit anti-β-catenin (Abcam), (1:1000) rabbit anti-Cav-1 (Abcam), or (1:1000) mouse-anti-transferrin receptor (Life Technology) followed by HRP-coupled secondary antibodies (Jackson ImmunoResearch), and developed with ECL-Plus (GE Healthcare). Protein bands were quantified by densitometry using Image Lab 5.1 (Bio-Rad) software. Plasma membrane connexin43 levels were normalized to plasma membrane transferrin receptor. This value was normalized to total β-catenin as a housekeeping reference protein.
2.10. BBB hypotonic treatment
Mice were anesthetized and perfused with a Gilson minipulse 3 pump (set to a speed of 2). We sequentially perfused i) 175 μl Alexafluor 488 WGA (1 mg/ml) (preloaded into the needle); ii) Hoechst 33258 (100 μg/ml in 1 mM CaCl2 hypotonic solution), injected for 2 min and 15 s; iii) saline, perfused for 30 s; iv) 4% PFA, perfused for 2 min. Each solution was passed through an IV drip tube to create continuous flow and pressure. After dissection and removal, brains were post-fixed in 4% PFA for 3 h, embedded in agar and set on ice to solidify. Solidified blocks were trimmed and further post-fixed in 4% PFA at 4 °C overnight. 50-μm vibratome sections were mounted onto slides, cover-slipped with Fluorogel and immediately imaged on a Leica Leitz DMRBE microscope or Zeiss LSM710 confocal microscope. Images were captured in the green (WGA), blue (Hoechst), and red (autofluorescence) channels and channels combined in Adobe Photoshop.
2.11. Astrocyte endfeet quantification
3 mice per group were perfused with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer using a Gilson minipulse 3 at ~1.3 ml/min flow ratio for maintaining BBB integrity. Motor cortex samples of 1mm2 were collected, and 50 μm vibrotome sections were obtained. Samples were then postfixed in 1% osmium tetroxide for 1 h., stained with uranyl acetate, dehydrated in a graded ethanol series, and embedded in Epon resin. Ultrathin 70 nm sections were cut on a Leica UC6 ultra-microtome (Leica Microsystems) and 12–23 cortical capillaries per animal analyzed with a 1230 JEOL-JEM transmission EM (Jeol USA) or a Zeiss Libra 120 transmission EM. Microvasculature was recognized by detecting the basal lamina which is localized between endothelial cells and astrocytic endfeet. Only long processes surrounding the basal lamina were measured to exclude neuronal terminals. The area surrounded by astrocytic endfeet plasma membrane was measured using ImageJ software (NIH). Endfeet data did not follow a normal distribution so it was normalized by lognormal transformation. Statistical significance of the normalized data was tested by two-tailed t-test.
2.12. Autofluorescence
Brains were collected as explained above in the BBB hypotonic treatment method. From each brain, 20 X images were taken from four different sections of medial cortex. Autofluorescence images were collected in the red channel and intensity calculated using ImageJ. Significance was calculated by a one-way ANOVA with Tukey post-hoc analysis.
2.13. Drug treatment
Carbenoxolone disodium salt (Abcam) was dissolved to 0.5 M in ethanol:water (1:1), and further diluted in cell culture media to obtain the following final concentrations and treatment durations: 50 μM for 2 h. (G-LISA), 25 μM for (30 min fluid-phase endocytosis; 2 h. FRAP, cholesterol, cav-1 trafficking), and 3 μM overnight (scratch assay). In the multidrug screening experiment, CBX was dissolved with DMSO and diluted in cell culture media to a final concentration of 3 μM for a 24 h. treatment. For in vivo experiments, a 30 mg/ml stock solution of carbenoxolone was prepared in saline (vehicle) and diluted to 2.0 mg/ml for gavage. Mice were administered 20 mg/kg CBX or vehicle by gavage, given at the same time each morning for 14 days.
Enoxenolone (Enx, Abcam) was dissolved in ethanol:water (1:1) and diluted in cell culture media to a final concentration of 3 μM.
Drugs tested in the multidrug screening test were dissolved in DMSO and then diluted in cell culture media for 24 h. treatment. Experimental concentrations matched those recommended on the LINCS database: Prednisolone (Prd, Sigma-Aldrich) and α-ketocholesterol (α-KC, Sigma-Aldrich) at 10 μM, BRD-K95814727 (K8, Broad Institute), -K30446755 (K4, Broad Institute), -K33258928 (K2, Broad Institute) at 5 μM, and BRD-K95985487 (K9, Broad Institute) at 0.5 μM.
2.14. LINCS database analyses
Cln3−/− MBE cells were treated with 3 μM CBX or no-treatment for 18 h., following which RNA was harvested from 6 wells per condition using the mirVana™ total RNA isolation kit procedure. Total RNA was tested for quality on an Agilent Model 2100 Bioanalyzer (Agilent Technologies), and gene expression profiles were generated using the MouseRef-8 V2 BeadChip Kit (Illumina).
Differential gene expression analysis was performed using the Partek® Genomics Suite®, Gene Expression workflow to identify the most significant up- and down-regulated genes. LINCS queries are based on a set of 1000 landmark genes which are representative of the whole transcriptome. To acquire the required number of genes we used a p value cutoff < 0.02 and > 1.2 gave roughly 200 genes in each category, which is required for reaching 150–200 up or down regulated landmark genes for the query. The fold changes of the genes were then uploaded onto the Apps.lincscloud.org/start (now https://clue.io/) to query the LINCS database.
Every reference signature in the database is compared with the query signatures and given a score termed the “connectivity score” based on the extent of similarity between the two. Scores range from +1 meaning higher similarity, to 0 meaning no similarity, to −1 meaning opposite similarity. We removed all non-compounds and negative connectivity scores, and sorted the remaining compounds by connectivity score alone. We prioritized the top 70 compounds with the highest connectivity score for further analysis.
2.15. Experimental design and statistical analysis
Data are presented as mean ± s.e.m. with p values, and sample size (n). Prism v6 (GraphPad Software) was used for statistical analyses. Comparisons between different datasets were made depending on data distribution tested by the D’Agostino normality test. Parametric tests were used for normally distributed data (two-tailed t-test for comparing two groups and ANOVA for comparing three or more groups). A two-way ANOVA was used to determine significance in experiments with multiple time points and groups. When a significant interaction was found between factors by two-way ANOVA, one-way ANOVA were used to determine significance between experimental groups. When the normality assumption was violated, nonparametric tests were performed (Mann-Whitney test for two groups and Kruskal-Wallis for three or more groups) unless the n was high enough to allow the transformation of the data to a normal distribution using lognormal function. Differences were considered statistically significant at p < 0.05. Different levels of significance between groups were denoted as * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
3. Results
3.1. An astrocyte endfeet phenotype in Cln3-null mice is corrected by CBX
At the BBB, astrocytes and brain endothelial cells are juxtaposed at astrocyte endfeet, which together serve to regulate BBB maintenance and function (Abbott et al., 2006). As we previously described structural alterations in mouse brain endothelia in vivo (Tecedor et al., 2013), we next tested cerebral sections for structural changes in Cln3−/− mice astrocytes. Transmission electron microscopy (TEM) analysis revealed astrocyte endfeet area in Cln3−/− mice was significantly increased at 0.2661 μm2 (± 0.03003) compared to 0.0003332 μm2 (± 0.02956) in Cln3+/+ littermates (Fig. 1A). Given prior work showing impaired GAP junction connectivity in JNCL astrocytes (Burkovetskaya et al., 2014) we subsequently tested if treating Cln3−/− mice with the GAP junction inhibitor carbenoxolone (CBX) could impact this phenotype. CBX was administered daily to Cln3−/− mice or normal littermates for two weeks by oral gavage, and endfeet area assessed as before by TEM. CBX reduced astrocyte endfeet area in Cln3−/− mice to 0.1291 μm2 (± 0.03120), which is similar to that found in control littermates (0.01833 ± 0.03310 μm2; Fig. 1B).
Fig. 1.
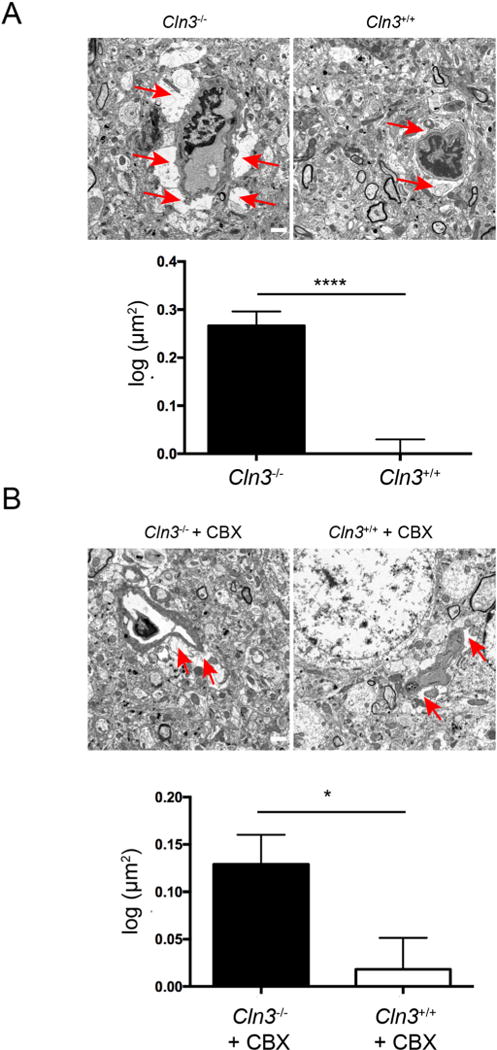
Astrocyte endfeet in Cln3−/− mice are enlarged. A, Astrocytic endfeet in the somatosensory cortex from brain sections harvested from Cln3+/+ or Cln3−/− mice. B Astrocyte endfeet in Cln3−/− mice can be corrected by CBX treatment. CBX was administered for two weeks by oral gavage and brain sections harvested and evaluated by TEM. For both (A) and (B) the red arrows indicate astrocyte endfeet. Scale bar 1 μm. Astrocytic endfeet area was quantified with ImageJ, n = 342, 327, 436, 398 for Cln3−/−, Cln3+/+, Cln3−/− + CBX, and Cln3+/+ + CBX respectively. Results were transformed to normal distribution by log normal function, and evaluated by t-test. *, p < 0.05, ****, p < 0.0001. Bars show mean ± s.e.m. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2. CBX modifies microdomain fluidity
Gap junctions are produced by coupling the plasma membrane-resident hemichannels of adjacent cells allowing the exchange of cytoplasmic molecules and electrical signals. Due to the crosstalk between astrocytes and brain endothelial cells, we investigated in vitro GAP junction activity in brain endothelial cells derived from Cln3−/− mice. We used our previously established mouse brain endothelial cell lines (MBECs) for this work; this includes a Cln3-null line (Cln3−/− MBECs), and a line in which Cln3 expression was restored (Cln3R MBECs). These cells behave similarly to primary MBECs from null or WT mice, respectively (Schultz et al., 2014; Tecedor et al., 2013). To study the impact of Cln3 deficiency on GAP junction connectivity, MBEC mono-layer cultures were incubated with calcein-AM, which moves between cells using GAP junctions and the cell-cell transfer rate quantified using whole cell fluorescence recovery after photobleaching (FRAP). For FRAP, the fluorescent calcein-AM in one cell is photobleached and then sequential images taken to determine the rate by which the dye enters the photobleached cell from neighboring cells. FRAP analysis revealed that Cln3−/− cells have a faster calcein-AM fluorescence recovery (one-way ANOVA, F(3,112) = 30.22, p < 0.0001) relative to Cln3R cells, indicating increased flow of calcium-AM through GAP junctions with neighboring cells (Fig. 2A). As expected, treating cells with the reported GAP junction inhibitor CBX blunted the rate of fluorescence recovery in the photobleached cells (Fig. 2A). Connexin 43 (Cx43) levels, which is a main component of GAP junctions in the brain, were similar between Cln3−/− and Cln3R MBECs (Fig. 2B; Kruskal-Wallis, p = 0.1085). This indicates that increased GAP junction activity is not caused by more Cx43 protein and suggests another mechanism for increased calcium-AM flow.
Fig. 2.
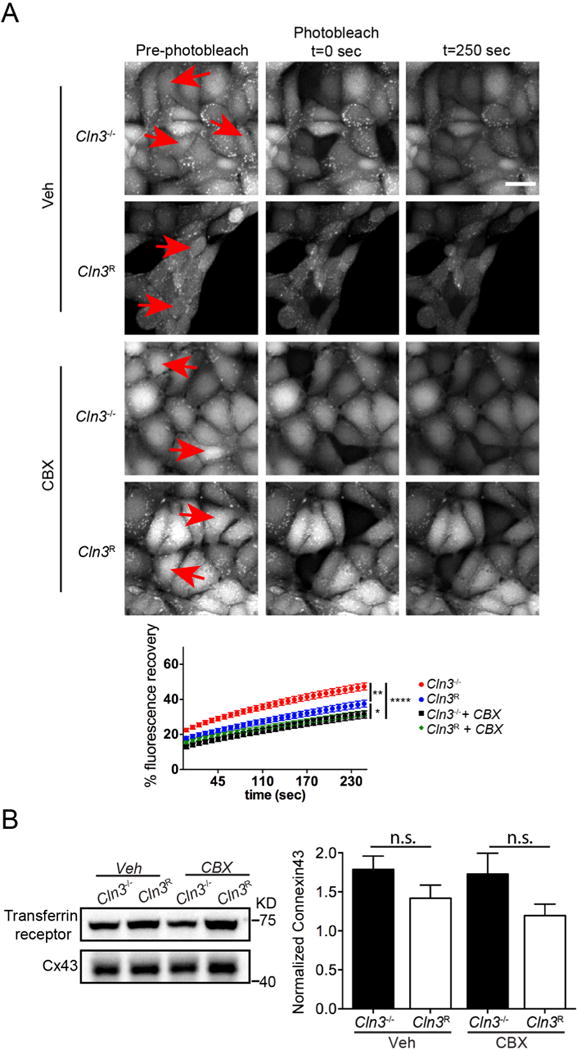
CBX corrects GAP junction communication in MBECs. A, After calcium-AM was loaded into cells, whole cell FRAP analysis was used to investigate GAP junction communication in Cln3R and Cln3−/− MBECs. Cells were treated for 2 h with vehicle or 25 μM CBX. ImageJ was used to quantify FRAP over time. Scale bar 20 μm. Data show mean ± s.e.m. from n = 76, 42, 52, 53 Cln3−/−, Cln3R, Cln3−/− + CBX, Cln3R + CBX cells respectively. Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparison test. Relative to Cln3R, Cln3−/− MBECs had significantly more calcium-AM recovery at all-time points. * p < 0.05, ** p < 0.01, **** p < 0.0001. B, Plasma membrane enriched fractions were analyzed by western blot for Connexin 43 levels. Bars show mean ± s.e.m. of six independent experiments. Analysis with Kruskal-Wallis test showed no significant differences among experimental groups.
Although CBX is commonly used as a GAP junction inhibitor, data also support the notion that it directly modifies cell membranes. Administration of CBX to purified lysosomes stabilizes membranes (Porta et al., 1986; Symons et al., 1978), and incubation of enveloped viral particles with CBX reduces adsorption (Dargan and Subak-Sharpe, 1986) and viral release (Dargan and Subak-Sharpe, 1986). As GAP junctions are associated with cholesterol-rich domains (Schubert et al., 2002) and regulated by membrane fluidity (Bastiaanse et al., 1993), we tested if CBX influences lipid microdomain dynamics by evaluating GM1 ganglioside movement in the plasma membrane. GM1 gangliosides were labeled in living MBECs with Alexa-488-cholera toxin subunit B (A488-CTB) and a high intensity laser was used to photobleach a A488-CTB-positive region of the plasma membrane. Sequential images were captured to assess the rate of A488-CTB movement into the photobleached area. Consistent with our previous report (Tecedor et al., 2013), Cln3−/− MBECs displayed significantly faster recovery compared to Cln3R cells, indicating enhanced membrane fluidity (Fig. 3A). When MBECs were tested after 2 h of CBX treatment, fluorescence recovery in Cln3−/− cells was normalized (one-way ANOVA F (3,108) = 18.10, p < 0.0001) (Fig. 3A).
Fig. 3.
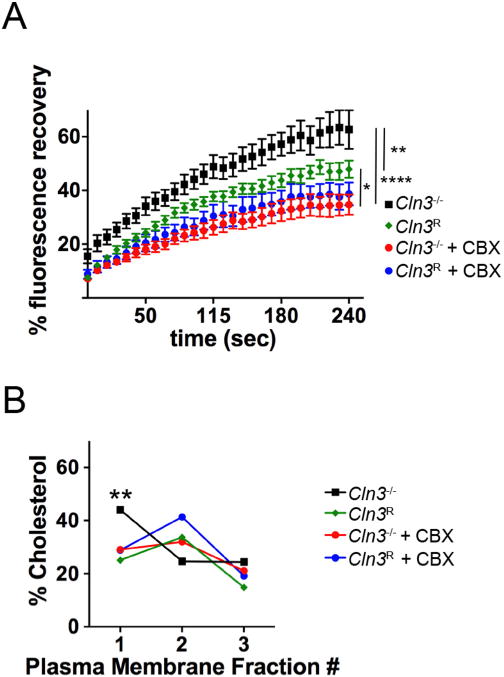
CBX treatment repairs membrane fluidity in Cln3−/− MBECs. A, MBECs were treated with CBX (25 μM) or vehicle for 2 h. The plasma membrane was labeled with Alexa-488-CTB and FRAP was performed using a live cell confocal microscope. ImageJ was used to quantify intensity on images collected over 300 s. Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparison test. Relative to Cln3R, Cln3−/− MBECs had significantly more Alexa-488-CTB recovery at all-time points. * p < 0.05, ** p < 0.01, **** p < 0.0001. B, Lipid microdomains were enriched by cell fractionation and the amplex red assay used to quantify cholesterol levels in each fraction. Data are from three independent experiments. Statistical significance was determined by two-way ANOVA with Tukey’s multiple comparison test. ** p < 0.01. Expanded data in Fig. S1. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To determine if CBX corrects the previously described abnormal distribution of microdomain lipids (Tecedor et al., 2013), plasma membranes were isolated and fractionated over sucrose gradients using a detergent-free extraction method and cholesterol levels quantified. Similar to our earlier data (Tecedor et al., 2013), Cln3−/− MBECs demonstrated cholesterol partitioning into more buoyant fractions, with the highest concentration found in the first (lightest) fraction compared to a cholesterol peak in the second fraction for Cln3R MBEC (two-way ANOVA of “membrane fraction” factor F(11,96) = 60.30, p < 0.0001; Fig. 3B). Pretreatment with CBX normalized the profile of Cln3−/− MBECs such that cholesterol concentration peaked in fraction 2. (Fig. 3B, Fig. S1).
Cumulatively, these data support a model where CBX is a novel modifier of lipid microdomains. Interestingly, CBX did not significantly alter microdomain dynamics or the partitioning of cholesterol in Cln3R MBECs.
3.3. CBX restores Cav-1 plasma membrane distribution
Since CBX corrected membrane fluidity dynamics in Cln3−/− MBECs, we subsequently investigated if CBX corrects caveolar defects. Caveolae are flask-shaped invaginations that form within distinct cholesterol/sphingolipid-enriched lipid microdomains on the plasma membrane. Caveolae are common in BBB endothelia, where they serve as signaling platforms, mediate endocytosis, and transcytosis. The primary structural protein of caveolae is caveolin-1 (Cav-1). Cav-1 inserts into membranes at the ER and traffics through the Golgi where high-order oligomerization drives transport to the plasma membrane (Chadda et al., 2007). We previously found enhanced microdomain fluidity in Cln3−/− MBECs and modified Cav-1 distribution as assessed by fractionation and immunofluorescence of the plasma membrane. Altered plasma membrane Cav-1 distribution causes reduced caveolae formation in vitro and in vivo (Tecedor et al., 2013). As shown in Fig. 4, CBX treatment of Cln3−/− MBECs increased Cav-1 immunosignal at the plasma membrane, evaluated by immunocytochemistry (Fig. 4A); western blot analysis of membrane enriched fractions showed no differences in Cav-1 protein levels (one-way ANOVA, F(3,20) = 0.3744, p = 0.7724; Fig. 4B) implying that trafficking not protein expression levels were impacted by CBX treatment. The corrective effect of CBX on these phenotypes is similar to our prior work where addition of exogenous lipids recovered Cav-1 plasma membrane localization (Tecedor et al., 2013). Cumulatively, the data support that CBX is acting to modify membranes in MBECs.
Fig. 4.
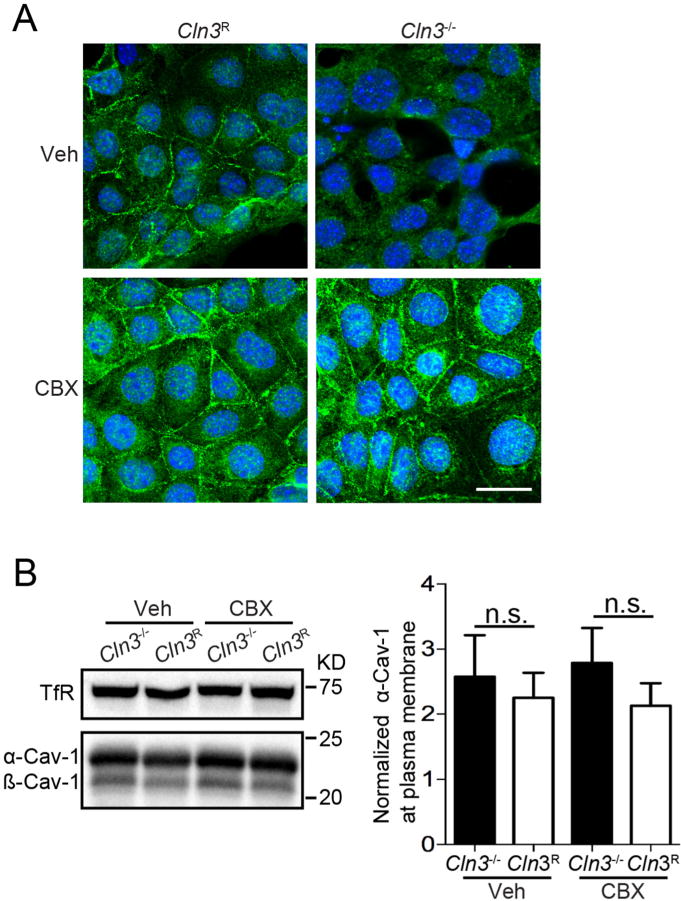
CBX restores Cav-1 subcellular localization in Cln3−/− MBECs. A, MBECs were treated with CBX (25 μM) or vehicle for 2 h, fixed, and Cav-1 intracellular distribution evaluated using immunocytochemistry and confocal microscopy. Photomicrographs are representative of 3 independent experiments. Scale bar = 20 μm.B, Western blotting of plasma membrane enriched fractions for Cav-1 levels and quantification. Data are from six independent experiments. Bars show mean ± s.e.m., No statistical differences were found by one-way ANOVA.
3.4. CBX corrects Cdc42-dependent defects in Cln3−/− MBECs
In addition to caveolar endocytosis, Cln3−/− MBECs have decreased levels of fluid-phase endocytosis (FPE) and increased GTP-loaded Cdc42 (Schultz et al., 2014). Cdc42 is a small GTPase that cycles between GTP-bound (active, effector-binding) and GDP-bound (inactive) states (Chadda et al., 2007; Nevins and Thurmond, 2006; Rojas et al., 2001). Cdc42 GTP-to-GDP cycling is required for fluid-phase endocytosis and cell migration, both of which are reduced in Cln3−/− cells (Schultz et al., 2014). This suggests that inefficient cycling of Cdc42 to the “off” state underlies multiple actin-related defects in MBEC, and suggests further that drugs able to promote cycling may have beneficial effects.
We incubated subconfluent MBECs with CBX and quantified Cdc42-GTP levels. As expected, in vehicle-treated cells, active Cdc42 levels were significantly higher in Cln3−/− compared to Cln3R MBECs (Fig. 5A). In Cln3R MBECs, CBX did not significantly alter Cdc42-GTP levels (Fig. 5A), likely due to the already low baseline levels of Cdc42-GTP as shown previously (Schultz et al., 2014). In contrast, CBX treatment significantly reduced the levels of Cdc42-GTP in Cln3−/− MBECs (Fig. 5A; ANOVA, F(3, 16) = 9.947, p = 0.0006).
Correction of Cdc42-GTP suggested that CBX may mitigate the downstream actin-dependent cell migration and fluid-phase endocytosis defects previously found in Cln3−/− cells (Schultz et al., 2014). To test for correction of cell migration, a scratch wound was made in confluent monolayers of Cln3R and Cln3−/− MBECs. Cells were subsequently treated with vehicle or CBX overnight and imaged using live cell microscopy to assess cell migration over time, and the percentage of wound remaining at each time point calculated. As in our former report (Schultz et al., 2014), Cln3R MBECs migrated significantly faster than Cln3−/− cells at all-time points past 6 h (Fig. 5B). Importantly, CBX treatment improved Cln3−/− MBEC migration, such that there was no significant difference between Cln3R, Cln3R + CBX or Cln3−/− + CBX groups (Fig. 5B, Fig. S2). Thus, CBX is effective at correcting the cell migration defect in Cln3−/− MBECs.
In addition to the role of Cdc42 on cell migration, Cdc42 is a key regulator of fluid-phase endocytosis (Kumari and Mayor, 2008; Schultz et al., 2014). Impaired fluid phase endocytosis has been reported for Cln3 mutant yeast, Cln3−/− mouse brain endothelial cells, Cln3−/− neuronal cells, and in fibroblasts harvested from patients with CLN3 deficiency (Codlin et al., 2008; Fossale et al., 2004; Luiro et al., 2004; Schultz et al., 2014; Vidal-Donet et al., 2013). We therefore measured fluid-phase endocytosis by uptake of fluorescently labeled dextran in cells treated with CBX or vehicle. CBX pretreatment of Cln3−/− MBECs for 30 min prior to dextran addition corrected Cln3−/− fluid-phase endocytic defects (Fig. 5C; (ANOVA, F(3, 8) = 10.68, p < 0.0036) to levels similar to Cln3R cells. Together these results show that in Cln3−/− MBECs, CBX improves Cdc42 regulation and the associated actin-dependent functions. To our knowledge, the impact of CBX on Cdc42 activity and Cdc42-dependent cellular processes has not been previously tested.
3.5. LINCS compounds correct fluid-phase and caveolar defects in Cln3−/− MBECs
We next employed a transcriptome-based discovery approach to identify drugs that may mimic the effects of CBX (Fig. 6A). We generated gene expression signatures from Cln3−/− MBEC cells treated with 3 μM CBX vs. vehicle treated cells and normal cells. These signatures were used to mine the NIH Library of Integrated Network-based Cellular Signatures (LINCS) (Lamb et al., 2006) to identify small molecules that induced gene expression profiles similar to that induced by CBX. We also analyzed the effect of enoxolone, a metabolite of CBX (Iveson et al., 1971). Of hits, we focused on hydrophobic, neutral molecules that could impact membrane fluidity, and screened these for their ability to correct FITC-dextran uptake. The top six molecules (Prd, α-KC, K8, K9, K4, and K2) significantly increased fluid-phase endocytosis in Cln3−/− MBEC (Fig. 6B; one-way ANOVA, F(9,128) = 15.58, p < 0.0001), similar to CBX.
For caveolar endocytosis to occur, plasma membrane Cav-1 is phosphorylated. In Cln3−/− MBECs, the altered distribution of Cav-1 at the plasma membrane coincides with a decrease of caveolae number and reduction in phosphorylated Cav-1 (P-cav-1) (Tecedor et al., 2013). We therefore tested if plasma membrane-resident phospho-cav-1 was corrected by the LINCS hit compounds. Interestingly, we found that P-cav1 levels significantly recovered after treatment with CBX, its metabolite enoxolone, α-KC, K8, or K9 (Fig. 6C; Mann-Whitney test). Treatment with Prd, K4, or K2 did not reach statistical significance but showed a positive trend (Fig. 6C).
3.6. In vivo CBX treatment increases BBB responses and reduces autofluorescence
Brain endothelial cells are vital for maintaining the integrity of the BBB. Interestingly, injecting hypotonic solutions into blood induces an osmotic gradient across the BBB endothelium (Tecedor et al., 2013). In wildtype mice, this osmotic gradient triggers endothelial cell swelling followed by over-shrinkage, allowing small molecule access to the underlying neuropil (Kozler and Pokorny, 2003; Reid and O’Neil, 2000; Tecedor et al., 2013). The in vivo response to hypotonic shock is defective in Cln3−/− mice, as demonstrated by impaired passage of small molecule reporters into the brain (Tecedor et al., 2013). Here we tested if short-term oral dosing of CBX corrects this defect in aged (8-month old) Cln3-null mice. For this, Cln3−/− and Cln3+/+ mice were gavaged daily with low-dose CBX or vehicle for two weeks, after which they were subjected to hypotonic challenge along with a dye that, unless the vasculature is compromised (e.g., by hypotonic challenge), remains restricted to the vascular lumen. As before, we used Hoechst dye penetration as a readout for the leakiness of the vasculature (Tecedor et al., 2013). For optimal detection of dye leaking into brain parenchyma, our procedure was modified to include sequential delivery of i) labeled Alexa488-conjugated wheat germ agglutinin (WGA) to label the luminal surfaces of the vasculature, ii) isotonic (control) or hypotonic solution containing Hoechst dye, and iii) fixative (Fig. 7A). As expected, Cln3+/+ mice treated with isotonic solution do not show extravasation of dye into the brain parenchyma but instead, the dye remains confined to the vessel lumen (Fig. 7A). In vehicle-gavaged mice, hypotonic-induced Hoechst penetration was evident in Cln3+/+ mice, but minimal in Cln3−/− mice (Fig. 7B), in line with our previous report (Tecedor et al., 2013). In contrast, CBX-treated Cln3−/− mice had substantially increased Hoechst signal in the parenchymal tissue, with the amount of Hoechst dye penetration similar to Cln3+/+ mice treated with CBX (Fig. 7B). These data indicate that CBX normalizes the impaired BBB response of Cln3−/− mice.
Fig. 7.
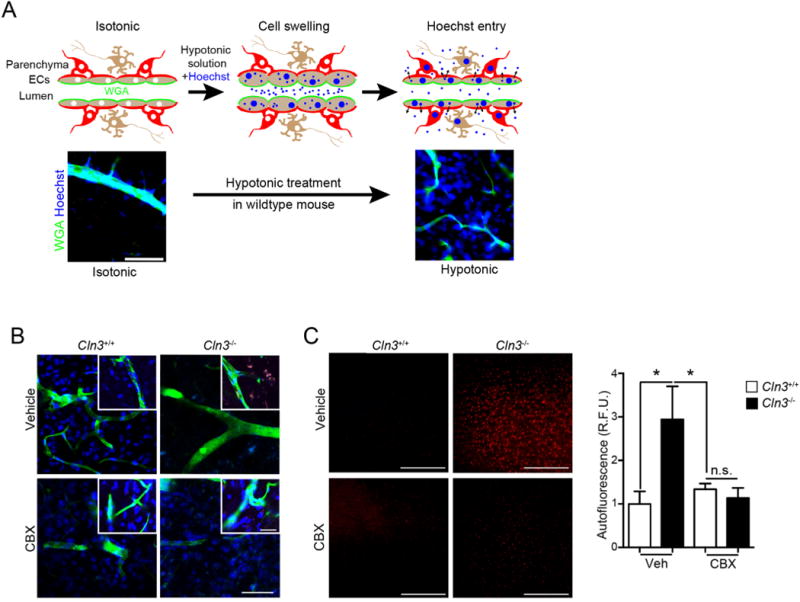
CBX corrects regulated volume defects and reduces Cln3−/− autofluorescent inclusions in vivo. A, (Top) Schematic representing Hoechst entry into the brain during hypotonic treatment. (Bottom) Cln3+/− mice exposed to isotonic or hypotonic treatments. Animals were anesthetized and wheat germ agglutinin (WGA) (green) infused intravascularly to label endothelial cells followed by infusion of Hoechst dye (blue) in a hypotonic solution, then saline infusion, followed by fixation, sectioning and imaging. The Hoechst signal outside the WGA-labeled vascular is indicative of regulated volume decrease and dye extravasation after hypotonicity exposure. Scale bar = 100 μm. B, Cln3+/+ or Cln3−/− mice were treated daily with vehicle or 20 mg/kg CBX for two weeks. After anesthetization, mice were perfused as in (A). Brain slices (50 μm) were imaged by confocal microscopy and show that CBX permits Hoechst dye extravasation in Cln3−/− mice brain. Images are representative of 5 mice per group performed on different days. Scale bar = 50 μm. C, Thin sections from fixed brains were imaged under low magnification in the red channel to detect autofluorescence. Scale bar = 200 μm. Cortical images (4 images/animal; 4 mice/group) were taken and fluorescence intensity averaged using ImageJ. Data are mean ± s.e.m., Kruska-Wallis test with Tukey’s multiple comparisons post-hoc, *, p < 0.05. Scale bar is 200 μm. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In addition to the various BBB defects, JNCL patients and Cln3 mutant mouse models display progressive accumulation of auto-fluorescence in multiple cell types (Cotman et al., 2002; Mole et al., 2005; Stein et al., 2010). Due to the close proximity of the BBB to brain neuropil, we next investigated if CBX alters the accumulation of parenchymal autofluorescence. For this study, aged (8 month old) Cln3+/+ and Cln3−/− mice were gavaged with CBX or vehicle for two weeks and cortical autofluorescence quantified. As expected, vehicle treated Cln3−/− mice showed extensive autofluorescence levels compared to wildtype mice. Of note, there was significant reduction of auto-fluorescence (Kruskal-Wallis, p = 0.0234) in CBX-treated Cln3−/− mice relative to those that received vehicle-treatment (Fig. 7C).
4. Discussion
We show that Cln3−/− mice have increased astrocytic endfeet area (Fig. 1A). Interestingly, enlarged astrocytic endfeet were lacking the notable organelles or membrane inclusions typically seen in these structures. Astrocyte endfeet swelling is an indicator of BBB dysfunction and is commonly found in stroke (Xiang et al., 2016). Post stroke, astrocyte endfeet swelling is hypothesized to prevent serum protein entry into the brain (Xiang et al., 2016). Enlarged astrocyte endfeet in Cln3−/− deficient mice may explain the decreased permeability of the Hoechst dye into the brain during hypotonic stress (Fig. 7). Interestingly, this phenotype improved with CBX treatment (Fig. 1B).
Additionally, we found increased GAP junction activity in MBECs which was corrected with CBX (Fig. 2). Although CBX is a GAP junction inhibitor, our data support that it also modifies lipid microdomains, affecting fluidity and correcting the regulation and function of caveolar and fluid-phase endocytosis (Figs. 2–5). CBX is hypothesized to alter hemi channel opening or synaptic transmission by directly intercalating into the plasma membrane (Davidson and Baumgarten, 1988; Goldberg et al., 1996; Tovar et al., 2009). This is consistent with previous reports where CBX was shown to have membrane stabilizing effects, which are dependent on lipid concentration (Symons and Parke, 1980). Interestingly, CBX application to wild type cells had little effect, suggesting that the microdomain modifying effect of CBX may be context dependent.
Although we demonstrate an effect on membrane dynamics, the exact mechanism of how CBX modifies lipid microdomains or corrects the in vitro and in vivo phenotypes tested here is not known. CBX can modify lysosomal permeability (Symons and Parke, 1980), reduce fatty acid induced reactive oxygen species, inhibit sterol regulatory element binding protein-1c (Rhee et al., 2012), alter calcium signaling (Liu et al., 2010), inhibit 11-beta hydroxysteroid dehydrogenase (Monder et al., 1989), and increase heat shock protein expression (Nagayama et al., 2001). One could argue that modulation of one or more of these pathways could influence the cellular phenotypes investigated in this study. However, we found that CBX restores most in vitro cell phenotypes quickly (within 1–2 h), and speculate that it has a direct influence on cell membranes, creating conformational shifts and altered functions of membrane-associated proteins.
We also found other small molecules that behaved similarly to CBX (Fig. 6). These were identified using an unbiased genomics-based drug-discovery strategy (NIH Library of Integrated Network-based Cellular Signatures, LINCS). Compound screening allowed us to prioritize six small molecules that rescued membrane dynamics to levels comparable to CBX. Interestingly, all six small drugs are uncharged and hydrophobic. These drugs have the potential to intercalate into membranes and produce physicochemical effects on lipid bilayers (Royer et al. 2009, Ragot et al. 2013, Heier et al. 2013). These drugs were effective at correcting fluid-phase endocytosis and Cav-phosphorylation in Cln3−/− cells, and support the hypothesis that correcting membrane deficiencies in JNCL may be therapeutically beneficial (Fig. 6).
CXB has a steroid-like structure and a blood half-life of 13–26 h (Baron et al., 1978; Davidson and Baumgarten, 1988; Hayes et al., 1977). Recently a BBB-permeable derivative of CBX (Leshchenko et al., 2006), INI-0602, was used to alleviate excessive hemichannel activity associated with Cln3−/− mouse glial cells, and this drug showed positive effects in Cln3−/− mouse slice culture models (Burkovetskaya et al., 2014). Using a novel BBB permeability assay, we found that CBX treatment restored the BBB response to hypotonic shock. Moreover, two-weeks of CBX treatment resolved autofluorescence in Cln3−/− mouse brain (Fig. 7).
Rodent gut microflora hydrolyzes CBX into sulfate conjugates of enoxolone (Iveson et al., 1971), which enters the blood stream with subsequent presence in the brain and CSF (Tabuchi et al., 2012). Interestingly, both CBX, and enoxolone corrected the in vitro phenotypes. As CBX does not cross the BBB (Leshchenko et al., 2006; Takeuchi et al., 2011), the metabolite enoxolone may be responsible for the reduced autofluorescent storage either by improving the astrocytic phenotype, the endothelial phenotype, or both. Studies show that amyloid-β accumulation in Alzheimer’s disease brain is due in part to faulty clearance by brain endothelial cells (Sagare et al., 2013), and recent work in the JNCL field shows that, in vitro, normalizing astrocytes can improve neuronal phenotypes (Parviainen et al., 2017). Regardless of the exact cell type and mechanism of action, CBX and the LINCS compounds provide promising therapeutic avenues for treatment of CLN3 deficiency disease.
Supplementary Material
Acknowledgments
This work was supported by: Beyond Batten Disease Foundation Fellowship, BDSRA Fellowship, the Roy J. Carver Trust, the UI Central Microscopy and Research Facility (1S10RR025439-01) and the Children’s Hospital of Philadelphia Research Institute. We thank the Bioimaging Center at the Delaware Biotechnology Institute for access to their Zeiss Libra 120 transmission EM (supported by grants from the NIH-NIGMs (P20 GM103446), the NSF (IIA-1301765) and the State of Delaware). The funding bodies were not involved in study design, data collection and analysis, writing, or decision to submit the article for publication.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2018.04.010.
Footnotes
Conflicts of interest
The data in this scientific report was used for patent application US14/776,558.
Data statement
All data is available upon request.
References
- Abbott NJ, et al. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Baron JH, et al. Serum carbenoxolone in patients with gastric and duodenal ulcer: absorption, efficacy and side-effects. Gut. 1978;19:330–335. doi: 10.1136/gut.19.4.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastiaanse EM, et al. Heptanol-induced decrease in cardiac gap junctional conductance is mediated by a decrease in the fluidity of membranous cholesterol-rich domains. J Membr Biol. 1993;136:135–145. doi: 10.1007/BF02505758. [DOI] [PubMed] [Google Scholar]
- Burkovetskaya M, et al. Evidence for aberrant astrocyte Hemichannel activity in juvenile neuronal ceroid Lipofuscinosis (JNCL) PLoS One. 2014;9:e95023. doi: 10.1371/journal.pone.0095023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, et al. Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J Biol Chem. 2006;281:20483–20493. doi: 10.1074/jbc.M602180200. [DOI] [PubMed] [Google Scholar]
- Chadda R, et al. Cholesterol-sensitive Cdc42 activation regulates actin poly-merization for endocytosis via the GEEC pathway. Traffic. 2007;8:702–717. doi: 10.1111/j.1600-0854.2007.00565.x. [DOI] [PubMed] [Google Scholar]
- Codlin S, Mole SE. S. pombe btn1, the orthologue of the Batten disease gene CLN3, is required for vacuole protein sorting of Cpy1p and Golgi exit of Vps10p. J Cell Sci. 2009;122:1163–1173. doi: 10.1242/jcs.038323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codlin S, et al. btn1 affects endocytosis, polarization of sterol-rich membrane domains and polarized growth in Schizosaccharomyces pombe. Traffic. 2008;9:936–950. doi: 10.1111/j.1600-0854.2008.00735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotman SL, Staropoli JF. The juvenile Batten disease protein, CLN3, and its role in regulating anterograde and retrograde post-Golgi trafficking. Clin Lipidol. 2012;7:79–91. doi: 10.2217/clp.11.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotman SL, et al. Cln3(Deltaex7/8) knock-in mice with the common JNCL mutation exhibit progressive neurologic disease that begins before birth. Hum Mol Genet. 2002;11:2709–2721. doi: 10.1093/hmg/11.22.2709. [DOI] [PubMed] [Google Scholar]
- Dargan DJ, Subak-Sharpe JH. The antiviral activity against herpes simplex virus of the triterpenoid compounds carbenoxolone sodium and cicloxolone sodium. J Antimicrob Chemother. 1986;18(Suppl B):185–200. doi: 10.1093/jac/18.supplement_b.185. [DOI] [PubMed] [Google Scholar]
- Davidson JS, Baumgarten IM. Glycyrrhetinic acid derivatives: a novel class of inhibitors of gap-junctional intercellular communication. Structure-activity relationships. J Pharmacol Exp Ther. 1988;246:1104–1107. [PubMed] [Google Scholar]
- Eliason SL, et al. A knock-in reporter model of Batten disease. J Neurosci. 2007;27:9826–9834. doi: 10.1523/JNEUROSCI.1710-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossale E, et al. Membrane trafficking and mitochondrial abnormalities precede subunit c deposition in a cerebellar cell model of juvenile neuronal ceroid lipofuscinosis. BMC Neurosci. 2004;5:57. doi: 10.1186/1471-2202-5-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geback T, et al. TScratch: a novel and simple software tool for automated analysis of monolayer wound healing assays. BioTechniques. 2009;46:265–274. doi: 10.2144/000113083. [DOI] [PubMed] [Google Scholar]
- Golabek AA, et al. CLN3 protein regulates lysosomal pH and alters intracellular processing of Alzheimer’s amyloid-beta protein precursor and cathepsin D in human cells. Mol Genet Metab. 2000;70:203–213. doi: 10.1006/mgme.2000.3006. [DOI] [PubMed] [Google Scholar]
- Goldberg GS, et al. Evidence that disruption of connexon particle arrangements in gap junction plaques is associated with inhibition of gap junctional communication by a glycyrrhetinic acid derivative. Exp Cell Res. 1996;222:48–53. doi: 10.1006/excr.1996.0006. [DOI] [PubMed] [Google Scholar]
- Hayes MJ, et al. Changes in the plasma clearance and protein binding of car-benoxolone with age, and their possible relationship with adverse drug effects. Gut. 1977;18:1054–1058. doi: 10.1136/gut.18.12.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holopainen JM, et al. Elevated lysosomal pH in neuronal ceroid lipofuscinoses (NCLs) Eur J Biochem. 2001;268:5851–5856. doi: 10.1046/j.0014-2956.2001.02530.x. [DOI] [PubMed] [Google Scholar]
- Iveson P, et al. The metabolism of carbenoxolone in the rat. Xenobiotica. 1971;1:79–95. doi: 10.3109/00498257109044381. [DOI] [PubMed] [Google Scholar]
- Kama R, et al. The yeast Batten disease orthologue Btn1 controls endosome-Golgi retrograde transport via SNARE assembly. J Cell Biol. 2011;195:203–215. doi: 10.1083/jcb.201102115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenworthy AK. Fluorescence recovery after photobleaching studies of lipid rafts. Methods Mol Biol. 2007;398:179–192. doi: 10.1007/978-1-59745-513-8_13. [DOI] [PubMed] [Google Scholar]
- Kozler P, Pokorny J. Altered blood-brain barrier permeability and its effect on the distribution of Evans blue and sodium fluorescein in the rat brain applied by intracarotid injection. Physiol Res. 2003;52:607–614. [PubMed] [Google Scholar]
- Kumari S, Mayor S. ARF1 is directly involved in dynamin-independent endocytosis. Nat Cell Biol. 2008;10:30–41. doi: 10.1038/ncb1666. [DOI] [PubMed] [Google Scholar]
- Lamb J, et al. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- Leshchenko Y, et al. Carbenoxolone does not cross the blood brain barrier: an HPLC study. BMC Neurosci. 2006;7:3. doi: 10.1186/1471-2202-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim MJ, et al. IgG entry and deposition are components of the neuroimmune response in Batten disease. Neurobiol Dis. 2007;25:239–251. doi: 10.1016/j.nbd.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Liu X, et al. Gap junctions/hemichannels modulate interkinetic nuclear migration in the forebrain precursors. J Neurosci. 2010;30:4197–4209. doi: 10.1523/JNEUROSCI.4187-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luiro K, et al. CLN3 protein is targeted to neuronal synapses but excluded from synaptic vesicles: new clues to Batten disease. Hum Mol Genet. 2001;10:2123–2131. doi: 10.1093/hmg/10.19.2123. [DOI] [PubMed] [Google Scholar]
- Luiro K, et al. Interconnections of CLN3, Hook1 and Rab proteins link Batten disease to defects in the endocytic pathway. Hum Mol Genet. 2004;13:3017–3027. doi: 10.1093/hmg/ddh321. [DOI] [PubMed] [Google Scholar]
- Monder C, et al. Licorice inhibits corticosteroid 11 beta-dehydrogenase of rat kidney and liver: in vivo and in vitro studies. Endocrinology. 1989;125:1046–1053. doi: 10.1210/endo-125-2-1046. [DOI] [PubMed] [Google Scholar]
- Metcalf DJ, et al. Loss of the Batten disease gene CLN3 prevents exit from the TGN of the mannose 6-phosphate receptor. Traffic. 2008;9:1905–1914. doi: 10.1111/j.1600-0854.2008.00807.x. [DOI] [PubMed] [Google Scholar]
- Mole SE, et al. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6:107–126. doi: 10.1007/s10048-005-0218-3. [DOI] [PubMed] [Google Scholar]
- Nagayama S, et al. Carbenoxolone, a new inducer of heat shock protein 70. Life Sci. 2001;69:2867–2873. doi: 10.1016/s0024-3205(01)01362-5. [DOI] [PubMed] [Google Scholar]
- Narayan SB, et al. CLN3P, the Batten’s disease protein, is a novel palmitoyl-protein Delta-9 desaturase. Ann Neurol. 2006;60:570–577. doi: 10.1002/ana.20975. [DOI] [PubMed] [Google Scholar]
- Nevins AK, Thurmond DC. Caveolin-1 functions as a novel Cdc42 guanine nucleotide dissociation inhibitor in pancreatic beta-cells. J Biol Chem. 2006;281:18961–18972. doi: 10.1074/jbc.M603604200. [DOI] [PubMed] [Google Scholar]
- Parviainen L, et al. Glial cells are functionally impaired in juvenile neuronal ceroid lipofuscinosis and detrimental to neurons. Acta Neuropathol Commun. 2017;5:74. doi: 10.1186/s40478-017-0476-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce DA, Sherman F. A yeast model for the study of Batten disease. Proc Natl Acad Sci U S A. 1998;95:6915–6918. doi: 10.1073/pnas.95.12.6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce DA, et al. Action of BTN1, the yeast orthologue of the gene mutated in Batten disease. Nat Genet. 1999;22:55–58. doi: 10.1038/8861. [DOI] [PubMed] [Google Scholar]
- Phillips SN, et al. CLN3, the protein associated with batten disease: structure, function and localization. J Neurosci Res. 2005;79:573–583. doi: 10.1002/jnr.20367. [DOI] [PubMed] [Google Scholar]
- Porta R, et al. Gastroprotection and lysosomal membrane stabilization by sulglicotide. Arzneimittelforschung. 1986;36:1079–1082. [PubMed] [Google Scholar]
- Reid JM, O’Neil RG. Osmomechanical regulation of membrane Trafficking in polarized cells. Biochem Biophys Res Commun. 2000;271:429–434. doi: 10.1006/bbrc.2000.2638. [DOI] [PubMed] [Google Scholar]
- Rhee SD, et al. Carbenoxolone prevents the development of fatty liver in C57BL/6-Lep ob/ob mice via the inhibition of sterol regulatory element binding protein-1c activity and apoptosis. Eur J Pharmacol. 2012;691:9–18. doi: 10.1016/j.ejphar.2012.06.021. [DOI] [PubMed] [Google Scholar]
- Rojas R, et al. Cdc42-dependent modulation of tight junctions and membrane protein Traffic in polarized Madin-Darby canine kidney cells. Mol Biol Cell. 2001;12:2257–2274. doi: 10.1091/mbc.12.8.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein CS, et al. Osmoregulation of ceroid neuronal lipofuscinosis type 3 in the renal medulla. Am J Physiol Cell Physiol. 2010;298:C1388–400. doi: 10.1152/ajpcell.00272.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagare AP, et al. Neurovascular defects and faulty amyloid-beta vascular clearance in Alzheimer’s disease. J Alzheimers Dis. 2013;33(Suppl 1):S87–100. doi: 10.3233/JAD-2012-129037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert AL, et al. Connexin family members target to lipid raft domains and interact with caveolin-1. Biochemistry. 2002;41:5754–5764. doi: 10.1021/bi0121656. [DOI] [PubMed] [Google Scholar]
- Schultz ML, et al. CLN3 deficient cells display defects in the ARF1-Cdc42 pathway and actin-dependent events. PLoS One. 2014;9:e96647. doi: 10.1371/journal.pone.0096647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons AM, Parke DV. The effects of sodium carbenoxolone on the stability of cellular membranes. Scand J Gastroenterol Suppl. 1980;65:3–10. [PubMed] [Google Scholar]
- Symons AM, et al. The effect of sodium carbenoxolone on lysosomal enzyme release. Biochem Pharmacol. 1978;27:2461–2463. doi: 10.1016/0006-2952(78)90361-1. [DOI] [PubMed] [Google Scholar]
- Tabuchi M, et al. The blood-brain barrier permeability of 18beta-glycyrrhetinic acid, a major metabolite of glycyrrhizin in Glycyrrhiza root, a constituent of the traditional Japanese medicine yokukansan. Cell Mol Neurobiol. 2012;32:1139–1146. doi: 10.1007/s10571-012-9839-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, et al. Blockade of gap junction hemichannel suppresses disease progression in mouse models of amyotrophic lateral sclerosis and Alzheimer’s disease. PLoS One. 2011;6:e21108. doi: 10.1371/journal.pone.0021108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecedor L, et al. CLN3 loss disturbs membrane microdomain properties and protein transport in brain endothelial cells. J Neurosci. 2013;33:18065–18079. doi: 10.1523/JNEUROSCI.0498-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, et al. Direct actions of carbenoxolone on synaptic transmission and neuronal membrane properties. J Neurophysiol. 2009;102:974–978. doi: 10.1152/jn.00060.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal-Donet JM, et al. Alterations in ROS activity and lysosomal pH account for distinct patterns of macroautophagy in LINCL and JNCL fibroblasts. PLoS One. 2013;8:e55526. doi: 10.1371/journal.pone.0055526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RE, Mole SE. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology. 2012;79:183–191. doi: 10.1212/WNL.0b013e31825f0547. [DOI] [PubMed] [Google Scholar]
- Xiang J, et al. Mechanisms underlying astrocyte end-feet swelling in stroke. Acta Neurochir Suppl. 2016;121:19–22. doi: 10.1007/978-3-319-18497-5_4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.