

ABSTRACT
Incidents that slow or stall replication fork progression, collectively known as replication stress, represent a major source of spontaneous genomic instability. Here, we determine the requirement for global protein biosynthesis on DNA replication and associated downstream signaling. We study this response side by side with dNTP deprivation; one of the most commonly used means to investigate replication arrest and replicative stress. Our in vitro interrogations reveal that inhibition of translation by cycloheximide (CHX) rapidly impairs replication fork progression without decoupling helicase and polymerase activities or inducing DNA damage. In line with this, protein deprivation stress does not activate checkpoint signaling. In contrast to the direct link between insufficient dNTP pools and genome instability, our findings suggest that replication forks remain stable during short-term protein deficiency. We find that replication forks initially endure fluctuations in protein supply in order to efficiently resume DNA synthesis upon reversal of the induced protein deprivation stress. These results reveal distinct cellular responses to replication arrest induced by deprivation of either nucleotides or proteins.
KEYWORDS: Cycloheximide, translation, stalled replication fork, replication stress, DNA damage
Introduction
Replication stress caused by slowing or stalling of replication fork progression, has a profound impact on genomic stability and diseases such as cancer [1]. There are many different sources of replication stress including physical obstacles that directly block progression of replication forks or inadequate supplies of factors required for DNA synthesis. While most studies concerning replication stress are focused on delineating the cellular response to limited dNTP pools or DNA polymerase stalling, it remains unclear how deficient protein supplies affect replication and downstream signaling events. It has previously been reported that incorporation of 3H-thymidine into DNA decreases upon cellular exposure to compounds that disrupt protein synthesis [2-4]. However, the underlying mechanism(s) and associated downstream signaling events remain to be precisely described.
Replication stress is commonly measured by assessing activation of the ataxia telangiectasia and Rad3-related protein (ATR) signaling pathway, including detection of single-stranded DNA (ssDNA), phosphorylation of checkpoint kinase 1 (Chk1) and replication protein A (RPA) or pan-nuclear phosphorylation of the histone variant H2AX (γH2AX) [5,6]. It is well established that dNTP deprivation results in rapid γH2AX induction, activation of the ATR/Chk1 checkpoint and increased single stranded DNA (ssDNA) exposure at active replication forks. Prolonged dNTP deprivation results in collapse of replication forks and generation of DNA double strand breaks (DSBs) [7-9].
Here, we characterize the replication stress response associated with protein depletion by exposing cells to the most commonly used protein biosynthesis inhibitor cycloheximide (CHX) [10]. We find that protein deprivation results in acute disruption of replication to a similar extent as dNTP deprivation, while interference of protein production does not induce checkpoint activation or significant amounts of DNA damage. This suggests that short-term protein deprivation stall replication through a distinct mechanism generating stalled replication forks not prone to collapse.
Results
Short-term protein biosynthesis blockage inhibits DNA replication
To investigate the requirement of ongoing protein synthesis for DNA replication and the kinetics of this response, we treated U2OS cells with the translation inhibitor cycloheximide (CHX) and examined its effect on DNA synthesis by measuring incorporation of the thymidine analogue 5-ethynyl-2’-deoxyuridine (EdU). The EdU-positive cell population was reduced by half already 10 minutes after CHX addition and completely abolished at 20 minutes post-treatment (Fig. 1A, B). The observed CHX-induced blockage of EdU incorporation was comparable to the inhibition of DNA replication by hydroxyurea (HU) (Fig. 1A, B). Next, we extended our studies to the non-transformed fibroblasts BJ-hTERT in order to determine whether the observed cessation of DNA replication upon translation inhibition was limited to cancer cells. Interestingly, a similar negative impact on DNA synthesis was observed in the hTERT-immortalized fibroblasts treated with CHX (Figure S1A). The inhibitory effect of protein synthesis blockage on DNA replication was dose- and time-dependent (Figure S1B) and not limited to CHX since puromycin, a structurally unrelated inhibitor of translation, also blocked DNA replication with similar kinetics as CHX (Figure S1C).
Figure 1.

Short-term CHX treatment blocks DNA replication. (A) Representative images of U2OS cells treated with 10 μg/ml CHX (36 μM) or 2 mM HU for the timepoints indicated. Cells were subsequently pulsed with 10 μM EdU for 20 minutes (in presence of inhibitors), followed by fixation and fluorescent labeling. (B) Quantification of (A) as the percentage of 100 cells counted in each experiment (n = 3). (C) Schematic representation of experimental setup of the fiber assay and representative images of DNA fibers. (D) Average replication speed of control or CHX (10 μg/ml) treated U2OS cells (n = 2). (E) U2OS cells were left untreated or treated with 0.4 μM aphidicolin (APH) for 24 hours. When indicated, cells were pre-treated with 10 μg/ml CHX or 2 mM HU 10 minutes prior to APH addition and kept in the media for the duration of the experiment. Cells were fixed and immunostained for 53BP1. Note that cells treated with HU and APH still induce 53BP1 foci (as seen in the image), whereas 53BP1 nuclear body formation is diminished. (F) Quantification of immunofluorescence in (E) as the percentage of 100 cells containing >10 53BP1 nuclear bodies (NBs) in each experiment (n = 2). The error bars depict standard deviation; *P ≤ 0.05, ** P < 0.01, ***P ≤ 0.001 as determined by Student's t-test. See also Figure S1.
To investigate if the reduction of replicating cells following CHX treatment was a consequence of reduced replication speed, we next performed DNA fiber assays to monitor the speed of individual replication forks upon CHX treatment. Following CHX exposure, replication rates were greatly reduced and a similar effect was observed upon inhibition of protein biosynthesis by puromycin treatment (Fig. 1C, D and S1D).
Slower replication fork speed can result in incomplete DNA replication of genomic loci such as common fragile sites. These under-replicated loci are marked by 53BP1-containing nuclear bodies (NBs) in the subsequent G1 cell cycle phase and serve to protect DNA lesions until they can be properly repaired [11]. According to previous studies, treatment of cells with low doses of the DNA polymerase inhibitor aphidicolin (APH) resulted in a clear induction of 53BP1 NBs in a fraction of cells (Fig. 1E, F) [11,12]. However, treating cells with CHX, or HU, 10 minutes prior to APH addition prevented the APH-dependent induction of 53BP1 NBs (Fig. 1E, F), although HU and APH treated cells still displayed 53BP1 foci formation (far right panel Fig. 1E). Notably, 53BP1 protein levels remained stable for the duration of the experiment (Figure S1E). This further supports that CHX treatment disrupts DNA replication and the subsequent formation of APH-induced replication-associated damage. Collectively, these findings reveal that continuously ongoing protein biosynthesis is required for progression of DNA replication in a similar manner as adequate supplies of nucleotides are required.
Transcription is incompletely blocked by short-term CHX treatment
To assess if CHX treatment disrupts all nuclear processes in a non-discriminatory manner, we investigated RNA synthesis by quantifying nascent RNA transcripts via 5-ethynyl uridine (EU) labeling following CHX treatment [13]. Upon cell incubation with EU following CHX treatment, the intensity and subcellular pattern of EU staining was reduced but not completely inhibited (Figure S2A, B). Upon prolonged exposures to CHX, transcription was still not completely inhibited with visible EU staining both in the nucleoli and nucleoplasm (Figure S2A, B), suggesting that both RNA polymerase I and II activities are decreased but not fully abolished. As expected, treating cells with the RNA polymerase II inhibitor 5,6-dichlorobenzimidazole 1-β-D-ribofuranoside (DRB), reduced EU incorporation in the nucleoplasm, but not in the nucleoli, at both shorter and longer timepoints (Figure S2A, B). However, since the total nuclear EU intensity, including nucleolar staining, was quantified in this experiment and not only nucleoplasmic intensity, the negative effect on transcription observed upon DRB treatment compared to CHX treatment is actually more pronounced than demonstrated in the quantification in Figure S2B (compare nucleoplasmic EU intensity following DRB and CHX treatment in Figure S2A).
Next we studied phosphorylation of the C-terminal domain (CTD) of RNA polymerase II, which is hyperphosphorylated upon transition from transcriptional initiation to transcriptional elongation [14,15]. RNA polymerase II CTD phosphorylation was slightly decreased at 1 hour and reduced by half following 24 hours of CHX treatment, whereas DRB, a direct inhibitor of CTD phosphorylation, inhibited the phosphorylation severely already 1 hour post-treatment (Figure S2C, D). Thus, contrary to DNA replication, ongoing protein synthesis is not entirely required for proper RNA synthesis at early timepoints of inhibition. However, upon extended CHX exposures, most proteins will be completely depleted, including total RNA polymerase II levels, and their associated processes will therefore be discontinued.
Deficient protein synthesis does not mimic the cellular response to dNTP depletion
Our findings establish that ongoing protein synthesis is directly required for DNA replication, where CHX induces stalling of replication with similar kinetics as HU-induced nucleotide depletion. To dissect the replication stress response activated during conditions of protein deprivation, we next assessed potential activation of the ATR signaling pathway following CHX treatment. In contrast to HU treatment, CHX did not induce RPA-coated ssDNA stretches (Fig. 2A, B), indicating that CHX does not induce uncoupling of helicase and polymerase activities [16]. In line with this, CHX did not promote Chk1 phosphorylation prior to depletion of total Chk1 levels (Fig. 2C) or activate the ATM-Chk2 pathway (Figure S3A) consistent with previous findings [17].
Figure 2.
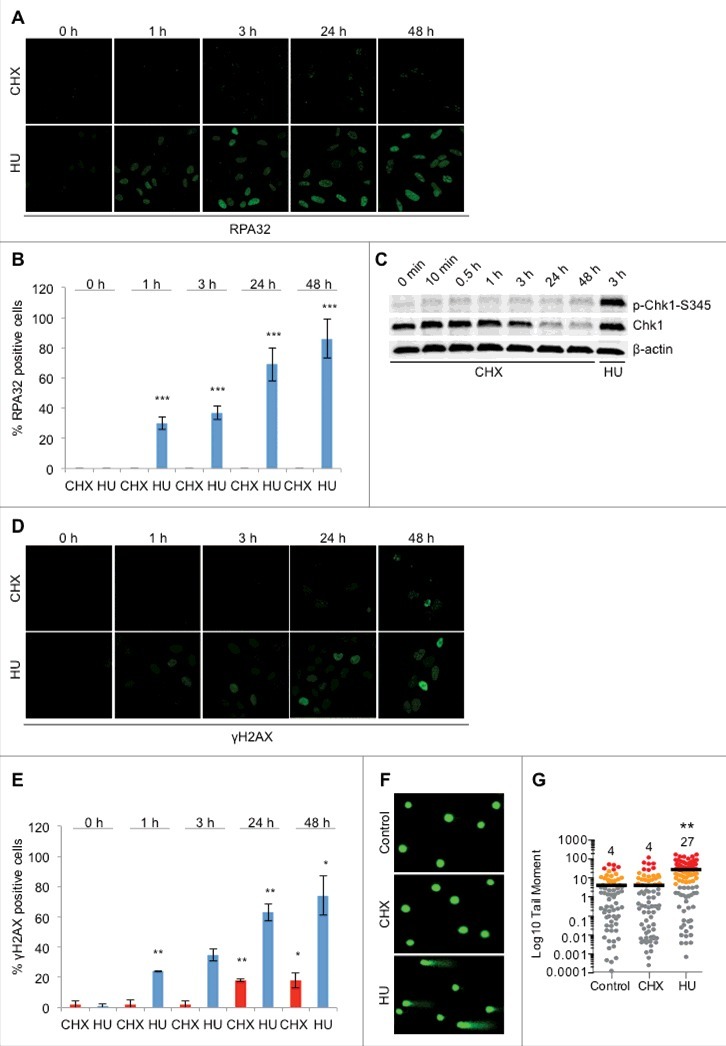
CHX treatment does not result in ssDNA formation, checkpoint activation or DNA damage. (A) Representative images of U2OS cells treated with 10 μg/ml CHX or 2 mM HU for the indicated timepoints and stained for RPA32. Cells were pre-extracted with CSK buffer (10 mM Pipes, pH 7.0, 100 mM NaCl, 300 mM sucrose, and 3 mM MgCl2, 0.7% Triton X-100) for 5 minutes prior to fixation. (B) Quantification of RPA32 positive cells (A), (n = 3). (C) U2OS cells were either left untreated or treated with CHX or HU for the timepoints indicated followed by Western blot probed with p-Chk1-S345, Chk1 and β-actin antibodies. (D) Representative images of U2OS cells treated with 10 μg/ml CHX or 2 mM HU for the indicated timepoints and stained for γH2AX. (E) Quantification of γH2AX positive cells (D), (n = 2). (F) and (G) DNA damage in U2OS assessed by the comet assay following treatment with 10 μg/ml CHX or 2 mM HU for 24 hours. Representative images (F) and quantification of tail moment (G). The error bars depict standard deviation; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 as determined by Student's t-test. See also Figure S3.
We continued by investigating the formation of DNA damage and found that CHX treatment did not result in accumulation of DNA breaks, as measured by γH2AX accumulation (Fig. 2D, E) and the comet assay (Fig. 2F, G). Formation of γH2AX foci by ionizing radiation was not affected by CHX pre-treatment (Figure S3B), indicating that CHX treatment does not generally inhibit formation of γH2AX foci. In addition, we assessed focal accumulation of the DNA damage marker 53BP1 and homologous recombination marker RAD51. Here, we found HU to induce high levels of foci whereas CHX exposure resulted in little or no foci induction (Figure S3C). Together this demonstrates that CHX exposure, in contrast to HU exposure, is not sufficient to induce replication fork collapse although both drugs stall replication fork progression. Importantly, we observed a global increase of γH2AX levels via Western blotting at later timepoints of CHX treatment, comparable to that of HU treatment (Figure S3D), which was attenuated by pre-treatment with the proteasome inhibitor velcade (bortezomib) (Figure S3D). This suggests that CHX-induced toxicity leads to secondary cell death-associated DNA fragmentation resulting in induction of total γH2AX levels, which also can be observed microscopically by means of apoptotic bodies fully stained with γH2AX following CHX treatment (Fig. 2D, far right panel). As previously reported, dNTP depletion by HU combined with ATR inhibition results in highly increased γH2AX levels compared to HU treatment alone (Figure S3D) [18]. Notably, combining protein biosynthesis inhibition by CHX with an ATR inhibitor only resulted in a slight induction of γH2AX (Figure S3D). Altogether, the cellular signaling response to protein deficiency differs substantially from the response to dNTP depletion.
Short term CHX treatment is rapidly reversed whereas long-term treatment is toxic
Replication forks arrested by short-term HU treatment are able to re-start through a RAD51-dependent mechanism, whereas long-term HU blocks result in replication fork collapse and global replication is rescued by new origin firing [8]. In order to gain mechanistic insight into CHX-induced replication arrest, we exposed cells to CHX or HU for 1 hour and then released the cells into fresh media prior to EdU pulsing. We observed that the 1 hour short-term treatment with either CHX or HU was readily reversible with restored replication at 5 hours after drug release as measured by EdU fluorescence intensity (Fig. 3A, B). Interestingly, replication forks stalled by CHX were able to re-start efficiently already after 1 hour of drug release which is in contrast to HU stalled replication that required more time for efficient restart. Importantly, long-term (24 hours) HU treated cells recommenced EdU incorporation upon release from treatment whereas cells exposed to long-term CHX treatment were unable to do so (Figure S4A), indicating a proliferative block. Consistently, CHX treatment resulted in a gradual accumulation of cells in the G1-phase of the cell cycle followed by an increasing non-viable sub-G1 population with fragmented DNA (Figure S4B).
Figure 3.

Short-term cellular exposure to CHX is rapidly reversed whereas long-term exposure is toxic. (A) U2OS cells were treated with 10 μg/ml CHX or 2 mM HU for 1 hour, washed and released into fresh media for 1 or 5 hours, pulsed with 10 μM EdU for 20 minutes, followed by fixation and fluorescent labeling. (B) Quantification of relative fluorescence intensities in (A) (n = 2). Viability as determined by resazurin in U2OS cells (C) or immortalized BJ-hTERT cells (D) exposed to 10 μg/ml CHX or 2 mM HU for 24, 48 or 72 hours (n = 3). (E) Clonogenic survival assays of U2OS cells treated with 10 μg/ml CHX or 2 mM HU for 48 hours, followed by trypsinization and re-seeding of 500 cells in a 6-well format. After 10 days, cells were fixed and stained in 4% methylene blue-MeOH and quantified (n = 2). The error bars depict standard deviation; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 as determined by Student's t-test. See also Figure S4.
Next, we investigated the long-term effects associated with chronic depletion of protein synthesis on cellular survival. Although continuous cellular exposure to CHX resulted in a larger decrease in cellular viability compared to HU treatment at early timepoints, the decrease in cellular viability was similar at 72 hours for CHX and HU treatment in both U2OS cells and BJ-hTERT cells (Fig. 3C, D). Furthermore, 48 hours exposure to the drugs followed by re-plating of cells in fresh media for clonogenic outgrowth revealed a greater toxicity of HU compared to that of CHX (Fig. 3E).
Altogether, replication forks stalled by short-term CHX exposure restart rapidly compared to HU stalled forks. In contrast, long-term CHX treated cells are unable to resume replication during short recovery but during more extensive releases, as used in the clonogenic survival assay, a fraction of cells are capable to proliferate and survive, whereas the opposite holds true for HU treatment. This suggests that the inhibitory effects of long-term CHX treatment are partially reversible.
CHX treatment inhibits the production of new histones
The observed CHX-induced replication arrest is best explained by hypothesizing that (1) one or several essential replication factors are depleted due to their intrinsic instability, (2) de novo protein synthesis is required for continuation of DNA replication, or (3) a combined effect of diminished expression levels of pre-existing proteins and suppression of new protein production. In order to clarify this, we performed CHX chase experiments and studied the expression of core replisome components. Interestingly, all of the components studied remained stable for the first hours of CHX exposure, indicating that their expression is preserved (Fig. 4A) at times when DNA replication is completely halted (Fig. 1A).
Figure 4.

CHX does not immediately deplete replicative polymerases but disrupts new production of histones. (A) U2OS cells were either left untreated or treated with CHX for the timepoints indicated followed by Western blot probed with the specified antibodies. (B) Schematic protocol and representative images for fluorescent SNAP-tag labeling of U2OS cells stably expressing histone H3.1 or H3.3. Cells were quenched by DMSO (panel 1) or 0.2 μM blocking agent for 20 minutes (panel 2 and 3), labeled with 1 μM TMR for 30 minutes after a 3 hour release into fresh media +/- 10 μg/ml CHX. The contrast of the images in the middle panel of this figure has been equally enhanced in order to visualize the production of new histones after release from the quenching agent (panel 2), whereas the upper panel is unaltered. Furthermore, figure S4B and C (panel 3) shows cells without enhanced contrast, where arrows instead indicate new histone production. (C) Effect of HU on new histone H3.1 and H3.3 production via quenching of pre-existing histones using 0.2 μM blocking agent for 20 minutes, labeling with 1 μM TMR for 30 minutes after a 3 hour release into fresh media +/- 2 mM HU. (D) U2OS cells were treated with 10 μg/ml CHX or 2 mM HU for 24 hours and stained for RPA32. (E) U2OS cells were treated with 10 μg/ml CHX for 24 hours and co-stained for RPA32 and PML. See also Figure S5.
Short-term CHX treatment has previously been described as an unspecific approach to inhibit histone biosynthesis [17,19]. In order to directly study the production of new histones following CHX treatment, we used U2OS cells stably expressing SNAP-tagged histone H3 variants H3.1 or H3.3. The SNAP-tag based technology allows for visualization of new proteins by the use of a non-fluorescent quenching agent, which binds and blocks the SNAP-tag thus requiring new SNAP-tagged fused proteins to be synthesized to enable fluorescent labeling [20]. We found the labeling to be specific since U2OS cells that do not express any SNAP-tagged fusion protein remained non-fluorescent after pulse labeling with the fluorescent TMR dye (Figure S5A).
Following quenching of the pre-existing histone pools (Figure S5B and S5C, panel 1–2) it took approximately 3 hours of release from the quenching agent into fresh media in order for the cells to synthesize detectable amounts of new replication-dependent H3.1 and replication-independent H3.321 (Figure S5B and S5C, panel 3 and Figure 4B, panel 2). In non-CHX treated cells, newly synthesized histone H3.3 distributed evenly across the nucleus, whereas the replication-dependent histone variant H3.1 re-formed in a punctuate pattern (Fig. 4B). Co-labeling new histone H3.1 with EdU confirmed that this histone variant is found at replication foci (Figure S5D). Upon continuous CHX exposure during the chase period, de novo H3.1 and H3.3 production was completely blocked (Fig. 4B, panel 3, S5B and S5C, panel 4), while CHX had no effect on the pre-existing pool of H3.1 or H3.3 (S5B and S5C, panel 5). On the contrary, HU treatment only affected the replication specific histone H3.1, whereas synthesis of the replication-independent replacement histone H3.3 was largely unaffected (Fig. 4C). Overall, these results support the hypothesis that the drastic inhibitory effect of CHX on DNA replication might be linked to its blockage of histone supplies in S-phase.
As a consequence of histone management dysfunction, RPA has been reported to accumulate at telomeres that maintain their chromosome ends via alternative lengthening [22], a telomerase-independent mechanism relying on recombination between telomeric sequences [23]. When treating the ALT cell line U2OS with CHX, we could not see a redistribution of RPA into microfoci representing extensive stretches of ssDNA as compared to HU treatment (Fig. 2A). However, we did observe a gradual accumulation of RPA into larger nuclear foci (Fig. 4D). These foci resembled ALT-associated PML nuclear bodies (APBs), and, indeed, RPA co-localized with the APB marker PML after CHX treatment (Fig. 4E and S5E), further supporting that the drastic inhibitory effect of CHX on DNA replication might be linked to its blockage of histone supplies in S-phase.
Discussion
Numerous reports from as early as the 1970's clearly demonstrate that CHX blocks replication [2],[4],[24-29]. However, less is known about the potential mechanism and the cellular context in terms of activation of replication-associated checkpoints or restart of replication forks upon protein deprivation. Clearly, the replication stress response associated with protein deprivation has not been extensively investigated. In this study, we confirm earlier results from the 1970's using the DNA fiber assay and observe that progression of replication forks is obstructed by protein deprivation. A variety of agents stall replication fork progression, including agents binding directly to DNA or agents interfering with dNTP metabolism[6]. Common for these agents is that they also induce a DNA damage response in the cell [5,6]. Nevertheless, we report distinct mechanistic differences between protein and nucleotide deprivation stress (Fig. 5). Although replication forks were rapidly arrested following both protein and nucleotide deprivation, it is obvious that forks stalled by nucleotide deprivation subsequently collapse, whereas forks stalled by CHX endure (Fig. 5A and 5B).
Figure 5.
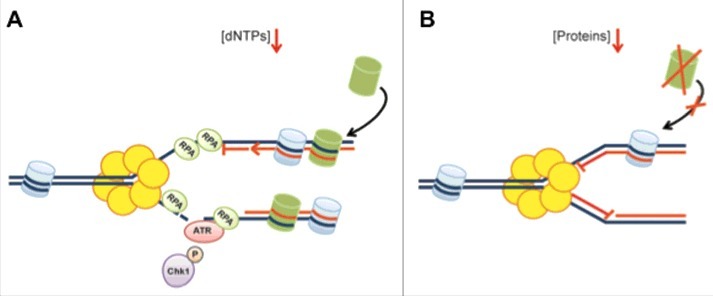
Overview of the cellular responses associated with protein and dNTP deprivation. (A) Replication fork arrest upon nucleotide deprivation is associated with replication fork uncoupling resulting in ssDNA generation, DNA damage and ATR-mediated checkpoint activation. (B) Replication fork arrest caused by protein deprivation occurs rapidly without subsequent exposure of ssDNA or DNA damage formation. This is likely caused by insufficient supply of newly synthesized histones (in green) and reduced DNA unwinding. Replication is readily resumed following short-term protein deprivation.
Helicase-polymerase uncoupling or resection at replication forks lead to the loading of RPA on ssDNA, which via ATR activates the DNA damage response pathway [30]. With ongoing resection and continuous RPA depletion, stalled replication forks become unprotected and substrates for cleavage by endonucleases generating collapsed replication forks, which are potentially highly toxic [18]. As expected, we show that HU treatment rapidly induces RPA foci, checkpoint signaling, γH2AX accumulation and DNA breaks as measured by the comet assay (Fig. 5A). Surprisingly, blockage of de novo protein production inhibits DNA replication without inducing the classical replication stress response including RPA foci, ATR signaling or DNA damage (Fig. 5B). Replication is also easily resumed following removal of short-term CHX treatment whereas HU stalled replication restart with slower kinetics. Overall, our notion is that replication fork uncoupling and exposure of ssDNA is prevented at high priority at replication forks arrested during conditions of protein deprivation.
Recent data from de Bruins lab investigating the role of E2F-dependent transcription in the replication stress response revealed that combining CHX and HU resulted in increased DNA damage as compared to HU treatment alone, while exposing cells to CHX alone, in line with our data, did not results in increased γH2AX signaling [31]. They hypothesized that de novo protein production is required to elicit a proper replication stress response following dNTP depletion by maintain levels of crucial proteins, and CHX pre-treatment therefore increases replication stress-associated DNA damage. In contrast to our study, Bertoli et al induced replication stress via HU and delineated the role of active protein translation in the associated stress response. Indeed, they find that active protein production is necessary in order to combat the threats posed by dNTP depletion and the succeeding DNA damage induction [31]. However, our study instead focuses on the finding that CHX treatment alone induces replication fork stalling and since we wanted to investigate the differences between dNTP and protein depletion, we did not combine HU and CHX. Therefore, our data are not contradictory, since we study different aspects of de novo protein production and the replication stress response.
In our study we make use of nucleoside analogues in order to examine the effect of CHX on DNA replication. One could speculate that proteins required for efficient nucleoside analogue incorporation into DNA could be adversely affected by short-term CHX treatment, including nucleoside transporter proteins and proteins of the nucleotide salvage pathway. However, we also used the nucleoside analogue EU in order to visualize nascent RNA transcription and did not observe an equivalent effect following CHX treatment as compared to EdU incorporation into DNA. EU also requires a functioning pyrimidine salvage pathway in order to be properly incorporated into nuclear RNA [13,32], suggesting that the diminished population of EdU positive cells observed after CHX treatment indeed reflects a replication defect and not an issue of low nuclear nucleoside pools available for incorporation. Nevertheless, the potential direct effect of CHX on the nuclear pools of EdU needs to be more carefully explored and measured.
There can be several potential explanations as to how CHX and other translation inhibitors stall replication fork progression. Diminished translation may generate a physical block on the template strand, but it is hard to envision how the non-nuclear translation process so rapidly would result in replication fork obstruction. Our observation that forks stalled by short-term exposures to CHX have the capacity to restart is also unlikely associated with any removal of physical obstructions. Another explanation is that proteins associated with the replication machinery or replication progression have such a short half-life that they would require constant translation. Although we only tested a handful of proteins we could not detect any direct effect by CHX treatment on factors directly associated with the replication machinery. A proteomic-wide analysis would be warranted to study all proteins. However, we did detect rapid cessation of histone biosynthesis. We therefore propose that it is the lack of histone loading that obstructs replication progression upon short-term translation inhibition, although the long-term effects following treatment with translation inhibitors likely represent toxicity linked to the complete halt in protein production
It is well established that replicating cells require synthesis of both DNA and histone proteins in order to faithfully package newly replicated DNA into chromatin [33]. Therefore, expression of the core histone genes, referred to as replication-dependent or canonical histones, is tightly coordinated with the rate of DNA synthesis. Consistently, reagents that interfere with DNA replication and dNTP metabolism result in a concomitant inhibition of histone synthesis [17,33,34]. Conversely, inhibition of histone synthesis results in impaired replication fork progression and DNA synthesis arrest, as has been demonstrated by depleting factors involved in histone gene expression and mRNA processing [17,35,36]. The importance of histones for DNA replication is also illustrated by disruption of de novo incorporation of H3.1/H4 by depletion of the histone chaperone chromatin assembly factor-1 (CAF-1), which results in stalled DNA replication and induction of cell death [37]. How the histone disposition mechanistically affects replication fork progression is still not established. However, one likely explanation is that it involves inhibiting DNA unwinding by the MCM2-7 replicative helicase as suggested before [19]. Histone H3 binds MCM2 with high affinity [38] and the histone chaperone anti-silencing function protein 1 (ASF1) form complex with MCF2-739 demonstrating the tight association of histones and the replicative helicase. In line with histones being the deprived proteins critical for replication, depletion of histone regulatory factors has previously been demonstrated to inhibit DNA replication without initially inducing checkpoint signaling or DNA damage [17]. The same is observed for short-term protein deprivation in our study. Moreover, we detect accumulation of RPA in ALT-associated PML bodies following CHX exposure. Localization of RPA to PML bodies has previously been observed following depletion of the histone chaperone ASF1, however why RPA localizes to APBs remains incompletely understood [22,39]. Also, in the case of histone deprivation, inhibiting checkpoint signaling might be beneficial, since activation of the ATR signaling pathway has been linked to the degradation of replication-dependent histone mRNAs after inhibition of DNA synthesis [33].
In conclusion, we here present distinct mechanistic responses to replication fork stalling induced by either nucleotide or protein deprivation stress (Fig. 5). Additionally, we suggest that histones are the critical factors for replication fork progression following protein deprivation (Fig. 5B). Without the supply of new histones it is probable that no further unwinding of DNA will take place and replication will consequently stall. It is tempting to speculate about the structure of a replication fork stalled by protein deprivation. However, the supply of nucleotides is likely sufficient for avoiding formation of ssDNA, triggering damage signaling and subsequent replication fork collapse during short-term protein deprivation. In light of this, further studies of protein deprivation is important since preserving intact replication forks during conditions of replication stress is critical for maintaining genome stability and cell survival.
Materials and methods
Cells and culture conditions
Human U2OS, BJ-hTERT, U2OS H3.1- and H3.3-SNAP were cultured in Dulbecco's modified Eagle's medium (Invitrogen, Paisley, UK) supplemented with 10% fetal bovine serum and Pen/Strep in a humidified CO2 atmosphere at 37°C. U2OS H3.1 and H3.3 cells [40] were kindly provided by Sophie Polo (Paris Diderot University) and maintained in 100 μg/ml G418.
Drug treatments
Compounds used in this study include: cycloheximide (Sigma-Aldrich; cat# C4859), hydroxyurea (Sigma-Aldrich; cat# H8627), 5,6-dichlorobenzimidazole 1-β-D-ribofuranoside (Sigma-Aldrich; cat# D1916), puromycin (Sigma-Aldrich; cat# P9620), aphidicolin (Sigma-Aldrich; cat# A0781), velcade (bortezomib; Selleckchem, cat# PS-341), ATR inhibitor (VE-821; Axon MedChem, cat# Axon 1893). If not otherwise specified, 10 μg/ml CHX, 2 mM HU, 1 μg/ml puromycin, 100 μM DRB, 0.4 μM aphidicolin, 10 nM velcade or 10 μM ATRi were added to the culture medium for the timepoints indicated.
Antibodies
The following antibodies were used in this study: rabbit anti-53BP1 (Abcam; cat# ab36823), mouse anti-β-actin (Abcam; cat# ab6276), mouse anti-Chk1 (Cell Singling; cat# 2360S), rabbit anti-p-Chk1-S345 (Cell signaling; cat# 2348S), mouse anti-Chk2 (Cell signaling; cat# 3440S), rabbit anti-p-Chk2-T68 (Cell Signaling; cat# 2661S), mouse anti-DNA polymerase δ, catalytic subunit (Abcam; cat# ab10362), mouse anti-DNA polymerase ε (Santa Cruz Biotechnology; cat# sc-56655), mouse anti-γH2AX-S139 (Millipore; cat# 05–636), rabbit anti-MCM7 (Abcam; cat# ab3732), mouse anti-p53 DO-1 (Santa Cruz Biotechnology; cat# sc-126), rat anti-p58 primase 8D3 (Cell Signaling; cat# 4726S), mouse anti-PCNA F-2 (Santa Cruz Biotechnology; cat# sc-25280), rabbit anti-PML (Santa Cruz Biotechnology; cat# sc-5621), mouse anti-PML (Santa Cruz Biotechnology; cat# sc-966), rabbit anti-RAD51 H-92 (Santa Cruz Biotechnology; cat# sc-8349), rabbit anti-RNA polymerase II CTD p-S2 (Abcam; cat# ab5095), rat anti-RPA32 (Cell Signaling; cat# 2208), rabbit anti-RPA70 (Cell Signaling; cat# 2267).
The secondary antibodies used were: donkey anti-rabbit Alexa Fluor® 555 (Life Technologies; cat# A-31572), goat anti-rabbit Alexa Fluor® 488 (Life Technologies; cat# A-11008), goat anti-mouse Alexa Fluor® 488 (Life Technologies; cat# A-11029), donkey anti-mouse Alexa Fluor® 555 (Life Technologies; cat# A-31570), goat anti-rat Alexa Fluor® 488 (Life Technologies; cat# A-11006), goat anti-rat Alexa Fluor® 568 (Life Technologies; cat# A-11077), IRDye® 800CW donkey anti-rabbit (LI-COR; cat# 926–32213), IRDye® 680RD donkey anti-rabbit (LI-COR; cat# 926–68073), IRDye® 800CW donkey anti-mouse (LI-COR; cat# 926–32212), IRDye® 680RD donkey anti-mouse (LI-COR; cat# 926–68072), IRDye® 800CW goat anti-rat (LI-COR; cat# 926–32219).
DNA fiber analysis
U2OS cells were left untreated or treated with 10 μg/ml CHX or 1 μg/ml puromycin for 1 hour, pulse-labeled with 25 μM CldU for 30 min, washed with medium and pulse-labeled with 250 μM for 30 min. Labeled cells were harvested and DNA fiber spreads were prepared as described elsewhere [41]. CldU was detected by incubating acid-treated fiber spreads with rat anti-BrdU monoclonal antibody (AbD Serotec; cat# MCA2060), whereas IdU was detected using mouse anti-BrdU monoclonal antibody (BD Biosciences; cat# 347580) for 1 hour in 37°C. Slides were fixed with 4% PFA and incubated with goat anti-rat Alexa Fluor 555 or goat anti-mouse Alexa Fluor 488 for 1.5-2 hours. Fibers were examined using a Zeiss (Jena, Germany) LSM710 confocal laser scanning microscope with a 63x oil immersion objective. For quantification of replication structures, at least 250 structures were counted per experiment. The lengths of red (AF 555) or green (AF 488) labeled patches were measured using the ImageJ software (National Institutes of Health; http://rsbweb.nih.gov/ij/) and arbitrary length values were converted into micrometers using the scale bars created by the microscope.
Immunofluorescence microscopy
Cells were grown on coverslips, fixed with 4% PFA for 15 min and permeabilized with 0.1% Triton-X-100 for 5 min. Cells were kept in blocking buffer for 1 hour (2% BSA, 5% glycerol, 0.2% Tween20, 0.1% NaN3), followed by 1 hour incubation in primary antibody and 30 minutes in secondary antibody. DNA was stained with DAPI and mounted using ProLong® Gold Antifade Mountant (Molecular Probes; cat# P36934). Imaging was carried out using a Zeiss LSM710 confocal laser scanning microscope and Zen software (2012).
For RAD51 or RPA32/70 immunostainings, cells were pre-extracted with CSK buffer (10 mM Pipes, pH 7.0, 100 mM NaCl, 300 mM sucrose, and 3 mM MgCl2, 0.7% Triton X-100) for 5 minutes prior to fixation.
Flow cytometry
Cells were harvested, washed in PBS and fixed in 70% ethanol for 60 min at -20°C or stored until analyzed. Cell were then stained in PBS containing 40 μg/ml propidium iodide, 100 μg/ml RNase A and 0.1% Triton X-100 for 1 hour in 4°C. Cell cycle profiles were analyzed using a Navios flow cytometer (Beckman Coulter) and Kaluza analysis software (version 1.2).
Western blotting
Cell were harvested, washed in PBS and proteins were extracted in lysis buffer containing 100 mM Tris-HCL at pH 8, 150 mM NaCl, 1% NP-40 supplemented with phosphatase and protease inhibitors, for 30 min on ice. Protein concentrations were determined using Pierce™ BCA protein assay kit (Thermo Fisher Scientific; cat# 23227) and Western blotting was carried out according to standard protocols.
Resazurin assay
Cells were seeded in 96-well plates (1500 cells/well) and treated with the indicated drugs and doses. 10 μg/ml resazurin was then added to the cells for 2 hours before analysis using a Hidex Sense microplate reader. The absorbance was normalized against background levels and the data processed in Microsoft Excel.
Clonogenic survival assays
Cells were seeded and treated with the indicated drugs and doses for 48 hours. Cells were then trypsinized, counted and re-seeded at a concentration of 250 cells/ml. After 7–10 days, cells were fixed and stained in 4% methylene blue-MeOH.
Ionizing radiation exposure
Cells were γ-irradiated in a Cs137 chamber with a dose rate of 0.5 Gy/min.
Comet assay
Cells were seeded in 6-well plates (150000 cells/well) and treated with indicated drugs and doses the following day. Treatment was removed after 24 hours and cells harvested by trypsinization and washed in PBS. Cell pellet was re-suspended in PBS to a concentration of 1 × 106 cells/μl, 50 μl was mixed with 250 μl of 1.2% low melting point agarose in PBS at 37°C. Cells were prepared by pipetting mixtures to fully frosted slides (Thermo-Fisher Scientific) precoated with a layer of 120 μl 1% normal temperature melting agarose. Slides were covered with coverslips and solidified on ice. After this, all processes were conducted at dark conditions. After gelling, coverslips was removed and slides were immerged in lysis buffer (10 mM Tris pH 10.0, 2.5 M NaCl, 0.1 mM EDTA, 10% DMSO and 1% Triton-X) at 4°C over night. Slides were moved to electrophoresis tank and immerged in alkaline electrophoresis buffer (0.3 N NaOH and 1 mM EDTA). Samples were denatured for 30 minutes and electrophoresis was performed at 300 mA, 25 V for 30 minutes. Slides were then washed with neutralization buffer (0.4 M Tris-HCl pH 7.5) and counterstained with 5 μM YOYO-1 dye (Invitrogen). Images were obtained using a 20X or 10X objective in a Zeiss LSM 510 confocal microscope and quantified using CometScore software. At least 100 comets per sample were analyzed and tail moment is calculated as per cent DNA in the tail multiplied by the tail length.
SNAP labeling of histones
Pre-existing SNAP-tagged histones were labeled by incubating cells with 1 μM SNAP-cell TMR star (New England Biolabs; cat# S9105S) for 30 minutes followed by 30 minutes incubation in fresh medium. For specific labeling of new histones, pre-existing histones were first quenched by incubating cells with 0.2 μM SNAP-cell Block (New England Biolabs; cat# S9106S) for 20 minutes, followed by wash in medium and release for 3 hours in fresh media (with or without CHX or HU) followed by TMR labeling as above. Cells were then fixed and counterstained with DAPI before image acquisition.
EdU incorporation
Cells were incubated in media supplemented with 10 μM EdU for 20 min, washed in PBS and then fixed using 4% PFA. EdU incorporation was visualized using Click-iT® EdU Alexa Fluor® 488 Imaging Kit (Life Technologies; cat# C10337) according to manufacturer's instructions. Cells were counterstained with DAPI before imaging.
EU incorporation
Cells were incubated in media supplemented with 0.2 mM EU for 45 min, washed in PBS and then fixed using 4% PFA. EU incorporation was visualized using Click-iT® RNA Alexa Fluor® 488 Imaging Kit (Life Technologies; cat# C10329) according to manufacturer's instructions. Cells were counterstained with DAPI before imaging. The mean EU fluorescence intensity per cell was obtained using ImageJ software by averaging the mean value of the EU signal measured in each nucleus.
Statistical analysis
Statistical significance was determined via two tailed Student's t-test using Microsoft Excel. The results originate from at least two independent experiments and are presented as mean ± standard deviation of the mean. Significance values were set at *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Supplementary Material
Funding Statement
This work was supported by the Swedish Children's Cancer Foundation (S.H, N.G), the Swedish Society for Medical Research (N.G), Swedish Research Council, the European Research Council, Göran Gustafsson Foundation, Swedish Cancer Society, the Swedish Pain Relief Foundation, and the Torsten and Ragnar Söderberg Foundation (T.H).
Acknowledgments
We thank members of the Helleday lab for discussions. The U2OS cells stably expressing H3.1 or H3.3 were a kind gift from Sophie Polo, Paris Diderot University, France.
Author summary
DNA replication is an essential nuclear process that ensures faithful duplication of the genome for its subsequent transfer to daughter cells during each round of the cell cycle. Accurate replication of genetic information must also be accompanied by restoration of the chromatin landscape on newly replicated DNA. Perturbations of these processes pose a potential threat to genome integrity as it is accompanied by DNA damage and subsequent mutations. A common way to investigate replication arrest is to disturb dNTP production and the cellular response to such a disturbance has been thoroughly studied. Here, we use another approach investigating the requirement of protein biosynthesis for functional DNA replication by utilizing the specific protein translation inhibitor cycloheximide. We find that replication is rapidly impaired by inhibition of protein biosynthesis, however, we find no evidence of DNA damage formation. This is surprising and implicate that this response is distinct from arresting DNA replication by inhibition of dNTP production.
References
- [1].Gaillard H, Garcia-Muse T, Aguilera A.. Replication stress and cancer. Nat Rev Cancer. 2015;15:276–89. doi: 10.1038/nrc3916. PMID:25907220 [DOI] [PubMed] [Google Scholar]
- [2].Hand R. DNA replication in mammalian cells. Altered patterns of initiation during inhibition of protein synthesis. J Cell Biol 1975;67:761–73. doi: 10.1083/jcb.67.3.761. PMID:1202023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Schulte D, Knippers R, Dreier T, Probst G, Probst H.. Cycloheximide inhibits cellular, but not SV40, DNA replication. FEBS Lett. 1992;299:149–154. doi: 10.1016/0014-5793(92)80235-9. PMID:1312038 [DOI] [PubMed] [Google Scholar]
- [4].Stimac E, Housman D, Huberman JA.. Effects of inhibition of protein synthesis on DNA replication in cultured mammalian cells. J Mol Biol. 1977;115:485–511. doi: 10.1016/0022-2836(77)90167-X. PMID:592371 [DOI] [PubMed] [Google Scholar]
- [5].Branzei D, Foiani M.. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11:208–19. doi: 10.1038/nrm2852. PMID:20177396 [DOI] [PubMed] [Google Scholar]
- [6].Zeman MK, Cimprich KA.. Causes and consequences of replication stress. Nat Cell Biol. 2014;16:2–9. doi: 10.1038/ncb2897. PMID:24366029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hanada K, Budzowska M, Davies SL, van Drunen E, Onizawa H, Beverloo HB, Maas A, Essers J, Hickson ID, Kanaar R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat Struct Mol Biol. 2007;14:1096–1104. doi: 10.1038/nsmb1313. PMID:17934473 [DOI] [PubMed] [Google Scholar]
- [8].Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T.. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. PMID:20188668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Saintigny Y, Delacôte F, Varès G, Petitot F, Lambert S, Averbeck D, Lopez BS. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 2001;20:3861–3870. doi: 10.1093/emboj/20.14.3861. PMID:11447127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, Liu JO. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol. 2010;6:209–217. doi: 10.1038/nchembio.304. PMID:20118940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grøfte M, Chan KL, Hickson ID, Bartek J, et al. . 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol. 2011;13:243–253. doi: 10.1038/ncb2201. PMID:21317883 [DOI] [PubMed] [Google Scholar]
- [12].Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, Jackson SP. Replication stress induces 53BP1-containing OPT domains in G1 cells. J Cell Biol. 2011;193:97–108. doi: 10.1083/jcb.201011083. PMID:21444690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Jao CY, Salic A.. Exploring RNA transcription and turnover in vivo by using click chemistry. Proc Natl Acad Sci U S A. 2008;105:15779–15784. doi: 10.1073/pnas.0808480105. PMID:18840688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bird G, Zorio DA, Bentley DL.. RNA polymerase II carboxy-terminal domain phosphorylation is required for cotranscriptional pre-mRNA splicing and 3'-end formation. Mol Cell Biol. 2004;24:8963–9. doi: 10.1128/MCB.24.20.8963-8969.2004. PMID:15456870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hsin JP, Manley JL.. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 2012;26:2119–37. doi: 10.1101/gad.200303.112. PMID:23028141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA.. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–52. doi: 10.1101/gad.1301205. PMID:15833913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mejlvang J, Feng Y, Alabert C, Neelsen KJ, Jasencakova Z, Zhao X, Lees M, Sandelin A, Pasero P, Lopes M, et al. . New histone supply regulates replication fork speed and PCNA unloading. J Cell Biol. 2014;204:29–43. doi: 10.1083/jcb.201305017. PMID:24379417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, Lukas J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. 2013;155:1088–1103. doi: 10.1016/j.cell.2013.10.043. PMID:24267891 [DOI] [PubMed] [Google Scholar]
- [19].Klimovskaia IM, Young C, Strømme CB, Menard P, Jasencakova Z, Mejlvang J, Ask K, Ploug M, Nielsen ML, Jensen ON, et al. . Tousled-like kinases phosphorylate Asf1 to promote histone supply during DNA replication. Nat Commun. 2014;5:3394. doi: 10.1038/ncomms4394. PMID:24598821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bodor DL, Rodriguez MG, Moreno N, Jansen L.E.. Analysis of protein turnover by quantitative SNAP-based pulse-chase imaging. Curr Protoc Cell Biol. 2012;Chapter 8, Unit8 8. doi: 10.1002/0471143030.cb0808s55. PMID:23129118 [DOI] [PubMed] [Google Scholar]
- [21].Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y.. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell. 2004;116:51–61. doi: 10.1016/S0092-8674(03)01064-X. PMID:14718166 [DOI] [PubMed] [Google Scholar]
- [22].O'Sullivan RJ, Arnoult N, Lackner DH, Oganesian L, Haggblom C, Corpet A, Almouzni G, Karlseder J. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat Struct Mol Biol. 2014;21, 167–74. doi: 10.1038/nsmb.2754. PMID:24413054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cesare AJ, Reddel RR.. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet. 2010;11:319–30. doi: 10.1038/nrg2763. PMID:20351727 [DOI] [PubMed] [Google Scholar]
- [24].Venkatesan N. Mechanism of inhibition of DNA synthesis by cycloheximide in Balb/3T3 cells. Biochim Biophys Acta. 1977;478:437–53. doi: 10.1016/0005-2787(77)90099-5. PMID:911842 [DOI] [PubMed] [Google Scholar]
- [25].Hershey H, Stieber J, Mueller GC. Effect of inhibiting the cellular synthesis of RNA, DNA and protein on DNA replicative activity of isolated S-phase nuclei. Biochim Biophys Acta. 1973;312:509–17. doi: 10.1016/0005-2787(73)90449-8. PMID:4724599 [DOI] [PubMed] [Google Scholar]
- [26].Gautschi JR, Kern RM. DNA replication in mammalian cells in the presence of cycloheximide. Exp Cell Res. 1973;80:15–26. doi: 10.1016/0014-4827(73)90270-X. PMID:4798835 [DOI] [PubMed] [Google Scholar]
- [27].Highfield DP, Dewey W.C. Inhibition of DNA synthesis in synchronized Chinese hamster cells treated in G1 or early S phase with cycloheximide or puromycin. Exp Cell Res. 1972;75:314–20. doi: 10.1016/0014-4827(72)90435-1. PMID:4674691 [DOI] [PubMed] [Google Scholar]
- [28].Nagata K, Enomoto T, Yamada MA. A system of DNA replication in HeLa nuclei treated with inhibitors of protein synthesis. Biochim Biophys Acta. 1981;653:316–30. doi: 10.1016/0005-2787(81)90188-X. PMID:7248294 [DOI] [PubMed] [Google Scholar]
- [29].Roufa DJ. Replication of a mammalian genome: the role of de novo protein biosynthesis during S phase. Cell. 1978;13:129–38. doi: 10.1016/0092-8674(78)90144-7. PMID:620420 [DOI] [PubMed] [Google Scholar]
- [30].Zou L, Elledge SJ.. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–8. doi: 10.1126/science.1083430. PMID:12791985 [DOI] [PubMed] [Google Scholar]
- [31].Bertoli C, Herlihy AE, Pennycook BR, Kriston-Vizi J, de Bruin RA. Sustained E2F-Dependent Transcription Is a Key Mechanism to Prevent Replication-Stress-Induced DNA Damage. Cell Rep. 2016;15:1412–22. doi: 10.1016/j.celrep.2016.04.036. PMID:27160911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Qu D, Zhou L, Wang W, Wang Z, Wang G, Chi W, Zhang B. 5-Ethynylcytidine as a new agent for detecting RNA synthesis in live cells by “click” chemistry. Anal Biochem. 2013;434:128–35. doi: 10.1016/j.ab.2012.11.023. PMID:23219562 [DOI] [PubMed] [Google Scholar]
- [33].Kaygun H, Marzluff WF.. Regulated degradation of replication-dependent histone mRNAs requires both ATR and Upf1. Nat Struct Mol Biol. 2005;12:794–800. doi: 10.1038/nsmb972. PMID:16086026 [DOI] [PubMed] [Google Scholar]
- [34].Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9. doi: 10.1158/0008-5472.CAN-04-2727. PMID:15604286 [DOI] [PubMed] [Google Scholar]
- [35].Zhao J, Dynlacht B, Imai T, Hori T, Harlow E. Expression of NPAT, a novel substrate of cyclin E-CDK2, promotes S-phase entry. Genes Dev. 1998;12:456–61. doi: 10.1101/gad.12.4.456. PMID:9472014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhao J, Kennedy BK, Lawrence BD, Barbie DA, Matera AG, Fletcher JA, Harlow E. NPAT links cyclin E-Cdk2 to the regulation of replication-dependent histone gene transcription. Genes Dev. 2000;14:2283–97. doi: 10.1101/gad.827700. PMID:10995386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nabatiyan A, Krude T. Silencing of chromatin assembly factor 1 in human cells leads to cell death and loss of chromatin assembly during DNA synthesis. Mol Cell Biol. 2004;24:2853–62. doi: 10.1128/MCB.24.7.2853-2862.2004. PMID:15024074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ishimi Y, Ichinose S, Omori A, Sato K, Kimura H. Binding of human minichromosome maintenance proteins with histone H3. J Biol Chem. 1996;271:24115–22. doi: 10.1074/jbc.271.39.24115. PMID:8798650 [DOI] [PubMed] [Google Scholar]
- [39].Groth A, Corpet A, Cook AJ, Roche D, Bartek J, Lukas J, Almouzni G. Regulation of replication fork progression through histone supply and demand. Science. 2007;318:1928–31. doi: 10.1126/science.1148992. PMID:18096807 [DOI] [PubMed] [Google Scholar]
- [40].Dunleavy EM, Almouzni G, Karpen GH. H3.3 is deposited at centromeres in S phase as a placeholder for newly assembled CENP-A in G(1) phase. Nucleus. 2011;2:146–57. doi: 10.4161/nucl.2.2.15211. PMID:21738837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Henry-Mowatt J, Jackson D, Masson JY, Johnson PA, Clements PM, Benson FE, Thompson LH, Takeda S, West SC, Caldecott KW. XRCC3 and Rad51 modulate replication fork progression on damaged vertebrate chromosomes. Mol Cell. 2003;11:1109–17. doi: 10.1016/S1097-2765(03)00132-1. PMID:12718895 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.