

Abstract
Limb remote ischemic preconditioning (RIPC) is an effective means of protection against ischemia/reperfusion (IR)–induced injury to multiple organs. Many studies are focused on identifying endocrine mechanisms that underlie the cross-talk between muscle and RIPC-mediated organ protection. We report that RIPC releases irisin, a myokine derived from the extracellular portion of fibronectin domain–containing 5 protein (FNDC5) in skeletal muscle, to protect against injury to the lung. Human patients with neonatal respiratory distress syndrome show reduced concentrations of irisin in the serum and increased irisin concentrations in the bronchoalveolar lavage fluid, suggesting transfer of irisin from circulation to the lung under physiologic stress. In mice, application of brief periods of ischemia preconditioning stimulates release of irisin into circulation and transfer of irisin to the lung subjected to IR injury. Irisin, via lipid raft–mediated endocytosis, enters alveolar cells and targets mitochondria. Interaction between irisin and mitochondrial uncoupling protein 2 (UCP2) allows for prevention of IR-induced oxidative stress and preservation of mitochondrial function. Animal model studies show that intravenous administration of exogenous irisin protects against IR-induced injury to the lung via improvement of mitochondrial function, whereas in UCP2-deficient mice or in the presence of a UCP2 inhibitor, the protective effect of irisin is compromised. These results demonstrate that irisin is a myokine that facilitates RIPC-mediated lung protection. Targeting the action of irisin in mitochondria presents a potential therapeutic intervention for pulmonary IR injury.
INTRODUCTION
Acute lung injury (ALI) is commonly encountered in hospital and outpatient settings and is associated with a high rate of morbidity and mortality. Many medical conditions, such as pulmonary embolism, thrombosis, acute respiratory distress syndrome (ARDS), and cardiopulmonary bypass surgery, can cause ischemia/reperfusion (IR)–induced lung injuries (1, 2). In infants born with neonatal respiratory distress syndrome (NRDS), insufficiency of pulmonary surfactant production and structural immaturity increase the susceptibility of the lung to IR injury(3). IR injury is often associated with activation of cellular inflammatory and oxidative stresses. IR-induced increase in reactive oxygen species (ROS) (4–6), in particular, can disrupt mitochondrial function and plasma membrane integrity and thus contribute to alveolar damage, lung edema, and hypoxemia that accompany ALI (7, 8). Preservation of mitochondrial function under IR stress represents a potential therapeutic target for treatment of ALI.
Mild exercise training has demonstrated beneficial effects in preventing pulmonary IR injury (9) and is associated with the reduced mortality in ARDS (10). Limb remote ischemic preconditioning (RIPC), defined as intermittent periods of ischemia applied at a remote limb muscle before prolonged IR injury of a target organ, is a noninvasive intervention that has substantial benefits in protection against tissue injuries (11). Humoral signaling pathways involving cross-talk between skeletal muscle and the target organ have been hypothesized to explain the beneficial effects of RIPC on ALI (12). However, the underlying mechanisms remain largely unknown.
Because of the unique vascular supply and the continuous physiologic demand for oxygen uptake and gas exchange, the lung is especially vulnerable to IR injury. Mitochondrial dysfunction is a key factor in lung IR injury (13, 14). Irisin, a type I membrane protein encoded by the Fndc5 gene, participates in mitochondrial biogenesis and oxidative metabolism (15, 16). Here, we tested the hypothesis that irisin functions as a myokine to protect against mitochondrial dysfunction during IR-induced pulmonary injury. We provide evidence that RIPC increases irisin in the bloodstream, and under IR stress, irisin transfers to injured alveolar cells. Through interaction with mitochondrial uncoupling protein 2 (UCP2), a homolog of UCP1, which is predominantly expressed in the lung tissue (17, 18), irisin preserves mitochondrial function and prevents IR-induced lung injury. We also found that intravenous administration of exogenous irisin can ameliorate IR-induced pulmonary injury in mouse models, and the pulmonary protective function of irisin is compromised in mice with genetic ablation of UCP2.
RESULTS
RIPC causes increase of irisin in the bloodstream and transfer to injured alveolar cells
We used Western blots to quantify the changes in serum concentration of irisin after IR-induced lung injury in a mouse model. Specificity of the irisin antibody was tested in fig. S1. Injury to the left lung was established by ischemia for 1 hour followed by reperfusion for another 1 hour in mice. As shown in Fig. 1A, mice subjected to RIPC, consisting of five cycles of 5-min ischemia/5-min reperfusion applied to the hindlimb, showed a time-dependent increase of irisin in the serum. RIPC rapidly raised irisin concentrations in the bloodstream (immediately after RIPC), and they reached about threefold of the basal level at 1 hour after RIPC (Fig. 1A). In mice subjected to lung IR injury, the serum concentration of irisin was significantly reduced when compared with sham-treated mice (Fig. 1B; P < 0.05). Moreover, under IR condition, the RIPC-induced increase in serum irisin was reduced, indicating the possibility of irisin transfer from the serum to the injured lung.
Fig. 1. Limb RIPC caused an increase in circulating irisin and translocation of irisin to injured lung alveoli.
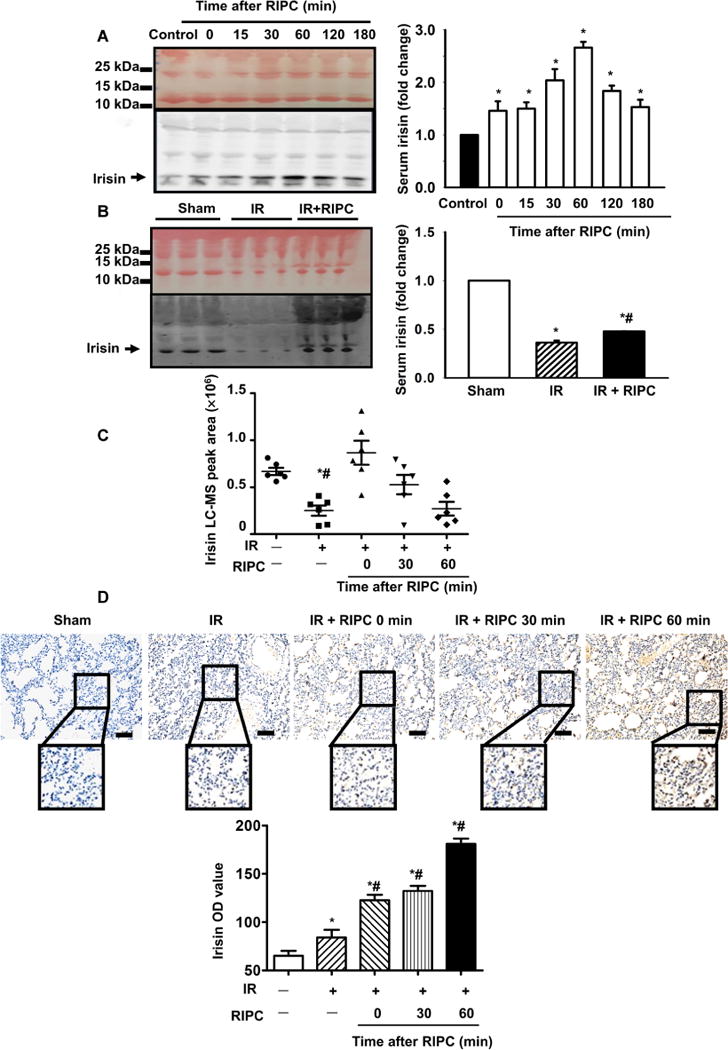
(A) Western blot of serum samples derived from mice subjected to remote ischemic preconditioning (RIPC) (20 μl of serum per lane). Colloidal staining served as loading control. Time course of RIPC-induced elevation of irisin in serum (*P < 0.05 versus control; n = 4). (B) Effect of ischemia/reperfusion (IR) on irisin concentration in mouse serum (*P < 0.05 versus sham and #P < 0.05 versus IR group; n = 6). (C) Time-dependent changes in irisin concentration in serum from mice after RIPC, with or without IR injury, determined by liquid chromatography–mass spectrometry (LC-MS) (*P < 0.05 versus non-IR sham mice and #P < 0.05 versus IR mice 0 min after RIPC; n = 6). (D) Immunohistochemical staining showed irisin expression in the IR-injured lung tissue from mice subjected to RIPC and lung IR injury (*P < 0.05 versus non-IR sham group and #P < 0.05 versus IR only group; n = 5; scale bars, 50 μm). OD, optical density.
In addition to Western blotting, we followed the protocol of Albrecht et al. (19) and established a label-free liquid chromatography–mass spectrometry (LC-MS) method to quantify the time-dependent changes in the serum concentration of irisin in mice subjected to IR and RIPC. The LC-MS demonstrated the presence of a short peptide, FIQEVNTTTR, which is present in irisin (fig. S2). Figure 1C showed that the irisin concentration in mouse serum is decreased upon lung IR injury. Although RIPC could initially raise the concentration of irisin, a time-dependent decline was observed (Fig. 1C).
Immunohistochemical (IHC) staining was used to quantify the changes in irisin protein expression in mouse lung tissue (Fig. 1D). IR injury resulted in accumulation of irisin in the lung, and there was a progressive increase of irisin protein in the injured lung tissue after RIPC treatment. RIPC applied to sham-treated mice did not induce irisin uptake into the alveolar cells (fig. S3). The reciprocal relationship of time-dependent decline of irisin in the serum (Fig. 1C) and concurrent accumulation of irisin in the lung tissue (Fig. 1D and fig. S4) support the notion IR makes irisin transfer from blood circulation to the injured lung tissue, which may have physiological consequences.
Infants with NRDS show reduced irisin concentrations in their serum
We conducted a pilot human study to establish the relationship between irisin and NRDS in newborn patients (see table S1). A total of 15 controls and 7 NRDS patients were enrolled in this study. The serum samples and characteristics of control infants and NRDS patients (including the samples and characteristics during acute illness and after recovery) were collected (Table 1). Using a protein liquid-chip assay, we found that the serum irisin concentrations derived from NRDS and recovery infants were considerably lower than those of healthy infants (Fig. 2A). On average, the serum of healthy infants contained 1113 ± 183 pg/ml irisin, the serum of patients with NRDS contained 331 ± 63 pg/ml irisin, and that of patients with NRDS after recovery from lung injury contained 553 ± 90 pg/ml irisin (Fig. 2B).
Table 1.
The characteristics of patients included in the study.
| Groups | n | Gender (male/female) | Gestational age (weeks) | Birth weight (kg) | 5-min Apgar score | SpO2 (%) | |
|---|---|---|---|---|---|---|---|
| Control | 15 | 7/8 | 33.5 ± 3.3 | 3.12 ± 0.12 | 9.3 ± 0.2 | 96.1 ± 1.8 | |
| NRDS | 7 | 3/4 | 31.6 ± 4.8 | 2.65 ± 0.29* | 6.9 ± 0.3* | Acute illness | 82.7 ± 5.1*† |
| Recovery | 92.9 ± 2.7 | ||||||
P < 0.05, versus control.
P < 0.05, versus neonatal respiratory distress syndrome (NRDS) infants after recovery.
Fig. 2. Infants with NRDS had reduced serum concentrations of irisin.
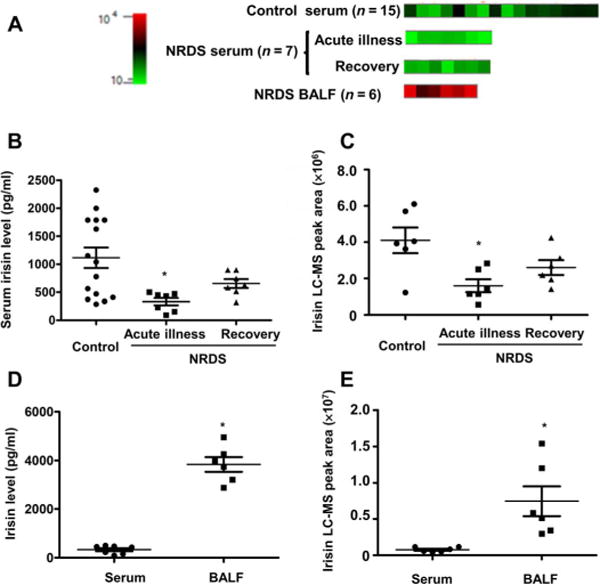
(A) Heat map of irisin expression in serum or bronchoalveolar lavage fluid (BALF) from each newborn patient or control infant was performed by protein liquid-chip assay (n = 15 in control, n = 7 in NRDS group for serum samples; six BALF samples were used in the study). Serum and BALF concentrations of irisin in NRDS infants were detected by an irisin protein liquid-chip assay and are provided as irisin concentration (B) or peak area (C) [*P < 0.05 versus control; n = 15 in control and n = 7 in NRDS group (B); *P < 0.05 versus the irisin concentration in serum; n = 7 for serum samples and n = 6 for BALF samples (C)]. Serum and BALF concentrations of irisin in NRDS infants were detected by LC-MS and are provided as irisin concentration (D) or peak area (E) [*P < 0.05 versus control; n = 6 in each group (D);*P < 0.05 versus the irisin concentration in serum; n = 6 in each group (E)].
Using the LC-MS method, we determined the serum concentrations of irisin in these infant populations (Fig. 2C; n = 6 in each group, partial samples from the chip study). Similar to the protein liquid-chip assay, there were significantly lower amounts of irisin in serum derived from NRDS patients compared with controls (P < 0.05). Although NRDS infants who recovered from lung injury appeared to have increased concentrations of irisin in the serum, because of the limited number of patient samples, we could not determine the statistical significance among the NRDS patients during acute illness and after recovery (Fig. 2C).
We also measured the irisin concentrations in bronchoalveolar lavage fluid (BALF) from the NRDS infants and found higher concentrations than those present in the serum (Fig. 2, D and E). Considering the lack of irisin expression in the lung under normal conditions (fig. S1), this indicates that irisin could transfer to lung from circulation in the condition of NRDS. Together, these findings suggest that the serum concentration of irisin may be a potential marker for human pulmonary diseases and that therapeutic means of increasing irisin concentrations in the serum may benefit treatment of lung injuries.
RIPC-mediated pulmonary protection is replicated by application of exogenous irisin
Previous studies demonstrated protective effects of RIPC against IR injury to the lung (20, 21). Because irisin is an important humoral factor released by skeletal muscle during RIPC, we also tested whether the protective effect of RIPC could be simulated by the application of exogenous irisin. Studies from other investigators have reported the basal serum concentrations of irisin to be in a range of 5 to 15 ng/ml in mice (22, 23). On the basis of this and our observation of about threefold increase in irisin after RIPC (Fig. 1A), we treated the mice with intravenous administration of irisin (1 μg/kg) given by bolus injection immediately after lung ischemia. Assuming a 2-ml blood volume with a mouse of 20-g body weight, intravenous administration of 1 μg/kg should increase irisin in the circulation by ~20 ng/ml and about two- to threefold increase over the basal concentration, as we observed with RIPC.
We found that implementation of RIPC improved the survival of mice that were subjected to IR treatment. Similarly, mice receiving irisin treatment also showed improvement in survival rate after IR treatment (Fig. 3A). To quantify the effect of irisin and RIPC on lung edema, we measured the wet/dry lung weight ratio after completion of the survival experiment. Exogenous irisin administration reduced edema in mice subjected to lung IR injury, to a similar degree as RIPC (Fig. 3B). Hypoxemia is another hallmark associated with lung injury. The arterial blood gas analysis showed that PaO2 was decreased and PaCO2 was increased in mice subjected to lung IR injury, but both irisin administration and RIPC ameliorated these hallmarks of lung function (Fig. 3, C and D). Moreover, both RIPC and exogenous irisin caused comparable reduction of proinflammatory cytokine concentrations, interleukin-1β (IL-1β) in Fig. 3E and IL-6 in Fig. 3F, in serum derived from mice subjected to lung IR injury. These results demonstrate that systemic administration of irisin has beneficial effects in preservation of lung function after IR injury.
Fig. 3. RIPC and exogenous irisin protected against IR injury in the mouse lung.
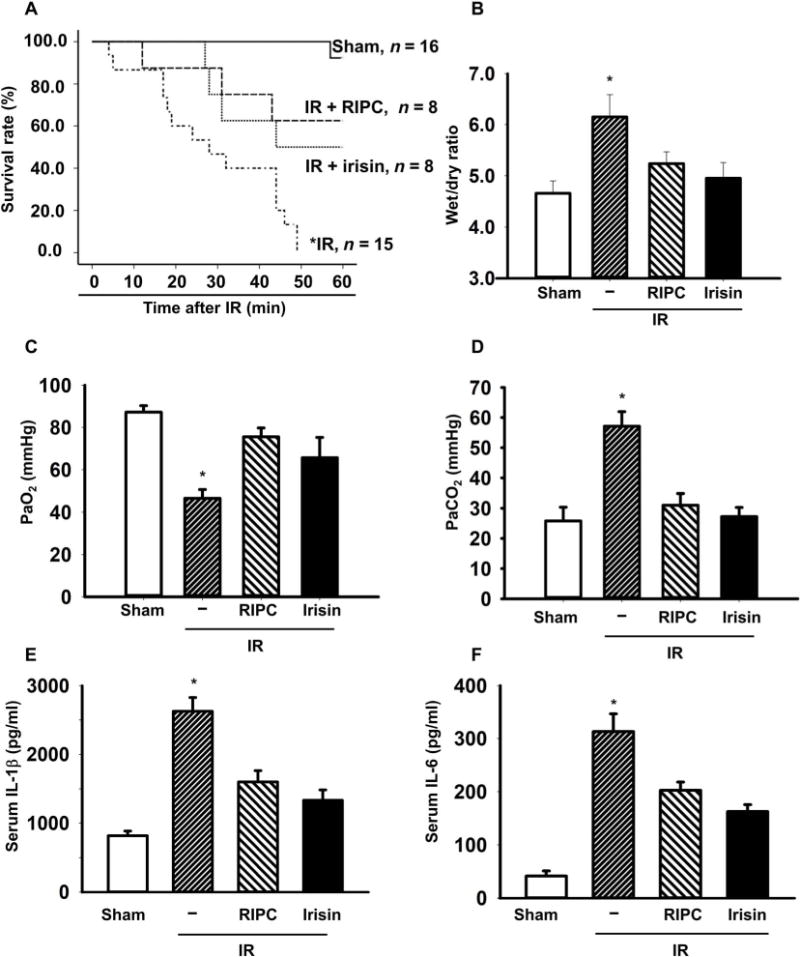
(A) The animal survival rate after lung IR injury with RIPC treatment or irisin administration (1 μg/kg) (*P < 0.05 versus others; n = 16 in sham group, n = 15 in IR group, and n = 8 in other groups). (B) Lung edema was evaluated as the wet/dry weight ratio of the excised lung from mice (*P < 0.05 versus others; n = 9 in sham group, n = 8 in IR group, and n = 4 in other groups). The plasma PaO2 (C) and PaCO2 (D) were measured (*P < 0.05 versus others; n = 8 in sham and IR group and n = 4 in other groups). A.U., arbitrary units. Serum concentrations of interleukin-1β (IL-1β) (E) and IL-6 (F) were measured using enzyme-linked immunosorbent assay (*P < 0.05 versus others; n = 4).
Irisin targets mitochondria to ameliorate apoptosis of lung epithelial cells during IR injury
Using IHC staining, we found that the intravenously administered exogenous irisin could target IR-injured lung tissue but not the sham-operated lung tissue (Fig. 4A). Irisin staining was not visible in lung tissue derived from sham-operated mice (Fig. 4B, top). Alveolar epithelial cells took up endogenous irisin after IR injury because the irisin signal was mostly surrounded by AT1a, an alveolar type I epithelial cell marker (Fig. 4B, middle). Intravenous administration of exogenous irisin (1 μg/kg) increased accumulation of irisin in the alveolar cells after IR injury (Fig. 4B, bottom).
Fig. 4. Irisin protected against oxidative stress and apoptosis in IR-injured lung tissue.
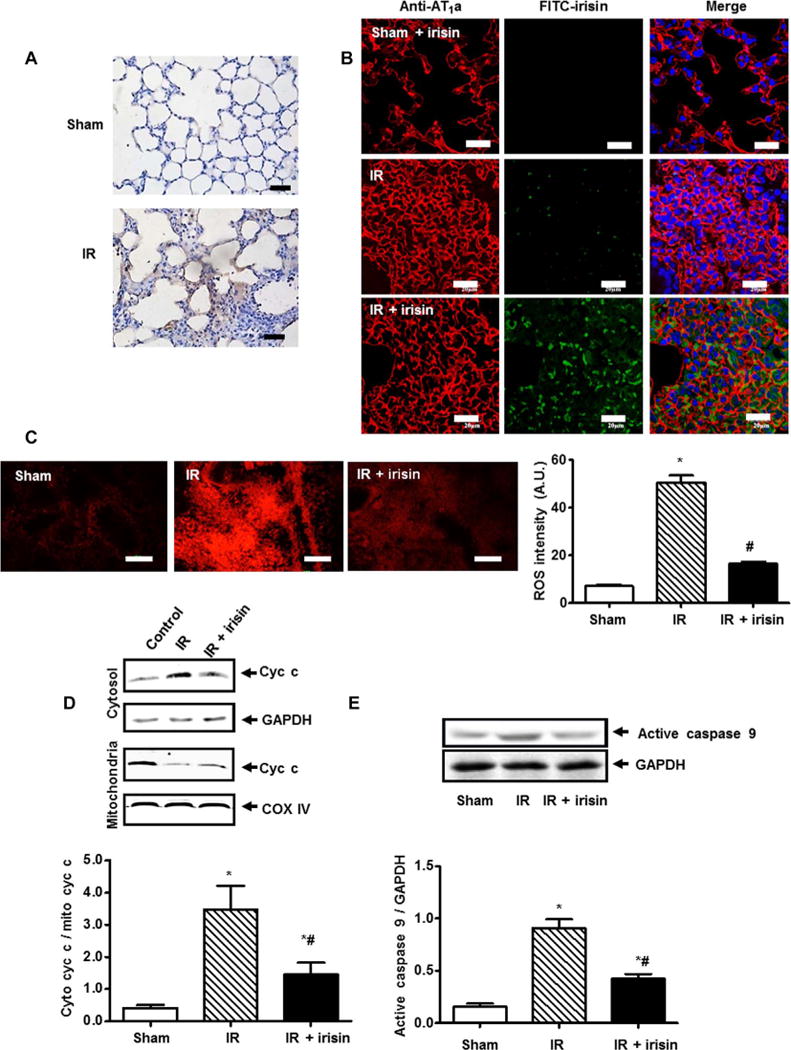
(A) Immunostaining showed that irisin was detected in the mouse lung tissue after IR injury with irisin treatment but not in sham-operated animals (scale bars, 50 μm). (B) Lung tissue sections were immunostained for irisin (green) and AT1a (red, alveolar type I epithelial cell marker) (scale bars, 20 μm). (C) The extent of reactive oxygen species (ROS) production in mouse lung tissue was determined by dihydroethidium staining (*P < 0.05 versus sham group and #P < 0.05 versus IR group; n = 5; scale bars, 100 μm). (D) Assessment of cytochrome c (cyc c) in mitochondria and cytosol derived from lung tissue subjected to IR (*P < 0.05 versus sham and #P < 0.05 versus IR; n = 4). COX IV, cyclooxygenase IV; GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (E) Caspase 9 activation in lung tissue subjected to IR (*P < 0.05 versus sham and #P < 0.05 versus IR; n = 4).
As a key factor in lung IR injury, mitochondria dysfunction causes cellular energy depletion, ROS generation, and mitochondria-induced cellular apoptosis (13, 14). Previous studies established a link between irisin and the metabolic function of mitochondria in multiple tissues (24). We found that mouse lung tissue subjected to IR injury showed enhanced dihydroethidium (DHE) fluorescence intensity, indicative of increased ROS, whereas irisin treatment significantly reduced ROS in IR-injured lung tissue (Fig. 4C; P < 0.05). A biochemical assay showed that IR injury to the lung caused release of cytochrome c from mitochondria, and systemic administration of exogneous irisin could reduce the release of cytochrome c (Fig. 4D). The effect of irisin in prevention of apoptotic cell death was evident in immunoblotting of caspase 9, a key enzyme involved in execution of apoptosis. Figure 4E showed that caspase 9 was activated in IR-treated lung (represented by the cleavage product of caspase 9), and administration of irisin reduced caspase 9 activity in IR-injured lung. Using Seahorse measurement of the mitochondrial metabolic function, we found that the oxygen consumption rate (OCR) of the basal respiration and adenosine 5′-triphosphate (ATP) production were decreased in IR-injured lung tissue, and irisin increased OCR and ATP production in lung tissue derived from IR-treated mice (fig. S5). Overall, these results suggest that irisin-mediated pulmonary protection involves mitochondria-dependent apoptosis.
In addition to the mouse studies, we conducted experiments with three different lung epithelial cell lines, including A549 cells (a human lung epithelial cell line), RLE-6TN cells (a rat lung alveolar epithelial cell line), and BEAS-2B cells (a human lung bronchus epithelial cell line). These lung cells do not express endogenous irisin (fig. S1), thus allowing convenient assessment of the exogenous irisin on the mitochondria function under stress conditions. Figure 5A showed that A549 cells subjected to anoxia/reoxygenation (AR; 2-hour anoxia and 2-hour reoxygenation) could take up exogenous irisin in a time-dependent manner. Similar observations were made with RLE-6TN and BEAS-2B cells (fig. S6 and movies S1 to S3).
Fig. 5. Irisin protected mitochondria from apoptosis in cultured lung epithelial cells subjected to AR injury.
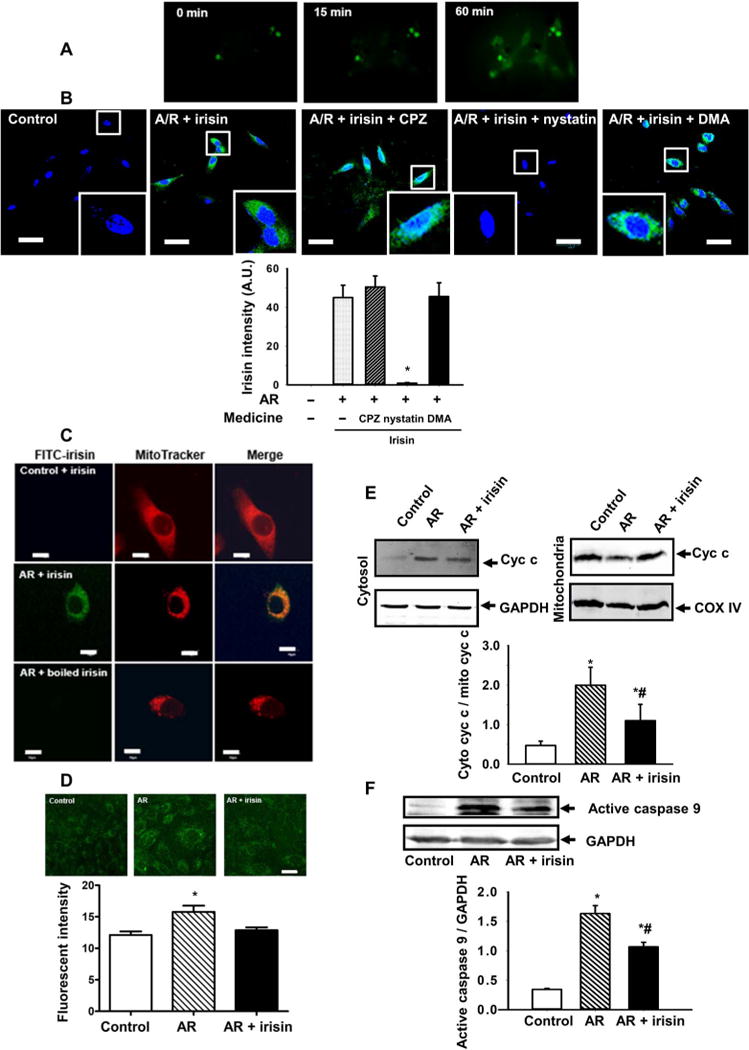
(A) Accumulation of exogenous irisin in A594 cells after anoxia recorded by a time-lapse live cell system (see also movies S1 to S3). (B) Pharmacological agents that disrupt endocytic pathways were used to test their effect on uptake of irisin into A594 cells. Representative images and quantification data are presented. CPZ (chloropyrazine) is an inhibitor of clathrin-mediated endocytosis; DMA (dimethylacetamide) is an inhibitor of large endocytic uptake pathway; and nystatin is an inhibitor of lipid raft–mediated endocytosis. The staining was repeated five times, and 15 cells in each slide were randomly chosen for fluorescence intensity analysis [*P < 0.05 versus other anoxia/reoxygenation (AR) groups; scale bars, 50 μm]. (C) A549 cells were incubated with MitoTracker and fluorescein isothiocyanate (FITC)–conjugated irisin for 60 min. Cells did not take up FITC-irisin at the normal condition (top), with measurable uptake of irisin after AR treatment for 2 hours (middle). FITC-boiled irisin was used as a negative control (bottom). Note that FITC-irisin shows overlap with MitoTracker in AR-treated A549 cells. Scale bars, 10 μm. (D) The amount of ROS in the A549 cells was determined by MitoSOX Red (green) (*P < 0.05 versus others; n = 4; scale bar, 5 μm). (E) Assessment of cyc c in mitochondria and cytosol derived from AR-treated A549 cells (*P < 0.05 versus control and #P < 0.05 versus AR; n = 4). (F) Caspase 9 activation in AR-treated A549 cells (*P < 0.05 versus control and #P < 0.05 versus AR; n = 4).
Because entry of exogenous irisin was only observed in lung tissues or cells subjected to either IR or AR, recognition of stress-induced extracellular signal plus receptor-mediated endocytosis are most likely required for penetration of irisin into the injured alveolar cells. We conducted confocal microscopic imaging of AR-treated A549 cells and tested the effects of several pharmacological agents that disrupt endocytic pathways for uptake of irisin into cells. As shown in Fig. 5B, AR treatment stimulated irisin uptake into A549 cells. Addition of either chloropyrazine (CPZ), a known inhibitor of clathrinmediated endocytosis (25), or dimethylacetamide (DMA), a known inhibitor of large endocytic uptake pathway (26), did not affect uptake of irisin into A549 cells. However, the application of 50μM nystatin, a known inhibitor of lipid raft–mediated endocytosis (27), blocked the uptake of irisin into A549 cells after AR treatment. This result supports a role for lipid raft–mediated endocytosis in facilitating entry of irisin into cells that are exposed to IR stress.
Confocal microscopy imaging showed that the exogenous irisin could target mitochondria in A549 cells, as shown by colocalization of MitoTracker Deep Red and fluorescein isothiocyanate (FITC)–labeled irisin (green) (Fig. 5C). As a control, denatured irisin (boiled for 10 min at 100°C) did not show intracellular entry in A549 cells under AR conditions. Similar observations were made with the RLE-6TN and BEAS-2B cells (fig. S7). Irisin targeting mitochondria significantly reduced ROS in AR-injured cells (Fig. 5D; P < 0.05). Increased release of cytochrome c was observed in A549 cells after AR treatment, and irisin could reduce cytochrome c release (Fig. 5E). Moreover, similar to what we observed with the IR-treated lung tissue, the activation of caspase 9 activity in AR-treated A549 cells was reduced by the addition of irisin (Fig. 5F). In fig. S8, we showed that cultured lung epithelial cells, including A549, RLE-6TN, and BEAS-2B, displayed reduced viability after AR treatment, and incubation with exogenous irisin could improve their survival. In addition, AR-induced increases in lactate dehydrogenase (LDH) activity in culture medium derived from all three types of lung epithelial cells were reduced when irisin was present in the solution.
Irisin interacts with UCP2 to preserve mitochondrial function during IR
UCPs control mitochondrial function in several tissues (28), and irisin has been found to modulate UCP1 expression and act on the mitochondrial biogenesis in white adipose tissue (29). As compared with the other members of the UCP family, UCP2 is the predominant isoform expressed in lung tissue (17, 28) and shares 55 to 60% homology with UCP1 (17, 18). We found that IR-treatment reduced the expression of UCP2 protein in mouse lung tissue, and administration of exogenous irisin could partially restore UCP2 expression (Fig. 6A). A similar result was also found in AR-injured A549 cells (Fig. 6B).
Fig. 6. The protective effect of irisin on mitochondrial function is associated with UCP2.
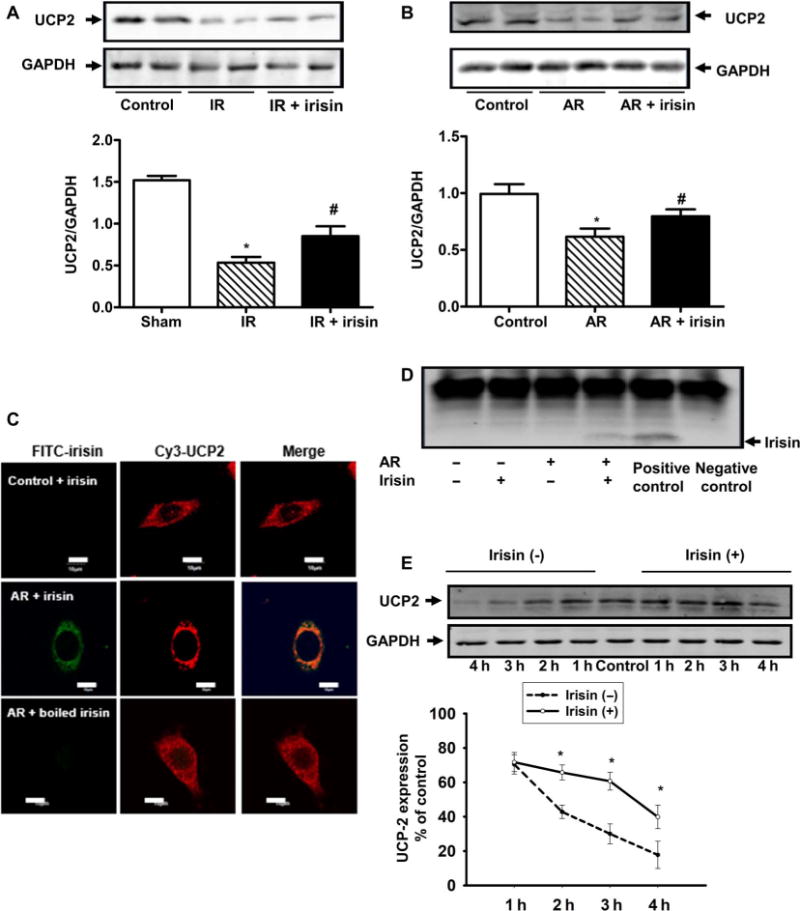
(A) Uncoupling protein 2 (UCP2) protein expression in mouse lung tissue after IR, with or without irisin treatment, was determined by immunoblotting. IR injury reduced the expression of UCP2, whereas irisin (1 μg/kg) treatment partially rescued UCP2 expression (*P < 0.05 versus sham control mice and #P < 0.05 versus lung IR injury mice; n = 5). (B) UCP2 protein expression in A549 cells after AR with or without irisin treatment was determined by immunoblotting (*P < 0.05 versus control group and #P < 0.05 versus AR group; n = 4). (C) Colocalization of irisin and UCP2 in A549 cells. A549 cell was first treated with FITC-irisin (0.1 μg/ml) or FITC-boiled irisin and then immunostained with anti-UCP2 antibody. Colocalization of irisin and UCP2 was observed after AR injury (scale bars, 10 μm). (D) Coimmunoprecipitation of irisin and UCP2 in A549 cells. Cell lysates were immunoprecipitated with anti-UCP2 antibody and then immunoblotted with anti-irisin antibody. For negative control, rabbit immunoglobulin G was used for immunoprecipitation. Anti-UCP2 antibody was used for both immunoprecipitation and immunoblot as a positive control. (E) Time course of UCP2 degradation in A549 cells during AR treatment. The cells were incubated with cycloheximide (10−5 M) to inhibit protein synthesis (*P < 0.05 versus without irisin treatment; n = 8).
To understand the potential role of UCP2 in irisin-mediated protection of mitochondrial function, we used confocal microscopy to examine the subcellular distribution of irisin and UCP2. Using FITC-labeled irisin, we observed a high degree of colocalization between FITC-irisin and Cy3-UCP2 in lung epithelial cells subjected to AR treatment, although there was no colocalization in control cells (Fig. 6C and fig. S9). The colocalization of irisin and UCP2 was lost if we used denatured irisin. We next performed coimmunoprecipitation studies and found that lung epithelial cells subjected to AR with supplementation of exogenous irisin displayed physical interaction between irisin and UCP2; the interaction was no longer noted in control cells with irisin treatment or AR-treated cells without irisin (Fig. 6D). To examine whether irisin treatment could prevent degradation of UCP2 in response to AR, we treated A549 cells with cycloheximide (CHX, 10−5M) to inhibit protein synthesis. As shown in Fig. 6E, in the presence of CHX, the half lifetime for UCP2 was increased from 1.5 ± 0.14 hours under control conditions to 3.51 ± 0.21 hours when irisin (0.1 μg/ml) was present in the culture medium. On the basis of these results, we conclude that functional interaction between irisin and UCP2 could protect UCP2 from degradation. These effects likely underlie the irisin-mediated amelioration of IR-induced oxidative stress in alveolar epithelial cells and improvement of mitochondrial function associated with pulmonary injury.
Genetic ablation of UCP2 abolishes the pulmonary protective function of irisin
To further elucidate the physiological role of UCP2 and its interaction with irisin in pulmonary protection, we used a mouse model with genetic ablation of Ucp2 (30, 31). The Ucp2−/− mice are viable, thus enabling us to examine their physiological function under stress conditions. In normal physiologic conditions, there are no significant differences in ROS and inflammation between wild-type (WT) and Ucp2−/− mice (fig. S10). Moreover, the Ucp2−/− micedonot show abnormal lung structure under normal conditions (Fig. 7A, left).
Fig. 7. Genetic ablation of UCP2 compromised the protective effect of exogenous irisin on lung IR injury.
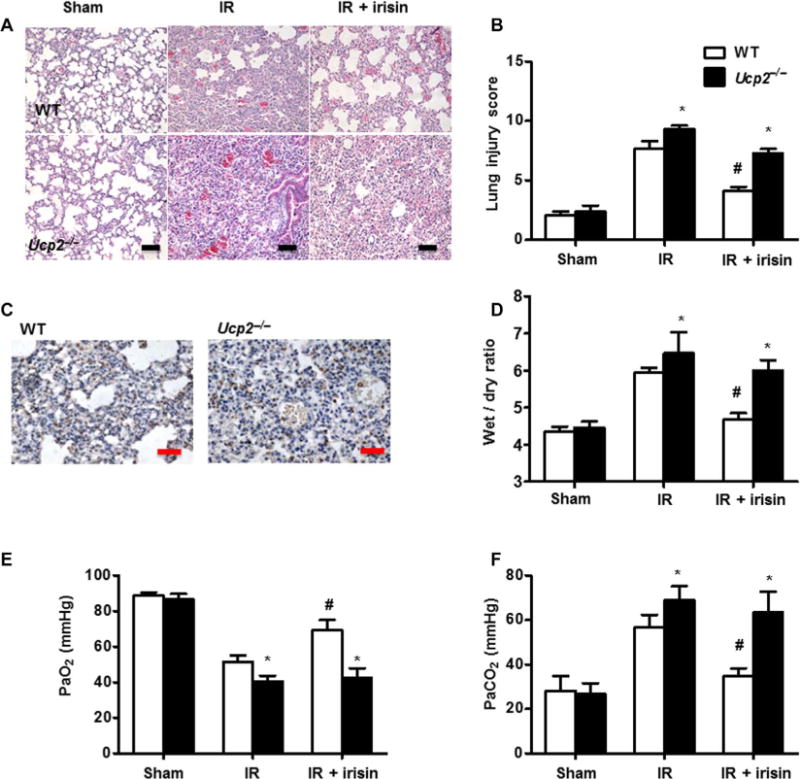
(A) Histopathological changes in IR-injured lung in WT or Ucp2−/− mice, with or without irisin administration (scale bars, 50 μm). (B) Histopathological assessment (lung injury score) of pulmonary tissue for each group is summarized. The staining for each group was repeated at least five times, and 15 fields of vision in each slide were chosen for histopathological assessment (*P < 0.05 versus WT mice under same conditions and #P < 0.05 versus IR group in WT mice). (C) Immunohistochemical staining shows targeting of irisin to the injured alveolar cells in WT and Ucp2−/− mice (scale bars, 50 μm). (D) Lung edema, determined by wet/dry ratio in WT and Ucp2−/− mice after IR injury (*P < 0.05 versus WT mice under same conditions and #P < 0.05 versus IR group in WT mice; n = 5). Measurement of PaO2 (E) and PaCO2 (F) in WT and Ucp2−/− mice after IR injury (*P < 0.05 versus WT mice under same conditions and #P < 0.05 versus IR group in WT mice; n = 5).
IR-induced lung injury was more severe in Ucp2−/− mice than in WT mice, with increased hemorrhage and inflammatory cell infiltration observed in the interstitium and alveoli of the Ucp2−/− mice (Fig. 7, A and B). When exogenous irisin was intravenously administered to the Ucp2−/− mice, we observed no significant difference in the targeting of irisin to the injured alveolar cells between WT and Ucp2−/− mice (Fig. 7C). In addition, arterial blood gases (PaO2 and PaCO2) and wet/dry ratio were more altered in IR-induced lung injury in Ucp2−/− mice than in WT mice (Fig. 7, D to F). Intravenous administration of irisin (1 μg/kg) protected the lungs from IR injury in WT mice. However, the protective effects of exogenous irisin were markedly reduced in Ucp2−/− mice (Fig. 7, A and D to F), indicating that UCP2 plays a role in the protective function of irisin in IR injury to the lung.
To further explore the contribution of UCP2 in irisin-mediated protection of IR-injured lung, we used genipin, a known inhibitor of UCP2 (32), to treat mice orally (50 mg/kg) 30 min before lung ischemia. The wet/dry ratio (Fig. 8A) and the gas exchange function (Fig. 8, B and C) were measured. The protective effects of irisin on lung edema and lung function were blocked by genipin. We further tested whether genipin affected the function of irisin in AR-injured lung epithelial cells. AR injury increased the amount of LDH in the supernatant of A549 cell culture (Fig. 8D). Similar to the data shown in fig. S8B, incubation of A549 cells with irisin reduced LDH. In the presence of genipin, the protective effect of irisin was partially lost. Moreover, the effect of irisin on caspase 9 activity and cytochrome c release was also largely blocked by genipin (Fig. 8, E and F).
Fig. 8. Irisin inhibitor eliminated the protective effect of irisin on IR-injured lung.
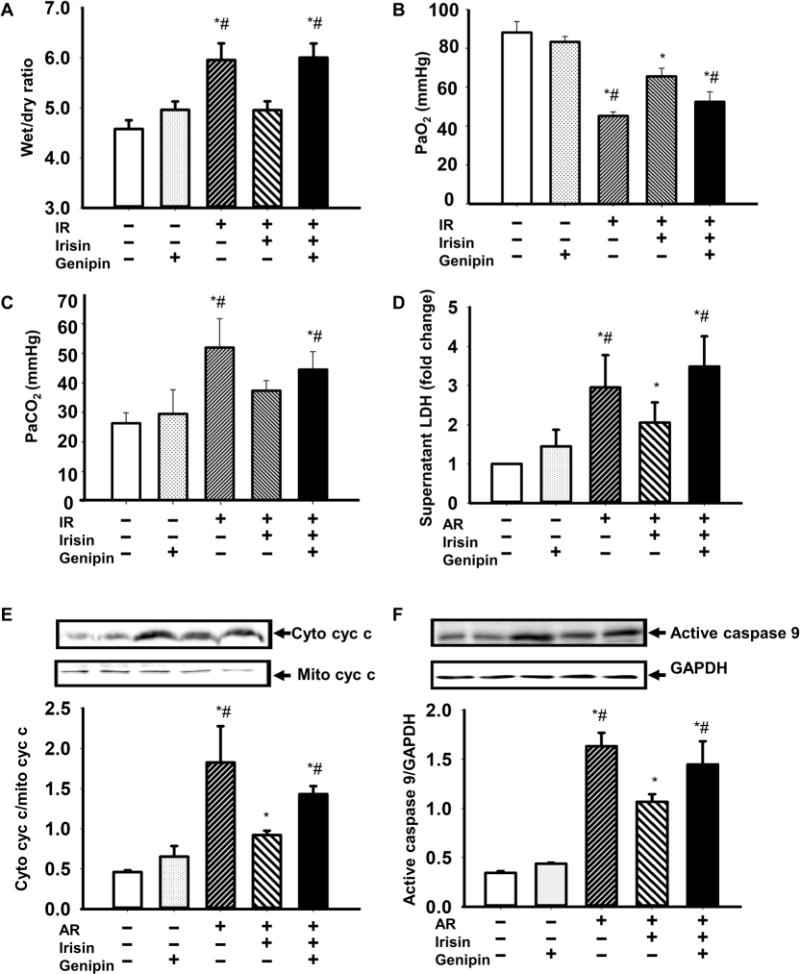
(A) Effect of irisin and genipin administration on lung edema in IR mice. Arterial blood samples were drawn from individual mice, and plasma PaO2 (B) and PaCO2 (C) were measured; n = 5 for control, 4 for genipin alone, 4 for IR, 4 for (IR + irisin), and 4 for (IR + irisin + genipin) (*P < 0.05 versus control and #P < 0.05 versus irisin-treated lung IR mice). (D) Effect of genipin on lactate dehydrogenase (LDH) concentration in supernatant from A549 cells after AR with or without irisin administration (*P < 0.05 versus control cells and #P < 0.05 versus irisin-treated AR cells; n = 4). (E and F) Effect of genipin on mitochondria-mediated apoptosis in A549 cells after AR, with or without irisin administration. Cyc c release (E) was expressed as the ratio of cyc c in cytosol and mitochondria, and caspase 9 activity (F) was expressed as the ratio of active caspase 9 and GAPDH (*P < 0.05 versus control cells and #P < 0.05 versus irisin-treated AR cells; n = 4).
DISCUSSION
Data presented in this study support the concept that circulating irisin alleviates lung IR injury through preservation of mitochondrial function. Although lung alveolar cells do not express endogenous irisin, irisin can transfer from blood circulation to the injured alveoli after IR treatment. In human infants with NRDS, the serum concentrations of irisin are lower than those present in control newborns. Ischemic preconditioning applied to the limb muscle ameliorates lung IR injury through elevation of irisin in circulation. Systemic administration of exogenous irisin protein protects the lung fromIR injury in mice through reduction of ROS activity and mitochondria-dependent apoptosis. Biochemical and cell imaging studies show that irisin interacts with UCP2 in mitochondria to preserve its function in controlling oxidative stress associated with lung injury. Mice with knockout of UCP2 display increased susceptibility to lung IR injury relative to the WT littermates, and the protective effect of irisin is compromised in the Ucp2−/− animals. On the basis of these data, we conclude that a functional interaction between irisin and UCP2 contributes to maintenance of pulmonary integrity under pathologic conditions of excessive oxidative stress.
Skeletal muscle has recently been recognized as an endocrine organ that not only contains metabolically important molecules but also communicates with other tissues through the secretion of hormones, known as myokines, that are released into circulation during or immediately after physical activity (33). Irisin is an exercise-induced hormone secreted by skeletal muscle that drives brown fat–like conversion of white adipose tissue, which improves systemic metabolism by increasing energy expenditure (29). Because of these properties, irisin has emerged as an appealing therapeutic target for treatment of metabolic diseases (29, 34–36). However, published research on irisin faced several controversies regarding the method for detection/quantification of irisin protein expression and deciphering the long-term versus acute action of irisin in physiological or pathological settings (19, 37, 38). Here, we used four complementary methods, including Western blotting, IHC staining, protein liquid-chip assay, and LC-MS, for quantification and subcellular localization of irisin in lung epithelial cells and tissues under stress conditions. We present data to show that human and mouse lung epithelial cells do not express endogenous irisin protein but can take up irisin from the extracellular environment under AR or IR conditions.
RIPC as a noninvasive intervention can be readily applied in a hospital setting or at home as a prophylactic means to protect against injury to the lung or other organs via different humoral factors (39). We showed that RIPC increased irisin in the bloodstream, whereas IR would make irisin transfer from blood circulation to the injured lung tissue. On the basis of the reciprocal relationship of time-dependent decline of irisin in the serum and the concurrent accumulation of irisin in mice subjected to RIPC and lung IR injury, we conclude that transfer of irisin from the bloodstream to the injured lung tissue underlies the pulmonary protective function of irisin. Under IR condition, we show that injury to the lung can stimulate transfer of endogenous irisin from blood circulation to alveolar cells. However, the amount of endogenous irisin accumulation inside the cells is unlikely to be sufficient for pulmonary protection during IR injury, especially in NRDS infants. Thus, systemic administration of exogenous irisin would supplement the endogenous irisin and offer a potential treatment modality for NRDS.
Mitochondrial dysfunction is a key factor in lung IR injury, and impaired mitochondrial integrity may predispose cells to energy depletion, free radical generation, and eventual cell death (13). One contributing factor for ALI is the excessive ROS generation (1, 6, 7), which could damage the alveolar-capillary interaction and disrupt integrity of the lung epithelia, leading to lung edema (40). Irisin participates in mitochondrial biogenesis and oxidative metabolism (29). Although previous studies by other investigators have demonstrated the myokine function of circulating irisin in regulating lipogenesis (41, 42), little is known on the role of irisin in pulmonary protection. Here, we present evidence that IR-induced elevation of ROS and inflammatory factors (such as IL-1b and IL-6) can be suppressed by irisin. We found that systemic administration of exogenous irisin before IR injury can preserve lung function and prevent lung edema. Targeting of exogenous irisin to the injured alveolar cells requires recognition of injury-associated extracellular signal plus vesicle- or receptor-mediated endocytosis. Through the use of pharmacological agents that disrupt endocytic pathways, we provide evidence that lipid raft–mediated endocytosis plays an important role in facilitating the entry of irisin into the lung epithelial cells under IR stress conditions. Thus, irisin can potentially be used as a prophylactic agent in the medical settings of anticipated lung injury.
The protective effect of irisin on ALI involves UCP2-related preservation of mitochondrium function. Mitochondrial UCP plays important roles in regulating cellular metabolic function and its response to physiologic or pathologic stresses (43–47). There are three UCP subtypes: UCP1, UCP2, and UCP3 (46). Irisin has previously been shown to up-regulate UCP1 expression in adipocytes (48). UCP2 shares 55 to 60% similarity with UCP1, and they have a comparable activity (17, 18, 45). Besides the spleen, the lung has the highest expression of UCP2 (17). UCP2 has been suggested to participate in attenuation of the ROS production (49–51) and to inhibit ROS-mediated apoptosis in lung epithelial cells (52). Our present study showed colocalization of irisin and UCP2 in mitochondria. The irisin/UCP2 interaction was able to prevent IR-induced degradation of UCP2 protein, thus facilitating the protection of lung mitochondria. A role of UCP2 on the protection of lung IR injury was further confirmed in the in vivo experiment, where knockout of UCP2 abolished the protective effect of irisin on lung and mitochondrial function during IR injury. Moreover, pharmacological interventions with genipin to inhibit UCP2 further support a role of irisin-UCP2 interaction in preservation of lung function under stress conditions.
The current study has several limitations. First, our pilot study with the human NRDS infants only included a limited number of serum and BALF samples. With the LC-MS and protein liquid-chip assay methods for quantification of irisin, we should be able to expand the sample size to help establish a causative relationship between the changes in irisin expression and pulmonary function in human diseases. Moreover, genome sequencing may be used to identify the genetic basis for the reduced irisin circulation in the bloodstream of human patients with pulmonary dysfunction. Second, our present study focused on the role of irisin in preservation of mitochondria function in the lung tissue, but IR-induced oxidative stress can also affect the integrity of the plasma membrane. We recently showed that MG53, a skeletal muscle–derived secretory protein, is a key component of the cell membrane repair machinery and plays a protective role after lung IR injury (53). Elevation of ROS in the cytosol can inhibit MG53’s function in cell membrane repair (54). Thus, IR-induced ROS elevation could, in principle, cause defective cell membrane repair and contribute to lung dysfunction associated with IR injury. Whether irisin can ameliorate cell membrane repair is another topic for future study. Third, irisin circulation in the bloodstream is regulated by exercise (37). Exercise shares some similarities with RIPC, and RIPC could be used as an adjunct to exercise for pulmonary protection (55). Further research will need to expand on the mechanisms that underlie the potential synergy between exercise training and RIPC in modulation of irisin secretion to treat pulmonary injury.
The identified functional interaction between irisin and UCP2 at the interface of mitochondria has important implications for cell signaling and the stress response. Therapeutic targeting of irisin/UCP2 to modulate mitochondria-dependent ROS signaling may have translational value for treating ALI or other human diseases associated with IR tissue injuries. Additional studies to elucidate the molecular motifs that directly control irisin/UCP2 interaction or the mechanisms that facilitate anchoring of irisin to mitochondria can advance our understanding of the biology of irisin and its translational applications. Because controlled elevation of irisin in the bloodstream has therapeutic benefits in treating ALI, development of noninvasive physiologic approaches, such as RIPC or pharmacological interventions that enhance secretion or ectodomain shedding of irisin from the fibronectin domain– containing 5 protein (FNDC5), may offer therapeutic potential for human diseases associated with oxidative stress.
MATERIALS AND METHODS
Study design
In light of the effect of RIPC on pulmonary protection, our study was designed to determine whether irisin, a myokine, could replicate RIPC’s effect. The experiments were carried out in mice with pulmonary IR injury and three different lung epithelial cell lines. To show the role of UCP2 in irisin-mediated protective action, we also used Ucp2−/− mice and a UCP2 inhibitor, genipin. Equal numbers of male and female mice were used for experimentation. Whenever possible, littermate control animals were used for comparison without randomization. To assess the clinical relevance of our findings, we conducted a pilot human study to establish the relationship between irisin and NRDS in newborn patients. Serum and BALF concentrations of irisin were determined by a protein liquid-chip assay and LC-MS method. Investigators for the survival study and lung injury scoring were blinded to the group assignment and treatment.
Clinical study of NRDS samples
Seven newborns with NRDS were recruited from the neonatal intensive care unit (NICU) at Daping Hospital, Third Military Medical University (Chongqing, China). Samples of serum and BALF from NRDS patients during acute illness and after recovery were collected. In addition, 15 control blood samples were collected from neonatal jaundice patients in the NICU at Daping Hospital, excluding those with NRDS, neonatal pneumonia, meconium aspiration syndrome, or other respiratory diseases. The inclusion criteria and NRDS diagnosis were described in a previous study (56). The irisin concentration in serum or BALF was measured by an irisin-specific Luminex bead-based multiplex detection system (Merck Millipore) and LC-MS. Because of the limited volume and quality of collected serum samples, six samples in each group were measured by LC-MS. All procedures were conducted in compliance with protocols approved by the Ethics Committee of Daping Hospital, Third Military Medical University, and informed consent was received from all participants’ legal representatives. Details of all participants are presented in tables S1 and S2.
LC-MS test
Irisin measurement by LC-MS was performed as previously described (19, 37, 57). The serum or BALF samples (50 μl) from control or NRDS newborns were collected for SDS–polyacrylamide gel electrophoresis (SDS-PAGE). Gels were stained by Coomassie, and the fragments were excised from the 10- to 15-kDa region. After destaining, dehydrating, resuspending, and desalting, the gel pieces were digested by trypsin. The digested peptides were eluted for LC-MS by an linear ion trap (LTQ) Orbitrap Velos Pro mass spectrometer and an LC pump (Thermo Fisher Scientific). Peptides were separated using a 90-min gradient of 4 to 50% acetonitrile in 0.1% formic acid with a flow rate of 0.5 μl/min. For each analysis, we loaded 5 μl onto the column. The MS data were generated in the LTQ linear ion trap. The full-scan MS spectra were recorded at a resolution of 100,000× with a scan range from 300 to 1800 m/z (mass/charge ratio) in the LTQ and acquired in the Orbitrap at m/z 604. The tryptic peptide (48FIQEVNTTTR57) was chosen from the central portion after collision-induced dissociation. The intensity of the y-ion series for peptides corresponds to the irisin sequence (fig. S2), confirming that these ions can be used for identification and quantification of irisin.
Mouse lung IR model
UCP2 knockout mice with C57BL/6J background and C57BL/6J WT mice of both sexes, weighing 20 to 25 g, were purchased from the Model Animal Research Center of Nanjing University in Nanjing, China. The UCP2 knockout mice, as a global knockout model, were created as described previously (30, 31). Mice were randomized to the sham-operated group and lung IR group, anesthetized with an intraperitoneal injection of sodium pentobarbital (50 mg/kg), and placed on a heating pad to maintain their body temperature. The right femoral arteries and veins of mice were then catheterized with polyethylene tubing for arterial blood sampling and irisin (or saline as control) injection. After intubation via tracheotomy, mice were connected to a Harvard ventilator (Hugo Sachs Elektronik) with an inspiratory pressure of 7 ml/kg. The respiratory rate was set at 100 breaths per minute. After thoracotomy, the left hilum of the IR group was clamped for 1 hour and released; reperfusion proceeded for 1 hour. IR groups were divided into the following groups: IR control group, RIPC group, and irisin treatment group. In RIPC group, RIPC was induced by five episodes of 5-min ischemia of the femoral artery with a small vascular clamp to occlude arterial blood flow followed by releasing the clamp for 5-min reperfusion, then 1-hour ischemia immediately, and then reperfusion. In the irisin treatment group, irisin was given by bolus injection immediately after lung ischemia. After reperfusion, blood samples were obtained immediately for arterial blood gas analysis (Gem primer 3000, Instrumentation Laboratory) or enzyme-linked immunosorbent assay (ELISA).
This study was approved by the Research Council and Animal Care and Use Committee of Daping Hospital, Third Military Medical University. All experiments conformed to the guidelines of the American Association for the Accreditation of Laboratory Animal Care.
Survival analysis
After 1-hour reperfusion, mice were ventilated and observed for additional time to evaluate the survival rate. The surviving animals were euthanized by overdosage of sodium pentobarbital after 1-hour observation. The definition of death was a combination of (i) cardiac arrest or cessation of regular cardiac activity, (ii) the collapse of the left atrium, and (iii) brief clonus activity (58). A researcher blinded to the group assignment (all groups were undergoing a thoracotomy) would record the time of death for survival analysis. Left lung tissues were taken for histological analysis or other experiments.
Wet/dry lung weight ratio
At the time of death, the left lungs of mice were taken and weighed to determine the wet weight, then placed in an oven at 80°C for 24 hours, and reweighed to determine the dry weight for calculation of the wet/dry weight ratio.
IL-1b and IL-6 concentrations in serum by ELISA
After 1-hour reperfusion, blood samples were collected from right femoral arteries by catheterizing, kept at room temperature for clotting, and centrifuged at 3000g for 15 min to obtain serum. The IL-1β and IL-6 concentrations were detected by ELISA (Jiancheng).
Histological study
Lung tissues, cleared of blood with phosphate-buffered saline (PBS) and kept in 4% paraformaldehyde for 1 to 2 days at 4°C, were embedded in paraffin, sectioned, and mounted on slides. After deparaffinizing and rehydrating by xylene and different concentrations of ethanol, the sections were stained with hematoxylin solution for 10 min, washed with running water for 5 min, soaked in 1% acid alcohol for 1 min, and counter-stained with eosin Y solution for 1 min; alternatively, the sections were treated with 0.1% Triton X-100 and 10 mM sodium citrate (pH 6.0) and blocked with 5% goat serum in PBS for 1 hour at 37°C for immunohistochemistry. The tissues were incubated with rabbit anti-irisin antibody (1:200; Phoenix Pharmaceuticals) overnight at 4°C.
DHE staining
Superoxide production in the mouse lung tissue was evaluated with the fluorescent dye DHE (Beyotime). Frozen section of the left lung tissue isolated from sham-treated and IR mice with vehicle (saline) or irisin treatment was stained with DHE (10−5 M) for 20 min. After the tissues were washed, images were taken with a fluorescence microscope at an excitation wavelength of 490 nm and an emission wavelength of 590 nm. All sections were processed under the same conditions. Settings for image acquisition were identical for all sections (exposure time, 30 ms). The DHE fluorescence intensity was quantified by ImageJ (National Institutes of Health).
OCR assay
To assess the OCR, lung tissues were obtained from mice after IR or irisin treatment and rinsed briefly in Seahorse XF medium supplemented with 1 mM sodium pyruvate, 2 mM L-glutamine, and 10 mM glucose to clear any blood. Lung tissues were cut into small pieces (2 to 5 mg), placed in the center of each well of an XF24 Islet Capture Microplate, and covered with a customized nylon mesh by an islet capture screen insert tool. The OCR was measured at 37°C in a Seahorse XF24 extracellular flux analyzer (Agilent) to assess effects of oligomycin (10 μg/ml), carbonyl cyanide p-trifluoromethoxyphenylhydrazone (16 μM), and rotenone/antimycin A (3:12 μM) on OCR. Once the XF experiment was completed, the lung tissue was homogenized to determine the protein concentration for normalization. Protein concentrations were measured using a bicinchoninic acid (BCA) protein assay kit (HyClone Pierce). The production of ATP and basal respiration rate were determined by observing how the OCR changes in response to drugs that modulate mitochondrial activity (59).
AR-induced injury in lung epithelial cells
Lung epithelial cell lines were obtained from the American Type Culture Collection. A549, a human lung epithelial cell line (60), was cultured in 10% fetal bovine serum (FBS) Dulbecco’s modified Eagle’s medium/F-12 culture medium. BEAS-2B, a clonal cell line derived from human lung bronchus epithelium, was cultured in bronchial epithelial cell basal medium supplemented with BEGM kit (Lonza/Clonetics Corporation) and 10% FBS. RLE-6TN, a rat lung alveolar type II epithelial cell line, was cultured in Ham’s F-12 medium with 2 mM L-glutamine supplemented with bovine pituitary extract (0.01 mg/ml), insulin (0.005 mg/ml), insulin-like growth factor (2.5 ng/ml), transferrin (0.125 μg/ml), epidermal growth factor (2.5 ng/ml), and 10% FBS. All three cell lines were cultured at 37°C in 95% air and 5% CO2 atmosphere.
Cells were exposed to an anoxic chamber with 5% CO2 and 95% N2 at 37°C for 2 hours followed by reoxygenation in 95% air and 5% CO2 atmosphere for 2 hours. Exogenous irisin was conjugated with FITC by a dye labeling kit (G-Biosciences) and given to cells immediately after anoxia. After reoxygenation, the cells were analyzed using MitoTracker Deep Red FM (a far-red fluorescent dye, working concentration of 200 nM in our study; Cell Signaling Technology) staining or immunofluorescence staining. The time-dependent FITC-irisin accumulation was observed with an Olympus IX83 inverted microscope. The time-lapse images were taken for 0 to 60 min in the stage of reoxygenation with 5% CO2 and 95% O2 at 37°C.
Confocal microscopy of irisin and UCP2 colocalization in lung epithelial cells
A549, RLE-6TN, or BEAS-2B cells, which were grown on coverslips and treated by AR and exogenous FITC-labeled irisin, were fixed with 4% paraformaldehyde (30 min) and stained with a rabbit anti-UCP2 antibody (1:100 dilution; Proteintech Group) overnight at 4°C, followed by Cy3-conjugated goat anti-rabbit immunoglobulin G (IgG) antibody (1:1000 dilution, green; Jackson ImmunoResearch Laboratory). Immunofluorescence images were acquired (Olympus AX70 laser confocal microscopy) at excitation wavelengths of 570 nm; emission was detected at 615 nm. Cells that were treated with only Cy3-conjugated goat anti-rabbit IgG antibody had no immunofluorescence, and omission of the anti-UCP2 antibody showed no green color after merging the images.
Immunoblotting
Lung tissues were washed twice with ice-cold PBS and lysed in lysis buffer [10mM tris-HCl (pH 8.0), 150 mM NaCl, 1% NP-40, 1mM phenylmethylsulfonyl fluoride, and leupeptin and aprotinin (10 mg/ml each)]. After centrifugation at 12,000g for 15 min, the supernatants were collected and their protein concentrations were measured using a BCA protein assay kit (HyClone Pierce). The tissue homogenates (50 μg of protein) were separated by 10% SDS-PAGE and transferred onto nitrocellulose membranes. The blots were then washed with tris-buffered saline with Tween 20 (TBST), blocked with 5% milk powder in TBST buffer for 1 hour, and incubated with the appropriate primary antibodies at appropriate dilutions. The blotted membranes were probed with the rabbit anti-irisin antibody (1:500; Phoenix Pharmaceuticals), the rabbit anti-UCP2 antibody (1:500; Sigma-Aldrich), rabbit anti-active caspase 9 antibody (1:500; Abcam), and rabbit anti–cytochrome c antibody (1:500, Abcam) at 4°C overnight. Then, the membranes were washed and primary antibodies were detected with fluorescent-labeled goat anti-rabbit IgG (1:15,000; IRDye, LI-COR), and the bands were visualized by Odyssey Western Blot Detection System (LI-COR). The amount of protein transferred onto the membranes was verified by immunoblotting for glyceraldehyde-3-phosphate dehydrogenase.
Immunoprecipitation
Equal amounts of cell lysates (300 mg of protein per milliliter of supernatant) were incubated with affinity-purified anti-UCP2 antibodies (3 mg) for 1 hour and protein G agarose at 4°C for 12 hours. The immunoprecipitates were suspended in sample buffer, boiled for 10 min, and subjected to immunoblotting with the irisin antibody. To determine the specificity of the bands found on the immunoblots, IgG (negative control) and irisin antibody (positive control) were used as the immunoprecipitants, instead of the UCP2 antibody.
Statistical analysis
The data are expressed as means ± SEM. Comparisons within groups were made by analysis of variance (ANOVA) for repeated measures, and comparisons among groups were made by ANOVA with Holm-Sidak test. The product limit (Kaplan-Meier) estimate of cumulative survival was assessed with the log-rank test to evaluate significance of differences. A value of P < 0.05 was considered significant. All original data and P values are provided in table S3.
Supplementary Material
Fig. S1. Specificity of irisin antibody and absence of endogenous irisin expression in lung epithelial cells.
Fig. S2. Mass spectrogram profile of irisin peptide.
Fig. S3. Irisin expression in lung tissues from sham-treated mice after RIPC.
Fig. S4. Irisin expression in mouse lung tissues at different time points after RIPC, with or without IR injury.
Fig. S5. OCR of lung tissue from IR-injured mice.
Fig. S6. Time-dependent accumulation of exogenous irisin in RLE-6TN and BEAS-2B cells.
Fig. S7. Localization of irisin in mitochondria of cultured lung epithelial cells.
Fig. S8. Measurement of cell viability and LDH concentration in culture medium derived from lung epithelial cells after AR.
Fig. S9. Colocalization of irisin and UCP2 in lung epithelial cells.
Fig. S10. Basal UCP2 expression, inflammation markers, and amount of ROS in WT and Ucp2−/− mice.
Table S1. The characteristics of control and NRDS newborn patients in Fig. 2B.
Table S2. The characteristics of control and NRDS newborn patients in Fig. 2C.
Table S3. Original data and P values (Excel file).
Movie S1. Uptake of exogenous irisin into BEAS-2B cells.
Movie S2. Uptake of exogenous irisin into RLE-6TN cells.
Movie S3. Uptake of exogenous irisin into A549 cells.
Acknowledgments
We thank Y. Wan, W. Sun, L.T. Wang, H.Y. Ran, Y. Liu, and Z. F. Tao from the Biomedical Analysis Center, Third Military Medical University; Y. Tang from Chongqing Medical and Pharmaceutical College; and L. Y. Chen, X. F. Tang, and H. Zhang from the Department of Anesthesiology, Daping Hospital for technical support.
Funding: This project was supported by grants from the National Natural Science Foundation of China (31430043 and 81600585 to Chunyu Zeng), China Postdoctoral Science Foundation funded project (2017M613431 to K.C.), National Basic Research Program of China (2013CB531104 to Chunyu Zeng), and Program for Changjiang Scholars and Innovative Research Team in University (IRT1216 to Chunyu Zeng) and by U.S. NIH grants (AR057404 to J.Z.; AR067766 to H.Z.; and DK106394, AR070752, and AR061385 to J.M.). K.C., Chunyu Zeng, H.Z., J.Z., and J.M. provided funding.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/9/418/eaao6298/DC1
Author contributions: K.C., H.Z., Y. Yang, J.M., and Chunyu Zeng conceived and designed the project; K.C., H.Z., T.T., Chunyu Zeng, J.Z., and J.M. wrote the paper; K.C., Z.X., Y. Liu, Z.W., Y. Li, X.X., C.C., T.X., Q.L., Y. Yao, J.Y., and Cindy Zeng acquired the data; and K.C., Z.X., Y. Liu, D.H., J.M., and Chunyu Zeng analyzed and interpreted the data.
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Ambrosio G, Tritto I. Reperfusion injury: Experimental evidence and clinical implications. Am Heart J. 1999;138:S69–S75. doi: 10.1016/s0002-8703(99)70323-6. [DOI] [PubMed] [Google Scholar]
- 2.de Perrot M, Liu M, Waddell TK, Keshavjee S. Ischemia–reperfusion–induced lung injury. Am J Respir Crit Care Med. 2003;167:490–511. doi: 10.1164/rccm.200207-670SO. [DOI] [PubMed] [Google Scholar]
- 3.Kamath BD, Macguire ER, McClure EM, Goldenberg RL, Jobe AH. Neonatal mortality from respiratory distress syndrome: Lessons for low-resource countries. Pediatrics. 2011;127:1139–1146. doi: 10.1542/peds.2010-3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ng CSH, Wan S, Arifi AA, Yim APC. Inflammatory response to pulmonary ischemia–reperfusion injury. Surg Today. 2006;36:205–214. doi: 10.1007/s00595-005-3124-2. [DOI] [PubMed] [Google Scholar]
- 5.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 6.Crimi E, Sica V, Williams-Ignarro S, Zhang H, Slutsky AS, Ignarro LJ, Napoli C. The role of oxidative stress in adult critical care. Free Radic Biol Med. 2006;40:398–406. doi: 10.1016/j.freeradbiomed.2005.10.054. [DOI] [PubMed] [Google Scholar]
- 7.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–1583. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med. 2011;17:1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delbin MA, Antunes E, Zanesco A. Role of exercise training on pulmonary ischemia/reperfusion and inflammatory response. Rev Bras Cir Cardiovasc. 2009;24:552–561. doi: 10.1590/s0102-76382009000500017. [DOI] [PubMed] [Google Scholar]
- 10.Files DC, Liu C, Pereyra A, Wang Z-M, Aggarwal NR, D’Alessio FR, Garibaldi BT, Mock JR, Singer BD, Feng X, Yammani RR, Zhang T, Lee AL, Philpott S, Lussier S, Purcell L, Chou J, Seeds M, King LS, Morris PE, Delbono O. Therapeutic exercise attenuates neutrophilic lung injury and skeletal muscle wasting. Sci Transl Med. 2015;7:278ra232. doi: 10.1126/scitranslmed.3010283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Birnbaum Y, Hale SL, Kloner RA. Ischemic preconditioning at a distance: Reduction of myocardial infarct size by partial reduction of blood supply combined with rapid stimulation of the gastrocnemius muscle in the rabbit. Circulation. 1997;96:1641–1646. doi: 10.1161/01.cir.96.5.1641. [DOI] [PubMed] [Google Scholar]
- 12.Weber C. Far from the heart: Receptor cross-talk in remote conditioning. Nat Med. 2010;16:760–762. doi: 10.1038/nm0710-760. [DOI] [PubMed] [Google Scholar]
- 13.Sommer SP, Sommer S, Sinha B, Wiedemann J, Otto C, Aleksic I, Schimmer C, Leyh RG. Ischemia-reperfusion injury–induced pulmonary mitochondrial damage. J Heart Lung Transplant. 2011;30:811–818. doi: 10.1016/j.healun.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 14.Sommer SP, Sommer S, Sinha B, Walter D, Aleksic I, Gohrbandt B, Otto C, Leyh RG. Glutathione preconditioning ameliorates mitochondria dysfunction during warm pulmonary ischemia–reperfusion injury. Eur J Cardiothorac Surg. 2012;41:140–148. doi: 10.1016/j.ejcts.2011.02.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu D, Wang H, Zhang J, Zhang X, Xin C, Zhang F, Lee Y, Zhang L, Lian K, Yan W, Ma X, Liu Y, Tao L. Irisin improves endothelial function in type 2 diabetes through reducing oxidative/nitrative stresses. J Mol Cell Cardiol. 2015;87:138–147. doi: 10.1016/j.yjmcc.2015.07.015. [DOI] [PubMed] [Google Scholar]
- 16.Lee P, Linderman JD, Smith S, Brychta RJ, Wang J, Idelson C, Perron RM, Werner CD, Phan GQ, Kammula US, Kebebew E, Pacak K, Chen KY, Celi FS. Irisin and FGF21 are cold-induced endocrine activators of brown fat function in humans. Cell Metab. 2014;19:302–309. doi: 10.1016/j.cmet.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Couplan E, del Mar Gonzalez-Barroso M, Alves-Guerra MC, Ricquier D, Goubern M, Bouillaud F. No evidence for a basal, retinoic, or superoxide-induced uncoupling activity of the uncoupling protein 2 present in spleen or lung mitochondria. J Biol Chem. 2002;277:26268–26275. doi: 10.1074/jbc.M202535200. [DOI] [PubMed] [Google Scholar]
- 18.Stuart JA, Cadenas S, Jekabsons MB, Roussel D, Brand MD. Mitochondrial proton leak and the uncoupling protein 1 homologues. Biochim Biophys Acta. 2001;1504:144–158. doi: 10.1016/s0005-2728(00)00243-7. [DOI] [PubMed] [Google Scholar]
- 19.Albrecht E, Norheim F, Thiede B, Holen T, Ohashi T, Schering L, Lee S, Brenmoehl J, Thomas S, Drevon CA, Erickson HP, Maak S. Irisin – a myth rather than an exercise-inducible myokine. Sci Rep. 2015;5:8889. doi: 10.1038/srep08889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tapuria N, Kumar Y, Habib MM, Abu Amara M, Seifalian AM, Davidson BR. Remote ischemic preconditioning: A novel protective method from ischemia reperfusion injury—A review. J Surg Res. 2008;150:304–330. doi: 10.1016/j.jss.2007.12.747. [DOI] [PubMed] [Google Scholar]
- 21.Li C, Xu M, Wu Y, Li YS, Huang WQ, Liu KX. Limb remote ischemic preconditioning attenuates lung injury after pulmonary resection under propofol–remifentanil anesthesia: A randomized controlled study. Anesthesiology. 2014;121:249–259. doi: 10.1097/ALN.0000000000000266. [DOI] [PubMed] [Google Scholar]
- 22.Yang Z, Chen X, Chen Y, Zhao Q. Decreased irisin secretion contributes to muscle insulin resistance in high-fat diet mice. Int J Clin Exp Pathol. 2015;8:6490–6497. [PMC free article] [PubMed] [Google Scholar]
- 23.Quinn LS, Anderson BG, Conner JD, Wolden-Hanson T. Circulating irisin levels and muscle FNDC5 mRNA expression are independent of IL-15 levels in mice. Endocrine. 2015;50:368–377. doi: 10.1007/s12020-015-0607-9. [DOI] [PubMed] [Google Scholar]
- 24.Zhou X, Li R, Liu X, Wang L, Hui P, Chan L, Saha PK, Hu Z. ROCK1 reduces mitochondrial content and irisin production in muscle suppressing adipocyte browning and impairing insulin sensitivity. Sci Rep. 2016;6:29669. doi: 10.1038/srep29669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwartz V, Krüttgen A, Weis J, Weber C, Ostendorf T, Lue H, Bernhagen J. Role for CD74 and CXCR4 in clathrin-dependent endocytosis of the cytokine MIF. Eur J Cell Biol. 2012;91:435–449. doi: 10.1016/j.ejcb.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Turner EJH, Jarvis HG, Chetty MC, Landon G, Rowley PS, Ho MM, Stewart GW. ATP-dependent vesiculation in red cell membranes from different hereditary stomatocytosis variants. Br J Haematol. 2003;120:894–902. doi: 10.1046/j.1365-2141.2003.04175.x. [DOI] [PubMed] [Google Scholar]
- 27.Schraw W, Li Y, McClain MS, van der Goot FG, Cover TL. Association of Helicobacter pylori vacuolating toxin VacA with lipid rafts. J Biol Chem. 2002;277:34642–34650. doi: 10.1074/jbc.M203466200. [DOI] [PubMed] [Google Scholar]
- 28.Alán L, Smolková K, Kronusova E, Santorová J, Ježek P. Absolute levels of transcripts for mitochondrial uncoupling proteins UCP2, UCP3, UCP4, and UCP5 show different patterns in rat and mice tissues. J Bioenerg Biomembr. 2009;41:71–78. doi: 10.1007/s10863-009-9201-2. [DOI] [PubMed] [Google Scholar]
- 29.Boström P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, Rasbach KA, Bostrom EA, Choi JH, Long JZ, Kajimura S, Zingaretti MC, Vind BF, Tu H, Cinti S, Højlund K, Gygi SP, Spiegelman BM. A PGC1-a-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. 2012;481:463–468. doi: 10.1038/nature10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu DQ, Guo YL, Bian Z, Chen YY, Chen X, Liu Y, Zhang CY, Zen K. Uncoupling protein-2 negatively regulates polymorphonuclear leukocytes chemotaxis via modulating [Ca2+] influx. Arterioscler Thromb Vasc Biol. 2010;30:575–581. doi: 10.1161/ATVBAHA.109.198374. [DOI] [PubMed] [Google Scholar]
- 31.Zhang CY, Baffy G, Perret P, Krauss S, Peroni O, Grujic D, Hagen T, Vidal-Puig AJ, Boss O, Kim YB, Zheng XX, Wheeler MB, Shulman GI, Chan CB, Lowell BB. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, b cell dysfunction, and type 2 diabetes. Cell. 2001;105:745–755. doi: 10.1016/s0092-8674(01)00378-6. [DOI] [PubMed] [Google Scholar]
- 32.Zhang C-Y, Parton LE, Ye CP, Krauss S, Shen R, Lin C-T, Porco JA, Jr, Lowell BB. Genipin inhibits UCP2-mediated proton leak and acutely reverses obesity- and high glucose-induced b cell dysfunction in isolated pancreatic islets. Cell Metab. 2006;3:417–427. doi: 10.1016/j.cmet.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 33.Pedersen BK. Muscles and their myokines. J Exp Biol. 2011;214:337–346. doi: 10.1242/jeb.048074. [DOI] [PubMed] [Google Scholar]
- 34.Perakakis N, Triantafyllou GA, Fernández-Real JM, Huh JY, Park KH, Seufert J, Mantzoros CS. Physiology and role of irisin in glucose homeostasis. Nat Rev Endocrinol. 2017;13:324–337. doi: 10.1038/nrendo.2016.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fukushima Y, Kurose S, Shinno H, Cao Thi Thu H, Tamanoi A, Tsutsumi H, Hasegawa T, Nakajima T, Kimura Y. Relationships between serum irisin levels and metabolic parameters in Japanese patients with obesity. Obes Sci Pract. 2016;2:203–209. doi: 10.1002/osp4.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu J, Han Y, Wang J, Liu Y, Zheng S, Zhou L, Jose PA, Zeng C. Irisin lowers blood pressure by improvement of endothelial dysfunction via AMPK-Akt-eNOS-NO pathway in the spontaneously hypertensive rat. J Am Heart Assoc. 2016;5:e003433. doi: 10.1161/JAHA.116.003433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jedrychowski MP, Wrann CD, Paulo JA, Gerber KK, Szpyt J, Robinson MM, Nair KS, Gygi SP, Spiegelman BM. Detection and quantitation of circulating human irisin by tandem mass spectrometry. Cell Metab. 2015;22:734–740. doi: 10.1016/j.cmet.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Erickson HP. Irisin and FNDC5 in retrospect: An exercise hormone or a transmembrane receptor? Adipocyte. 2013;2:289–293. doi: 10.4161/adip.26082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gill R, Kuriakose R, Gertz ZM, Salloum FN, Xi L, Kukreja RC. Remote ischemic preconditioning for myocardial protection: Update on mechanisms and clinical relevance. Mol Cell Biochem. 2015;402:41–49. doi: 10.1007/s11010-014-2312-z. [DOI] [PubMed] [Google Scholar]
- 40.Chen KH, Chao D, Liu CF, Chen CF, Wang D. Ischemia and reperfusion of the lung tissues induced increase of lung permeability and lung edema is attenuated by dimethylthiourea (PP69) Transplant Proc. 2010;42:748–750. doi: 10.1016/j.transproceed.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 41.Cereijo R, Giralt M, Villarroya F. Thermogenic brown and beige/brite adipogenesis in humans. Ann Med. 2015;47:169–177. doi: 10.3109/07853890.2014.952328. [DOI] [PubMed] [Google Scholar]
- 42.Park MJ, Kim DI, Choi JH, Heo YR, Park SH. New role of irisin in hepatocytes: The protective effect of hepatic steatosis in vitro. Cell Signal. 2015;27:1831–1839. doi: 10.1016/j.cellsig.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 43.Pecqueur C, Alves-Guerra C, Ricquier D, Bouillaud F. UCP2, a metabolic sensor coupling glucose oxidation to mitochondrial metabolism? IUBMB Life. 2009;61:762–767. doi: 10.1002/iub.188. [DOI] [PubMed] [Google Scholar]
- 44.Pecqueur C, Couplan E, Bouillaud F, Ricquier D. Genetic and physiological analysis of the role of uncoupling proteins in human energy homeostasis. J Mol Med. 2001;79:48–56. doi: 10.1007/s001090000150. [DOI] [PubMed] [Google Scholar]
- 45.Boss O, Hagen T, Lowell BB. Uncoupling proteins 2 and 3: Potential regulators of mitochondrial energy metabolism. Diabetes. 2000;49:143–156. doi: 10.2337/diabetes.49.2.143. [DOI] [PubMed] [Google Scholar]
- 46.Bouillaud F, Alves-Guerra MC, Ricquier D. UCPs, at the interface between bioenergetics and metabolism. Biochim Biophys Acta. 2016;1863:2443–2456. doi: 10.1016/j.bbamcr.2016.04.013. [DOI] [PubMed] [Google Scholar]
- 47.Boss O, Samec S, Paoloni-Giacobino A, Rossier C, Dulloo A, Seydoux J, Muzzin P, Giacobino JP. Uncoupling protein-3: A new member of the mitochondrial carrier family with tissue-specific expression. FEBS Lett. 1997;408:39–42. doi: 10.1016/s0014-5793(97)00384-0. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y, Li R, Meng Y, Li S, Donelan W, Zhao Y, Qi L, Zhang M, Wang X, Cui T, Yang LJ, Tang D. Irisin stimulates browning of white adipocytes through mitogen-activated protein kinase p38 MAP kinase and ERK MAP kinase signaling. Diabetes. 2014;63:514–525. doi: 10.2337/db13-1106. [DOI] [PubMed] [Google Scholar]
- 49.Lee FYJ, Li Y, Zhu H, Yang S, Lin HZ, Trush M, Diehl AM. Tumor necrosis factor increases mitochondrial oxidant production and induces expression of uncoupling protein-2 in the regenerating rat liver. Hepatology. 1999;29:677–687. doi: 10.1002/hep.510290320. [DOI] [PubMed] [Google Scholar]
- 50.Alves-Guerra MC, Rousset S, Pecqueur C, Mallat Z, Blanc J, Tedgui A, Bouillaud F, Cassard-Doulcier AM, Ricquier D, Miroux B. Bone marrow transplantation reveals the in vivo expression of the mitochondrial uncoupling protein 2 in immune and nonimmune cells during inflammation. J Biol Chem. 2003;278:42307–42312. doi: 10.1074/jbc.M306951200. [DOI] [PubMed] [Google Scholar]
- 51.Jabůrek M, Ježek J, Zelenka J, Ježek P. Antioxidant activity by a synergy of redox-sensitive mitochondrial phospholipase A2 and uncoupling protein-2 in lung and spleen. Int J Biochem Cell Biol. 2013;45:816–825. doi: 10.1016/j.biocel.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 52.Deng S, Yang Y, Han Y, Li X, Wang X, Zhang Z, Wang Y. UCP2 inhibits ROS-mediated apoptosis in A549 under hypoxic conditions. PLOS ONE. 2012;7:e30714. doi: 10.1371/journal.pone.0030714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jia Y, Chen K, Lin P, Lieber G, Nishi M, Yan R, Wang Z, Yao Y, Li Y, Whitson BA, Duann P, Li H, Zhou X, Zhu H, Takeshima H, Hunter JC, McLeod RL, Weisleder N, Zeng C, Ma J. Treatment of acute lung injury by targeting MG53-mediated cell membrane repair. Nat Commun. 2014;5:4387. doi: 10.1038/ncomms5387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hwang M, Ko J-k, Weisleder N, Takeshima H, Ma J. Redox-dependent oligomerization through a leucine zipper motif is essential for MG53-mediated cell membrane repair. Am J Physiol Cell Physiol. 2011;301:C106–C114. doi: 10.1152/ajpcell.00382.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sprick JD, Rickards CA. Cyclical blood flow restriction resistance exercise: A potential parallel to remote ischemic preconditioning? Am J Physiol Regul Integr Comp Physiol. 2017;313:R507–R517. doi: 10.1152/ajpregu.00112.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li W, Long C, Renjun L, Zhangxue H, Yin H, Wanwei L, Juan M, Yuan S. Association of SCNN1A single nucleotide polymorphisms with neonatal respiratory distress syndrome. Sci Rep. 2015;5:17317. doi: 10.1038/srep17317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Castro-Gamero AM, Izumi C, Rosa JC. Biomarker verification using selected reaction monitoring and shotgun proteomics. Methods Mol Biol. 2014;1156:295–306. doi: 10.1007/978-1-4939-0685-7_20. [DOI] [PubMed] [Google Scholar]
- 58.Okada K, Fujita T, Minamoto K, Liao H, Naka Y, Pinsky DJ. Potentiation of endogenous fibrinolysis and rescue from lung ischemia/reperfusion injury in interleukin (IL)-10-reconstituted IL-10 null mice. J Biol Chem. 2000;275:21468–21476. doi: 10.1074/jbc.M002682200. [DOI] [PubMed] [Google Scholar]
- 59.Bugge A, Dib L, Collins S. Measuring respiratory activity of adipocytes and adipose tissues in real time. Methods Enzymol. 2014;538:233–247. doi: 10.1016/B978-0-12-800280-3.00013-X. [DOI] [PubMed] [Google Scholar]
- 60.Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, Parks WP. In vitro cultivation of human tumors: Establishment of cell lines derived from a series of solid tumors. J Natl Cancer Inst. 1973;51:1417–1423. doi: 10.1093/jnci/51.5.1417. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Specificity of irisin antibody and absence of endogenous irisin expression in lung epithelial cells.
Fig. S2. Mass spectrogram profile of irisin peptide.
Fig. S3. Irisin expression in lung tissues from sham-treated mice after RIPC.
Fig. S4. Irisin expression in mouse lung tissues at different time points after RIPC, with or without IR injury.
Fig. S5. OCR of lung tissue from IR-injured mice.
Fig. S6. Time-dependent accumulation of exogenous irisin in RLE-6TN and BEAS-2B cells.
Fig. S7. Localization of irisin in mitochondria of cultured lung epithelial cells.
Fig. S8. Measurement of cell viability and LDH concentration in culture medium derived from lung epithelial cells after AR.
Fig. S9. Colocalization of irisin and UCP2 in lung epithelial cells.
Fig. S10. Basal UCP2 expression, inflammation markers, and amount of ROS in WT and Ucp2−/− mice.
Table S1. The characteristics of control and NRDS newborn patients in Fig. 2B.
Table S2. The characteristics of control and NRDS newborn patients in Fig. 2C.
Table S3. Original data and P values (Excel file).
Movie S1. Uptake of exogenous irisin into BEAS-2B cells.
Movie S2. Uptake of exogenous irisin into RLE-6TN cells.
Movie S3. Uptake of exogenous irisin into A549 cells.