

Abstract
Synaptic inhibition in the adult brain is primarily mediated by the γ-aminobutyric acid (GABA) type A receptor (GABAAR). The distribution, properties, and dynamics of these receptors are largely determined by their subunit composition. Alteration of subunit composition after a traumatic brain injury (TBI) may result in abnormal increased synaptic firing and possibly contribute to injury-related pathology. Several studies have shown that the Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) signaling pathway can alter GABAAR subunit expression. The present study investigated changes in JAK/STAT pathway activation after two different severities of experimental TBI in the mouse using the controlled cortical impact (CCI) model. It also investigated whether modulating the activation of the JAK/STAT pathway after severe controlled cortical impact (CCI-S) with a JAK/STAT inhibitor (WP1066) alters post-traumatic epilepsy development and/or neurological recovery after injury. Our results demonstrated differential changes in both the activation of STAT3 and the expression of the GABAAR α1 and γ2 subunit levels that were dependent on the severity of the injury. The change in the GABAAR α1 subunit levels appeared to be at least partly transcriptionally mediated. We were able to selectively reverse the decrease in GABAAR α1 protein levels with WP1066 treatment after CCI injury. WP1066 treatment also improved the degree of recovery of vestibular motor function after injury. These findings suggest that the magnitude of JAK/STAT pathway activation and GABAAR α1 subunit level decrease is dependent on injury severity in this mouse model of TBI. In addition, reducing JAK/STAT pathway activation after severe experimental TBI reverses the decrease in the GABAAR α1 protein levels and improves vestibular motor recovery.
Keywords: Traumatic brain injury, GABAA receptor, JAK/STAT pathway, STAT3
Introduction
Traumatic brain injury (TBI) spans a spectrum of injury severities. The extent of cerebral injury has been shown to positively correlate with the extent of neurological morbidity including the degree of cognitive dysfunction, motor impairment and the occurrence of posttraumatic epilepsy (PTE) (Annegers et al., 1998, Gaetz, 2004, Saatman et al., 2006, Willmore and Ueda, 2009, Kharatishvili and Pitkanen, 2010).
TBI activates a number of signaling pathways that are important for recovery but may also exacerbate injury-related pathology. One of the pathways that has been shown to be activated after several types of cerebral insults, including TBI, is the Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) pathway (Wen et al., 2001, Lund et al., 2008b, Oliva et al., 2011, Zhao et al., 2011a, Zhao et al., 2011b, Raible et al., 2012, Grabenstatter et al., 2013). The JAK/STAT pathway has been shown to regulate the γ-aminobutyric acid (GABA) type A receptor (GABAAR) subunit α1 expression in the hippocampus by increasing the phosphorylation of STAT3 after the cerebral injury status epilepticus (SE) (Lund et al., 2008a). The decrease in α1 subunit-containing GABAARs is thought to contribute to the increased hyperexcitability observed in the hippocampus of animals following SE and to contribute to subsequent development of temporal lobe epilepsy (TLE) (Brooks-Kayal et al., 1998, Peng et al., 2004, Zhang et al., 2004, Raol et al., 2006). Decreases in the GABAAR α1 subunit have also been reported in injured hippocampus after experimental TBI (Satriotomo et al., 2006, Gibson et al., 2010, Raible et al., 2012), but whether the severity of injury modulates JAK/STAT pathway activation and regulates GABAAR α1 levels is not known. It is also not known if inhibiting pathway activation after a TBI prevents these changes in GABAAR subunit protein levels.
In this manuscript, we examined the levels of GABAAR subunits after two different severities of CCI in the mouse and found that the α1 and γ2 subunit levels are decreased 1 week and 16 weeks after severe experimental TBI (CCI-S) but not moderate CCI (CCI-M) and that δ, β2 and β3 subunit levels remain unchanged. We also found that the JAK/STAT pathway (which is known to regulate GABAAR subunit composition) is differentially activated depending on the severity of the TBI. CCI-S (but not CCI-M) injured animals exhibited decreased performance in memory and motor tasks and infrequently developed epilepsy after TBI. Lastly, the degree of JAK/STAT pathway activation after severe experimental TBI can be modulated with WP1066, resulting in a selective rescue in the GABAAR α1 protein levels and an improvement in vestibular motor performance after severe CCI.
Materials and Methods
Controlled Cortical Impact (CCI) and WP1066 administration
Adult CD-1 male mice (25–30g) were subjected to a severe cortical injury by CCI, similar to previous descriptions (Hunt et al., 2009, Guo et al., 2013). Mice were anesthetized with 2–3% isoflurane and placed in a stereotaxic head frame. After scalp incision and reflection, a 4-mm diameter craniotomy was created lateral to the sagittal suture and centered between bregma and lambda. The skull cap was removed without damage to the exposed underlying dura. The contusion device consisted of a computer controlled, pneumatically driven impactor fitted with a beveled stainless steel tip 3 mm in diameter (Precision Systems and Instrumentation, Fairfax, VA). Brain injury was generated using this device to compress the cortex to a depth of 2.0 mm at a velocity of 5.0 m/s and 199 ms duration (CCI-S) or to a depth of 1.0 mm at a velocity of 5.0 m/s and 400 ms duration (CCI-M). After the injury, the resulting bleeding was controlled and incision sutured.
The commercially available cell-permeable inhibitor WP1066 (Selleck Chemicals LLC) has been shown to be an effective STAT3 pathway inhibitor (Hussain et al., 2007, Iwamaru et al., 2007, Kupferman et al., 2009, Grabenstatter et al., 2013). This inhibitor has been shown to block the JAK2/STAT3 interaction and subsequent phosphorylation of STAT3 at tyrosine 705 (Faderl et al., 2007). WP1066 has also been shown to cross the blood-brain barrier (BBB) and effectively block STAT3 activation after intraperitoneal (i.p.) administration in rodent models of cerebral injuries (Iwamaru et al., 2007, Statler et al., 2009, Grabenstatter et al., 2013). For this study, all operated mice were give i.p. injections of 0.05 ml of dimethyl sulfoxide (DMSO) or 0.05 ml of 75 mg/kg of WP1066 in DMSO 30 and 90 minutes after experimental injury.
A total of 433 animals underwent surgery of which 103 were sham injured animals, 105 were CCI-S injured animals, 98 were CCI-M injured animals and 127 were CCI-S + WP1066 injured animals. None of the animals that underwent Sham, CCI-M or CCI-S injury died due to the injury while of the 127 mice that underwent CCI-S injury and received 75 mg/kg of WP1066 30 and 90 minutes after injury 104 lived; thus, we had a survival rate of 82% for this group which is statistically different from all other groups (Chi-square test). The University of Colorado Institutional Animal Care and Use Committee approved all procedures described.
After the CCI injury, the animals were returned to their cages and allowed to recover for 6 hours, 24 hours, 48 hours, 72 hours, 1 week, 2 weeks or 16 weeks after injury (all timepoints for analysis are post CCI). At one of the above time points, animals were deeply anesthetized with isoflurane in an induction chamber, decapitated, and their brains were rapidly removed. The hippocampi were dissected and flash frozen for storage in a −80° C freezer.
Western blot
Western blot was performed with modifications of published protocols (Raol, et al., 2006). Protein was extracted from whole hippocampus (intact hippocampus in sham-injured controls and the entire remaining hippocampus in injured mice). Since after severe CCI injury a large portion of the anterior hippocampus is absent (see Figure 1), for a subset of sham injured mice (1 week post operation), we transected the hippocampus after dissection, then extracted and measured protein separately from the anterior and posterior halves to determine if there was a regional difference in the levels of GABAAR subunit proteins that changed after CCI-S (see Suppl Fig 7). Protein lysate (10–20 μg depending on type of blot) was loaded into 8% SDS-polyacrylamide gels and run for 1.5 hours at 115 V. Blots were then transferred to nitrocellulose membranes and blocked in 5% milk/trisphosphate-buffered saline with Tween-20 (TBS-T). Membranes were incubated with rabbit polyclonal antibodies raised against STAT3 phosphorylated at Tyr705 (anti-pSTAT3) (Cell Signaling Technologies; 1:8,000, α1 (Millipore; 1:7000), β2 (PhosphoSolutions;1:1,000), β3 (PhosphoSolutions; 1:1,000), γ2 (PhosphoSolutions; 1:1,000) or δ (Millipore; 1:600) overnight at 4°C in 5% bovine serum albumin/TBS-T (for pSTAT3) or 1% milk/TBS-T (for GABAAR), then washed and incubated with anti-rabbit antibody (1:15,000) conjugated to horseradish peroxidase for 1 hour. Protein bands were detected with the use of chemiluminescent solution (Pierce), then membranes were stripped and reprobed with rabbit polyclonal antibody raised against total STAT3 (anti-STAT3) (Cell Signaling Technologies; 1:6,000) or β-actin (Sigma 1:40,000) and bands were quantified using Image J software (NIH) and expressed as percent or fold change with respect to mean control values in the same run (defined as 100 or 1, respectively).
Figure 1.

Progressive tissue loss following CCI-S and CCI-M injuries. (A) A series of representative images of CV stained coronal sections from sham, CCI-S and CCI-M injured mice. Sections show cerebral damage at three different levels posterior from bregma. Calibration bar, 5 mm (B) Quantitation of cerebral extent by measuring the area of the ipsilateral hemisphere and normalizing it to the area of the contralateral hemisphere for sham, CCI-S and CCI-M injured mice. Each group is statistically different from each other group with CCI-S having the largest lesion. * P < 0.05, ** P < 0.01, P < 0.001 (n = 4 for sham, n = 7 for CCI-M and n = 8 for CCI-S)
RT-PCR
RNA was extracted from whole hippocampus with the use of Trizol reagent protocol (Invitrogen, Carlsbad, CA). To synthesize complementary DNA (cDNA), 1 μg of RNA was separated and processed with the SuperScript II reverse transcription kit (Invitrogen) according to the manufacturer’s instructions and then diluted 1:4 for storage and subsequent RT-PCR. For RT-PCR reactions, each sample was run in triplicate and each 25-μl reaction contained 1.25 μl GABAAR subunit α1 or beta-2 microglobulin (B2M) Taqman gene expression primer/probe sets from Applied Biosystems (Foster City, CA), 12.5 μl of Taqman Master mix, and 10 μl of sample cDNA. RT-PCR formed on the SDS-7500 PCR machine (Applied Biosystems). The RT-PCR runs consisted of 1 cycle of 50°C for 2 min, then 1 cycle of 95°C for 10 min, and 40 cycles of 95°C for 15 s and 60°C for 1 min. All values were normalized to B2M expression in the same samples to control for loading variability, and then expressed as fold change with respect to mean controls values in the same run (defined as 1).
Cresyl violet (CV) staining and analysis
Mice were sacrificed two weeks following CCI by transcardial perfusion with cold phosphate buffered saline (0.01M) and 4% paraformaldehyde (in 0.1 M). After the brain was perfused, it was removed and post fixed overnight in 4% paraformaldehyde (in 0.1M) at 4°C, then cryoprotected in 30% sucrose. The brain was then embedded (in Tissue-Tek OCT Compound, Sakura Finetek, Torrance, CA), frozen, and then was serially sectioned in the coronal plane throughout the entire extent of the lesion. Every 6th coronal brain section (30 μm each) was mounted, stained with cresyl violet (CV) and imaged for quantitation of lesion area. Tissue sections were briefly mounted on gelatin-coated slides and dehydrated at room temperature for 5 minute immersion in 70% ethanol, followed by two 5 minute immersions in 95% ethanol, and then immersed for 5 minutes in xylenes. The sections were then rehydrated by immersion for 5 minutes in 95% ethanol, 5 minutes in 70% ethanol, and 5 minutes in distilled H2O. The sections were then stained with cresyl violet acetate (1.5 grams of cresyl echt violet in 300 ml H2O plus 1 ml 10% glacial acetic acid) for 4 minutes then rinsed with H2O for 2 minutes, and differentiated by immersion for 1 minute in acidified distilled H2O (1 ml of glacial acetic acid to 300 ml of H2O) and 1 minute in H2O. The sections were dehydrated by immersion for 3 minutes in 70% ethanol, 3 minutes 95% ethanol twice and 3 minutes 100% ethanol twice. Lastly the sections were cleared by immersion for 5 minutes in xylenes twice and cover-slipped with permount. Sections were imaged using a 2X objective (Nikon Eclipse TE2000-U) and quantitated using ImageJ software. To best approximate the amount of volume loss associated with the lesion, the total area of the contralateral (unlesioned) hemisphere as well as the ipsilateral (lesioned) hemisphere was measured in every 6th section throughout the A-P extent of the lesion for each animal. The area of the lesioned side was summated from all sections from a given animal and the same was done for the unlesioned side from the same animal, then the summated area of the unlesioned side was divided by the summated area of the lesioned side for each animal, converted to a percent value, and subtracted from 100% to calculate the total percentage of tissue loss within the region of the lesion in the ipsilateral hemisphere compared to the contralateral hemisphere. The technician who conducted the analysis was blinded to injury severity and treatment group.
EEG acquisition and analysis
To accurately analyze electrographic seizure frequency, two bilateral subdural stainless steel screws (0.25 mm posterior, 1.5 mm lateral relative to the bregma for the ipsilateral side and 1 mm posterior, 1.5 mm from lateral relative to the bregma for the contralateral) were placed over the temporolimbic cortices. Additional stainless steel screws were placed on each side of the brain behind lambda (i.e., over the cerebellum) and were used as reference and ground electrodes. Animals were allowed to recover from surgery for 1 week before proceeding with any further experimentation. Implanted mice were video-EEG monitored 24 h/day for six consecutive weeks using Triangle BioSystems International and Stellate digital video-EEG systems. Mice were placed in the recording chamber and flexible cables were attached to a commutator (i.e., electric swivel) system, which allowed the animal to move freely. EEG signals were sampled at 1 kHz, amplified by 800 ×, and band-pass filtered between 0.3 Hz and 60 Hz.
Off-line data analyses were performed by trained technicians blinded to all experimental parameters. Electrographic seizures were differentiated from background noise by the appearance of large-amplitude (at least twice times baseline), high frequency (minimum of 5 Hz) activity, with progression of the spike frequency that lasts for a minimum of 10 s. Electrographic seizures were manually detected in EEG recordings based on the above criteria and correlated with behavioral manifestations in continuous video recordings. Motor seizures were scored by standard behavioral classes (Racine, 1972) as follows: (1) behavioral arrest, eye closure, vibrissae twitching, sniffing: (2) facial clonus and head bobbing; (3) forelimb clonus; (4) rearing with continued forelimb clonus; and (5) rearing with loss of motor control and falling. Electrographic seizures class 3 and above were classified as convulsive and seizures scored as class 2 and below were classified as non-convulsive.
Novel Object Recognition (NOR)
To test memory performance, Novel Object Recognition (NOR) testing was used 2 and 16 weeks post CCI. The test was performed in a 30cm by 30cm by 30cm box where the mouse was video recorded during each phase of the test. On the first testing day, each mouse was placed in the box for a 5 minute session and left to explore and acclimatize to the arena. Approximately 5 minutes after the acclimatization period, the mouse was again placed into the same arena with two copies of the same object placed systematically in the box and allowed to explore the object freely for 5 minutes. After the habituation period elapsed, the mouse was returned to its home cage. 24 hours later, the mouse was again placed into the same arena, but this time one of the objects was replaced with a new object of similar size. The mouse was again allowed to freely explore the objects for 5 minutes. During each phase of the test, the entire box and the object were cleaned to avoid the presence of olfactory cues.
Off-line data analyses were performed by trained technicians blinded to all experimental parameters to identify (1) if the mouse was able to freely move around the arena and (2) measure the amount of time the mouse spent exploring each object (for the first 3 minutes of each phase of the test).
RotaRod (RR)
The rotarod test has been shown to be a sensitive index of injury-induced vestibular motor dysfunction following experimental TBI. The rotarod device consists of a metal and plastic frame, which suspends a rotating rod large enough in diameter for adult mice to stand on. Plastic wafers split the rod into four slots, and prevent the animals from seeing each other or coming into contact while on the rod. Each animal was removed from its home cage and placed on the rod. Then, the animal’s task was to walk on the rod as it rotates, to avoid falling off. If the animal did fall off, he only fell a short distance (about 8 inches) and activated a magnetic timer that recorded the duration that the animal was on the rod. The rod’s rotation was programmed to accelerate from 4 to 40 rpm over the course of 5 minutes (300s.) The duration the animal lasts on the rod, up to the maximum of 300s, was recorded. Immediately after testing, the animal was returned to its home cage. After a brief (1–2 minute) rest, the next trial began. A pre-injury period of learning trials was completed over the four days prior to injury, with four trials per day. After injury, four trials were run at each selected time point following injury (3 days post CCI, 7 days post CCI, 12 weeks post CCI, and 16 weeks post CCI) and the individual trial and mean times for each animal were recorded.
Statistical Analysis
Statistical significance was defined as a P value of less than 0.05. All calculations were done using GraphPad Prism 6 software. Comparisons between sham injured mice, CCI-S injured mice and CCI-S + WP injured mice were done by using one-way analysis of variance (ANOVA) with a Tukey’s HSD test for multiple comparisons or with Fisher’s exact testing.
Results
CCI-S generates larger lesions 2 weeks after injury
Figure 1A shows representative CV-stained coronal sections throughout the lesion in the rostral to caudal direction in sham injured, CCI-M injured and CCI-S injured animals 2 weeks after injury. Figure 1B shows the mean area of the ipsilateral hemisphere compared to the contralateral hemisphere. This analysis showed statistical differences between sham, CCI-S and CCI-M injured mice (Figure 1B).
GABAAR subunits levels are differentially altered after varying severities of CCI
Figure 2A&B show mean GABAAR α1 subunit protein levels in whole hippocampus in sham injured, CCI-M injured and CCI-S injured animals. Protein levels of the α1 subunit showed no statistical difference between sham injured and CCI-M injured mice at any of the time points assessed, but there was a significant decrease at 48 hours, 72 hours, 1 week and 16 weeks post injury in the ipsilateral hippocampus in the CCI-S injured animals compared to both sham injured and CCI-M injured mice (Figure 2 A&B). There was no significant difference in the α1 subunit levels at any time point in the hippocampus contralateral to the CCI when compared to sham injured controls (data not shown).
Figure 2.

Temporal profile of GABAA α1 receptor subunit after CCI. (A) Representative western blots form whole hippocampus of mice 24 hours, 48 hours, 72 hours, 1 week and 16 weeks after CCI probed with anti-GABAAR α1 and β-actin antibodies. (B) Quantification of α1 blots showed a significant decrease at 48 hours through 16 weeks after CCI-S injury relative to CCI-M and sham injured controls. α1 levels were normalized to β-actin levels and expressed as a percent change compared to sham injured controls. (C) mRNA levels of α1 were quantified using RT-PCR analysis and represented as histograms showing the fold change of α1 6 hours, 24 hours and 48 hours after CCI in CCI-S, CCI-M and sham injured controls. α1 mRNA levels were normalized to B2M mRNA levels in the same samples and expressed as a fold change compared to sham injured controls (defined as 1). * P < 0.05, ** P < 0.01, P < 0.001 (for A and B n = 7 for sham, n = 7 for CCI-S and n = 6 for CCI-M at 24 hours and n = 7 for sham, n = 7 for CCI-S and n = 7 for CCI-M at 48 hours and n = 6 for sham, n = 6 for CCI-S and n = 6 for CCI-M at 72 hours and n = 6 for sham, n = 7 for CCI-S and n = 7 for CCI-M at 1 week and n = 7 for sham, n = 10 for CCI-S and n = 9 for CCI-M at 16 weeks) (for C n = 7 for sham, n = 7 for CCI-S and n = 6 for CCI-M at 6 hours and n = 7 for sham, n = 7 for CCI-S and n = 7 for CCI-M at 24 hours and n = 7 for sham, n = 7 for CCI-S and n = 7 for CCI-M at 48 hours).
To investigate whether changes in the α1 subunit levels associated with transcriptional regulation, we used quantitative RT-PCR on samples of whole hippocampus 6 hours, 24 hours and 48 hours after injury to determine the mRNA levels for the α1 subunits. Figure 2C shows that in the ipsilateral hippocampus 6 hours after CCI-S, there was a statistically significant decrease in α1 subunit transcripts (~0.6 fold relative to sham injured mice). 24 hours after experimental TBI there was a statistically significant difference between CCI-S vs CCI-M, CCI-S vs sham injured controls and CCI-M vs sham injured controls (Figure 2C). At 48 hours post injury, however, there was no statistical difference between α1 mRNA levels in CCI-S vs CCI-M injured mice, and both of the CCI injured groups had significantly decreased levels of α1 subunit mRNAs compared to sham injured controls (Figure 2C).
Figure 3 shows that γ2 subunit protein levels were significantly decreased 1 week and 16 weeks only in the CCI-S injury group. The δ, β2 and β3 GABAAR subunits were unchanged for both CCI-S and CCI-M injured groups when compared to sham injured controls (Figure 3).
Figure 3.

GABAA receptor subunit γ2 is decreased while δ, β2 and β3 are unchanged 1 and 16 weeks post CCI. (A) Representative western blots from whole hippocampus of mice 1 week after CCI probed with anti-GABAAR γ2 and β-actin antibodies. (B) Representative western blots from whole hippocampus of mice 16 weeks after CCI probed with anti-GABAAR γ2 and β-actin antibodies. (C) Quantification of γ2 blots showed that the γ2 subunit is significantly decreased 1 and 16 weeks after CCI-S relative to CCI-M and sham injured controls. (D) Representative western blots from whole hippocampus of mice 1 week after CCI probed with anti-GABAAR δ and β-actin antibodies. (E) Representative western blots from whole hippocampus of mice 16 weeks after CCI probed with anti-GABAAR δ and β-actin antibodies. (F) Quantification of δ blots showed that the δ subunit was unchanged 1 and 16 weeks after CCI-S relative to CCI-M and sham injured controls. (G) Representative western blots from whole hippocampus of mice 1 week after CCI probed with anti-GABAAR β2 and β-actin antibodies. (H) Representative western blots from whole hippocampus of mice 16 weeks after CCI probed with anti-GABAAR β2 and β-actin antibodies. (I) Quantification of β2 blots shows that the β2 subunit was unchanged 1 and 16 weeks after CCI-S relative to CCI-M and sham injured controls. (J) Representative western blots from whole hippocampus of mice 1 week after CCI probed with anti-GABAAR β3 and β-actin antibodies. (K) Representative western blots from whole hippocampus of mice 16 weeks after CCI probed with anti-GABAAR β3 and β-actin antibodies. (L) Quantification of β3 blots showed that the β3 subunit was unchanged 1 and 16 weeks after CCI-S relative to CCI-M and sham injured controls. * P < 0.05, ** P < 0.01, P < 0.001 (n = 6 for sham, n = 7 for CCI-S and n = 7 for CCI-M at 1 week and n = 10 for sham, n = 10 for CCI-S and n = 9 for CCI-M at 16 weeks).
CCI transiently activates the JAK/STAT pathway
As shown is Figure 4, there was a statistically significant increase in the protein levels of pSTAT3 in the ipsilateral hippocampus after both CCI-S and CCI-M injury when compared to sham injured controls at multiple time points after experimental traumatic brain injury. There was also a statistically significant lower amount of STAT3 phosphorylation in the CCI-M injured animals compared to the CCI-S injured group at 6 hours post CCI. This differential activation was only seen at the 6 hour time point. The elevated levels of pSTAT3 in both CCI-S and CCI-M injuries declined over time, and at one week, there was no statistically significant difference in the levels of pSTAT3 in either the CCI-S or CCI-M injured mice when compared to sham injured controls (Figure 4). pSTAT3 levels were investigated in the contralateral hippocampus, and there was no statistically significant difference at any time point after either CCI injury when compared to sham injured controls (data not shown).
Figure 4.

Phosphorylated STAT3 levels in injured hippocampus after CCI. (A) Representative western blots of protein homogenates from whole ipsilateral hippocampus (relative to side of CCI) of mice 6 hours, 24 hours, 48 hours, 1 week and 16 weeks after CCI probed with pSTAT3 and STAT3 antibodies. (B) Quantification of pSTAT3 levels from CCI-S, CCI-M and sham injured controls showed that from 6 hours to 72 hours post injury the pSTAT3 levels were higher in both the CCI-S and CCI-M injured group when compared to sham injured controls. Levels of pSTAT3 in the CCI-S groups were statistically higher than the levels in the CCI-M group at 6 hours post injury but, at all other time points there was no statistical difference between CCI-S and CCI-M groups in pSTAT3 protein levels. * P < 0.05, ** P < 0.01, P < 0.001 (n = 9 for sham, n = 9 for CCI-S and n = 9 for CCI-M at 6 hours, n = 10 for sham, n = 9 for CCI-S and n = 10 for CCI-M at 24 hours and n = 7 for sham, n = 7 for CCI-S and n = 7 for CCI-M at 48 hours and n = 6 for sham, n = 8 for CCI-S and n = 6 for CCI-M at 72 hours and n = 6 for sham, n = 7 for CCI-S and n = 7 for CCI-M at 1 week and n = 6 for sham, n = 7 for CCI-S and n = 6 for CCI-M at 16 weeks).
Total STAT3 protein levels were also analyzed post injury, and surprisingly, both CCI-S and CCI-M groups had a statistically significant increase in total STAT3 protein levels at 1 week post injury when compared to sham injured controls (Supplemental Figure 1). At 16 weeks post injury, however, there was no statistical difference in STAT3 protein levels in any of the groups (Supplemental Figure 1).
CCI-S injured mice have memory and vestibular motor impairment
Memory was assessed using novel object recognition (NOR) testing. During NOR testing, CCI-S injured mice spent significantly less time exploring the novel object compared to sham injured controls (Figure 5A). The amount of time exploring the novel object was not different between CCI-M injured mice when compared to either the CCI-S injured mice or sham injured controls (Figure 5A). All of the injury groups performed similarly during the open field and habituation portions of the test.
Figure 5.

Memory and vestibular motor performance is significantly reduced after CCI-S. (A) Quantification of average time spent exploring the new object during Novel Object Recognition testing shows that the CCI-S injured mice performed statistically worse than the sham injured controls or CCI-M injured mice. (B) Quantification of average time spent on the Rotarod apparatus showed that the CCI-S injured mice performed significantly worse than the CCI-M injured mice or sham injured controls. * P < 0.05, ** P < 0.01, P < 0.001 (for A n = 19 for sham, n = 18 for CCI-S and n = 12 for CCI-M at 2 weeks and n = 11 for sham, n = 12 for CCI-S and n = 10 for CCI-M at 16 weeks) (for B n = 20 for sham, n = 28 for CCI-S and n = 19 for CCI-M at 3 days and n = 20 for sham, n = 28 for CCI-S and n = 19 for CCI-M at 7 days and n = 12 for sham, n = 13 for CCI-S and n = 9 for CCI-M at 16 weeks).
Vestibular motor performance after experimental TBI was investigated using rotarod testing. CCI-S injured mice spent significantly less time on the rotarod apparatus at 3 days, 7 days and 16 weeks after CCI when compared to CCI-M injured mice and sham injured controls. There was no significant difference in the amount of time spent on the apparatus for CCI-M injured mice compared to sham injured controls (Figure 5B).
CCI-S injury causes a small percentage of mice to develop epilepsy
To examine the development of PTE after CCI, the animals were continuously video-EEG recorded for 6 consecutive weeks starting at either 8 weeks or 10 weeks post experimental traumatic brain injury. Table 1 shows the frequencies of epilepsy development after sham, CCI-S and CCI-M injury. The only group which contained animals that developed epilepsy was the CCI-S injured group; however, due to the small number of CCI-S injured animals that developed epilepsy, there was no significant statistical difference of PTE development between any of the groups (Table 1). Of the two animals that developed epilepsy, one of them had several seizures that lasted over a minute in duration and had stage 5 behavioral correlates (Supplemental Figure 2).
Table 1.
Comparison of epilepsy development between all groups examined. No statistically significant differences in PTE development were found between any of the groups. The only group that had epileptic animals was the CCI-S and CCI-S + WP injury groups.
| Sham | Sham + WP | CCI-M | CCI-S | CCI-S + WP | |
|---|---|---|---|---|---|
| Number of Epileptic Mice | 0 | 0 | 0 | 2 | 4 |
| Total Number of Mice Recorded | 5 | 4 | 10 | 15 | 12 |
| Percentage of Mice that Became Epileptic | 0% | 0% | 0% | 13% | 33% |
Administration of WP1066 modulates STAT3 phosphorylation after CCI-S
WP1066 is a small molecule STAT3 inhibitor designed from the caffeic acid benzyl ester/AG490 scaffold (Hussain et al., 2007; Iwamaru et al., 2007; Kupferman et al., 2009) to optimally inhibit the JAK2/STAT3 interaction and subsequent phosphorylation of STAT3 at tyrosine 705 (Faderl et al., 2007).
As shown in Figure 6, there was a statistically significant elevation in the levels of pSTAT3 in the ipsilateral hippocampus in both CCI-S and CCI-S + WP injury groups when compared to sham injured controls at multiple time points after injury. The addition of 75 mg/kg of WP1066 30 and 90 minutes post injury significantly reduced STAT3 phosphorylation in the injured hippocampus from 6 hours to 48 hours post CCI-S. The increase in pSTAT3 levels for both CCI-S and CCI-S + WP injuries declined temporally, and at one week, there was no statistically significant difference in the levels of pSTAT3 when compared to sham injured controls (Figure 6). pSTAT3 levels were investigated in the contralateral hippocampus, and there was no statistically significant difference at 6 hours or 24 hours after either CCI injury (data not shown).
Figure 6.
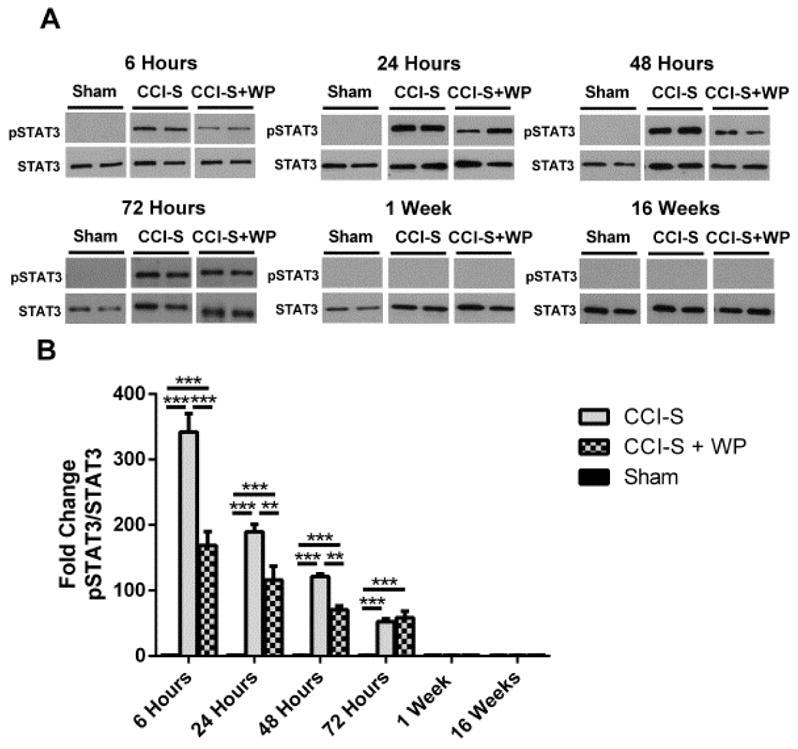
WP1066 administration after CCI-S reduces phosphorylated STAT3 levels in injured hippocampus. (A) Representative western blots of protein homogenates from whole ipsilateral hippocampus (relative to side of CCI) of mice 6 hours, 24 hours, 48 hours, 1 week and 16 weeks after CCI-S treated with vehicle or WP1066 probed with pSTAT3 and STAT3 antibodies. (B) Quantification of pSTAT3 levels from CCI-S, CCI-S + WP and sham injured controls showed that from 6 hours to 48 hours post injury that pSTAT3 levels were statistically lower than CCI-S injured mice treated with WP1066 compared to vehicle-treated CCI-S mice. * P < 0.05, ** P < 0.01, P < 0.001 (n = 9 for sham, n = 9 for CCI-S and n = 6 for CCI-S + WP at 6 hours, n = 10 for sham, n = 9 for CCI-S and n = 7 for CCI-S + WP at 24 hours and n = 7 for sham, n = 7 for CCI-S and n = 7 for CCI-S + WP at 48 hours and n = 6 for sham, n = 8 for CCI-S and n = 6 for CCI-S + WP at 72 hours and n = 6 for sham, n = 7 for CCI-S and n = 6 for CCI-S + WP at 1 week and n = 6 for sham, n = 7 for CCI-S and n = 7 for CCI-S + WP at 16 weeks).
Total STAT3 levels were also analyzed post injury in mice treated with WP1066. WP1066 administration did not have an effect on total STAT3 protein levels; thus, both CCI-S and CCI-S + WP groups had a statistically significant increase in STAT3 protein levels 1 week post injury when compared to sham injured controls (Supplemental Figure 3). At 16 weeks post injury, however, there was no statistically significant difference in STAT3 protein levels in any of the groups (Supplemental Figure 3).
Lesion size is unchanged with administration of WP1066
Supplemental Figure 4A shows CV stained coronal sections throughout the entire lesion in sham injured, CCI-S injured and CCI-S + WP injured animals 2 weeks post injury. Analysis of the extent of the injury showed statistical differences between sham injured and both CCI-S, CCI-S + WP injured groups. There was no statistical difference between CCI-S and CCI-S + WP injured mice (Supplemental Figure 4B).
GABAAR α1 subunit decline after CCI-S is selectively inhibited by WP1066 administration
Figure 7A&B shows GABAAR α1 subunit protein levels in the hippocampus in sham injured, CCI-S injured and CCI-S + WP injured animals. Protein levels of the α1 subunit are significantly lower in CCI-S injured mice at 48 hours, 72 hours, 1 week and 16 weeks post injury in the ipsilateral hippocampus when compared to sham-injured animals (Figure 7A&B). The CCI-S injured mice that were treated with WP1066 had GABAAR α1 protein levels that were statistically higher than vehicle-treated CCI-S injured mice at multiple time points post injury (Figure 7A&B). There was no significant differences between groups in the α1 subunit levels at 16 weeks post injury in the hippocampus contralateral to the CCI when compared to sham injured controls regardless of whether they received WP1066 (data not shown).
Figure 7.

GABAA α1 subunit receptor protein levels after CCI-S are rescued by administration of WP1066. (A) Representative western blots from whole hippocampus of mice 24 hours, 48 hours, 72 hours, 1 week and 16 weeks after CCI-S probed with anti-GABAAR α1 and β-actin antibodies. (B) Quantification of α1 blots showed a significant decrease at 48 hours through 16 weeks after CCI-S injury relative to CCI-S + WP and sham injured controls. α1 levels were normalized to β-actin levels and expressed as a percent change compared to sham injured controls. (C) mRNA levels of α1 were quantified using RT-PCR analysis and represented as histograms showing the fold change of α1 at 6 hours, 24 hours and 48 hours after CCI in CCI-S, CCI-S + WP and sham injured controls. α1 mRNA levels were normalized to B2M mRNA levels in the same samples and expressed as a fold change compared to sham injured controls (defined as 1). * P < 0.05, ** P < 0.01, P < 0.001 (for A&B n = 7 for sham, n = 7 for CCI-S and n = 6 for CCI-S + WP at 24 hours and n = 7 for sham, n = 7 for CCI-S and n = for CCI-S + WP at 48 hours and n = 6 for sham, n = 6 for CCI-S and n = 6 for CCI-S + WP at 72 hours and n = 6 for sham, n = 7 for CCI-S and n = 7 for CCI-S + WP at 1 week and n = 10 for sham, n = 10 for CCI-S and n = 12 for CCI-S + WP at 16 weeks) (for C n = 7 for sham, n = 7 for CCI-S and n = 7 for CCI-S + WP at 6 hours and n = 7 for sham, n = 7 for CCI-S and n = 7 for CCI-S + WP at 24 hours and n = 7 for sham, n = 7 for CCI-S and n = 7 for CCI-S + WP at 48 hours).
To investigate whether there was evidence that the α1 subunit changes might be due to transcriptional regulation, we used quantitative RT-PCR on whole hippocampus samples 6 hours, 24 hours and 48 hours after injury to determine the mRNA levels for the α1 subunits. Figure 7C shows that in the ipsilateral hippocampus 6 hours after CCI-S there was significantly lower expression of the α1 subunit (~0.6 fold relative to sham injured mice) relative to sham injured controls, but no statistical difference between the CCI-S vs CCI-S + WP nor between CCI-S + WP vs sham injured controls. 24 hours and 48 hours after experimental TBI there was a statistically significant decrease in α1 subunit levels between CCI-S vs sham injured controls and between CCI-S + WP vs sham injured controls (Figure 7C).
Supplemental Figure 5 shows that GABAAR γ2 protein subunit levels were significantly decreased 1 week and 16 weeks after CCI-S injury and that WP1066 administration did not affect this change. The δ, β2 and β3 GABAAR subunits were unchanged for both CCI-S treatment groups when compared to sham injured controls (Supplemental Figure 5).
WP1066 administration reduces vestibular motor impairment 16 weeks after CCI-S injury while memory performance is unchanged
Figure 8A shows that CCI-S injured mice spent significantly less time exploring the novel object compared to sham injured controls. In contrast, the amount of time exploring the novel object was not different between CCI-S + WP injured mice compared to either the sham injured controls or the CCI-S injured mice (Figure 8A). All of the injury groups performed similarly during the open field and habituation sessions of the NOR testing paradigm (data not shown).
Figure 8.

Memory performance is not significantly changed while vestibular motor performance is partially rescued with administration of WP1066. (A) Quantification of average time spent exploring the new object during Novel Object Recognition testing shows that the CCI-S + WP injured mice did not perform statistically differently than either the sham injured controls or CCI-S injured mice. (B) Quantification of average time spent on the Rotarod apparatus shows that the CCI-S injured mice performed significantly worse than the CCI-S + WP injured mice and sham injured controls. * P < 0.05, ** P < 0.01, P < 0.001 (for A n = 19 for sham, n = 18 for CCI-S and n = 14 for CCI-S + WP at 2 weeks and n = 11 for sham, n = 12 for CCI-S and n = 11 for CCI-S + WP at 16 weeks) (for B n = 20 for sham, n = 28 for CCI-S and n = 29 for CCI-S + WP at 3 days and n = 20 for sham, n = 28 for CCI-S and n = 28 for CCI-S + WP at 7 days and n = 12 for sham, n = 13 for CCI-S and n = 12 for CCI-S + WP at 16 weeks).
Figure 8B displays that at 3 and 7 days post injury, CCI-S + WP injured mice performed no differently than CCI-S injured animals that received vehicle. At 16 weeks post injury, however, the WP1066 treated mice performed significantly better than vehicle treated CCI-S injured mice but still performed more poorly than sham injured controls (Figure 8B).
WP1066 administration after CCI-S dose not significantly the development of epilepsy
Table 1 shows the frequencies of epilepsy development after sham, CCI-S and CCI-S + WP injury. A fraction of CCI-S injured mice in both the WP1066 and vehicle treated groups developed epilepsy, and the percentage that developed epilepsy was not statistically different between groups (Table 1). The animals that did develop epilepsy after experimental TBI had seizures lasting approximately 30 seconds with stage 1 or 2 behavioral correlates (Supplemental Figure 6).
Discussion
In this study, we demonstrated that the degree of impact generated by CCI affects post-injury lesion size, levels of several GABAAR subunits, activation of the JAK/STAT pathway, and functional neurological outcomes. We found a positive correlation between the severity of injury, the extent of cerebral damage, and memory and vestibular motor dysfunction as assessed by NOR and RR, suggesting that the severity of the injury is directly related to the amount of subsequent macrostructural cerebral damage and functional impairment. Our findings are similar to Saatman et al., which suggests that there is a direct positive correlation between the severity of CCI impact, extent of cerebral injury and neurological outcomes (Saatman et al., 2006).
This study also examined the alterations in the GABAAR α1subunit levels following CCI. The GABAAR α1subunit protein levels were lower at 48 hours after injury and remained lower than sham injured controls for at least 16 weeks following CCI-S, but the CCI-M injured group did not have significant alterations of the GABAAR α1 subunit protein levels at any of the time points explored. We further found that mRNA expression of the α1 subunit was significantly decreased in the post CCI-S mice compared to controls from as early as 6 hours post injury to as late as 48 hours post injury, and was also lower than CCI-M injured mice. These results suggest that the differential changes in the GABAAR α1 subunit protein levels seen between the CCI-S and CCI-M injury may be due at least in part to transcriptional regulation.
To further examine if the decreased levels of GABAAR α1 protein were due to differential gene regulation or were a result of overall GABAAR loss associated with cell death, we looked at several other GABAAR subunits to determine if their levels in the surviving tissue changed after injury. The γ2 subunit level was found to decrease at 1 week and 16 weeks post injury in the severely injured animals, but the δ, β2 and β3 subunit levels showed no significant difference in either CCI injury group and sham injured controls. In addition, since after CCI-S injury a large portion of the anterior hippocampus is absent, we wanted to ensure that the decreases seen in α1 levels were not an artifact of normal gradients of the level of this subunit from anterior to posterior. We thus transected the hippocampus of a subset of sham injured mice 1 week post sham-injury, measured the protein levels for the α1 subunit separately in the anterior and posterior portions, and found no differences (Supplemental Figure 7). Taken together, these findings suggest that the α1 subunit changes are likely due at least in part to differential transcriptional regulation and are less likely to be exclusively due to trauma-related cell loss or specific preferential ablation of the anterior hippocampus. The α1 subunit most commonly pairs with γ2- and a β-subunit to produce synaptically localized receptors that mediate phasic currents with rapid kinetics, and reduced α1 and γ2 expression following CCI-S would thus be expected to reduce synaptic inhibition and contribute to hyperexcitability following brain injury.
Previous studies utilizing CCI in the rat showed a reduction in γ2 subunit levels in the contralateral hippocampus ~5–9 months after injury, similar to the reduction in γ2 found in ipsilateral hippocampus found in the current study, but showed no change in α1 and an increase in δ (Kharlamov et al., 2011). The γ2 and δ subunit levels have been shown to influence tonic and phasic synaptic inhibition and GABAAR location (Somogyi et al., 1996, Nusser et al., 1998, Sur et al., 1999, Sassoe-Pognetto et al., 2000, Wei et al., 2003, Semyanov et al., 2004, Sun et al., 2004, Jia et al., 2005, Liang et al., 2006, Zhang et al., 2007, Goodkin et al., 2008). Alterations in these subunit levels after TBI may also contribute to altered GABAergic signaling, hyperexcitability and/or altered motor/memory function after head trauma.
A number of changes in inhibitory transmission, in addition to changes in GABAAR subunit expression, have previously been reported. Consistent with inhibitory neuron loss, synaptic inhibition of granule cells ipsilateral to the injury is reduced several weeks after CCI (Hunt et al., 2012) as has been shown in models of temporal lobe epilepsy (Kobayashi et al., 2003, Shao and Dudek, 2005). Altered IPSC kinetics have also been observed in granule cells after CCI (Hunt et al., 2011) and in temporal lobe epilepsy models (Kobayashi and Buckmaster, 2003; Shao and Dudek, 2005; Sun et al., 2007). Interestingly, after severe CCI in rats, the amplitude of IPSCs in granule cells contralateral to the injury displayed reduced responsiveness to diazepam 90 days post-injury, and tonic GABA-A receptor-mediated currents were increased after CCI, suggesting altered GABA-A receptor pharmacology in the contralateral hemisphere (Mtchedlishvili et al., 2010). Collectively, these findings are consistent with our current results suggesting changes in GABA-A receptor subunits in granule cells lasting for weeks after injury.
In the current study, we found that pSTAT3 levels were differentially increased in the injured hippocampus 6 hours after CCI depending of the severity of injury, but both injury severities returned to baseline pSTAT3 levels by one week after injury. Our data is consistent with data from several TBI studies suggesting that the JAK/STAT pathway is transiently activated in the ipsilateral hippocampus following cerebral injury (Oliva et al., 2011, Raible et al., 2012).
STAT3 levels were also examined 1 and 16 weeks after injury, and there was a statistical increase in protein levels for both injury groups at the one week time point. This result is different from Oliva et al. and Raible et al. that showed no statistically significant difference in STAT3 protein levels after other types of experimental TBI (Oliva et al., 2011, Raible et al., 2012). The variability in findings between this study and previous studies could potentially be due to differences in the injury model (FPI vs CCI) and species (rat vs mouse). The transient increase in STAT3 protein levels after CCI suggests that there could be more STAT3 available to be phosphorylated in the days following initial brain injury and raise the concern of whether a second cerebral insult occurring shortly after the first TBI could potentially result in even greater STAT3 activation and further exacerbate adverse neurological outcomes.
Since PTE development post CCI has been shown previously by several investigators (Pitkanen and McIntosh, 2006, Hunt et al., 2009, 2010, Bolkvadze and Pitkanen, 2012, Guo et al., 2013), long-term continuous video-EEG recording was used to determine if differences in injury severity following CCI altered epilepsy development. Previous studies have shown a positive correlation between injury severity and PTE development. Although our results did not show a statistical difference in epilepsy development between CCI-S and CCI-M injury, the only animals which became epileptic were the CCI-S injured mice. Thus, our findings were similar to results using FPI in that they displayed a positive correlation between the extent of cortical damage and the rate of developing epilepsy (Kharatishvili and Pitkanen, 2010). The differences between our findings could be attributed to differences in the injury method (CCI for our studies vs FPI for their studies) and the larger number of animals recorded by Kharatishvili et al.
Administration of WP1066 48 hours after injury rescued GABAAR α1subunit protein levels and levels remained higher than vehicle treated CCI-S animals for at least 16 weeks after injury. This result is similar to Lund et al. which showed that inhibiting STAT3 activation after status epilepticus (SE) induced cerebral injury prevents the expected decrease in the GABAAR α1 subunit levels (Lund et al., 2008a). To determine if this rescue was transcriptionally mediated, we examined the mRNA levels of the α1subunit acutely after injury, but found no difference in α1 mRNA expression between groups to suggest this. This result differs from the results of Lund et al., which showed that inhibition of STAT3 activation by pyridone 6 (a pSTAT3 inhibitor) after SE injury prevents the decrease in transcription of Gabra1 (Lund et al., 2008a). Although the precise mechanism underlying the rescue of GABAAR α1 protein levels by WP1066 administration remains to be determined, it is notable that the rescue is selective for the α1 subunit, as the decreases in the γ2 subunit seen at 1 week and 16 weeks post CCI-S injury were not affected by WP1066 treatment. The δ, β2 and β3 subunit levels showed no significant difference between either treatment group and sham injured controls. These findings suggest that modulating STAT3 phosphorylation after TBI selectively alters the GABAAR α1 subunit.
Since WP1066 was originally developed for cancer therapeutics to increase apoptosis of malignant cells (Iwamaru et al., 2007), the extent of cerebral injury was examined to determine if this was altered with administration of WP1066 after experimental TBI. Analysis of the lesion area showed no statistical difference in the extent of brain damage between WP1066 treated and vehicle-treated mice after severe CCI, but interpretation of this finding is limited by the fact that stereological cell counts were not performed and the lesion volume was estimated based on measurements taken from only one out of every six serial 30uM sections throughout the A-P extent of the lesion, and thus very subtle differences in lesion volume could have been missed. This finding is similar to that of Grabenstatter et al., which found that after SE-induced cerebral injury there was not a significant increase in neuronal cell death in the animals treated with WP1066 (Grabenstatter et al., 2013). Taken together, these results suggest that cerebral cell death is not worsened with the use of WP1066 after experimental cerebral insults.
Since STAT3 protein levels were increased 1 week after CCI, we wanted to investigate whether WP1066 administration affected this outcome. We found that regardless of WP1066 treatment STAT3 protein levels were elevated when compared to sham injured controls. Thus, WP1066 is not reducing pSTAT3 levels by decreasing total STAT3 protein levels. This result supports the findings of Faderl et al., which showed that WP1066 inhibits STAT3 phosphorylation by preventing the interaction of JAKs with STAT3 (Faderl et al., 2007).
To examine if there are any differences in neurological performance following treatment with WP1066 after CCI-S injury, we used two well established tests of memory and motor performance (NOR and RR, respectively). WP1066 treatment after CCI-S did not significantly affect memory performance but did improve vestibular motor recovery 16 weeks post injury. Our results differed from those of Zhao et al., which showed that neurological recovery was worsened by administration of a JAK/STAT inhibitor after experimental TBI (Zhao et al., 2011b). A variety of factors could account for the differences between their results and our results. The first being the differences in the TBI model (CCI for ours vs FPI for theirs) and animals used (mouse for ours and rat for theirs). Also, they used a different STAT inhibitor (AG490) and a different dosing schedule (pretreatment 20 minutes before injury) than we utilized in the current study (WP1066 at 75 mg/kg given 30 and 90 minutes after injury). Lastly, to determine neurological recovery, they used a modified version of the neurological severity score (NSS), which examines several neurological functions but does not measure memory function (Shohami et al., 1995, Chen et al., 1996, Chen et al., 2001, Li et al., 2002), and we used NOR and RR. Due to these differences, it is difficult to compare results directly. There was no statistically significant difference in epilepsy between treatment groups, however, very few of the CCI-S injured mice became epileptic, and thus the sample size may have been too small to assess differences between treatment groups. It must be noted that we were unable to completely inhibit the phosphorylation of STAT3 after CCI using WP1066, likely due to its very short half-life and limited brain penetration (Grabenstatter et al., 2014). We therefore do not know if the reason that we did not see a difference between treatment groups was because of insufficient extent or duration of pSTAT3 inhibition, and if more complete or prolonged pSTAT3 inhibition might have resulted in a greater effect on memory function and epileptogenesis. Since initiation of the studies contained in this report, newer JAK/STAT inhibitors with increased stability and better brain penetration have become available, and have the potential to more completely inhibit STAT3 phosphorylation in brain. Further research utilizing these newer inhibitors should further elucidate the effects of JAK/STAT pathway inhibition on neurological recovery.
The studies conducted in this paper have several limitations in addition to those already mentioned. Firstly, we only examined two injury severities. Therefore, we could not determine if there is a linear association between the degree of injury and subsequent molecular and functional changes. Secondly, the depth, velocity and duration for each of the injury severities were different. Thus, it is impossible to attribute any of the molecular or functional changes observed to a specific injury parameter. However, Hunt et al. examined two different injury severities generated by CCI with all variables the same between injury groups, except for injury depth, and found that increased injury depth positively correlated with histological cerebral damage and epilepsy development (Hunt et al., 2009, Hunt et al., 2012). Saatman et al. also examined two different injury severities by only changing the variable of injury depth, and their results also showed that increased injury depth positively correlates with histological damage and decreased neurological recovery (Saatman et al., 2006). Our results agree with their findings, which demonstrate that changing the injury depth is a critical variable for altering the subsequent cerebral injury.
The reason that we changed the depth, velocity, and duration concurrently for our studies is that that the above mentioned CCI parameters have shown the highest seizure development. The CCI-M injury (1 mm depth) has been shown to generate PTE at a rate of 36 and 40% (Hunt et al., 2009, 2010) while the CCI-S (2 mm depth) injury generated PTE at a rate of 50% (Guo et al., 2013). Interestingly, we were unable to reproduce these rates of PTE development, but our results do support the notion that increased severity of brain trauma is positively associated with PTE development.
Another limitation is that we only modulated the JAK/STAT pathway activation with a single pharmacological agent (WP1066), and a single dosing paradigm, and as noted above did not obtain complete inhibition of pSTAT3. Therefore, we could not determine if the degree or specificity of pharmacologic inhibition correlated with the molecular and functional changes. Our results however, are similar to Lund et al., who showed a rescue in GABAAR α1 subunit after a cerebral insult (SE) by inhibiting the JAK/STAT pathway activation pharmacologically (Lund et al., 2008a). Also, although we demonstrated that inhibiting JAK/STAT pathway activation rescued GABAAR α1 subunit and improved vestibulomotor performance after CCI, we did not establish that there was a causal relationship between these two outcomes. The JAK/STAT pathway regulates numerous genes in many regions of brain, and we have previously shown in the rat pilocarpine model that JAK/STAT inhibition impacts expression of multiple genes in addition to GABAAR α1, any of which could be important for motor recovery (Grabenstatter et al., 2014). Understanding the mechanisms underlying the effect of JAK/STAT inhibition on vestibulomotor function, and what role if any GABAAR α1 subunit rescue plays in this effect, will be the focus of future studies in the lab.
Finally, using area analysis to determine if WP1066 administration after CCI affected cerebral injury is not as precise as stereological cell counts or full volumetric analysis and thus we could be missing a small difference in the extent of cerebral damage. However, our results show that there is very little difference in cortical area ablated after injury and if there is a difference we are missing due to the method we chose to analysis lesion extent we expect it would be very minimal. Lastly, the WP1066 treatment paradigm used increased post injury mortality. For future studies, newer more potent and brain permeable JAK/STAT inhibitors should be examined to determine if these agents may be equally or more efficacious and associated with less toxicity.
Conclusions
In summary, we found that following CCI, the levels of the α1 and γ2 GABAAR subunits levels are decreased selectively and differentially based on the severity of injury, and that these changes appear to be at least partially due to transcriptional regulation. We further found that the JAK/STAT pathway was differentially activated depending on the extent of CCI injury, and that inhibition of this pathway after severe CCI prevented downregulation of the GABAAR α1 subunit expression, as has been previously shown following SE-induced cerebral injury. Finally, we identified that JAK/STAT inhibition after injury resulted in improvement of vestibular motor function chronically after experimental TBI, but did not improve memory function nor reduce the development of post-traumatic epilepsy.
Supplementary Material
Highlights.
We investigated the role of GABAAR and JAK/STAT activation after CCI in mice.
The severity of CCI in mice causes differential changes in JAK/STAT pathway activation, GABAAR levels and neurological recovery.
The JAK/STAT inhibitor (WP1066) rescues the decreased in GABA α1 and vestibular motor function after CCI.
Acknowledgments
The funding for this research was provided by the Department of Defense award number W81XWH-11-1-0501 (to ABK, LCF & BNS), NIH/NCRR Colorado CTSI Grant Number TL1 RR025778 (to DJR), and National Institutes of Neurological Disorders and Stroke award number R01NS051710 (to ABK and SJR). Contents are the authors’ sole responsibility and do not necessarily represent official DOD or NIH views
Footnotes
Author Disclosure Statement
No conflicting financial interests exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Annegers JF, Hauser WA, Coan SP, Rocca WA. A population-based study of seizures after traumatic brain injuries. The New England journal of medicine. 1998;338:20–24. doi: 10.1056/NEJM199801013380104. [DOI] [PubMed] [Google Scholar]
- Bolkvadze T, Pitkanen A. Development of post-traumatic epilepsy after controlled cortical impact and lateral fluid-percussion-induced brain injury in the mouse. Journal of neurotrauma. 2012;29:789–812. doi: 10.1089/neu.2011.1954. [DOI] [PubMed] [Google Scholar]
- Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA. Selective changes in single cell GABA(A) receptor subunit expression and function in temporal lobe epilepsy. Nat Med. 1998;4:1166–1172. doi: 10.1038/2661. [DOI] [PubMed] [Google Scholar]
- Chen J, Li Y, Wang L, Zhang Z, Lu D, Lu M, Chopp M. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke; a journal of cerebral circulation. 2001;32:1005–1011. doi: 10.1161/01.str.32.4.1005. [DOI] [PubMed] [Google Scholar]
- Chen Y, Constantini S, Trembovler V, Weinstock M, Shohami E. An experimental model of closed head injury in mice: pathophysiology, histopathology, and cognitive deficits. Journal of neurotrauma. 1996;13:557–568. doi: 10.1089/neu.1996.13.557. [DOI] [PubMed] [Google Scholar]
- Faderl S, Ferrajoli A, Harris D, Van Q, Kantarjian HM, Estrov Z. Atiprimod blocks phosphorylation of JAK-STAT and inhibits proliferation of acute myeloid leukemia (AML) cells. Leukemia research. 2007;31:91–95. doi: 10.1016/j.leukres.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Gaetz M. The neurophysiology of brain injury. Clinical neurophysiology: official journal of the International Federation of Clinical Neurophysiology. 2004;115:4–18. doi: 10.1016/s1388-2457(03)00258-x. [DOI] [PubMed] [Google Scholar]
- Gibson CJ, Meyer RC, Hamm RJ. Traumatic brain injury and the effects of diazepam, diltiazem, and MK-801 on GABA-A receptor subunit expression in rat hippocampus. J Biomed Sci. 2010;17:38. doi: 10.1186/1423-0127-17-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodkin HP, Joshi S, Mtchedlishvili Z, Brar J, Kapur J. Subunit-specific trafficking of GABA(A) receptors during status epilepticus. J Neurosci. 2008;28:2527–2538. doi: 10.1523/JNEUROSCI.3426-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabenstatter HL, Del Angel YC, Carlsen J, Wempe MF, White AM, Cogswell M, Russek SJ, Brooks-Kayal AR. The effect of STAT3 inhibition on status epilepticus and subsequent spontaneous seizures in the pilocarpine model of acquired epilepsy. Neurobiol Dis. 2013;62C:73–85. doi: 10.1016/j.nbd.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Zeng L, Brody DL, Wong M. Rapamycin attenuates the development of posttraumatic epilepsy in a mouse model of traumatic brain injury. PloS one. 2013;8:e64078. doi: 10.1371/journal.pone.0064078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt RF, Haselhorst LA, Schoch KM, Bach EC, Rios-Pilier J, Scheff SW, Saatman KE, Smith BN. Posttraumatic mossy fiber sprouting is related to the degree of cortical damage in three mouse strains. Epilepsy Res. 2012;99:167–170. doi: 10.1016/j.eplepsyres.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt RF, Scheff SW, Smith BN. Posttraumatic epilepsy after controlled cortical impact injury in mice. Exp Neurol. 2009;215:243–252. doi: 10.1016/j.expneurol.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt RF, Scheff SW, Smith BN. Regionally localized recurrent excitation in the dentate gyrus of a cortical contusion model of posttraumatic epilepsy. J Neurophysiol. 2010;103:1490–1500. doi: 10.1152/jn.00957.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain SF, Kong LY, Jordan J, Conrad C, Madden T, Fokt I, Priebe W, Heimberger AB. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer research. 2007;67:9630–9636. doi: 10.1158/0008-5472.CAN-07-1243. [DOI] [PubMed] [Google Scholar]
- Iwamaru A, Szymanski S, Iwado E, Aoki H, Yokoyama T, Fokt I, Hess K, Conrad C, Madden T, Sawaya R, Kondo S, Priebe W, Kondo Y. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene. 2007;26:2435–2444. doi: 10.1038/sj.onc.1210031. [DOI] [PubMed] [Google Scholar]
- Jia F, Pignataro L, Schofield CM, Yue M, Harrison NL, Goldstein PA. An extrasynaptic GABAA receptor mediates tonic inhibition in thalamic VB neurons. J Neurophysiol. 2005;94:4491–4501. doi: 10.1152/jn.00421.2005. [DOI] [PubMed] [Google Scholar]
- Kharatishvili I, Pitkanen A. Association of the severity of cortical damage with the occurrence of spontaneous seizures and hyperexcitability in an animal model of posttraumatic epilepsy. Epilepsy Res. 2010;90:47–59. doi: 10.1016/j.eplepsyres.2010.03.007. [DOI] [PubMed] [Google Scholar]
- Kharlamov EA, Lepsveridze E, Meparishvili M, Solomonia RO, Lu B, Miller ER, Kelly KM, Mtchedlishvili Z. Alterations of GABA(A) and glutamate receptor subunits and heat shock protein in rat hippocampus following traumatic brain injury and in posttraumatic epilepsy. Epilepsy Res. 2011;95:20–34. doi: 10.1016/j.eplepsyres.2011.02.008. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Wen X, Buckmaster PS. Reduced inhibition and increased output of layer II neurons in the medial entorhinal cortex in a model of temporal lobe epilepsy. J Neurosci. 2003;23:8471–8479. doi: 10.1523/JNEUROSCI.23-24-08471.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupferman ME, Jayakumar A, Zhou G, Xie T, Dakak-Yazici Y, Zhao M, Ju J, Mandal M, Jasser S, Madden T, Myers JN, Priebe W. Therapeutic suppression of constitutive and inducible JAK\STAT activation in head and neck squamous cell carcinoma. Journal of experimental therapeutics & oncology. 2009;8:117–127. [PubMed] [Google Scholar]
- Li Y, Chen J, Chen XG, Wang L, Gautam SC, Xu YX, Katakowski M, Zhang LJ, Lu M, Janakiraman N, Chopp M. Human marrow stromal cell therapy for stroke in rat: neurotrophins and functional recovery. Neurology. 2002;59:514–523. doi: 10.1212/wnl.59.4.514. [DOI] [PubMed] [Google Scholar]
- Liang J, Zhang N, Cagetti E, Houser CR, Olsen RW, Spigelman I. Chronic intermittent ethanol-induced switch of ethanol actions from extrasynaptic to synaptic hippocampal GABAA receptors. J Neurosci. 2006;26:1749–1758. doi: 10.1523/JNEUROSCI.4702-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund IV, Hu Y, Raol YH, Benham RS, Faris R, Russek SJ, Brooks-Kayal AR. BDNF selectively regulates GABAA receptor transcription by activation of the JAK/STAT pathway. Science signaling. 2008a;1:ra9. doi: 10.1126/scisignal.1162396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund IV, Hu Y, Raol YH, Benham RS, Faris R, Russek SJ, Brooks-Kayal AR. BDNF selectively regulates GABAA receptor transcription by activation of the JAK/STAT pathway. Science signaling. 2008b;1:ra9. doi: 10.1126/scisignal.1162396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Sieghart W, Somogyi P. Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J Neurosci. 1998;18:1693–1703. doi: 10.1523/JNEUROSCI.18-05-01693.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva AA, Jr, Kang Y, Sanchez-Molano J, Furones C, Atkins CM. STAT3 Signaling after Traumatic Brain Injury. J Neurochem. 2011 doi: 10.1111/j.1471-4159.2011.07610.x. [DOI] [PubMed] [Google Scholar]
- Peng Z, Huang CS, Stell BM, Mody I, Houser CR. Altered expression of the delta subunit of the GABAA receptor in a mouse model of temporal lobe epilepsy. J Neurosci. 2004;24:8629–8639. doi: 10.1523/JNEUROSCI.2877-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkanen A, McIntosh TK. Animal models of post-traumatic epilepsy. Journal of neurotrauma. 2006;23:241–261. doi: 10.1089/neu.2006.23.241. [DOI] [PubMed] [Google Scholar]
- Raible DJ, Frey LC, Cruz Del Angel Y, Russek SJ, Brooks-Kayal AR. GABA(A) receptor regulation after experimental traumatic brain injury. Journal of neurotrauma. 2012;29:2548–2554. doi: 10.1089/neu.2012.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raol YH, Lund IV, Bandyopadhyay S, Zhang G, Roberts DS, Wolfe JH, Russek SJ, Brooks-Kayal AR. Enhancing GABA(A) receptor alpha 1 subunit levels in hippocampal dentate gyrus inhibits epilepsy development in an animal model of temporal lobe epilepsy. J Neurosci. 2006;26:11342–11346. doi: 10.1523/JNEUROSCI.3329-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saatman KE, Feeko KJ, Pape RL, Raghupathi R. Differential behavioral and histopathological responses to graded cortical impact injury in mice. Journal of neurotrauma. 2006;23:1241–1253. doi: 10.1089/neu.2006.23.1241. [DOI] [PubMed] [Google Scholar]
- Sassoe-Pognetto M, Panzanelli P, Sieghart W, Fritschy JM. Colocalization of multiple GABA(A) receptor subtypes with gephyrin at postsynaptic sites. J Comp Neurol. 2000;420:481–498. doi: 10.1002/(sici)1096-9861(20000515)420:4<481::aid-cne6>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Satriotomo I, Bowen KK, Vemuganti R. JAK2 and STAT3 activation contributes to neuronal damage following transient focal cerebral ischemia. J Neurochem. 2006;98:1353–1368. doi: 10.1111/j.1471-4159.2006.04051.x. [DOI] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM, Silver RA. Tonically active GABA A receptors: modulating gain and maintaining the tone. Trends Neurosci. 2004;27:262–269. doi: 10.1016/j.tins.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Shao LR, Dudek FE. Changes in mIPSCs and sIPSCs after kainate treatment: evidence for loss of inhibitory input to dentate granule cells and possible compensatory responses. J Neurophysiol. 2005;94:952–960. doi: 10.1152/jn.01342.2004. [DOI] [PubMed] [Google Scholar]
- Shohami E, Novikov M, Bass R. Long-term effect of HU-211, a novel non-competitive NMDA antagonist, on motor and memory functions after closed head injury in the rat. Brain Res. 1995;674:55–62. doi: 10.1016/0006-8993(94)01433-i. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Fritschy JM, Benke D, Roberts JD, Sieghart W. The gamma 2 subunit of the GABAA receptor is concentrated in synaptic junctions containing the alpha 1 and beta 2/3 subunits in hippocampus, cerebellum and globus pallidus. Neuropharmacology. 1996;35:1425–1444. doi: 10.1016/s0028-3908(96)00086-x. [DOI] [PubMed] [Google Scholar]
- Statler KD, Scheerlinck P, Pouliot W, Hamilton M, White HS, Dudek FE. A potential model of pediatric posttraumatic epilepsy. Epilepsy Res. 2009;86:221–223. doi: 10.1016/j.eplepsyres.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Sieghart W, Kapur J. Distribution of alpha1, alpha4, gamma2, and delta subunits of GABAA receptors in hippocampal granule cells. Brain Res. 2004;1029:207–216. doi: 10.1016/j.brainres.2004.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur C, Farrar SJ, Kerby J, Whiting PJ, Atack JR, McKernan RM. Preferential coassembly of alpha4 and delta subunits of the gamma-aminobutyric acidA receptor in rat thalamus. Mol Pharmacol. 1999;56:110–115. doi: 10.1124/mol.56.1.110. [DOI] [PubMed] [Google Scholar]
- Wei W, Zhang N, Peng Z, Houser CR, Mody I. Perisynaptic localization of delta subunit-containing GABA(A) receptors and their activation by GABA spillover in the mouse dentate gyrus. J Neurosci. 2003;23:10650–10661. doi: 10.1523/JNEUROSCI.23-33-10650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen TC, Peng H, Hata R, Desaki J, Sakanaka M. Induction of phosphorylated-Stat3 following focal cerebral ischemia in mice. Neurosci Lett. 2001;303:153–156. doi: 10.1016/s0304-3940(01)01711-6. [DOI] [PubMed] [Google Scholar]
- Willmore LJ, Ueda Y. Posttraumatic epilepsy: hemorrhage, free radicals and the molecular regulation of glutamate. Neurochemical research. 2009;34:688–697. doi: 10.1007/s11064-008-9841-3. [DOI] [PubMed] [Google Scholar]
- Zhang G, Raol YH, Hsu FC, Coulter DA, Brooks-Kayal AR. Effects of status epilepticus on hippocampal GABAA receptors are age-dependent. Neuroscience. 2004;125:299–303. doi: 10.1016/j.neuroscience.2004.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Wei W, Mody I, Houser CR. Altered localization of GABA(A) receptor subunits on dentate granule cell dendrites influences tonic and phasic inhibition in a mouse model of epilepsy. J Neurosci. 2007;27:7520–7531. doi: 10.1523/JNEUROSCI.1555-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Li G, Zhang Y, Su X, Hang C. The potential role of JAK2/STAT3 pathway on the anti-apoptotic effect of recombinant human erythropoietin (rhEPO) after experimental traumatic brain injury of rats. Cytokine. 2011a;56:343–350. doi: 10.1016/j.cyto.2011.07.018. [DOI] [PubMed] [Google Scholar]
- Zhao JB, Zhang Y, Li GZ, Su XF, Hang CH. Activation of JAK2/STAT pathway in cerebral cortex after experimental traumatic brain injury of rats. Neurosci Lett. 2011b;498:147–152. doi: 10.1016/j.neulet.2011.05.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.