

Abstract
Hydrogel based scaffolds for neural tissue engineering can provide appropriate physico-chemical and mechanical properties to support neurite extension and facilitate transplantation of cells by acting as ‘cell delivery vehicles’. Specifically, in situ gelling systems such as photocrosslinkable hydrogels can potentially conformally fill irregular neural tissue defects and serve as stem cell delivery systems. Here, we report the development of a novel chitosan based photocrosslinkable hydrogel system with tunable mechanical properties and degradation rates. A two-step synthesis of amino-ethyl methacrylate derivitized, degradable, photocrosslinkable chitosan hydrogels is described. When human mesenchymal stem cells were cultured in photocrosslinkable chitosan hydrogels, negligible cytotoxicity was observed. Photocrosslinkable chitosan hydrogels facilitated enhanced neurite differentiation from primary cortical neurons and enhanced neurite extension from dorsal root ganglia (DRG) as compared to agarose based hydrogels with similar storage moduli. Neural stem cells (NSCs) cultured within photocrosslinkable chitosan hydrogels facilitated differentiation into tubulin positive neurons and astrocytes. These data demonstrate the potential of photocrosslinked chitosan hydrogels, and contribute to an increasing repertoire of hydrogels designed for neural tissue engineering.
1. Introduction
In the United States 300 000 people suffer from injuries of the peripheral and central nervous systems.1–4 The most common approach to address segmental defects in the peripheral nervous system (PNS) is to use an autologous nerve graft,5,6 while, for the central nervous system (CNS), the astroglial scar, and limited inherent regenerative ability limit repair. Autografts used in PNS repair have several drawbacks including loss of function at the donor site, size mismatch between the damaged nerve and the nerve graft and the inability to fill irregularly shaped nerve cavities.7 Also, neural stem cells (NSCs) are potentially well suited for tissue engineered therapies in the CNS, such as trauma, stroke and Parkinson’s disease.8 Several studies have recently demonstrated that NSCs transplanted into injured CNS tissues promote regeneration and repair of motor and sensory functions, albeit with limited success.9,10 Also, controlled and local delivery of stem cells to the site of injury has been challenging owing to sub-optimal gelation times of hydrogels, microenvironment and mechanical stiffness mismatch, leading to reduced cell availability and viability at the site of injury.
Several tissue engineering strategies using biomaterials to create or spur regeneration are currently being evaluated as alternatives to autografts.3,11,12 One such class of materials, hydrogels, are of particular interest due to their inherent ability to facilitate presentation of growth factors and extracellular matrix (ECM) like cues, as well as their ability to provide physical and structural environments that mimic hydrated tissues. Also, importantly for soft tissue repair, hydrogels can be engineered to ‘gel’ in situ, allowing them to fill defects conformally and also facilitate their serving as ‘carriers’ for spatially controlled, local delivery of cells including stem cells.9,13,14 Several natural and synthetic materials have been explored as hydrogel scaffolds to repair nerve injuries.15–21 Chitosan has been widely used as a matrix for tissue engineering applications, in the form of hydrogels,22–26 nanofibers27,28 and phase separated scaffolds.29,30 Chitosan scaffolds have demonstrated excellent biocompatibility, enzyme-mediated degradation profiles, and non-toxic degradation products.31 Also, the positive surface charge of chitosan promotes cell attachment and differentiation, and the active end groups of the polymer lend themselves to coupling of bioactive proteins making it an attractive scaffolding material.32–35
Widespread application of chitosan for neural tissue engineering has nevertheless been limited by issues of solubility at physiological pH, lack of control over its gelation time, and inability to tune its mechanical properties to support 3 dimensional (3D) neurite extension. Increasing the degree of deacetylation or introduction of carboxylation26,36 increases chitosan’s water solubility and in situ gelation with optimal gelling times and control of its physico-chemical properties continues to be challenging. The use of photocrosslinkable chitosan hydrogels may be advantageous for use in neural tissue engineering due to the minimal use of cytotoxic chemicals in the fabrication process, the potential ease of encapsulating cells or bioactive agents within the gel,25,36,37 and their ability to fill complex 3D voids created due to an injury. Photocrosslinkable chitosan hydrogels have been fabricated before, but some shortcomings persist such as those evident in azo-derivitized chitosan hydrogels which have a short gelation time and high storage moduli or pore sizes that are not ideal for neurite outgrowth.38–40 Conversely, photocrosslinkable chitosan synthesized using methacrylic anhydride yields chitosan that is soluble at acidic pH. However, due to the relatively long gelation times of these gels (approximately 10 to 30 min), encapsulated cells risk being exposed to extended UV radiation.22,25,36,37
Jeon et al. reported a functionalization of alginic acid resulting in photocrosslinked alginate based hydrogels41 derivitized with 2-amino ethyl methacrylate (AEMA). This approach yielded gels with controllable degradation rate, high water content, optimal and controllable mechanical stiffness and no cytotoxicity. Using similar methods, we fabricated photocrosslinkable water-soluble chitosan using AEMA to introduce functional methacrylate groups. Here, this hydrogel’s physical properties such as gelation time, swelling and rheological characteristics as well as its ability to support 3D neurite extensions and encapsulate cells were investigated. We report photocrosslinkable chitosan’s ability to support primary cortical neuron differentiation, neurite outgrowth of rat dorsal root ganglia (DRGs) cultured within photocrosslinked hydrogel networks. In addition, we report the ability of these gels to support 3D cultures of both mesenchymal stem cells and NSCs.
Several in vitro studies have explored the culture and behavior of stem cells when cultured on top of hydrogels, in a 2 dimensional (2D) culture environment.25 However, it is likely that success of NSC transplantation in vivo will depend on delivering them in 3D gels in vivo. Here, we demonstrate the viability, differentiation and phenotypic stability of NSCs when cultured in 3D chitosan hydrogels in addition to their ability to support 3D neurite extensions from primary neurons. Using RT-PCR analysis, we also examine the phenotypic stability of NSCs cultured in chitosan hydrogels.
2. Experimental
Materials
Chitosan, medium molecular weight (degree of acetylation 70%), chloroacetic acid, AEMA, EDC, NHS and other chemical reagents were obtained from Sigma-Aldrich (St. Louis, MO) and were used without further purification. The photoinitiator, Irgacure was a generous gift from Ciba and was used without further purification as well. Agarose (Seaprep®) was obtained from Lonza Technologies and was used without further modifications. Dialysis bags (10 000 Da) were obtained from Pierce (Thermo Fisher Corporation, NJ). Cell culture reagents were obtained from Mediatech (CellGro Technologies). Primary and secondary antibodies against tubulin, glial fibrillar acidic protein (GFAP) and DAPI were obtained from Invitrogen (Carlsbad, CA). All RNA extraction and purification kits were obtained from Qiagen and the primers for qRT-PCR were obtained from Integrated DNA Technologies (Coralville, IA) (Table 1). Neural stem cells and dorsal root ganglia (DRGs) were purchased from Brain Bits LLC (Springfield, IL).
Table 1.
Primer sets for qRT-PCR
| Gene | Forward primer | Reverse primer |
|---|---|---|
| GFAP (NM_017009.2) | 5′-GCTTCCTGGAACAGCAAAACA-3′ | 5′-CGAAGTTCTGCCTGGTAAACG-3′ |
| Olig1 (NM_021770.3) | 5′-TTACAGGCAGCCACCCATCT-3′ | 5′-GAGCGGAGCTTCAGGCTTCT-3′ |
| βIII Tubulin (Tubb3) (NM_139254.2) | 5′-AAGCCCTCTACGACATCTGCTT-3′ | 5′-GTGGTGACTCCGCTCATGGT-3′ |
| Nestin (Nes) (NM_012987.1) | 5′-AGGCCACAGTGCCTAGTTCTTC-3′ | 5′-TGTGGCTAAGGAGGTCAGATCA-3′ |
| GAPDH (NM_017008.3) | 5′-GGTGGACCTCATGGCCTACA-3′ | 5′-CAGCAACTGAGGGCCTCTCT-3′ |
Methods
2.1 Preparation of carboxylmethyl-chitosan
Carboxymethyl-chitosan (CM-chitosan) was prepared by a modification of a previously described method.26,42–46 Chitosan (1.5 g) was added to a round bottom flask and allowed to swell in 33% solution of aqueous sodium hydroxide at room temperature for 24 h. The hydrated chitosan was then vacuum filtered and transferred to a clean round bottom flask, incubated in a 50 °C water bath for 1 h. To this, 20% (w/v) monochloroacetic acid, dissolved in 20 mL isopropanol, was added drop wise over a 30 min period. The solution was stirred over 6 h at 50 °C and then allowed to cool to room temperature. The precipitate was filtered and rinsed with 100% ethanol to remove the salts and isopropanol and vacuum dried at room temperature. The product was the sodium salt form of CM-chitosan.
The CM-chitosan (1 g) was dissolved in 50 mL deionized (dI) water and homogenized for 2 h. The solution was centrifuged at 5000 rpm for 15 min to remove the insoluble chitosan. The supernatant was collected and purified CM-chitosan sodium salt was precipitated by adding chilled absolute ethanol. The fibrous chitosan was centrifuged and collected, and then dissolved in 100 mL of dilute (0.02 M) hydrochloric acid and stirred for 30 min. The solution was centrifuged at 1000 rpm for 10 min to remove the insoluble compounds and was reprecipitated in chilled absolute ethanol. The solution was filtered, and the solute was collected, rinsed in 100% ethanol, and then vacuum dried. The chitosan was used for further functionalization with methacrylate groups to make them photocrosslinkable.
2.2 Grafting of AEMA to CM-chitosan
CM-chitosan (1.0 g) was dissolved in 80 mL 0.1 M morpholinoethano sulfonic acid (MES, pH 6.0–8.0) at room temperature. To this 10 mM EDC and 10 mM n-hydroxy succinimide (NHS) were added to activate the carboxylic groups. After 1 h, 1.2 g of AEMA was added and allowed to stir for 24 h at 4 °C. The solution was then dialyzed, against water, with a molecular weight cut of 10 000 Da to remove the salts and the MES buffer for 48 h with frequent water change. The product, a methacrylate functionalized chitosan, as shown in Fig. 1, was lyophilized overnight to obtain photocrosslinkable chitosan.
Fig. 1.

Schematic of synthesis of carboxymethyl chitosan and methacrylated chitosan in a 2 step process.
2.3 Evaluation of degree of methacrylation
The degree of methacrylate modification of chitosan was determined using 1H NMR spectroscopy according to a modified published procedure.41,43,47 Briefly, a 0.1% (w/v) solution of chitosan–methacrylate was dissolved in deuterated water (D2O) by vortexing. As a control, carboxymethyl-chitosan was also prepared at the same concentration in the same solvent. The 1H-NMR spectrum was recorded using a 400 Hz mercury spectrometer (Varian). The degree of methacrylation was calculated based on the ratio of the integrated area of the H3–H6 peaks of CM-chitosan moiety between 3.3 and 3.8 ppm (5 protons) to that of the CH2 peaks of the methacrylate group at 5.6 and 6.1 ppm (2 protons).
2.4 Fabrication of photocrosslinked chitosan hydrogels
The photocrosslinkable chitosan was dissolved in either media or sterile phosphate buffered saline (PBS) to prepare concentrations ranging from 0.5 to 2% (w/v) of chitosan. The solutions were stirred overnight and degassed prior to application. The precursor solution was sterilized using ultraviolet (UV) light (Blakray, 160 W, 365 nm) for 30 min. The 0.1% (w/v) photo-initiator (Irgacure, I2959) was dissolved in PBS and sterile filtered via a 0.2 μm filter. Depending on the application and size of hydrogels needed, solutions from 200 μL to 1 mL were used. To the required amounts of hydrogel solution, 10% (v/v) of the photoinitiator solution was added and vortexed. This solution was then transferred to multi-well plates and exposed to UV light for 3–5 min. The gelling time was estimated based on the time needed for the solutions to resist gravitational flow.
2.5 Characterization of photocrosslinked hydrogels
2.5.1 Morphology of hydrogels by optical and scanning electron microscopy (SEM)
The scaffold integrity and morphology were examined using optical microscopy. The scaffolds were transparent, and would therefore allow for simple optical phase-contrast imaging of encapsulated cells within them. Also, surface and interior morphologies of photocrosslinked chitosan hydrogels were probed using a Hitachi variable pressure scanning electron microscope on uncoated hydrogel samples. The swollen hydrogel samples, after reaching their maximum swelling ratio in distilled water at room temperature after 24 h, were used for interior morphology observation with a scanning electron microscope instrument. High (1000×) and low (100×) magnification images were obtained to study the pore structures and morphologies.
2.5.2 Swelling and degradation of photocrosslinked chitosan hydrogels
2.5.2.1 Degradation rates
Immediately after fabrication, the sterile chitosan scaffolds were weighed (Wo) and transferred to 6 well plates and filled with PBS supplemented with 0.2% (w/v) lysozyme and left at 37 °C in an incubator. For the first week, weights were measured every day and then once every week after that for 8 weeks. In order to determine weights, the gels were blotted dry and were weighed (Wt) (Mettler Toledo). The percentage mass loss was calculated based on the ratio of change in weight to the original weight of the gels.
| (1) |
2.5.2.2 Characterization of chitosan swelling
Five millimetre diameter chitosan hydrogel (0.5% w/v) disks were lyophilized postfabrication, weighed (Wd, dry weight) and placed in PBS at 37 °C in a 12 well plate to swell. After certain time points, swollen samples were blotted dry and weighed (Ws, swollen weight). The swelling ratio of the hydrogels was calculated using the following equation:
| (2) |
2.6 Rheological studies on hydrogels
The storage modulus, loss modulus and complex modulus of the hydrogel system were determined using a ARES rheometer (Rheometrics Inc.) at 25 °C. Chitosan hydrogel samples of different concentrations (0.5%, 1% and 2%) were prepared as described above. As a control 1% Seaprep® agarose hydrogels were also evaluated. We have earlier shown that Seaprep hydrogels (1%) are known to support neurite outgrowth with implanted dorsal root ganglia.6,33,34,48 In order to evaluate the rheological parameters, the hydrogels were pre-swollen for 24 h in PBS and then discs of about 1 cm diameter were cut, using biopsy punches, and placed in a parallel plate rheometer. Frequency sweep experiments were performed in the range of 0.1 Hz to 100 Hz (corresponding to the linear response range of the hydrogels). At least 6 samples per sample type were evaluated for analyzing statistical significance.
2.7 Cytotoxity of photocrosslinked chitosan hydrogels
2.7.1 Direct contact method
In vitro cytotoxicity testing was conducted using the direct contact method previously established by Jiang et al.35 Hydrogel samples (photocrosslinkable chitosan and agarose) were placed in triplicate on the sub-confluent monolayer of human mesenchymal stem cells (hMSCs) and evaluated for cytotoxicity. Briefly, hMSCs, maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum and 1% Penicillin–Streptomycin, were sub-cultured by trypsinization and seeded onto 12-well tissue culture plates (Nunc). After incubation of cells with hydrogel samples at 37 °C for 48 h, the cells were incubated with a LIVE/DEAD assay solution (Invitrogen) according to manufacturer’s protocol and washed with PBS twice prior to analysis. Three samples per condition were imaged using a microscope equipped with an epifluorescence set-up, excitation/emission setting of 488/530 nm to detect green (live) fluorescence and 530/580 nm to detect red (dead) cells (Axiovert-Zeiss).
2.7.2 Human mesenchymal stem cell proliferation in hydrogels
Viability of encapsulated cells was analyzed after days 1, 4 and 7 in photocrosslinked chitosan hydrogels (0.5% v/v) obtained by exposure to 365 nm UV light in the presence of 0.1% (w/v) Irgacure 2959 and agarose (1% Seaprep®) hydrogels. Also, as a control, cells were cultured on the bottom of 12 well plates, in triplicates. In order to quantify the cell number, a cell counting kit (CCK, Dojindo) was used as per manufacturer’s protocols. Briefly, at days 1, 4 and 7, the gels were washed once in sterile PBS and were incubated with medium containing 10% (v/v) CCK reagent. After 2 h, the absorbance of the solution was measured at 490 nm using a microplate reader (Biotek Instruments).
2.7.3 3 Dimensional cultures of cortical neurons in chitosan hydrogels
Cortical neurons were obtained from embryonic day 18 (E-18) rats (Brain Bits LLC) as per previously published protocols.32,49,50 Cerebral hemispheres were stripped of meninges, cut into small pieces, and enzymatically dissociated with 0.25% trypsin in Ca2+/Mg2+-free PBS for 20 min at 37 °C. The reaction was quenched by addition of medium supplemented with 10% fetal bovine serum. After a brief centrifugation, the cells were resuspended in neurobasal medium, supplemented with 1% B-27 and L-Glutamine (Glutamax®). Sixty thousand cells were cultured within either 250 μL of (0.5%) chitosan or 1% Seaprep® agarose. Postgelation the chitosan gels were washed thoroughly in neurobasal medium to remove residual photoinitiator and the gels were transferred to the incubator after addition of fresh medium. 72 h postseeding, the cells were fixed in 4% paraformaldehyde. The cells were then incubated with β-tubulin primary antibody (1 : 250) and then stained with Texas red secondary antibody (1 : 220). The gels were then washed three times to remove unbound antibodies and then imaged with a confocal microscope (Zeiss Axiovert) at 10× magnification. The percentage of cells that differentiated (extended neurons) was evaluated by quantification of the ratio of cells that differentiated to the total number of cells present, in the field of view. In order to evaluate this, at least 5 images were obtained from 3 hydrogel samples of agarose and chitosan cells with and without neurites were counted manually. A cell was considered differentiated if the neurite length (extension) was greater than one cell diameter as described earlier.23,51
2.7.4 Dorsal root ganglion (DRG) explant studies
Embryonic day 18 (E18) rat DRGs were cultured with chitosan for 3D culturing. The solution was then exposed to UV radiation as described earlier to obtain hydrogels. As controls, 1% Seaprep® agarose hydrogels were used. Neurobasal medium, supplemented with 50 ng mL−1 nerve growth factor, 1% (v/v) B-27 and 0.1% (v/v) Glutamax® were added on top of the hydrogels. Quantification of neurite extension on the chitosan and agarose hydrogels was performed as previously described.16,52 Briefly, after 72 h culturing, the explants were fixed in 4% paraformaldehyde and then imaged using a digital image analysis system that consisted of a Zeiss Axiovert Model 35 microscope with a Nikon CCD camera. Images of neurites on different planes were captured and the lengths of the 10 longest neurites from each explant, obtained from the images, were measured using ImagePro.48
2.7.4.1 Confocal microscopy
Immunohistochemistry was performed on the above DRG as described in previous studies.52 Briefly, the fixed DRG were first incubated for one hour at room temperature in a blocking solution (4% goat serum (Invitrogen) in PBS containing 0.5% Triton X-100 (Sigma)), followed by an overnight incubation of primary rabbit anti mouse Neurofilament 200 (NF-200) antibody in blocking solution at 4 °C. After three washes, the dishes were incubated once more for 1 hour at room temperature in a solution of rhodamine anti-rabbit secondary antibodies, diluted 1 : 220 in PBS. After three final washes, the dishes were maintained in PBS at 4 °C until they were imaged using a Zeiss inverted confocal microscope equipped with necessary optics.
2.7.5 NSC isolation and encapsulation within chitosan hydrogels
NSCs were obtained from E18 rat cortex/hippocampus including subventricular zone (Brainbits LLC). NSCs were expanded in growth medium containing neurobasal media, B27 neural supplement, Glutamax, 100 μg mL−1 penicillin–streptomycin, 20 ng mL−1 epidermal growth factor (EGF—recombinant human) and 20 ng mL−1 basic fibroblastic growth actor (bFGF—recombinant human bFGF; Invitrogen) in ultra-low adhesion corning flasks, to form uniform neurospheres. The resulting neurospheres were passaged at least 3–4 times before encapsulation within hydrogels.
Prior to implantation, the NSC neurospheres were dissociated in Accutase (Stem Cell Technologies) for 5 min at 37 °C and triturated by mixing with a 1 mL pipette to obtain a uniform suspension. The single cells were then counted with a hemocytometer and were suspended in chitosan (0.5% w/v), prepared in Neurobasal medium. At least 1 million cells were present per 500 μL of gel solution and were transferred to 24 well plates and gelled as described in previous sections. The gels were then supplemented with differentiation medium containing neurobasal media, Glutamax® 100 μg mL−1 penicillin–streptomycin, without growth factors. The cells were transferred to an incubator at 37 °C and 5% CO2. The cells were cultured for 7 and 14 days and at these specific time points, cell-laden scaffolds were either fixed for imaging or the RNA was extracted for real time PCR analysis. At least 3 samples per time point per type of gel were used for analysis.
2.7.5.1 Immunohistochemistry of NSCs within hydrogels
At specific time points, on days 7 and 14, the gels were washed in PBS twice, fixed in 4% paraformaldehyde overnight at 4 °C. The gels and cells were then permeabilized with 0.1% Triton X-100, supplemented with 4% goat serum in PBS for an hour. The gels were then washed thrice and the following primary antibodies were used to stain for neurons and astrocytes; monoclonal mouse anti-β-III tubulin (1 : 500) and monoclonal mouse anti-glial fibrillary acidic protein (GFAP, 1 : 100) respectively were added and incubated for 24 h at 4 °C. After washing with PBS thrice, samples were incubated in appropriate secondary antibodies (1 : 150) for 2 h at room temperature and washed with PBS twice, prior to imaging.
2.7.5.2 RNA isolation from NSCs in hydrogels and real-time PCR
Cells within the gels were lysed using Trizol (Invitrogen) at days 7 and 14 and the total RNA was isolated using the RNeasy mini kit as described by Wang C et al.53 After RNA isolation and purification, total RNA concentration and purity were assessed using Quant-iT RiboGreen RNA Reagent Kit, as per the manufacturer’s protocols. For the reverse transcription (RT) reaction, 40 ng of the total RNA was converted to cDNA using a high-capacity cDNA reverse transcription kit (Applied Bio-systems). Specific primers of interest were designed using Primer Express® software (Applied Biosystems) and obtained from Integrated DNA Technologies. Primer validation and qRT-PCR using SYBR green mix (Applied Biosystems) were conducted on a StepOnePlus real-time PCR machine according to methods previously published.54,55 All qRT-PCR reactions were performed in triplicates and fold differences for each target gene were calculated against normalized controls (day 7 chitosan), and the relative fold change was calculated by normalizing against the endogenous reference gene GAPDH.
2.8 Statistical analysis
The data for hydrogel characterization, neurite extension and percentage differentiation are reported as means ± standard deviations (SD) for triplicate samples, unless otherwise described in the Experimental section. Two-way analysis of variance (ANOVA) was performed using GraphPad to assess the statistical significance of the results.
3. Results
3.1 Synthesis and characterization of photocrosslinkable chitosan
The synthesis of photocrosslinkable chitosan precursors required a two-step process including the preparation of a water-soluble chitosan derivative, followed by the incorporation of photocrosslinkable moieties into the derivative, as chitosan itself is not soluble in a neutral pH aqueous medium. The water-soluble precursor, carboxymethyl-chitosan, was prepared using a onestep chemical reaction between medium molecular weight chitosan (50 000–500 000 Da) and chloroacetic acid.42 The yield of the final product, as estimated by dry mass measurement after freeze drying, was 69 ± 7%. This acid functionalized water soluble chitosan was then modified to contain methacrylate groups as described earlier, using carbodiimide chemistry to couple AEMA. Fig. 2 shows the NMR spectra of carboxymethyl chitosan (Fig. 2A) and chitosan methacrylate (Fig. 2B). The incorporation of double bonds into the chitosan backbone was confirmed by 1H NMR, as indicated by the appearance of proton signals from –C(CH3)=CH2– at 5.6, 6.1 and 1.7 ppm (Fig. 2B). From the corresponding spectra, the degree of methacrylation was calculated based on the ratios of integrated area of the peaks from 3.3 to 3.8 and 5.6 to 6.1 ppm and was estimated to be approximately 28.14%.
Fig. 2.

(A) 1H-NMR spectrum of carboxymethyl chitosan (CMC). The CH2 peaks of (a) and (d) are overlapped with the solvent signal around 4.8 ppm. (B) 1H-NMR spectrum of methacrylated carboxymethyl chitosan.
3.2 In situ gelation and gelling time
This gelation phenomenon of aqueous photocrosslinkable chitosan solution upon exposure to 365 nm irradiation in the presence of photoinitiator led to the formation of subsequent network structure formation. Fig. 3A shows the solution prior to exposure to UV radiation and Fig. 3B shows the hydrogel formed, postexposure to UV radiation, after 3 min. In order to confirm the gelation, the hydrogel precursor solution was also exposed to UV radiation for 3 min, but in the absence of photoinitiator, Irgacure (I-2959). As shown in Fig. 3C, in the absence of the photoinitiator, the network formation was inhibited and no significant gelation was observed, as shown by the change in angle of the solution level in the tube in which it was held.
Fig. 3.

In situ gelling of chitosan based hydrogels. (A) Lack of gelation prior to UV induced photocrosslinking of chitosan precursor and (B) gelation occurs after exposure to UV radiation for 3 min. (C) Role of photocrosslinker in gelation, top shows gel formation due to photocrosslinker and bottom shows no gelation in the absence of photocrosslinker postexposure to UV radiation, after 3 min.
3.3 Hydrogel morphology and scanning electron microscopy
Fig. 4A shows the gross morphology of 1% (v/v) concentration of photocrosslinked chitosan hydrogels and Fig. 4B shows the morphology of 0.5% (v/v) chitosan hydrogels. Photocrosslinked chitosan hydrogels were transparent. However, opacity increased with increasing concentrations of chitosan in the gels. In order to investigate the micro-structure of the hydrogels, we examined the morphology of the hydrogels using SEM. From Fig. 4D we can observe the pore morphology of the hydrogel as well as the pore interconnectivity of the hydrogel hence formed. The hydrogels had a porous structure, with pore sizes ranging from 5 to 25 μm.
Fig. 4.
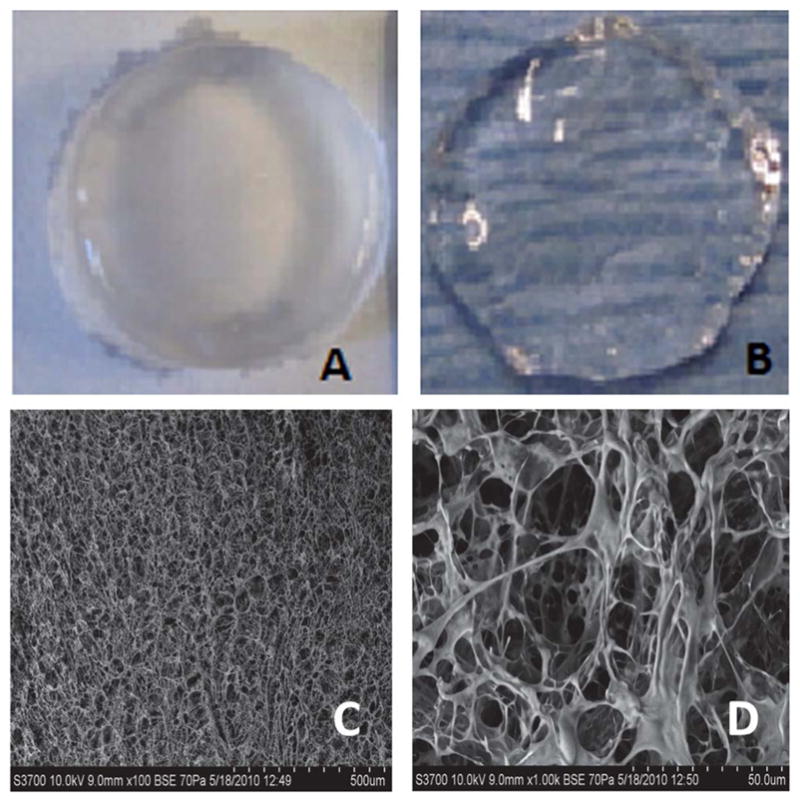
Optical microscopy and scanning electron micrographs of chitosan hydrogels. (A) 1% photocrosslinked chitosan hydrogels, (B) 0.5% photocrosslinked chitosan hydrogels, (C) low magnification (100×) scanning electron micrograph of chitosan hydrogels (0.5% w/v), and (D) high magnification (1000×) image of chitosan hydrogels (0.5% w/v).
3.4 Equilibrated swelling ratio of photocrosslinked chitosan hydrogels
The swelling kinetics of the photocrosslinked chitosan hydrogels were investigated over a period of 4 days using DI water. As shown in Fig. 5A, the photocrosslinked hydrogels generally showed a high swelling rate during the initial 3 h, then plateaued, and reached a swelling equilibrium after about 2 days. The photocrosslinked chitosan hydrogels absorbed over 12 times their dry weight in water over the first 24 h. After 48 h, they absorbed water content to about 20 times the initial dry weight and remained constant for the rest of the study period.
Fig. 5.

Swelling ratio and degradation of photocrosslinked chitosan hydrogels.
3.5 Degradation of photocrosslinked hydrogels
The mass loss (%) of photocrosslinked hydrogels over time was determined as a measure of degradation (Fig. 5B). The degradation studies were carried out in lysozyme supplemented PBS, as chitosan does not undergo uniform or complete degradation in the absence of enzymes. Due to the swelling properties of the hydrogel, no significant mass loss could be detected during the first week of the study; measureable differences in masses were observed after the first week. Approximately 50% of the mass was lost after the first 3 weeks and at the end of the study (6 weeks) 80% of the gel mass was lost. Due to the lack of gel integrity owing to degradation and the difficulty in handling of the gel, the study could not be continued beyond six weeks.
Covalently photocrosslinked hydrogels, like the ones described here, will produce extractables, usually crosslinkers or polymer chains of chitosan that were not polymerized or converted to methacrylated chitosan and therefore are not a part of the network formed. However, they are kept in the gel structure because of physical entanglements within the hydrogel. When the gels swell in a solvent, these polymer chains slowly diffuse into the medium. This could explain the mass gained in the initial phase of the degradation period, as described earlier.56,57 Approaches to determine the percentage of extractables could not be performed in this study as these hydrogels degrade significantly in physiological conditions in the solvents or medium used.
3.6 Rheological properties of chitosan hydrogels
The rheological properties of three concentrations of photocrosslinkable chitosan hydrogels were characterized using a rheometer as described earlier. Also, as a control, 1% agarose hydrogel was analyzed. Fig. 6A shows the angular frequency dependence of storage modulus (G′) and loss modulus (G″) for 0.5% (v/v) chitosan hydrogels after 3 min of UV irradiation. From these data, it is evident that the G′ value is fairly constant throughout the entire frequency region, although a slight increase is observed with the increase of frequency. This phenomenon indicates that the system after irradiation displays a predominantly gel like behavior, which can be attributed to UV-induced crosslinking. It is also evident from Fig. 6B that 0.5% chitosan methacrylate gels had similar mechanical properties to that of 1% Seaprep hydrogels in the range between 50 and 100 Hz in the frequency sweep studies, and therefore have been used for cytotoxicity and neurite extension studies. We also investigated the storage modulus as a function of frequency for two other photocrosslinked hydrogels (1% and 0.25%) formed by the same precursor solution and photocrosslinking process, as shown in Fig. 6C. We observed that the hydrogels with a higher chitosan content (1%) had a higher G′ value (404.08 ± 23.45 Pa) in the entire frequency region when compared to hydrogels with a lower chitosan concentration (0.25% w/v) (10.12 ± 4.73 Pa), as expected. This fact may be attributed to the formation of a denser network structure owing to higher availability of functionalized methacrylate groups.
Fig. 6.

Rheological assessment of chitosan and agarose scaffolds. (A) Frequency sweep of chitosan (0.5%) and agarose (1%) hydrogels. (B) Storage modulus comparison of chitosan versus agarose hydrogels, * shows significantly higher (p < 0.05) storage modulus as compared to chitosan scaffolds. (C) Storage moduli of various chitosan percentages, * indicates significant higher storage modulus of 1% chitosan as compared to 0.5 and 0.25% hydrogels, ** indicates significantly higher storage modulus of 0.5% hydrogels as compared to 0.25% hydrogels (p < 0.05).
3.7 Cytotoxicity of photocrosslinked chitosan hydrogels
The cytotoxicity of photocrosslinkable chitosan hydrogels was estimated using two techniques: the direct contact method, which allows for evaluation of cytotoxicity owing to degradation products, and by incorporation of cells within the hydrogels. For the first study, live-dead assay and imaging were used to qualitatively evaluate the cytotoxicity of the hydrogels, meanwhile, the effect on incorporated cells within the hydrogels was assessed using a cell counting kit, as described earlier. As shown in Fig. 7A, no significant change in morphology of the human mesenchymal stem cells was detected 2 days after incubation with chitosan hydrogels. There were also no visible signs of cell rounding or increased vacuolization, which are typical indicators of cell death. Similar results were observed with agarose based hydrogels as well at the same time points (Fig. 7B).
Fig. 7.
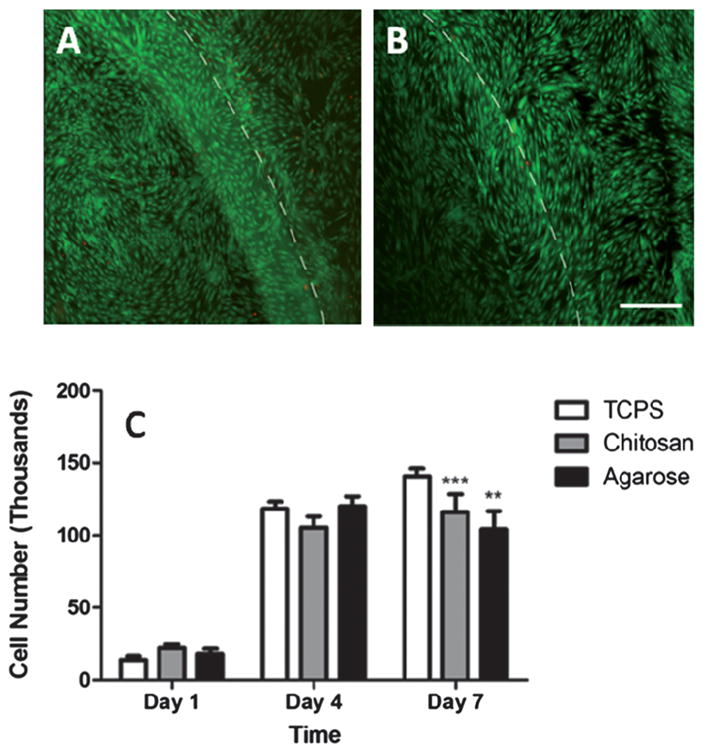
Human mesenchymal stem cytotoxicity studies with chitosan and agarose hydrogels. (A) Micrograph showing live (green) and dead (red) cells using the direct contact method, with chitosan hydrogels. Dashed lines indicate the gel interface. (B) Agarose hydrogels and (C) quantification of cell proliferation within hydrogels. ** and *** show a significantly lower number of cells as compared to tissue culture plate (TCPS) on day 7 (p < 0.05). Scale bar = 50 μm.
The results from the CCK assay (Fig. 7C) indicated that no significant differences in cell numbers were observed in the photocrosslinked chitosan hydrogels as compared to the blank controls (tissue culture plate wells, at earlier time points, days 1 and 4, in this study). However, both chitosan and agarose hydrogel showed a significant (p < 0.05) reduction in cell numbers as compared to control plates at day 7, but no significant reduction was observed between agarose and chitosan hydrogels at any time point. The significant reduction in cell numbers can be attributed to the reduced penetration depth of the analyte (CCK) into the scaffolds or its ability to leave the gel system, therefore not allowing for complete quantification.
3.8 3D cortical neuronal cultures
E-18 cortical neurons were immobilized within 0.5% chitosan and 1% agarose hydrogels as described earlier, to provide a preliminary assessment of neuronal survival and differentiation. By day 3 of culture, differences in cell morphology of the neurons in the two gels were apparent. In the photocrosslinked chitosan hydrogel, the cells appeared to be clumped into groups of about 5–10 cells and extended neuronal processes (Fig. 8A). Meanwhile, in 1% (v/v) agarose gel, at day 3, not much cell clumping was observed. Also, a very small percentage of the immobilized cells extended processes, which were usually short (Fig. 8B). Quantification of the number of cells extending processes greater in length than one cell diameter (10 μm) revealed that 38.02 ± 13.29% of the cells implanted in chitosan exhibited neurite outgrowth, meanwhile in agarose neurite outgrowth was limited to 6.66 ± 4.17%. Based on these observations, we can conclude that the chitosan hydrogels significantly enhanced neurite outgrowth, meanwhile the agarose based system does not support cortical neuron growth to the same extent as that of chitosan gels.
Fig. 8.
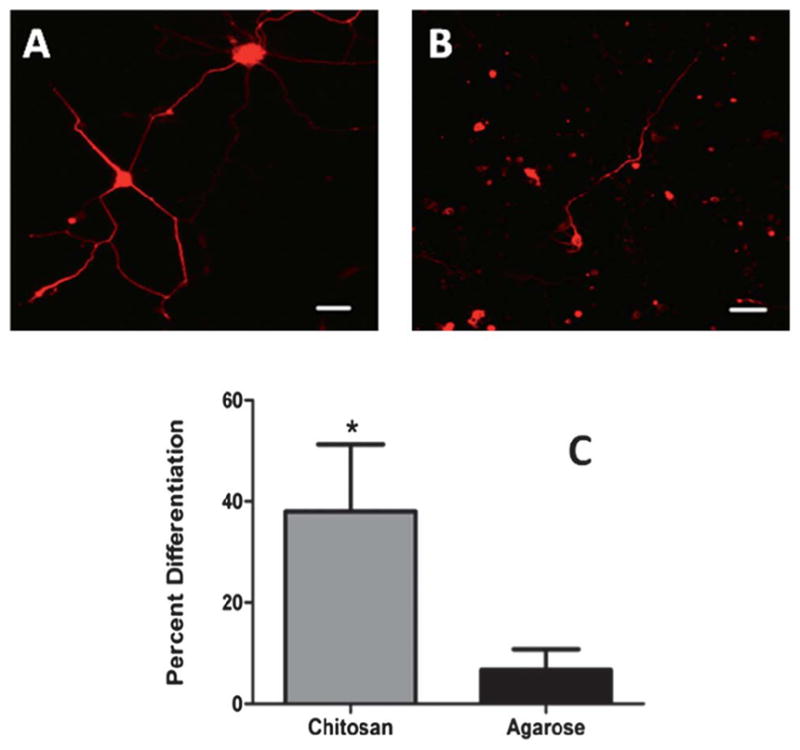
Cortical neuron differentiation within hydrogels. (A) Cortical neuron showing extensive neurite outgrowth within chitosan hydrogels (B) neurite extension within agarose hydrogels indicating low percentage differentiation. Scale bar = 50 μm. (C) Quantification of neurite outgrowth within chitosan (0.5%) and agarose hydrogels (1% Seaprep®). * indicates statistically significant higher percentage (p < 0.05) of neurite extension from chitosan hydrogels as compared to agarose hydrogels.
3.9 Dorsal root ganglion neurite extension studies
Chitosan and agarose hydrogels supported the growth of DRG explants dissected from rat embryos. Fig. 9A shows that DRG explants cultured within chitosan hydrogels were active and extended neurites. Also, 1% Seaprep hydrogels also supported neurite outgrowth in 3D, as shown previously.6,16,34 Image analysis was used to quantify the neurite outgrowth on chitosan and agarose hydrogels. DRG neurite lengths on different hydrogel formulations are compared in Fig. 9B, which shows that neurite lengths on chitosan based scaffolds (1062 ± 174.1 μm, n = 4) were significantly higher than the neurite lengths on 1% agarose based scaffolds (544.2 ± 23.26 μm, n = 4) after 72 h of culture.
Fig. 9.
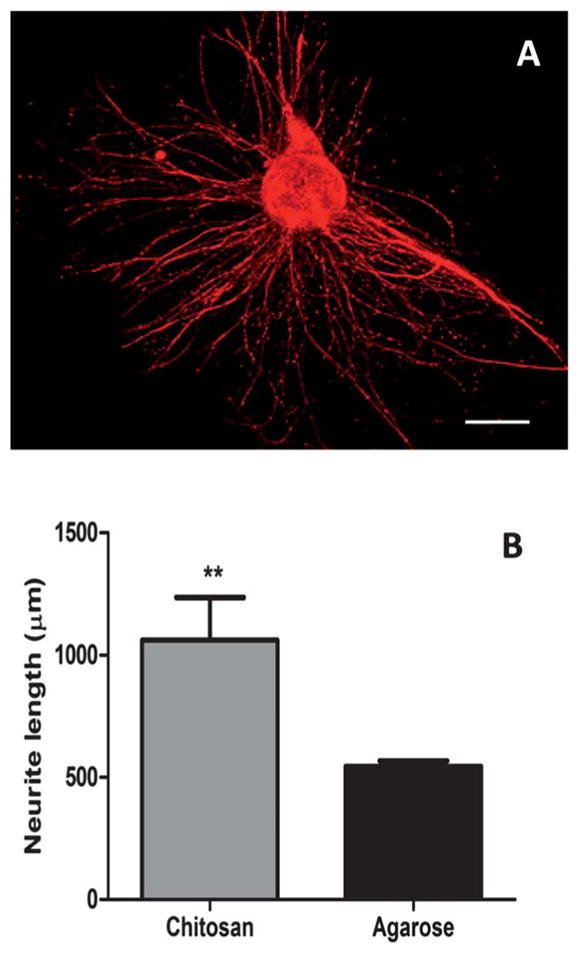
DRG neurite extension within photocrosslinked chitosan hydrogels. (A) Confocal micrograph of 3D neurite extension in hydrogels. Scale bar = 500 μm. (B) Quantification of neurite extension in chitosan (0.5%) hydrogels and 1% Seaprep hydrogels. ** indicates significantly higher neurite length (p < 0.05) within chitosan gels as compared to agarose.
3.10 3D culture of NSCs within chitosan hydrogels
Neural Progenitor Cells (NSCs) showed differentiation when cultured on chitosan hydrogels. Fluorescent microscopy showed differentiation of NSCs into tubulin positive cells (neurons), as well as GFAP positive cells (astrocytes), within the spheroids. However, nestin positive colonies were observed only at day 7 within chitosan hydrogels, as shown in Fig. 10A. Even though tubulin positive neurons were interspersed within the colonies in the hydrogels, some hydrogels showed migration of neurons from within the spheroids. This can be attributed to optimal mechanical stiffness of chitosan hydrogels to support cell migration and differentiation. At longer time points (day 14) significant expression of tubulin and GFAP is visible within the hydrogel cluster, indicative of successful differentiation within them, as shown in Fig. 10B. Nestin expression within these hydrogels was not very detectable at these time points (data not shown), indicative of complete differentiation of neurospheres to either neurite or astrocyte like colonies.
Fig. 10.
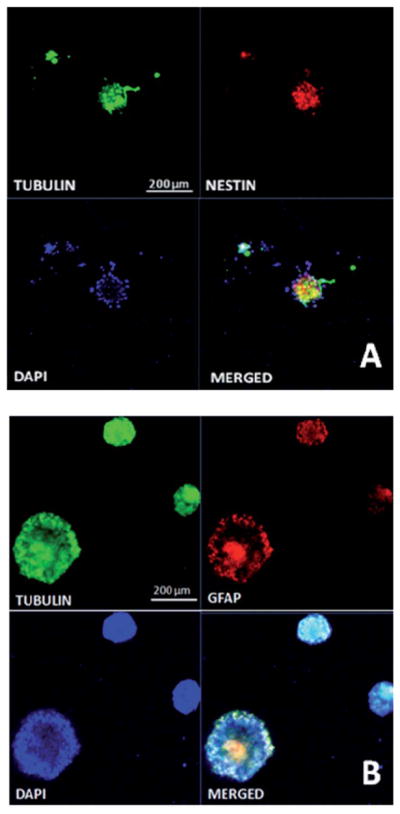
Characterization of NSCs in chitosan hydrogels. (A) NSC differentiation showing stem cell differentiation into tubulin positive neurite outgrowth at day 7 within chitosan hydrogels. (B) NSC differentiation in chitosan scaffolds at day 14, showing tubulin and glial fibrillary acidic protein expression, indicative of neurite and astrocytic differentiation of stem cells. Scale bar = 200 μm.
3.11 Quantitative RT-PCR on NSCs
In order to verify the effect of chitosan hydrogel microenvironment on NSC differentiation at longer time points (days 7 and 14) quantitative RT-PCR (qRT-PCR) was performed on the extracted RNA from the implanted cells. Based on qRT-PCR analysis, significant expression for all markers was observed in chitosan hydrogels at both time points. Analysis for GFAP reveals that chitosan hydrogel at day 14 yielded a significantly higher fold increase in mRNA levels as compared to chitosan hydrogels at day 7, as shown in Fig. 11. However, no significant differences were observed for OLIG-1, an oligodendrocyte marker and neurite marker tubulin, at the 2 time points. The NSC marker nestin decreased over time in chitosan hydrogels; however, no significant differences were observed demonstrating relative stability of the neuronal phenotype in 3D chitosan cultures.
Fig. 11.
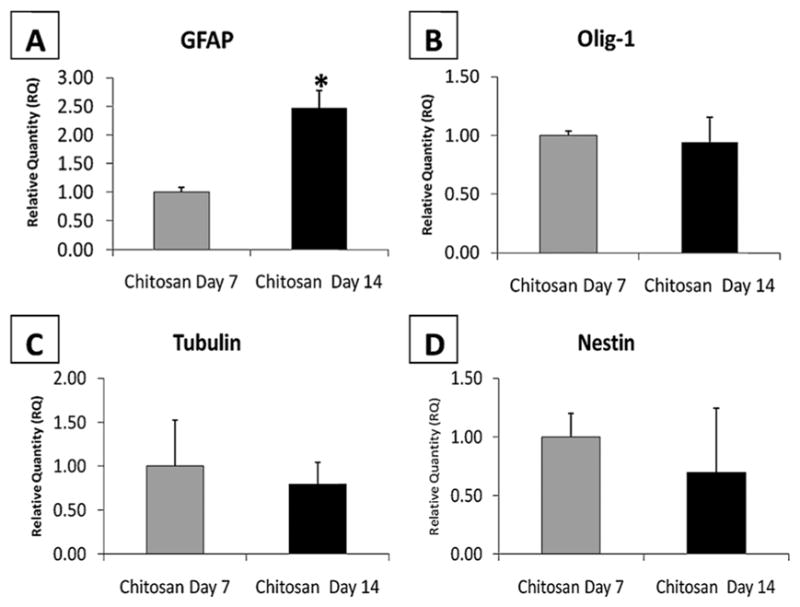
qRT-PCR data showing stem cell differentiation into 3 lineages, (A) astrocytic, (B) oligodendrocyte, (C) neuronal expression from cells within chitosan hydrogels at 2 different time points and (D) reduced stem cell marker nestin in day 14 scaffolds as compared to day 7. * indicates significantly higher (p < 0.01) expression of GFAP at day 14 as compared to day 7 within chitosan hydrogels scaffolds.
4. Discussion
Over the last decade, hydrogel based scaffolds have been explored for several tissue engineering applications.3,13,16,17,21,23,24,58–61 Traditional applications of hydrogels in neural tissue engineering are twofold: (A) they provide an ideal matrix for regeneration by enhancing endogenous cell attachment, proliferation, migration; and (B) they promote neurite extension. Hydrogels also serve as carriers of stem cells to enhance survival and function of transplanted cells. To achieve this, the cellular microenvironment and physical properties of the hydrogel matrices play a critical role in the success of the implanted hydrogel for repair and regeneration of tissues.12,17,23,33,34,36,48,49,59,60,62 Of particular interest for neural tissue engineering is the impact of gel stiffness, pore size and charge properties of the gels in promoting neurite extension. It has been hypothesized that materials whose mechanical properties closely mimic the native ECM of tissues in the central and peripheral nervous systems would provide the optimal environment for differentiation of cortical neurons, dorsal root ganglia, and even neural progenitor cells into the mature neuronal phenotypes that can regenerate lost tissues.11,24 For successful application as a carrier for stem cells, properties such as ease of application in situ, conformal filling of 3 dimensional spaces and non-cytotoxic byproducts are critical.
Several studies involving positively charged hydrogels have been shown to support increased cell attachment, survival and neurite elongation.23,32,33,63 Chitin and its derivative chitosan are natural polymers that are positively charged and possess functionalizable end groups; however, they are not usually soluble at physiological pH.26,31,36 Several techniques have been previously reported to render them soluble at neutral pH by incorporation of acid moieties to the chitosan backbone.26 Addition of photocrosslinkable end groups to the water soluble chitosan has been reported prior to our investigation, such as Az-CH-LA39,40 and methacrylated chitosan.22,25,36,37 All these processes involve reaction components that are not soluble in an aqueous medium; they also need drastic reduction in pH, which would cause rapid degradation of chitosan and would also cause them to be insoluble at neutral pH postmodification. Also, Az-CH-LA or the azido chitosan does not lend control over the degree of conversion as well as the degree of crosslinking, leading to uncontrollable mechanical properties and incompatible pore geometries.39
Yu et al. have shown the functionalization of amine groups on the chitosan with maleic anhydride to render them photocrosslinkable.36 This approach, while successful in fabricating hydrogels, decreases the potential for further functionalization as amine groups are utilized for the first reaction, even though the degree of conversion was not reported. The reaction was carried out by dissolving chitosan in a 2% acetic acid aqueous solution, implying insolubility at neutral pH and a gelation reaction time of about 30 min, which is significantly higher than that reported for CM-chitosan gels reported here. Also, the crosslinking step involves the use of ammonium persulfate (APS) and sodium metabisulfite (SMBS) as initiators, which could be cytotoxic to encapsulated cells and host tissues, and would also leave behind inorganic minerals within the gels after gelation. Yu et al. also showed that hydrogels produced by this approach yield matrices with increased stiffness and therefore neurite extension may be compromised due to the inability to tune stiffness. Also, hydrogels derivitized by methacrylic anhydride, which reacts at the amine terminal of chitosan, lack the positive charge to influence neurite outgrowth.
In another study, Rickett et al. reported the fabrication of UV crosslinkable chitosan hydrogels by derivitizing chitosan with azobenzoic acid.39 This approach also uses the amine groups to react with the azobenzoic acid, therefore rendering them uncharged, with increasing reaction efficiencies. Also, it is evident that this approach does not yield a chitosan whose stiffness properties can be tailored, with the lowest possible storage moduli of the hydrogel to be 400 Pa. Several studies demonstrate that a modulus in this range does not support neurite extension,16,23 as substantiated by the neurite outgrowth studies performed with PC12 cells. The photocrosslinkable chitosan reported here overcomes some of these shortcomings and its mechanical and gelation properties supported robust neurite outgrowth from DRG, enabled cortical neuron differentiation and demonstrated little or no cytotoxicity as evident from human MSC cultures.
Here we report the fabrication of novel water soluble photocrosslinkable chitosan based hydrogels that overcome some of these limitations so that they can be applied for neural tissue engineering. The novel methacrylate derivative of chitosan reported here is advantageous over previously reported water soluble chitosan derivatives because this methodology lends itself well to hydrogel scaffolds with tunable mechanical stiffness, and potentially lesser cytotoxicity as compared to previously reported photocrosslinkable chitosan derivatives.
Although several approaches to fabricate photocrosslinkable hydrogels from natural polymers such as hyaluronic acid, dextran, gelatin and chitosan with methacrylate moieties have been reported previously,15,22,40,64–66 it has been challenging to achieve high degrees of substitution using aqueous conditions.47,60,66 Therefore, most of the acrylated derivatives have been fabricated in organic media or in mixtures of organic and aqueous media, leading to a slightly improved degree of conversion, but loss of solubility at neutral pHs leads to difficulties in incorporation of cells within the hydrogels. In order to overcome these limitations, the ratio of photoinitiator to the gel precursor has been altered to fabricate hydrogels with different properties.15,22,64 The photoinitiator, Irgacure 2959 (I-2959), has been used to fabricate hydrogels using photochemistry, by exposure to ultraviolet radiation at 365 nm, and is known to have minimal cytotoxicity.66,67 However, the range of concentrations over which this can be used as a photocrosslinker is narrow owing to its insolubility in aqueous medium and its toxicity at higher concentrations, therefore having an effect on the quality of gels.
Earlier studies from our group have shown that mechanical stiffness of hydrogels plays a significant role in the neurite regeneration process from chicken dorsal root ganglia.48 We had reported that 1% Seaprep hydrogels have optimal mechanical properties to facilitate neurite extension from DRG. Other studies that have reported the role of mechanical stiffness on neurite extension have shown that with increasing stiffness, the neurite lengths are reduced.24,47 Therefore in this study, we synthesized hydrogels with storage moduli in the range of 1% Seaprep® agarose (100–150 Pa). In order to achieve a storage modulus in this range, we fabricated and evaluated 3 different concentrations (w/v %) of chitosan hydrogels, keeping the gelling time and the concentration of photoinitiator constant. From the data shown in Fig. 6B, we can see that the 0.5% chitosan hydrogel storage modulus of 104.56 ± 13.46 Pa matched the previously determined agarose hydrogel storage modulus and therefore were used for cytotoxicity and neurite extension studies.
Neurons are anchorage dependent and their migration and differentiation is dependent on their physical and biochemical interaction with the ECM.19,68 When cortical neurons lose ECM contacts, they do not differentiate to form neurites.49,51 Although DRG extend long processes in agarose hydrogel cultures, cortical neurite extension in agarose gels is minimal, perhaps due to lower production of ECM by these neurons as shown in Fig. 8B. O’Connor et al. have shown similar results, wherein agarose based scaffolds showed enhanced cortical neuron apoptosis and reduced differentiation as compared to collagen based scaffolds51 with similar pore sizes and mechanical stiffness. As demonstrated in Fig. 8C photocrosslinkable chitosan demonstrated significantly greater cortical neuron differentiation, likely due to chitosan’s positive charge, thereby reducing their dependence on cellular ECM production. This is consistent with other studies demonstrating positively charged, 3D hydrogels supporting enhanced neurite extension and outgrowth. Enhanced neurite extension in agarose based hydrogels from incorporation of ECM protein laminin or positively charged molecules such as chitosan has been demonstrated by our lab previously.33,34 Other studies by Dadsetan et al. have reported that oligo-propylene fumarate hydrogels containing a positive charge showed enhanced neurite outgrowth as compared to uncharged hydrogels, which did not support any neurite outgrowth.63 Charged nerve guidance channels demonstrated enhanced neurite outgrowth in a transected mice sciatic nerve model, as compared to uncharged channels.69 We have also reported that chitosan hydrogels functionalized with poly-lysine (PLL), which enhances the extent of positive charge, showed extensive neurite outgrowth in a 2D culture model.23
NSCs continue to show tremendous promise for repair of injuries and degenerative diseases in the central nervous system.8–10,70 It has been reported that when NSCs are cultured in 3D microenvironments that mimic native brain tissue mechanically, they promote differentiation down a neuronal pathway.25,36,37 However, most of these studies were performed by plating stem cells in 2D environments, either on hydrogel surfaces or with coatings and other growth factors that promoted differentiation. Leipzig and Shoichet have earlier studied the effect of substrate stiffness on NSC differentiation, albeit on 2D substrates,25 and reported that NSCs respond to the substrate and its moduli at earlier time points as compared to later. They hypothesized that this can be attributed to the fact that larger colony size, at later time points, hinders direct cell interaction with matrices, thereby preventing controlled differentiation. Their studies were performed at time points very early in the differentiation profile and do not discuss the impact of longer time points on cellular differentiation. We hypothesize that at longer time points, where cell clusters are larger and are hindered by diffusion limitations, differentiation is limited due to shortage of nutrients. In another instance, Banerjee et al. have demonstrated that when NSCs are cultured within 3D alginate hydrogels, increased gel moduli showed reduced β-III tubulin expression, quantitatively with RT-PCR and qualitatively using microscopy.17 They reported that the relative expression of β-tubulin III was 20-fold greater in cells cultured in alginate hydrogels having the lowest value of modulus, and that this expression decreases with increasing hydrogel modulus.
In the study by Leipzig et al., NSCs were cultured on photocrosslinkable chitosan hydrogels, in the presence of fetal bovine serum (FBS) in the medium leading to increased differentiation into neural and glial lineages.37,71 However, all our studies were performed in the absence of any FBS or differentiation factor, therefore allowing for endogenous differentiation mechanisms to occur. Several other studies illustrate the role of mechanical stimuli on the differentiation and lineage of stem cells.17,25 Stiffer surfaces have been shown to promote glial specific differentiation; meanwhile softer surfaces show neurite like differentiation. Our qRT-PCR data suggests increasing glial differentiation with time, while no significant differences in tubulin expression were observed. Similar results have been observed by Hynes et al.,72 who reported that with increasing positive charge within PLL containing polyethylene glycol (PEG) hydrogels of the same mechanical stiffness, reduced neurite outgrowth was observed in neural stem cells. Similarly, Pan et al. have shown that positively charged PLL grafted hyaluronic acid hydrogels showed enhanced glial cell proliferation and reduced tubulin staining as compared to uncharged hydrogels.73 Also, several studies have suggested that cell–ECM interactions direct specific cell migration and differentiation. It has earlier been reported that hydrogels with matrix proteins such as laminin have shown to promote neurite outgrowth, as evidenced by increased tubulin expression, as compared to blank scaffolds.14,74 In another study, Leipzig et al. showed that when recombinant interferon-gamma (IFN-γ) was present in the microenvironment of the hydrogels, increased tubulin expression and minimal glial expression was observed.71 Several other neutrophic factors have also been shown to promote neurite differentiation in 2D and 3D culture microenvironments.37,70 As described earlier, chitosan provides an ideal template for further modification, which could allow for crosslinking proteins such as laminin and therefore direct differentiation along specific pathways. The AEMA derivitized chitosan hydrogels, described above, offer an in situ gelling 3D system that can be tuned to support neurite outgrowth via altering its mechanical properties and other ECM cues. Also, these hydrogels could serve as carriers for NSC transplantation and facilitate controlled stem cell proliferation, migration and differentiation, as demonstrated above.
5. Conclusions
In this study, we report the synthesis and characterization of a novel photocrosslinkable chitosan derivative. This approach allows for fabrication of degradable, in situ gelling hydrogels with moderate gelling times and controllable physical properties. The transparency of the hydrogel facilitates direct light microscopic visualization of encapsulated cortical neurons and dorsal root ganglia. The chitosan based hydrogel showed enhanced neurite differentiation from cortical neurons and robust and longer neurite lengths from dorsal root ganglia (DRG) as compared to agarose based hydrogels with similar mechanical stiffness. Also, in this study we showed the differentiation of NSCs within chitosan hydrogels. We have demonstrated that chitosan hydrogels supported neuronal differentiation of encapsulated NSCs. We observed significant enhancement in expression of the glial marker, GFAP, within chitosan hydrogels at day 14 as compared to chitosan hydrogels at day 7. Also, the functionalization chemistry of chitosan gels did not alter the active end groups and therefore could allow for binding of proteins and growth factors for enhancing the applicability of hydrogels for any cell delivery application or controlled differentiation of NSCs into specific lineages depending on the application. Overall, photocrosslinkable chitosan hydrogels could potentially be applicable for peripheral and central nervous system repair after injuries, primarily due to their ability to support neurite outgrowth and promote survival and differentiation of neural stem cells into neurons and glia.
Acknowledgments
We would like to thank Sanjay Anand for assistance with microscopy and imaging. We acknowledge support from NIH RO1s NlS65109 NS44409, Georgia Cancer Coalition, and Institute of Bioscience and Bioengineering’s Core Facilities at Georgia Tech for support. The authors would like to acknowledge editorial assistance and suggestions of Dr S. Balakrishna Pai, Georgia Tech, in the preparation of this manuscript.
References
- 1.T.N.S.C.I.S. Center. SPINALCORDINJURY, Facts and Figures at a Glance. http://images.main.uab.edu/spinalcord/pdffiles/Facts08.pdf.
- 2.Noble J, Munro CA, Prasad VS, Midha R. J Trauma: Inj Infect Crit Care. 1998;45:116–122. doi: 10.1097/00005373-199807000-00025. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt CE, Leach JB. Annu Rev Biomed Eng. 2003;5:293–347. doi: 10.1146/annurev.bioeng.5.011303.120731. [DOI] [PubMed] [Google Scholar]
- 4.Yu X, Bellamkonda RV. Tissue Eng. 2003;9:421–430. doi: 10.1089/107632703322066606. [DOI] [PubMed] [Google Scholar]
- 5.Bellamkonda RV. Biomaterials. 2006;27:3515–3518. doi: 10.1016/j.biomaterials.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 6.Dodla MC, Bellamkonda RV. J Biomed Mater Res, Part A. 2006;78A:213–221. doi: 10.1002/jbm.a.30747. [DOI] [PubMed] [Google Scholar]
- 7.Belkas JS, Munro CA, Shoichet MS, Midha R. Restor Neurol Neurosci. 2005;23:19–29. [PubMed] [Google Scholar]
- 8.Snyder EY, Macklis JD. Clin Neurosci. 1995;3:310–316. [PubMed] [Google Scholar]
- 9.Kim H, Tator CH, Shoichet MS. J Biomed Mater Res, Part A. 2011;97A:395–404. doi: 10.1002/jbm.a.33070. [DOI] [PubMed] [Google Scholar]
- 10.Kim H, Zahir T, Tator CH, Shoichet MS. PLoS One. 2011;6:e21744. doi: 10.1371/journal.pone.0021744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoffman-Kim D, Mitchel JA, Bellamkonda RV. Annu Rev Biomed Eng. 2010;12:203–231. doi: 10.1146/annurev-bioeng-070909-105351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tibbitt MW, Anseth KS. Biotechnol Bioeng. 2009;103:655–663. doi: 10.1002/bit.22361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jain A, Kim YT, McKeon RJ, Bellamkonda RV. Biomaterials. 2006;27:497–504. doi: 10.1016/j.biomaterials.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 14.Stabenfeldt SE, Garcia AJ, LaPlaca MC. J Biomed Mater Res, Part A. 2006;77A:718–725. doi: 10.1002/jbm.a.30638. [DOI] [PubMed] [Google Scholar]
- 15.Baier Leach J, Bivens KA, Patrick CW, Jr, Schmidt CE. Biotechnol Bioeng. 2003;82:578–589. doi: 10.1002/bit.10605. [DOI] [PubMed] [Google Scholar]
- 16.Balgude AP, Yu X, Szymanski A, Bellamkonda RV. Biomaterials. 2001;22:1077–1084. doi: 10.1016/s0142-9612(00)00350-1. [DOI] [PubMed] [Google Scholar]
- 17.Banerjee A, Arha M, Choudhary S, Ashton RS, Bhatia SR, Schaffer DV, Kane RS. Biomaterials. 2009;30:4695–4699. doi: 10.1016/j.biomaterials.2009.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dodla MC, Bellamkonda RV. Biomaterials. 2008;29:33–46. doi: 10.1016/j.biomaterials.2007.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Georges PC, Miller WJ, Meaney DF, Sawyer ES, Janmey PA. Biophys J. 2006;90:3012–3018. doi: 10.1529/biophysj.105.073114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stalling SS, Akintoye SO, Nicoll SB. Acta Biomater. 2009;5:1911–1918. doi: 10.1016/j.actbio.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 21.Zheng Shu X, Liu Y, Palumbo FS, Luo Y, Prestwich GD. Biomaterials. 2004;25:1339–1348. doi: 10.1016/j.biomaterials.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 22.Amsden BG, Sukarto A, Knight DK, Shapka SN. Biomacromolecules. 2007;8:3758–3766. doi: 10.1021/bm700691e. [DOI] [PubMed] [Google Scholar]
- 23.Crompton KE, Goud JD, Bellamkonda RV, Gengenbach TR, Finkelstein DI, Horne MK, Forsythe JS. Biomaterials. 2007;28:441–449. doi: 10.1016/j.biomaterials.2006.08.044. [DOI] [PubMed] [Google Scholar]
- 24.Zuidema JM, Pap MM, Jaroch DB, Morrison FA, Gilbert RJ. Acta Biomater. 2011;7:1634–1643. doi: 10.1016/j.actbio.2010.11.039. [DOI] [PubMed] [Google Scholar]
- 25.Leipzig ND, Shoichet MS. Biomaterials. 2009;30:6867–6878. doi: 10.1016/j.biomaterials.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 26.Lu GY, Kong LJ, Sheng BY, Wang G, Gong YD, Zhang XF. Eur Polym J. 2007;43:3807–3818. [Google Scholar]
- 27.Jayakumar R, Prabaharan M, Nair SV, Tamura H. Biotechnol Adv. 2010;28:142–150. doi: 10.1016/j.biotechadv.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 28.Wang YY, Lu LX, Feng ZQ, Xiao ZD, Huang NP. Biomed Mater. 2010;5:054112. doi: 10.1088/1748-6041/5/5/054112. [DOI] [PubMed] [Google Scholar]
- 29.Chen JD, Wang Y, Chen X. J Biomater Sci, Polym Ed. 2009;20:1555–1565. doi: 10.1163/092050609X12464345036768. [DOI] [PubMed] [Google Scholar]
- 30.Ro IJ, Kwon IC. J Biomater Sci, Polym Ed. 2002;13:769–782. doi: 10.1163/156856202760197401. [DOI] [PubMed] [Google Scholar]
- 31.Madihally SV, Matthew HWT. Biomaterials. 1999;20:1133–1142. doi: 10.1016/s0142-9612(99)00011-3. [DOI] [PubMed] [Google Scholar]
- 32.Cao Z, Gilbert RJ, He W. Biomacromolecules. 2009;10:2954–2959. doi: 10.1021/bm900670n. [DOI] [PubMed] [Google Scholar]
- 33.Dillon GP, Yu X, Bellamkonda RV. J Biomed Mater Res. 2000;51:510–519. doi: 10.1002/1097-4636(20000905)51:3<510::aid-jbm28>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 34.Dillon GP, Yu X, Sridharan A, Ranieri JP, Bellamkonda RV. J Biomater Sci, Polym Ed. 1998;9:1049–1069. doi: 10.1163/156856298x00325. [DOI] [PubMed] [Google Scholar]
- 35.Jiang T, Kumbar SG, Nair LS, Laurencin CT. Curr Top Med Chem. 2008;8:354–364. doi: 10.2174/156802608783790974. [DOI] [PubMed] [Google Scholar]
- 36.Yu LM, Kazazian K, Shoichet MS. J Biomed Mater Res, Part A. 2007;82A:243–255. doi: 10.1002/jbm.a.31069. [DOI] [PubMed] [Google Scholar]
- 37.Leipzig ND, Wylie RG, Kim H, Shoichet MS. Biomaterials. 2011;32:57–64. doi: 10.1016/j.biomaterials.2010.09.031. [DOI] [PubMed] [Google Scholar]
- 38.Obara K, Ishihara M, Ozeki Y, Ishizuka T, Hayashi T, Nakamura S, Saito Y, Yura H, Matsui T, Hattori H, Takase B, Kikuchi M, Maehara T. J Controlled Release. 2005;110:79–89. doi: 10.1016/j.jconrel.2005.09.026. [DOI] [PubMed] [Google Scholar]
- 39.Rickett TA, Amoozgar Z, Tuchek CA, Park J, Yeo Y, Shi R. Biomacromolecules. 2011;12:57–65. doi: 10.1021/bm101004r. [DOI] [PubMed] [Google Scholar]
- 40.Yeo Y, Burdick JA, Highley CB, Marini R, Langer R, Kohane DS. J Biomed Mater Res, Part A. 2006;78A:668–675. doi: 10.1002/jbm.a.30740. [DOI] [PubMed] [Google Scholar]
- 41.Jeon O, Bouhadir KH, Mansour JM, Alsberg E. Biomaterials. 2009;30:2724–2734. doi: 10.1016/j.biomaterials.2009.01.034. [DOI] [PubMed] [Google Scholar]
- 42.Rinaudo M, Le Dung P, Gey C, Milas M. Int J Biol Macromol. 1992;14:122–128. doi: 10.1016/s0141-8130(05)80001-7. [DOI] [PubMed] [Google Scholar]
- 43.Delben F, Lapasin R, Pricl S. Int J Biol Macromol. 1990;12:9–13. doi: 10.1016/0141-8130(90)90075-l. [DOI] [PubMed] [Google Scholar]
- 44.Kennedy R, Costain DJ, McAlister VC, Lee TD. Surgery. 1996;120:866–870. doi: 10.1016/s0039-6060(96)80096-1. [DOI] [PubMed] [Google Scholar]
- 45.Wang G, Lu G, Ao Q, Gong Y, Zhang X. Biotechnol Lett. 2010;32:59–66. doi: 10.1007/s10529-009-0123-1. [DOI] [PubMed] [Google Scholar]
- 46.Song Q, Zhang Z, Gao J, Ding C. J Appl Polym Sci. 2011;119:3282–3285. [Google Scholar]
- 47.Seidlits SK, Khaing ZZ, Petersen RR, Nickels JD, Vanscoy JE, Shear JB, Schmidt CE. Biomaterials. 2010;31:3930–3940. doi: 10.1016/j.biomaterials.2010.01.125. [DOI] [PubMed] [Google Scholar]
- 48.Yu X, Bellamkonda RV. J Neurosci Res. 2001;66:303–310. doi: 10.1002/jnr.1225. [DOI] [PubMed] [Google Scholar]
- 49.Cullen DK, Lessing MC, LaPlaca MC. Ann Biomed Eng. 2007;35:835–846. doi: 10.1007/s10439-007-9292-z. [DOI] [PubMed] [Google Scholar]
- 50.He W, Bellamkonda RV. Biomaterials. 2005;26:2983–2990. doi: 10.1016/j.biomaterials.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 51.O’Connor SM, Stenger DA, Shaffer KM, Ma W. Neurosci Lett. 2001;304:189–193. doi: 10.1016/s0304-3940(01)01769-4. [DOI] [PubMed] [Google Scholar]
- 52.Kim YT, Haftel VK, Kumar S, Bellamkonda RV. Biomaterials. 2008;29:3117–3127. doi: 10.1016/j.biomaterials.2008.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang C, Hao J, Zhang F, Su K, Wang DA. Anal Biochem. 2008;380:333–334. doi: 10.1016/j.ab.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 54.Karathanasis E, Chan L, Karumbaiah L, McNeeley K, D’Orsi CJ, Annapragada AV, Sechopoulos I, Bellamkonda RV. PLoS One. 2009;4:e5843. doi: 10.1371/journal.pone.0005843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karumbaiah L, Anand S, Thazhath R, Zhong Y, McKeon RJ, Bellamkonda RV. Glia. 2011;59:981–996. doi: 10.1002/glia.21170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Georgiou TK, Patrickios CS. Biomacromolecules. 2008;9:574–582. doi: 10.1021/bm701123s. [DOI] [PubMed] [Google Scholar]
- 57.Kali G, Georgiou TK, Ivan B, Patrickios CS, Loizou E, Thomann Y, Tiller JC. Langmuir. 2007;23:10746–10755. doi: 10.1021/la7012478. [DOI] [PubMed] [Google Scholar]
- 58.Burdick JA, Ward M, Liang E, Young MJ, Langer R. Biomaterials. 2006;27:452–459. doi: 10.1016/j.biomaterials.2005.06.034. [DOI] [PubMed] [Google Scholar]
- 59.Dadsetan M, Hefferan TE, Szatkowski JP, Mishra PK, Macura SI, Lu L, Yaszemski MJ. Biomaterials. 2008;29:2193–2202. doi: 10.1016/j.biomaterials.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smeds KA, Hatchell DL, Saloupis P, Pfister-Serres A, Grinstaff MW. Abstr Pap Am Chem Soc. 1998;216:U333. [Google Scholar]
- 61.Smeds KA, Pfister-Serres A, Hatchell DL, Saloupis P, Grinstaff MW. Abstr Pap Am Chem Soc. 1998;216:U879. [Google Scholar]
- 62.Smeds KA, Pfister-Serres A, Hatchell DL, Grinstaff MW. J Macromol Sci, Part A: Pure Appl Chem. 1999;36:981–989. [Google Scholar]
- 63.Dadsetan M, Knight AM, Lu L, Windebank AJ, Yaszemski MJ. Biomaterials. 2009;30:3874–3881. doi: 10.1016/j.biomaterials.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Benton JA, DeForest CA, Vivekanandan V, Anseth KS. Tissue Eng A. 2009;15:3221–3230. doi: 10.1089/ten.tea.2008.0545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reza AT, Nicoll SB. Acta Biomater. 2010;6:179–186. doi: 10.1016/j.actbio.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 66.Smeds KA, Grinstaff MW. J Biomed Mater Res. 2001;54:115–121. doi: 10.1002/1097-4636(200101)54:1<115::aid-jbm14>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 67.Fedorovich NE, Oudshoorn MH, van Geemen D, Hennink WE, Alblas J, Dhert WJ. Biomaterials. 2009;30:344–353. doi: 10.1016/j.biomaterials.2008.09.037. [DOI] [PubMed] [Google Scholar]
- 68.Lu YB, Franze K, Seifert G, Steinhauser C, Kirchhoff F, Wolburg H, Guck J, Janmey P, Wei EQ, Kas J, Reichenbach A. Proc Natl Acad Sci U S A. 2006;103:17759–17764. doi: 10.1073/pnas.0606150103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Valentini RF, Sabatini AM, Dario P, Aebischer P. Brain Res. 1989;480:300–304. doi: 10.1016/0006-8993(89)90196-0. [DOI] [PubMed] [Google Scholar]
- 70.Ahmed S, Reynolds BA, Weiss S. J Neurosci. 1995;15:5765–5778. doi: 10.1523/JNEUROSCI.15-08-05765.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leipzig ND, Xu C, Zahir T, Shoichet MS. J Biomed Mater Res, Part A. 2010;93:625–633. doi: 10.1002/jbm.a.32573. [DOI] [PubMed] [Google Scholar]
- 72.Hynes SR, Rauch MF, Bertram JP, Lavik EB. J Biomed Mater Res, Part A. 2009;89A:499–509. doi: 10.1002/jbm.a.31987. [DOI] [PubMed] [Google Scholar]
- 73.Pan L, Ren Y, Cui F, Xu Q. J Neurosci Res. 2009;87:3207–3220. doi: 10.1002/jnr.22142. [DOI] [PubMed] [Google Scholar]
- 74.Stabenfeldt SE, Laplaca MC. Acta Biomater. 2011;7:4102–4108. doi: 10.1016/j.actbio.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]