

Abstract
Nanotechnology has demonstrated tremendous clinical utility, with potential applications in cancer immunotherapy. Although nanoparticles with intrinsic cytotoxicity are often considered unsuitable for clinical applications, such toxicity may be harnessed in the fight against cancer. Nanoparticle associated toxicity can induce acute necrotic cell death, releasing tumor-associated antigens which may be captured by the antigen presenting cells to initiate or amplify tumor immunity. To test this hypothesis, cytotoxic cationic silica nanoparticles (CSiNPs) were directly administered into B16F10 melanoma implanted in C57BL/6 mice. CSiNPs caused plasma membrane rupture and oxidative stress of tumor cells, inducing local inflammation, tumor cell death and release of tumor-associated antigens. CSiNPs were further complexed with Bis-(3′–5′)-cyclic dimeric guanosine monophosphate (c-di-GMP), a molecular adjuvant which activates the stimulator of interferon genes (STING) in antigen presenting cells. Compared with unformulated c-di-GMP, delivery of c-di-GMP with CSiNPs markedly prolonged its local retention within the tumor microenvironment and activated tumor-infiltrating antigen presenting cells. Combination of CSiNPs and STING agonist showed dramatically increased expansion of antigen-specific CD8+ T cells, and potent tumor growth inhibition in murine melanoma. These results demonstrate that cationic nanoparticles can be used as an effective in situ vaccine platform which simultaneously cause tumor destruction and immune activation.
Graphical Abstract
Potent antitumor immunity is induced by introtumoral injection of cytotoxic silica nanoparticles complexed with STING agonist.

Introduction
After many year’s developments, immunotherapy has become a clinically validated treatment for many cancers.1–3 An immune-responsive tumor microenvironment is critical for all forms of cancer immunotherapy. Ideally, both innate and adaptive immunity are required to polarize an effective antitumor response.1, 3 Toward this goal, localized therapies with engineered three-dimensional scaffolds,4, 5 nanoparticles,6 or immune stimulatory molecules,7, 8 and systemic treatment with immune checkpoint blockade antibodies,9 adoptive T cell transfer,10 or vaccination11 have shown considerable promise in the induction of antitumor immunity in treating local and metastatic cancer.
Among these strategies, vaccination represents a viable option for active immunotherapy of cancers that aims to treat late-stage diseases by harnessing the power of a patient’s own immune system. Historically, vaccine is one of the most successful and cost-effective medical interventions to prevent infectious diseases, saving millions of lives every year via pediatric and adult immunizations.12 However, the effectiveness of traditional vaccine approaches has not been translated to therapeutic settings such as cancer, due to the difficulties in eliciting CD8+ T cell responses as well as the complex coevolution of tumor and host immune cells.1, 6 A number of challenges must be overcome for a successful cancer vaccine. For example, although the repertoire of T cells in human can recognize self-antigens, cancer cells frequently undergo high rates of mutation, allowing them to escape the recognition by T lymphocytes.13, 14 Furthermore, a number of defense mechanisms appeared to have evolved to maintain a severely immunosuppressive microenvironment, including suppression of antigen presentation, recruitment of regulatory T cells, as well as up-regulation of inhibitory molecules such as PD-L1, adding an extra layer of protection against the host immune response.1–3
An emerging alternative strategy is “in situ vaccination” which exploits local intratumoral treatment to simultaneously destruct tumor cells and provides the immune system with an antigen source for the induction of antitumor immunity.15, 16 Unlike traditional vaccines where selected tumor-associated antigens are used, in situ vaccination exploits complete tumor-related antigenic repertoire, including tumor-specific neoantigens derived from non-synonymous mutations.17 Further, in situ vaccines can set the stage for potent antitumor immunity by inducing inflammation and facilitating the recruitment and activation of immune cells to the tumor. Thus, in situ vaccine approach provides opportunities for broad, more effective and less toxic treatment strategies to overcome tumor-related tolerance and promote systemic antitumor immunity.15, 16 A variety of intratumoral treatments (e.g., radiation, cryotherapy) have been delivered directly to the tumors to induce tumor cell death, release tumor antigens while providing pro-inflammatory signals, which result in systemic activation of anti-tumor T cell responses, followed by inflammatory infiltration of T lymphocytes into the tumor.7, 8, 17–19 While these early studies demonstrated the potential of in situ tumor destruction in promoting both T cell and humoral responses, the efficacy and wide-spread adoption of in situ vaccination have been limited. The major challenge lies in the relatively weak antitumor immunity following primary tumor destruction. For example, radiofrequency ablation or cryotherapy allows in situ tumor destruction and releases a large amount of tumor antigens, but only induces a weak and transient immune response which fails to prevent tumor relapse.19, 20 Preclinical and clinical studies combining tumor ablation with local administration of CpG-containing oligonucleotides (single-stranded oligonucleotides containing unmethylated cytosine-guanine motifs that bind Toll-like receptor-9 and serve as potent molecular adjuvants) can boost the induction of systemic antitumor effects.19 However, rapid dissemination of unformulated CpG from injection site often leads to systemic toxicity.21 Immobilizing CpG ODNs or other immunostimulants22, 23 in synthetic scaffolds at the tumor site blocks the systemic toxicity, but this approach lacks a mechanism for in situ tumor destruction, which is required to generate an antigen source for T cell priming.
Nanoparticles have found broad applications in vaccines, transforming many aspects of cancer immunotherapy.6, 24–27 Despite the exceptional ability to deliver vaccines, many nanoparticles exhibit non-specific cytotoxicity (induces both necrosis and apoptosis), causing damage to healthy cells when administrated systemically.28–30 Such nanotoxicity is often the limiting factor in their clinical applications. However, nanotoxicity delivered in a controlled manner may cause tumor cell death and function as cancer therapeutics.31, 32 The acute cytotoxicity of nanoparticles has promoted us to harness the intrinsic cytotoxic effects for tumor destruction following local treatment, which subsequently releases tumor-associated antigens and potentially initiates or amplifies antitumor immunity. Furthermore, engineered nanoparticles are widely used to deliver immunostimulatory signals to antigen presenting cells, amplifying tumor immunity, as shown in various clinical trials and pre-clinical animal studies.3, 4, 12 In this study, we test the efficacy of in situ vaccination by combining the cytotoxic effects of cationic silica nanoparticles (CSiNPs) with a cyclic dinucleotide (CDN) which activates stimulator of IFN genes (STING) pathways.33, 34 Intratumoral injection of CSiNPs loaded with a STING agonist cyclic dimeric guanosine monophosphate (c-di-GMP) induced acute necrotic death of tumor cells through membrane disruption and production of intracellular reactive oxygen species (ROS), and elicited strong local inflammation in the tumor microenvironment of mice, leading to markedly improved tumor antigen-specific T cell response and enhanced antitumor efficacy in murine melanoma (Figure 1). To the best of our knowledge, this is the first time the intrinsic cytotoxicity of nanoparticles was utilized for “in situ vaccination” to initiate an antitumor immune response, which was further amplified via the co-delivery of c-di-GMP activating STING pathways. Our results may find applications in the treatment of patients with solid tumors, particularly for those ineligible for surgical resection.
Fig. 1. Schematic diagram of in situ vaccination with cytotoxic nanoparticles.

Intratumoral injection of cationic silica nanoparticles (CSiNPs) loaded with STING agonist causes simultaneous tumor destruction and immune activation in the tumor microenvironment, leading to inflammation and systemic antitumor immune response.
Results and Discussion
Preparation and characterization of c-di-GMP loaded CSiNPs
Cationic silica nanoparticles (CSiNPs) offer several attributes in local delivery of immunostimulatory signals to antigen presenting cells (APCs),35, 36 including enhanced cellular and/or tissue interaction/uptake and prolonged retention within the tumor microenvironment.36 To prepare immunostimulatory signals loaded CSiNPs, c-di-GMP, a stimulator of IFN genes (STING) agonist was complexed with amine-modified SiNPs (30 nm). c-di-GMP, or bis-(3′–5′)-cyclic dimeric guanosine monophosphate, is a cyclic dinucleotide produced by bacteria and is sensed by STING in mammals, inducing a robust type-I interferon production in antigen presenting cells.33, 34 c-di-GMP interacts with CSiNPs via electrostatic interaction (Fig. 2a). To maintain the cationic surface charge and colloidal stability, an optimized mass ratio of 120 (CSiNPs : c-di-GMP) was adopted. To quantify the loading of c-di-GMP on CSiNPs, DY547 labeled c-di-GMP and FITC labeled CSiNPs were mixed and probe-sonicated. After a brief centrifugation, the c-di-GMP loaded CSiNPs (c-di-GMP/CSiNPs) formed a pellet at the bottom of 1.5 mL microcentrifuge tubes (Fig. 2b), emitting orange fluorescence under ultraviolet (UV) illumination (Fig. 2b), implying that the interaction between CSiNPs and c-di-GMP was sufficient to promote a strong adsorption of c-di-GMP on CSiNPs. Measuring the c-di-GMP in the supernatant revealed about 65 % loading efficiency (Fig. 2c). The c-di-GMP/CSiNPs showed a mean hydrodynamic diameters of 35 nm, which was slightly bigger than CSiNPs (30 nm). Zeta-potential measurements showed a decrease in surface potential from 25 mV for bare CSiNPs to 18 mV after loading with c-di-GMP, suggesting a highly charged surface after complexation.
Fig. 2. Preparation of c-di-GMP loaded cationic silica nanoparticles (CSiNPs) and local distribution of c-di-GMP and CSiNPs delivered into tumor sites.

a) Illustration of c-di-GMP loaded CSiNPs (c-di-GMP/CSiNPs) preparation via electrostatic charge interactions between CSiNPs and c-di-GMP. b) Optical images of c-di-GMP/CSiNPs before and after centrifugation (5,000 rpm for 10 s), illuminated under white light (left) or under ultra-violate (UV) light (right). The desired amount of DY547-c-di-GMP and FITC-CSiNPs were mixed in saline with a mass ratio of 120 (CSiNPs : c-di-GMP) and were briefly centrifuged before images were taken. c) c-di-GMP concentration measured from the supernatant of soluble c-di-GMP or c-di-GMP/CSiNPs samples. d) Fluorescence images of 10 μm tissue sections showing the distribution of c-di-GMP and CSiNPs 24 h after intratumoral injection into melanoma tumors. Cell nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole, blue). DY547-labeled c-di-GMP (red) and FITC-labeled CSiNPs (green) were used to detect c-di-GMP and CSiNPs distribution in the tumor microenvironment.
Next, the retention of c-di-GMP and CSiNPs in tumor microenvironment was analyzed 24 h after intratumoral injection into established B16F10 dermal melanoma. c-di-GMP/CSiNPs appeared to be stable in aqueous buffer, showing no sign of aggregation and releasing about 30% of the c-di-GMP at 37 °C for 3 days (Fig. S1) The local retention was measure in vivo. Briefly, mice received a single intratumoral injection of c-di-GMP (labeled with DY547), CSiNPs (labeled with FITC), or c-di-GMP/CSiNPs and 24 h after injection, tumors were isolated, sectioned, and imaged to visualize c-di-GMP and CSiNPs distribution (Fig. 2d). Due to its small molecular weight, c-di-GMP signal was not seen at the tumor site 24 h after injection of free c-di-GMP, suggesting a rapid diffusion or degradation of c-di-GMP after injection. In contrast, c-di-GMP delivered by CSiNPs were clearly visible and appeared to be largely and uniformly distributed in the tumor (Fig. 2d), demonstrating c-di-GMP delivered with CSiNPs prolonged its local retention at the injection site. Complexation of c-di-GMP to CSiNPs prevents it from diffusing into blood circulation and possibly protects it from enzymatic degradation.11 Additionally, cationic nanoparticles might strongly interact with the net anionic phospholipid membrane and extracellular matrix (ECM) at the injection site through electrostatic interactions, leading to enhanced local adherence.37
CSiNPs induce potent tumor cell necrosis in vitro
Distinct types of cell death induce different types of immune responses: physiological cell death (apoptosis) is found during normal cell turnover and is intrinsically tolerogenic, whereas pathological cell death (necrosis) is characterized by intensive tissue destruction, inflammation and is immunogenic.38, 39 We envision a potent necrotic toxicity can be achieved by intratumoral injection of cationic nanoparticles. The surface charge is one of the main influencing factors for cytotoxicity, as positively charged nanoparticles act more cytotoxically than their negative counterparts of similar size in nonphagocytic cells.40 The strong binding between cationic nanoparticles and tumor cells initiated by electrostatic interactions has been shown to disrupt the integrity of plasma and vesicular membranes of living cells, causing cell death.37, 41 Toxicity of cationic silica nanoparticles has also been correlated with enhanced generation of reactive oxygen species (ROS), leading to oxidative stress which in turn may trigger proinflammatory responses.29, 30 We tested whether a high rate of toxicity in CSiNPs can result in acute necrosis of tumor cells through membrane disruption and oxidative stress (Fig. 3a). Mouse melanoma B16F10 cells were incubated for 24 h at 37 °C with varying concentrations of CSiNPs, or negatively charged SiNPs (NSiNPs, hydroxyl-terminated). Though dose-dependent toxicity was observed in cells treated with both types of nanoparticles, as measured by mitochondrial metabolic activity, positively charged SiNPs showed stronger cytotoxic effects than their anionic counterpart (Fig. 3b). Treatment of CSiNPs led to an apparent increase in the number of necrotic cells which were stained positively by vital dye propidium iodide (PI) and enumerated by flow cytometry (Fig. 3c and Figure S2). Necrosis was further confirmed by confocal microscopy after 2 h or 24 h incubation of B16F10 cells with CSiNPs (Fig. 3d). The increase of necrotic cells was more evident over time as shown after 24 h incubation (Fig. 3d).
Fig. 3. Cell necrosis mediated by cationic silica nanoparticles in vitro.
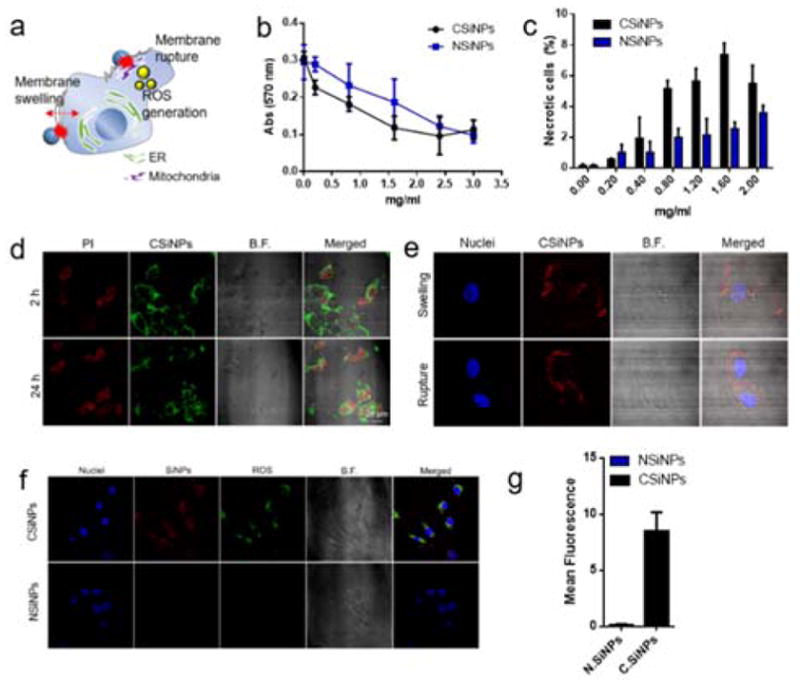
a) The illustration of cell necrosis induced by CSiNPs that lead to oxidative stress with their morphological changes such as membrane swelling and rupture. b) Cell viability detection by AlarmaBlue assay after 24 h exposure of B16-F10 cells with CSiNPs or negatively charged SiNPs (NSiNPs) at doses of 0.2–3.0 mg/mL. Cells were incubated with the AlarmaBlue agent for 4 h at 37 oC and the absorbance was measured at 570 nm, using 600 nm as a reference wavelength. c) The detection of the necrotic cells by flow cytometry with propidium iodide (PI) which enters cells with the disrupted membrane. Percentages of necrotic cells in the PI-positive region are shown after 24 h incubation of B16-F10 cells with CSiNPs or NSiNPs at 37 oC. Representative confocal microscope images, showing d) the detection of the necrotic cells with PI staining after 2 h or 24 h treatment of B16F10 cells with CSiNPs, and e) the observation of morphological changes of necrotic cells through swelling or membrane rupture. f) Confocal microscope images, showing intracellular ROS production measured by cell-permeant 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) in B16F10 after 24 h exposure to CSiNPs (labeled with Rhodamine) or NSiNPs at 37 oC. g) The mean fluorescent intensity measured by flow cytometry. ***P<0.001, **P<0.01, *P<0.05. NS, not significant by student’s test. Data represent mean ± standard error of the mean (s.e.m.) of 2–3 independent experiments.
It has been reported that nanocarriers with positive surface charges caused a rapid appearance of necrotic cells with characteristic morphological features40 and induced oxidative stress via the generation of reactive oxygen species (ROS).29, 40, 42 B16F10 cells incubated with CSiNPs were morphologically characterized by cell swelling and plasma membrane rupture (Fig. 3e). The intracellular ROS production which potentially leads to cell death by directly oxidizing or triggering various downstream pathways in the mitochondria43 was also quantified. B16F10 cells were treated with CSiNPs or NSiNPs for 24 h at 37°C and stained with 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) that reacts with multiple ROS species in the cells. Intracellular ROS levels were then determined by measuring the fluorescence of H2DCFDA with flow cytometry and confocal microscopy (Fig. 3f, g). Confocal microscopy analysis indicated a significant colocalization between H2DCFDA and CSiNPs (Fig. 3f), suggesting CSiNPs were the major source of ROS in these cells. Flow cytometry quantification of the H2DCFDA fluorescence showed a significant increase in ROS generation when cells were treated with CSiNPs, as compared with NSiNPs (Fig. 3g). CSiNPs were confined in the intracellular membrane structures, with a portion of them accumulated in mitochondria after 24 h exposure, confirmed by colocalization analysis of CSiNPs with a commercially available mitochondria-specific dye, Mitotracker (Figure S3). All considered, our data suggest CSiNPs disrupt plasma membrane and induce the production of reactive oxygen species, which lead to acute necrosis of tumor cells.
Intratumoral injection of cationic silica nanoparticles (CSiNPs) induces acute cell necrosis and inflammation in the tumor microenvironment
To test the cytotoxic effect of CSiNPs in vivo, 5.0 × 105 B16F10 tumor cells were inoculated in the flank of C57BL/6 mice. When tumor sizes were 45–50 mm2, mice were injected with a single intratumoral dose of NSiNPs or CSiNPs. At 24 h post injection, tumor tissues were removed, dissociated, and necrotic cells were analyzed. CSiNPs administered into the tumor resulted in a significant increase in the frequency of primary and secondary necrotic cells as detected by flow cytometry with PI and Annexin-V staining (Fig. 4a, b). Cell necrosis or death was also observed in the tumor tissue treated with CSiNPs, as shown by 0Hematoxylin-eosin (HE) stained histological sections (Fig. 4c). Interactions between CSiNPs and tumor cells resulted in significant tumor destruction characterized as a dark purple region surrounded by damaged tissues (Fig. 4c). In contrast, mice injected with PBS or soluble STING agonist c-di-GMP showed minimal necrotic cell death when examined sections stained by H&E (Fig. 4c). These observations suggest an intratumoral injection of CSiNPs results in tumor necrosis in vivo. Necrotic cell death stimulates a host inflammatory response, characterized by rapid infiltration of neutrophils, followed by monocytic phagocytes. CSiNPs, but not c-di-GMP treated tumors showed abundant infiltration of neutrophil-like cells surrounding the necrotic area and were Gr-1 positive by immunohistochemical staining (Fig. 4d). To determine the frequency and activation status of antigen presenting cells within the tumor milieu, tumors were removed 24 hours post injection, and dissociated cells were stained with anti-MHC II and anti-CD80 monoclonal antibodies. Intratumoral injection of soluble c-di-GMP induced minimal activation of antigen presenting cells in tumor (Fig. 4e, f), showing a basal level of CD80+ MHC II+ cell populations indistinguishable from those with PBS. In contrast, CSiNPs treated mice showed a significant increase in the frequency of CD80+ MHC II+ cells among total tumor infiltrates (Fig. 4e, f). However, the strongest APC activation was elicited by c-di-GMP complexed with CSiNPs (Fig. 4e, f). The increased activation of APCs within the tumor milieu, combined with prolonged intratumoral retention (Fig. 2d) and increased induction of tumor necrosis (Fig. 4a, b), suggest that CSiNPs have the potential to be a new in situ vaccine platform for simultaneously tumor destruction and immune activation.
Fig. 4. Intratumoral injection of cationic silica nanoparticles (CSiNPs) leads to acute cell necrosis and inflammation in the tumor microenvironment.

5.0×105 B16F10 tumor cells were inoculated in the flank of C57BL/6 mice (n=3/group) on day 0. a) When the size of tumor is 50 mm2 (on day 8), the direct intratumoral injection of c-di-GMP (5 μg) or the same amount of c-di-GMP complexed with CSiNPs (1.2 mg) was given. After 24 h, tumor tissues were isolated and analyzed by flow cytometry. b) A representative experiment of the detection of the necrotic cells at 24 h induced by the intratumoral injection of c-di-GMP formulated with NSiNPs or CSiNPs in vivo by flow cytometry with Annexin-V and PI staining (n=3/group). c) Representative micrographs of Hematoxylin-eosin (HE) stained histological sections. V, non-necrotic viable zone; N, necrotic zone) d) immunohistochemistry staining of Gr-1+ neutrophil cells. e) CD80 expressing MHC-II+ cells in the tumor microenvironment detected by flow cytometry. Mice were directly injected into tumor with c-di-GMP (5 μg), CSiNPs (1.2 mg) or c-di-GMP loaded CSiNPs. After 24 h, mice were euthanized and tumor tissues were isolated. Collected tumor cells were stained with CD80-APC and MHC-II-FITC before flowing cells with flow cytometry. Percentages of MHC-II+ CD80+ cells were counted out of total cells including tumor cells. ***P<0.001, **P<0.01, *P<0.05. ns, not significant by one-way analysis of variance (ANOVA) with Bonferroni post-test (b). Data represent mean ± standard error of the mean (s.e.m.) of 3 independent experiments.
CSiNPs in combination with c-di-GMP exhibits potent anti-tumor activity in a murine melanoma model
To elucidate and exploit the therapeutic impact of CSiNPs as in situ tumor vaccines, we evaluated the antitumor efficacy in mouse melanoma. C57BL/6 mice were inoculated with 5 × 105 B16F10 cells in the right flank, when tumor volumes reached ~50 mm2 (day 8), mice were injected with a single intratumoral injection of 5 μg soluble c-di-GMP or the same dose of c-di-GMP formulated with CSiNPs (Fig. 5a). Intratumoral injection of CSiNPs complexed with c-di-GMP triggered sustained regression of B16F10 tumors that was only modestly impacted by soluble c-di-GMP, or CSiNPs loaded with a control linear dinucleotide (inactive in STING stimulation, Fig. 5b–f). Mice treated with c-di-GMP/CSiNPs also showed greatly prolonged survival, with 3 out of 8 animals remained tumor free for at least 60 days (Fig. 5b, c). To access the induction of immunological memory after c-di-GMP/CSiNPs induced tumor regression, surviving mice from the c-di-GMP/CSiNPs treatment group were rechallenged with B16F10 cells in the opposite flank 60 d after primary challenge (Fig. 5a). Surviving mice previously treated with c-di-GMP/CSiNPs showed 37.5 % survival at 60 d (Fig. 5c, d), and the cured mice were fully protected against subsequent tumor rechallenge (Fig. 5d). Resistance to secondary tumor cell challenge confirms the development of adaptive memory after treatment induced tumor regression. Notably, antitumor activity was dependent on the combination of CSiNPs and c-di-GMP and correlated with increased activation of antigen presenting cells in the tumor microenvironment.
Fig. 5. Local c-di-GMP delivery via cationic silica nanoparticle exhibits potent anti-tumor activity in a melanoma model.

C57BL/6 mice (n=8 per group) were subcutaneously injected with 5 × 105 B16-F10 cells in the flank 8 d prior to treatment. Mice were treated with various c-di-GMP formulations at a dose of 5 μg administered intratumorally on day 8. a) Treatment scheme for therapeutic B16-F10 melanoma model. Three formulations of c-di-GMP, c-di-GMP control plus CSiNPs, or c-di-GMP loaded CSiNPs (c-di-GMP/CSiNPs) were injected directly into the tumors at day 8, and the b) tumor size and c) mice survival (Kaplan-Meier curves) were monitored. d) Mice that cleared their tumors were injected subcutaneously on the opposite side of the flank on day 60 with B16-F10 cells and monitored for survival. e) Individual tumor growth curves of mice in various treatment groups. f) Mice images represent the progression in tumor on day 11, 15, and 25. ***P<0.001, **P<0.01, *P<0.05. NS, not significant by one-way analysis of variance (ANOVA) with Bonferroni post-test. Data represent mean ± standard error of the mean (s.e.m.) of 2–3 independent experiments.
In order to determine whether necrotic destruction and immune stimulation by c-di-GMP/CSiNPs could promote antigen-specific immune response, we utilized the B16F10 melanoma cell line transduced to express the model antigen ovalbumin (B16-OVA).44 Due to the very low level of OVA expression in this tumor cell line,45, 46 a small amount of OVA protein was injected to increase the antigen dose. Briefly, C57BL/6 mice were inoculated with B16-OVA tumor cells and on day 8 mice received a single intratumoral dose of 10 μg soluble OVA followed by 5 μg c-di-GMP, or OVA plus c-di-GMP/CSiNPs at the same dose as the soluble counterparts (Fig. 6a). Injection of OVA plus c-di-GMP/CSiNPs induced tumor regression with complete tumor rejection in 50% of mice (Fig. 6b, c). Local treatment with c-di-GMP combined with OVA inhibited tumor growth over two weeks, after which time tumors rapidly progressed with all animals succumbing by day 31, a time when all the c-di-GMP/CSiNPs treated animals were still alive (Fig. 6c). A marked increase in the frequency of OVA-specific CD8+ T cells was detected on day 18 by SIINFEKL/H-2Kb tetramer in mice received c-di-GMP/CSiNPs + OVA, but not c-di-GMP + OVA (Fig. 6d). Similarly, functional analysis of blood T cells after antigen restimulation indicated that treatment with c-di-GMP/CSiNPs + OVA, but not c-di-GMP + OVA induced higher frequencies of IFN-γ and TNF-α producing CD8+ T cells (Fig. 6e–g). Immunofluorescent analysis of tumor-infiltrating CD8+ lymphocytes at tumor tissue collected 10 days after treatment revealed increased CD8+ T cell infiltration in mice treated with c-di-GMP/CSiNPs (Figure S4). Together, these data suggest that complexing c-di-GMP with CSiNPs induced the highest frequencies of antigen-specific CD8+ T cells capable of secreting pro-inflammatory cytokines IFN-γ and TNF-α, leading to potent anti-tumor activities and inhibition of tumor growth in murine melanoma.
Fig. 6. c-di-GMP formulated with CSiNPs triggers potent antigen-specific immune response.

a) C57BL/6 mice (n=8/group) were inoculated with 5 × 105 B16-OVA cells subcutaneously on day 0 and direct intratumoral injections of soluble OVA (10 ug/mice) plus c-di-GMP (5 ug/mice) or GS plus OVA (10 μg OVA, 5 μg c-di-GMP and 1.2 mg CSiNPs/mice) was given on days 8. 10 days after the intratumoral injections (day 18), mice were bled and peripheral blood mononuclear cells were evaluated by SIINFEKL/H-2Kb peptide-MHC tetramers staining. b) Tumor growth in C57BL/6 mice (n=8/group) inoculated with 5 × 105 B16-OVA cells. c) Kaplan-Meier survival curves. d) Mean percentages of OVA-specific CD8+ T cell measured in blood on day 18 after tumor incubation. e) Mean percentages of intracellular cytokines (TNF-α+ or IFN-γ+) secreting CD8+ T cells in blood after peptide restimulation. f) Representative flow cytometry plots of OVA-specific CD8+ T cells. g) Representative flow cytometry plots of TNF-α+ CD8+ T cells. ***P<0.001, **P<0.01, *P<0.05. NS, not significant by one-way analysis of variance (ANOVA) with Bonferroni post-test. Data represent mean ± standard error of the mean (s.e.m.) of 2–3 independent experiments.
Conclusions
Nanoparticles are emerging as a powerful tool in biomedicine, providing a practical solution to the challenges of traditional therapies in diseases especially cancer.6, 23–27 A wide variety of nanocarriers have been designed and tested in preclinical and clinical studies.6, 23–27 However, many nanocarriers exhibited intrinsic cytotoxicity, causing cellular stress, inflammation, and tissue destruction.25, 28–30 Such cytotoxicity is often considered unsuitable for clinical applications. Here we designed an in situ vaccine strategy based on cytotoxic silica nanoparticles. We showed that the intrinsic cytotoxicity of cationic silica nanoparticles can be harnessed for tumor destruction and immune activation. In addition to the intrinsic toxicity, CSiNPs can be easily complexed with STING agonist, enhancing its cellular uptake and retention within the tumor microenvironment. In mice, intratumoral injection of CSiNPs caused tumor cell destruction, releasing tumor-associated antigen and inflammation cues. When CSiNPs were combined with c-di-GMP which activates STING pathway, a markedly enhanced anti-tumor immune response and therapeutic efficacy in murine melanoma were achieved. While further studies will be needed to determine potential long-term toxicity, our results, along with the recent success in STING activation in clinical trials, support that in an appropriate combination, nanotoxicity can be harnessed to improve clinical outcomes in cancer immunotherapy.
Materials and Methods
All chemicals including SiNPs were purchased from Sigma-Aldrich unless noted otherwise. Bis-(3′–5′)-cyclic dimeric guanosine monophosphate (c-di-GMP) and c-di-GMP control were purchased from InvivoGen (San Diego, CA) and dissolved in ddH2O. DY547-c-di-GMP was purchased from BIOLOG Life Science Institute. Murine MHC class I tetramers were obtained from MBL International Corporation (Woburn, MA). All other antibodies were purchased from eBioscience (San Diego, CA) or BD Bioscience (San Jose, CA).
Animals and cells
Animals were housed in the United States Department of Agriculture (USDA)-inspected Wayne State University (WSU) animal facility under federal, state, local and NIH guidelines for animal care. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of WSU and approved by the Institutional Animal Care and Use Committee (IACUC) of WSU. Female C57BL/6 mice (6–8 weeks) were obtained from the Jackson Laboratory. B16-F10 cells were purchased from ATCC. Cells were cultured in complete medium (MEM, 10% fetal bovine serum (Greiner Bio-one), 100 U/mL penicillin G sodium and 100 μg/mL streptomycin (Pen/Strep).
Preparation of c-di-GMP loaded CSiNPs
Positively charged SiNPs were purchased from Sigma-Aldrich that were initially functionalized by triethoxypropylaminosilane. The size of purchased CSiNPs was about 30 nm and was confirmed by dynamic light scattering (DLS) measurements (Malvern Zetasizer). The CSiNPs solution was diluted 10 times using saline to decrease the nanoparticle density (0.116 g/mL) and mixed with the desired amount of c-di-GMP to prepare c-di-GMP-loaded CSiNPs. To facilitate the complexation process, probe-sonication was performed for 1 min with 2/2s on/off working cycle at a power output of 4 joules. c-di-GMP loaded CSiNPs were purified by repeated centrifugation and resuspension in PBS.
Size and zeta potential measurements
To measure the size and zeta potential of nanoparticles, dynamic light scattering (DLS, Zetasizer, Malvern) was used with He-Ne laser (633 nm) at 90° collecting optics at 25 °C.
Cell viability assay
Cellular viability was measured by the AlamarBlue assay (Thermo Fisher). Briefly, 5 × 104 B16-F10 cells were plated onto 96 multiwell plates (Costa, Corning, NY). After incubation with the indicated dose of nanoparticles for 24 h at 37 °C, cells were incubated with the AlarmaBlue agent for 4 h at 37 °C and the absorbance was measured at 570 nm, using 600 nm as a reference wavelength. The mean absorbance of non-exposed cells was the reference value for calculating 100 % cellular viability.
In vitro cellular uptake
To determine the intracellular uptake capacity of nanoparticles, the B16-F10 cells were seeded on glass coverslips in 6-well microscopy chamber at a density of 2 × 104 cells per well for 8 h at 37 °C. After 8 h, 2 μl of Mitochondria-Red Fluorescent Protein (RFP) (CellLight™ BacMam 2.0) per 10,000 cells was added into media to label mitochondria with RFP and cells were incubated for another 16 h at 37 °C. After total 24 h incubation, cells were treated with 10 μg/ml of fluorescence-labeled silica nanoparticles for 2 h or 24 h, and then washed with saline. For fixation, the glass that cells adhered to was immersed in 4 % paraformaldehyde in saline for 10 min at room temperature. Following fixation, the glass was washed with saline and mounted on a slide with nuclei staining by DAPI. Fluorescence images were obtained using a confocal microscope (Zeiss LSM-510) with a filter set of DAPI, FITC, and Mitochondria-RFP excitation/emission.
Cellular staining with Fluorescent Probes
The intracellular ROS production was measured by cell-permeant 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA, Thermo Fisher) staining following manufacturer’s instructions in B16-F10 after 2 h or 24 h exposure to nanoparticles at 37 °C.
Sectioning and Immunohistochemistry
Eight days after injection with 5 × 105 B16-F10 cells subcutaneously, mice were injected with formulations containing 5 μg of c-di-GMP in soluble or nanoparticle form directly into the tumor. Tumors were harvested 24 h later, placed in optimum cutting temperature (OCT) formulation (Sakura Finetek), and frozen in liquid nitrogen. Three-micrometer sections of the selected formalin-fixed, optimal cutting temperature (OCT)-embedded specimens were prepared and Gr-1 staining was performed using an anti-mouse Gr-1 Ab (BD Biosciences, 1:50). Stained histological sections were then imaged using an EVOS AMF4300 microscope.
Sectioning and Immunofluorescence staining
Eight days after injection with 5 × 105 B16-F10 cells subcutaneously into C57BL/6 mice, mice received a single intratumoral dose with formulations containing 5 μg c-di-GMP in soluble or nanoparticle form (5 μg c-di-GMP and 1.2 mg CSiNPs). Tumors were harvested 10 d later, placed in optimum cutting temperature (OCT) formulation (Sakura Finetek), and frozen in liquid nitrogen. For CD8+ T cell infiltration assessment, samples were cryosectioned and stained with CD8-APC antibody (eBioscience). Slides were imaged using a Zeiss LSM 510 confocal microscope.
Detection of necrosis in vivo by PI and Annexin V
5.0×105 B16F10 tumor cells were inoculated in the flank of C57BL/6 mice (n=3/group) on day 0. When the size of tumor is 50 mm2 (on day 8), the direct intratumoral injection of c-di-GMP (5 μg) or CSiNPs (1.2 mg) was given. After 24 h, mice were sacrificed and tumor tissues were isolated. Collected tumor tissues were then digested with 1.5 mL of freshly prepared enzyme mix composed of RPMI-1640 containing 0.8 mg/mL Collagenase/Dispase (Roche Diagnostics) and 0.1 mg/mL DNase (Roche Diagnostics) and cells were stained with PI and Annexin-V dye following manufacturer’s instructions. Percentages of PI and Annexin-V positive cells were determined by flow cytometry.
Detection of necrosis by confocal microscopy
B16-F10 cells were seeded on glass coverslips in 6-well microscopy chamber at a density of 2 × 104 cells per well for 8 h at 37 °C. After 8 h, cells were treated with 10 μg/ml of fluorescence-labeled CSiNPs for 2 h or 24 h, and then washed with saline without a fixation step. The desired amount of PI dye dissolved in saline of 1 mL was added to each well for 10 min at 37 °C and then washed with saline. Following PI staining, the glass was washed with saline and mounted on a slide with nuclei staining by DAPI. Fluorescence images were obtained using a confocal microscope (Zeiss LSM-510) with a filter set of DAPI, FITC, and PI excitation/emission.
Tumor model
B16-F10 or B16.OVA cells (5.0 × 105 cells) were subcutaneously inoculated into the right flank of 5–6-week-old C57BL/6 mice. When the tumor mass became palpable (7–8 mm, typically 8 days later), mice were divided into several treatment groups (n=8) and the tumor-bearing mice were intratumorally injected with 5 μg c-di-GMP in soluble or in nanoparticle form (5 μg c-di-GMP and 1.2 mg CSiNPs). For B16.OVA tumor study, c-di-GMP and OVA in soluble or in nanoparticle form (5 μg c-di-GMP, 5 μg OVA, and 1.2 mg CSiNPs) were intratumorally injected into B16.OVA tumor-bearing C57BL/6 mice. Survival and tumor size were measured every day using a sliding caliper. Ten days after the intratumoral injection of vaccine formulations to B16.OVA bearing mice (day 18), mice were bled and peripheral blood mononuclear cells were evaluated by SIINFEKL/H-2Kb peptide-MHC tetramer staining and intracellular cytokines (IFN-gamma and TNF-alpha) staining.
Statistical analysis
Comparisons of mean values of two groups were performed using unpaired Student’s t-tests. To analyze the statistical difference between groups, a one-way analysis of variance (ANOVA) with Bonferroni post-test was used. All of the values were expressed as means ± standard deviations. GraphPad Prism software was used for all the statistical analyses. ***P<0.001, **P<0.01, *P<0.05. NS, not significant.
Supplementary Material
Acknowledgments
This work is supported in part by American Cancer Society (11-053-01-IRG to Liu), Wayne State University President’s Research Enhancement Program (To Liu, Mao and Wei) and NIH (5R01CA076340-18 to Wei).
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
References
- 1.Mellman I, Coukos G, Dranoff G. Nature. 2011;480:480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. The New England journal of medicine. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. Nature reviews Clinical oncology. 2016;13:273–290. doi: 10.1038/nrclinonc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang C, Ye Y, Hochu GM, Sadeghifar H, Gu Z. Nano letters. 2016;16:2334–2340. doi: 10.1021/acs.nanolett.5b05030. [DOI] [PubMed] [Google Scholar]
- 5.Weiden J, Tel J, Figdor CG. Nature reviews. Immunology. 2017 doi: 10.1038/nri.2017.89. [DOI] [PubMed] [Google Scholar]
- 6.Irvine DJ, Hanson MC, Rakhra K, Tokatlian T. Chemical reviews. 2015;115:11109–11146. doi: 10.1021/acs.chemrev.5b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geary SM, Lemke CD, Lubaroff DM, Salem AK. Cancer immunology, immunotherapy : CII. 2011;60:1309–1317. doi: 10.1007/s00262-011-1038-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, Metchette K, Dubensky TW, Jr, Gajewski TF. Cell reports. 2015;11:1018–1030. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korman AJ, Peggs KS, Allison JP. Advances in immunology. 2006;90:297–339. doi: 10.1016/S0065-2776(06)90008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalos M, June CH. Immunity. 2013;39:49–60. doi: 10.1016/j.immuni.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu H, Irvine DJ. Bioconjugate chemistry. 2015;26:791–801. doi: 10.1021/acs.bioconjchem.5b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scully T. Nature. 2014;507:S2–3. doi: 10.1038/507s2a. [DOI] [PubMed] [Google Scholar]
- 13.Desrichard A, Snyder A, Chan TA. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22:807–812. doi: 10.1158/1078-0432.CCR-14-3175. [DOI] [PubMed] [Google Scholar]
- 14.Yarchoan M, Johnson BA, 3rd, Lutz ER, Laheru DA, Jaffee EM. Nature reviews Cancer. 2017;17:209–222. doi: 10.1038/nrc.2016.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pierce RH, Campbell JS, Pai SI, Brody JD, Kohrt HE. Human vaccines & immunotherapeutics. 2015;11:1901–1909. doi: 10.1080/21645515.2015.1049779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hammerich L, Binder A, Brody JD. Molecular oncology. 2015;9:1966–1981. doi: 10.1016/j.molonc.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marabelle A, Kohrt H, Caux C, Levy R. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20:1747–1756. doi: 10.1158/1078-0432.CCR-13-2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Formenti SC, Demaria S. Journal of the National Cancer Institute. 2013;105:256–265. doi: 10.1093/jnci/djs629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Veenstra JJ, Gibson HM, Littrup PJ, Reyes JD, Cher ML, Takashima A, Wei WZ. Cancer research. 2014;74:5409–5420. doi: 10.1158/0008-5472.CAN-14-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.den Brok MH, Sutmuller RP, van der Voort R, Bennink EJ, Figdor CG, Ruers TJ, Adema GJ. Cancer research. 2004;64:4024–4029. doi: 10.1158/0008-5472.CAN-03-3949. [DOI] [PubMed] [Google Scholar]
- 21.Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, Van Egeren DS, Park C, Irvine DJ. Nature. 2014;507:519–522. doi: 10.1038/nature12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hori Y, Stern PJ, Hynes RO, Irvine DJ. Biomaterials. 2009;30:6757–6767. doi: 10.1016/j.biomaterials.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ali OA, Lewin SA, Dranoff G, Mooney DJ. Cancer immunology research. 2016;4:95–100. doi: 10.1158/2326-6066.CIR-14-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi J, Kantoff PW, Wooster R, Farokhzad OC. Nature reviews Cancer. 2017;17:20–37. doi: 10.1038/nrc.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Min Y, Caster JM, Eblan MJ, Wang AZ. Chemical reviews. 2015;115:11147–11190. doi: 10.1021/acs.chemrev.5b00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shao K, Singha S, Clemente-Casares X, Tsai S, Yang Y, Santamaria P. ACS nano. 2015;9:16–30. doi: 10.1021/nn5062029. [DOI] [PubMed] [Google Scholar]
- 27.Bogart LK, Pourroy G, Murphy CJ, Puntes V, Pellegrino T, Rosenblum D, Peer D, Levy R. ACS nano. 2014;8:3107–3122. doi: 10.1021/nn500962q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yildirimer L, Thanh NT, Loizidou M, Seifalian AM. Nano today. 2011;6:585–607. doi: 10.1016/j.nantod.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khanna P, Ong C, Bay BH, Baeg GH. Nanomaterials (Basel, Switzerland) 2015;5:1163–1180. doi: 10.3390/nano5031163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Santos A, Evdokiou A, Losic D. Journal of Materials Chemistry B. 2015;3:7153–7172. doi: 10.1039/c5tb00956a. [DOI] [PubMed] [Google Scholar]
- 31.Mishra PK, Mishra H, Ekielski A, Talegaonkar S, Vaidya B. Drug discovery today. 2017;22:1825–1834. doi: 10.1016/j.drudis.2017.08.006. [DOI] [PubMed] [Google Scholar]
- 32.Li H, Li Y, Jiao J, Hu HM. Nature nanotechnology. 2011;6:645–650. doi: 10.1038/nnano.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corrales L, McWhirter SM, Dubensky TW, Jr, Gajewski TF. The Journal of clinical investigation. 2016;126:2404–2411. doi: 10.1172/JCI86892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barber GN. Nature reviews Immunology. 2015;15:760–770. doi: 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.An M, Li M, Xi J, Liu H. ACS applied materials & interfaces. 2017;9:23466–23475. doi: 10.1021/acsami.7b06024. [DOI] [PubMed] [Google Scholar]
- 36.Liberman A, Mendez N, Trogler WC, Kummel AC. Surface science reports. 2014;69:132–158. doi: 10.1016/j.surfrep.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leroueil PR, Berry SA, Duthie K, Han G, Rotello VM, McNerny DQ, Baker JR, Jr, Orr BG, Holl MM. Nano letters. 2008;8:420–424. doi: 10.1021/nl0722929. [DOI] [PubMed] [Google Scholar]
- 38.Green DR, Ferguson T, Zitvogel L, Kroemer G. Nature reviews Immunology. 2009;9:353–363. doi: 10.1038/nri2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weiner LM, Lotze MT. The New England journal of medicine. 2012;366:1156–1158. doi: 10.1056/NEJMcibr1114526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frohlich E. International journal of nanomedicine. 2012;7:5577–5591. doi: 10.2147/IJN.S36111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nel AE, Madler L, Velegol D, Xia T, Hoek EM, Somasundaran P, Klaessig F, Castranova V, Thompson M. Nature materials. 2009;8:543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 42.Bhattacharjee S, de Haan LH, Evers NM, Jiang X, Marcelis AT, Zuilhof H, Rietjens IM, Alink GM. Particle and fibre toxicology. 2010;7:25. doi: 10.1186/1743-8977-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. The Journal of biological chemistry. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 44.Mu Q, Jiang G, Chen L, Zhou H, Fourches D, Tropsha A, Yan B. Chemical reviews. 2014;114:7740–7781. doi: 10.1021/cr400295a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aranda F, Llopiz D, Diaz-Valdes N, Riezu-Boj JI, Bezunartea J, Ruiz M, Martinez M, Durantez M, Mansilla C, Prieto J, Lasarte JJ, Borras-Cuesta F, Sarobe P. Cancer research. 2011;71:3214–3224. doi: 10.1158/0008-5472.CAN-10-3259. [DOI] [PubMed] [Google Scholar]
- 46.Lund AW, Duraes FV, Hirosue S, Raghavan VR, Nembrini C, Thomas SN, Issa A, Hugues S, Swartz MA. Cell reports. 2012;1:191–199. doi: 10.1016/j.celrep.2012.01.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.