

Abstract
A variety of enzymes have been found to interact with double-stranded RNA (dsRNA) in order to carry out its functions. We have endeavored to prepare the covalently crosslinked native-like duplex RNA, which could be useful for biochemical studies and RNA nanotechnology. In this study, the interstrand covalently linked duplex RNA was formed by a crosslinking reaction between vinylpurine (VP) and the target cytosine or uracil in RNA. We measured melting temperatures and CD spectra to identify the properties of the VP crosslinked duplex RNA. The crosslinking formation increased the thermodynamic stability without disturbing the natural conformation of dsRNA. In addition, a competitive binding experiment with the duplex RNA binding enzyme, ADAR2, showed the crosslinked dsRNA bound the protein with nearly the same binding affinity as the natural dsRNA, confirming that it has finely preserved the natural traits of duplex RNA.
Keywords: RNA, Crosslinking reaction, 6-Vinylpurine, ADAR, Native-like
1. Introduction
Double-stranded RNA (dsRNA) plays an important role in many biological processes.1,2 Various enzymes recognize dsRNA and function at specific positions. For example, adenosine deaminases that act on RNA (ADARs) are RNA editing enzymes that recognize dsRNA substrates and convert adenosine (A) to inosine (I) at specific locations in the duplex. Inosine (I) is decoded as guanosine (G) which can lead to alteration of the codon meaning in RNAs.3,4 RNA editing leads to various functional consequences including regulation of the neurotransmitter receptor activity to modulating the functionality of small dsRNA molecules.5 It has also been reported that misregulation of the ADAR activity is related to numerous known diseases including cancer,6 Aicardi-Goutieres syndrome,7 and HIV.8 As another example, dsRNA is also known for its critical role in gene expression through RNA interference (RNAi) and as a biosynthetic precursor to miRNAs.9 Dicer and Drosha are the enzymes responsible for initiating these processes through their nuclease activity. RNAi is started by cleavage of long dsRNA by Dicer to create short dsRNA fragments known as siRNAs.10 Through different pathways, siRNA and miRNA perform gene silencing by intercepting the translation of mRNA in the cell. RNA helicases also recognize dsRNA. Brr2 is an RNA helicase that plays an essential role in pre-mRNA splicing.11 It catalyzes the ATP-dependent unwinding of the U4/U6 dsRNA structure. This is an essential step in the assembly of a catalytically-active spliceosome.
For the study of the interaction of dsRNA-modifying enzymes and for the development of new tools for biochemical studies and RNA nanotechnology,12–14 chemical tools to create crosslinked dsRNA are of great importance. However, the study of the RNA-RNA crosslink formation15–18 is still limited due to the complicated synthesis and the instability of RNA, while the DNA-DNA and DNA-RNA crosslink formation has been well studied and developed.19–27 An effective RNA-RNA crosslink formation is mostly achieved using photo-affinity reagents that require activation by UV light. Thionucleosides such as 6-thioguanosine16 and 4-thiouridine17 were reported to be photo crosslinking agents for RNA-RNA crosslinking. Crosslinking using thionucleotides has been mainly applied in the study of complex RNA such as tRNA or ribosomal RNA. Since the thionucleotides can crosslink only with the close contact-base by [2+2] photocycloaddition onto the 5, 6 double bond of pyrimidines, they have been used to identify the close contact-sites in folded RNA. However, due to this reaction site, this crosslinking method is unsuitable to form less distorted natural-like dsRNA. In recent developments, covalent bond formation at the terminus of siRNA was reported using p-cyanostilbenes or p-stilbazoles as the photo crosslinking moiety.18 Exposure of the oligonucleotides containing this group to UV light will induce [2+2] photocycloaddition, resulting in a crosslinked siRNA that is resistant toward nuclease activity.
In our previous study, we developed interstrand crosslinking bases, i.e., 2-amino-6-vinylpurine (AVP) and 6-vinylpurine (VP). Oligonucleotides conjugated with AVP or VP can undergo a selective crosslinking reaction with cytosine (C) or uracil (U) on the complementary strand.28–32 We expected that RNA containing VP will undergo an efficient crosslinking reaction with uracil (U) or cytosine (C) base of the complementary strand when at the proximal position (Fig. 1). Importantly, our designed VP can react with a chemically unmodified natural base and form a simple, two-carbon crosslink with no attachment of other bulky groups, thus reducing the potential of significant distortion of the native dsRNA structure. In this paper, we report on the synthesis of a less distorted native-like crosslinked duplex RNA using VP-containing RNA and further discuss the properties of the crosslinked dsRNA.
Figure 1.

RNA containing crosslinking base 6-vinylpurine (VP). Expected crosslinking reaction of RNA containing VP with cytosine (C) (top), and with uracil (U) (bottom) on the complementary RNA strand.
2. Results and discussion
2.1. Synthesis of 6-vinylpurine (VP)-containing RNA
The synthesis of SMe-protected VP controlled pore glass (CPG) precursor and a phosphoramidite precursor is shown in Scheme 1. The 5′-hydroxyl group of the commercially-available 6-chloropurine riboside was protected with a DMTr group. Vinylation by Stille coupling followed by S-methylation was performed to produce compound 3. For incorporating VP at the 3′ end position of the RNA, a CPG precursor was synthesized by coupling compound 3 with CPG that had previously been succinylated to give 4 and 5 (Scheme 1A).33 As for incorporating VP in the middle and at the 5′ end position, from compound 3 TBS protection at the 2′ position continued with phosphitylation to give the phosphoramidite precursor 7 (Scheme 1B).
Scheme 1.

Synthesis of CPG (4 and 5) and phosphoramidite (7): (a) DMTrCl, pyridine, 88%; (b) tributylvinyltin, PdCl2(PPh3)2, DMF, 77%; (c) NaSMe, H2O, DMF, 84%; (d) Succinylated CPG support, EDC, DMAP, DIPEA, pyridine, 56%; (e) TBSCl, AgNO3, THF, 56%; (f) iPr2NP(Cl)OCH2CH2CN, DIPEA, CH2Cl2, 92%.
Using CPG 4 and 5 and the phosphoramidite precursor 7, 6 different oligoribonucleotides (ORNs) were synthesized on a DNA/RNA synthesizer. The ORN sequences used in this study are shown in Table 1. After incorporation of the VP precursor into the oligonucleotides, deprotection and CPG cleavage were carried out under ultramild conditions using ammonium hydroxide/ethanol (3:1) at 30 °C for 12 h to avoid undesirable amination of the S-methyl group (Scheme 2). The 2′-O-TBS group was then removed with TEA-3HF. Oligonucleotide purification was performed using 20% polyacrylamide gel electrophoresis.
Table 1.
ORN sequences used in this study
| entry | Sequences (5′-3′) |
|---|---|
|
| |
| ORN1 | ACAGCAGGCGCX |
| ORN2 | ACAGCXGGCGCG |
| ORN3 | XCAGCAGGCGCG |
| ORN4 | ACUGAACCACCAAUGUGGGGAX |
| ORN5 | ACUXAACCACCAAUGUGGGGAA |
| ORN6 | XCUGAACCACCAAUGUGGGGAA |
| ORN7 | ACAGCAGGCGCG |
| ORN8 | ACUGAACCACCAAUGUGGGGAU |
| ORN9 | FAM-CGCGCCUGCUGU |
| ORN10 | FAM-UGCGCCUGCUGU |
| ORN11 | FAM-CGCGCCCGCUGU |
| ORN12 | FAM-CGCGCCUGCUGC |
| ORN13 | FAM-UUCCCCACAUUAGACGUUCAGU |
| ORN14 | UUCCCCACAUUAGACGUUCAGU |
Scheme 2.

RNA synthesis and purification. (a) RNA synthesizer, (b) NH4OH/EtOH (3:1), 30 °C, 12h; (c) TEA-3HF, 65 °C, 2.5 h.
After gel purification, HPLC analysis was performed on each of the synthesized ORNs. The HPLC profiles in Figure 2 show three types of compounds confirmed by MALDI-TOF mass spectroscopy (Table S1). ORN-B having a sulfoxide group was obtained by the auto oxidation of the S-Me group in ORN-A. ORN-C is a byproduct resulting from amination of the S-Me group that occurs due to ammonium hydroxide used in the deprotection step. ORN-C was not clearly observed when VP was incorporated at the middle position.
Figure 2.
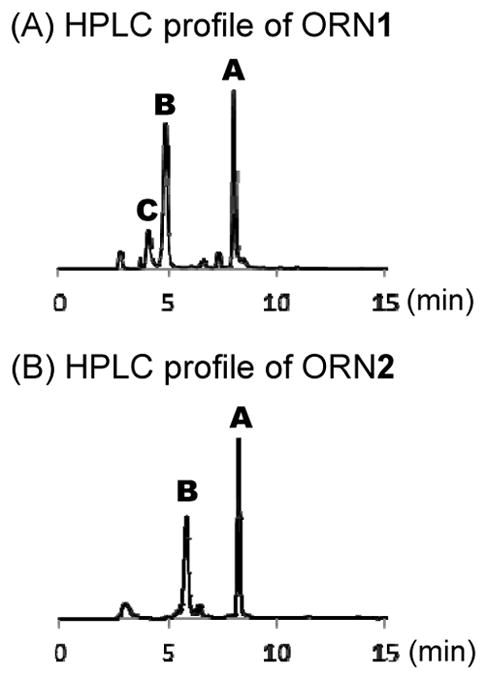
HPLC profiles of (A) ORN1 and (B) ORN2.
ORN-A having an S-Me group was converted into ORN-B by oxidation using magnesium monoperoxyphtalate (MMPP) (Scheme 3), then confirmed by HPLC and MALDI-TOF mass spectroscopy. The HPLC profile and MALDI-TOF mass spectroscopy have also confirmed the byproduct sulfonyl compound resulting from over oxidation of the S-Me by MMPP (Fig. S1). VP conversion was performed using 50% acetic acid at 37 °C for 3 h. The product was confirmed by MALDI-TOF mass spectroscopy (Fig. S2). The ORN-D was used for the crosslinking reaction without further purification.
Scheme 3.
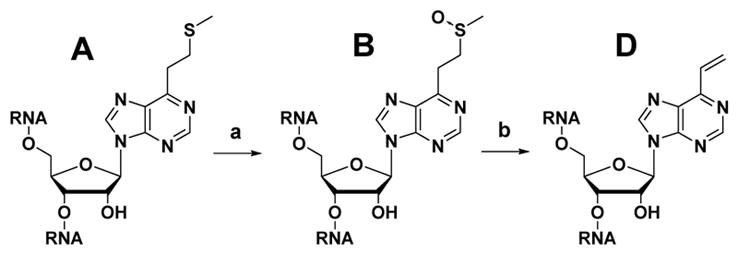
Synthesis of VP-containing ORN-D. (a) MMPP, H2O, RT, 30 min, (b) 50% AcOH, 37°C, 3h.
2.2. Crosslinking reaction in RNA duplex
First, the crosslinking reaction condition screening was performed using four different conditions by altering the pH and temperature of the reaction; (i) pH 7, 37 °C; (ii) pH 7, 50 °C; (iii) pH 5, 37 °C; (iv) pH 5, 50 °C (Fig. 3, 4). The synthesized VP-containing oligonucleotides (2 μM) and complementary RNA strand (1 μM) were dissolved in 50 mM of MES (pH 7.0) or MES (pH 5.0) containing 100 mM NaCl, then incubated at 37 or 50 °C for 24 h. The crosslink formation in the duplex RNA was analyzed by polyacrylamide gel electrophoresis (PAGE). The condition giving the best crosslinking yield was used for a time course experiment (Fig. 3, 4).
Figure 3.

Crosslinking reaction with the model sequence ORN1–3. Condition screening with (A) ORN1, (B) ORN2 and (C) ORN3. The reaction was performed in MES buffer (50 mM. pH 7.0 or 5.0) containing NaCl (100 mM) at 37 or 50 °C for 24 h. Time course of crosslinking reaction of (D, blue square) ORN1-ORN10(U), pH 7, 50 °C, (D, red circle) ORN1-ORN9(C), pH 5, 37 °C, (E, blue square) ORN1-ORN9(U), pH 5, 50 °C, (E, red circle) ORN1-ORN11(C), pH 5, 50 °C. (F) Gel image of crosslinking formation of ORN2-ORN9(U).
Figure 4.

Crosslinking reaction with the ADAR substrate sequence ORN4–6. Condition screening with (A) ORN4, (B) ORN5 and (C) ORN6. The reaction was performed in MES buffer (50 mM. pH 7.0 or 5.0) containing NaCl (100 mM) at 37 or 50 °C for 24 h. Time course of crosslinking reaction of (D) ORN4-ORN13, pH 7, 50 °C, (E) ORN5-ORN13, pH 5, 37 or 50 °C, (F) ORN6-ORN13, pH 5 or 7, 50 °C.
The results of the crosslinking reaction condition screening and time course experiments for the model sequence ORN1–3 are shown in Figure 3. For ORN1-ORN10 having U as the target, the conditions that gave a relatively high yield were pH 7, 50 °C and pH 5, 50 °C; whereas for ORN1-ORN9 having C as the target, the best yield was observed at pH 5, 37 °C (Fig. 3A). In 251661312251660288251663360251662336251665408251664384251666432251667456251668480251669504251670528251671552251672576251673600the time course experiment for ORN1 with the U target, we used milder conditions (pH 7, 50 °C) for the crosslinking. The time course showed that the crosslinking yield after 96 h for ORN1-ORN10(U) was at 52% and ORN1-ORN9(C) was at 46% (Fig. 3D). For ORN2, a 12 mer sequence of ORN having VP at the middle position, both U and C as the target gave a high yield at pH 5 and 50 °C (Fig. 3B). The time course showed that ORN2 rapidly undergoes a crosslinking reaction for both the target U and C. The crosslinking yield after 96 h was at 77% for ORN2-ORN9(U), while ORN2-ORN11(C) was at 80% (Fig. 3E). The gel image for the crosslinking reaction of ORN2-ORN9(U) is shown in Figure 3F and one clear new band was observed. As for ORN3 having VP at the 5′ position, the crosslinking yield was not more than 10% for any tested condition (Fig. 3C). Since the yield was insufficiently high, a time course experiment was not performed for this sequence.
For providing further information in the application of this crosslinking method, we next performed the same crosslinking formation toward the longer 22 mer sequences of ORN having VP at 3′, the middle, and 5′ end positions (Fig. 4). The sequence of ORN is reported as the substrate with the highest turnover rate by ADAR2.34,35 The crosslinking reaction result for ORN4 and ORN5 showed a similar tendency with the model sequences ORN1 and ORN2. However, the maximum yields were considerably low, even after 96 h of reaction, the yields were only 27% for ORN4 and 25% for ORN5 (Fig. 4D, E). On the other hand, the crosslinking formation at ORN6 gave the best yield among all the sequences. Condition screening has shown that ORN6 effectively crosslinked at pH 5, 50 °C followed by pH 7, 50 °C (Fig. 4C). An incredibly satisfying yield of 87% was obtained at pH 7, 50 °C and 75% yield at pH 5 after 96 h of reaction (Fig. 4F).
From the obtained data, we concluded that, in general, an effective crosslinking reaction with a cytosine base requires acidic conditions (pH 5) whereas for U, heat is required (50 °C). However, the crosslinking yields strongly depend on the sequence. In our previous study with the 2′-OMe type ORN, we observed a similar sequence dependency.32 We hypothesized that one of the reasons for the sequence dependency was due to the different lifetime of VP in each sequence. Since the decomposition of VP was not observed from the MALDI-TOF mass measurements (Table S1), we considered that VP would become inactive by an intramolecular reaction. In order to investigate the ratio of inactive VP, the reaction of VP with glutathione was pursued by MALDI-TOF mass spectroscopy. ORN1, 3, and 6 were investigated because they showed considerably different yields regardless of the similarity of VP position. The inactive products cannot react with glutathione, that is, the reaction yield with glutathione should be low. The yield for ORN3 was quite low compared to the other two sequences, suggesting that ORN3 has less active VP than the others, which is probably due to an unexpected intramolecular reaction within ORN3 (Fig. S3). More effort is required in the development of this crosslinking approach to overcome these sequence effects. This may be accomplished with effective additives36 or modification of the complimentary base, for example, using thiouracil instead of uracil.30
2.3. Alkali mediated foot-printing of crosslinked dsRNA
The position of the crosslink formation inside the dsRNA was determined by performing alkali mediated foot-printing. In order to obtain the crosslinking products of ORN2-ORN9(U), ORN2-ORN11(C) and ORN6-ORN13 on a large scale, we purified the crosslinking products by HPLC. The HPLC profile showed that the crosslinked dsRNA had a longer retention time compared to the complementary ORN, and the VP-containing ORN was almost completely consumed (Fig. S4). The crosslinking products were confirmed by MALDI-TOF mass spectroscopy.
To the crosslinked dsRNA was added a solution of K2CO3 in MeOH with 5% H2O (45 mM). The reaction was carried out at 30 °C for 4 h. Random cleavage of the ORN will reveal the crosslinking site in the dsRNA. The alkali mediated foot-printing result with the crosslinked ORN2-ORN9(U) is shown in Figure 5. Based on the PAGE analysis, short fragments of 1 nt to 6 nt and long fragments of more than 12 nt were observed, but not 7 nt-11 nt fragments. These results indicate that VP is crosslinked to the 7th base in the complementary stand. The crosslinked ORN2-ORN11(C) alkali mediated foot-printing analysis showed a similar result as with ORN2-ORN9(U) (Fig. S5). Therefore, the crosslinking site of ORN2-ORN11(C) was also at the 7th base, which is the expected C base target.
Figure 5.
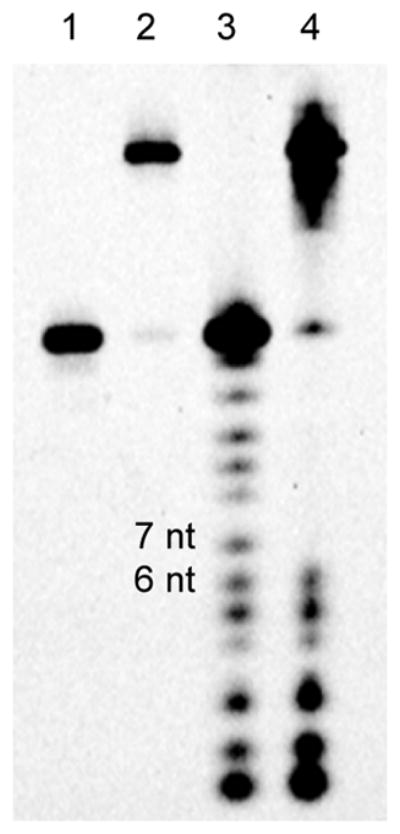
Alkali mediated foot printing of crosslinked ORN2-ORN9(U). 1: ORN9, 2: crosslinked duplex ORN2-ORN9(U), 3: after reaction with ORN9, 4: after reaction with crosslinked duplex ORN2-ORN9(U).
As for the crosslinked ORN6-ORN13, the alkali mediated foot-printing experiment showed that the crosslinking occurred at the expected position (Fig. S6). PAGE analysis revealed all the possible fragments except for the 22nd fragment. This result indicated that crosslinking of ORN6 occurred at the 22nd base, which is the expected U base target.
2.4. Melting temperature and CD spectra of crosslinked dsRNA
Melting temperature measurements were performed to provide insight into the crosslinked dsRNA thermodynamic stability compared to the natural dsRNA. The melting temperature measurements of ORN2-ORN9(U) and ORN2-ORN11(C) are shown in Figures 6A and 6B. Curves shown with a dotted line on the graph are the melting temperature curve of the natural duplex with their melting temperature values obtained as 70.5 °C and 59.6 °C, respectively, for ORN2(X=A)-ORN9(U) and ORN2(X=A)-ORN11(C). It should be noted that the natural duplex with ORN11(C) has a lower Tm than that with ORN9(U) because it contains one base mismatch at the C target position. Interestingly, no absorbance changes characteristic of the duplex melting were observed for the crosslinked duplex ORN2-ORN9(U) and ORN2-ORN11(C) (solid lines, Figures 6A and 6B). One possible explanation is that denaturation of the crosslinked dsRNA to single stranded RNA does not occur at temperatures below 95 °C. These results imply that crosslinking the dsRNA has significantly increased its thermodynamic stability.
Figure 6.

Melting temperature measurement of dsRNA. A: ORN2-ORN9(U) duplex, B: ORN2-ORN11(C) duplex, C: ORN6-ORN13 duplex. Natural duplex and crosslinked duplex are shown as dotted line and solid line, respectively.
Melting temperature measurements for ORN6-ORN13 are shown in Figure 6C. This duplex is an ADAR2 substrate RNA with an internal loop near the reactive adenosine and with a crosslink formed from VP at the 5′ end position. The natural duplex, ORN6(X=A)-ORN13, had a melting temperature value of 55.6 °C. After the crosslink was formed, the melting temperature value of the dsRNA significantly increased to 82.6 °C. A melting temperature increase by a value of 27 °C shows that the crosslink formation, regardless of the position, can significantly increase the thermodynamic stability of the dsRNA.
Circular dichroism (CD) spectra were obtained to provide information regarding the conformational properties of the crosslinked dsRNA. Prior to the measurement, both the natural and crosslinked dsRNA samples were reannealed by heating at 90 °C for 5 min, then gradually cooling to room temperature. The CD spectra of the typical dsRNA with its A conformation is characterized by a dominant positive band at 260 nm and a negative band at 210 nm.37 For the crosslinked duplexes, ORN2-ORN9(U), ORN2-ORN11(C), and ORN6-ORN13, the CD spectra showed almost no difference from those of the natural dsRNA, indicating that the crosslinking formation does not disturb the dsRNA natural conformational properties (Figs. 7A, B, C).
Figure 7.

CD measurement of dsRNA. A: ORN2-ORN9(U) duplex, B: ORN2-ORN11(C) duplex, C: ORN6-ORN13 duplex. Natural duplex and crosslinked duplex are shown as dotted line and solid line, respectively.
2.5. Competitive binding assay with ADAR2
In order to validate our hypothesis that the crosslink formation does not disrupt the nature of the dsRNA, a competitive binding assay was performed with the dsRNA-binding enzyme, ADAR2. The complex of the human ADAR2 bound to a 32P-labeled natural duplex ORN6(X=A)-ORN14 was resolved from the free duplex RNA using a non-denaturing PAGE gel. Either the unlabeled natural or crosslinked duplex, ORN6-ORN14, was then used as a competitor for formation of the labeled complex, and the IC50 for inhibition of the complex formation was measured for each duplex (Fig. S7).
The results are plotted in Figure 8 and the IC50s for the two duplexes were calculated from these data. The result shows that the natural duplex RNA had an IC50 = 330 ± 55 nM whereas the crosslinked IC50 = 270 ± 73 nM. Thus, the crosslinked dsRNA has nearly the same binding affinity for ADAR as the natural one, showing that the crosslinked dsRNA is well tolerated by the enzyme. This experiment confirms that our crosslinked duplex preserved the natural traits of the dsRNA.
Figure 8.
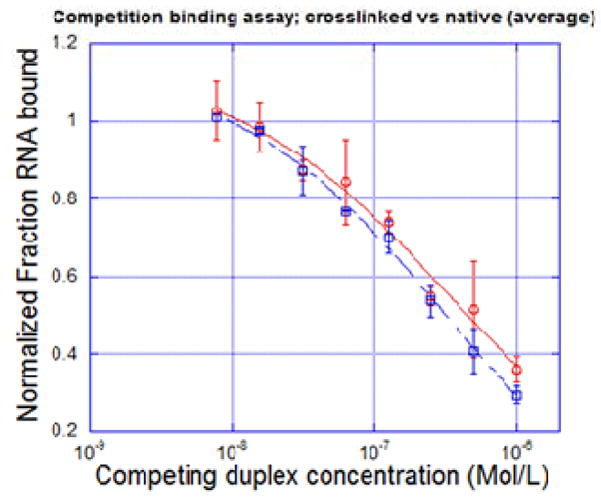
Competitive binding assay result with ADAR2 enzyme. red: native dsRNA; blue: crosslinked dsRNA.
3. Conclusion
We have successfully developed a native-like crosslinked dsRNA with our designed reactive base vinylpurine (VP). Two types of VP precursors, a phosphoramidite and a CPG precursor, were successfully synthesized and both VP precursors were incorporated into the RNA sequences at several different positions. We observed effective crosslinking with cytosine under acidic conditions, whereas for U, heating was required. We confirmed that the crosslinking occurred at the expected target by performing alkali facilitated foot printing.
The crosslinked duplex melting temperature (Tm) and CD spectra were obtained and compared to that of the natural oligonucleotides. The melting temperature of the crosslinked dsRNA showed a substantial increase compared to that of the natural duplex suggesting that by inducing crosslinking via VP, a much more stable dsRNA can be obtained. Moreover, a great similarity was found in the CD spectra of the crosslinked dsRNA and natural dsRNA implying that even after the crosslink formation, the native conformation of dsRNA is maintained. To further strengthen this point, a competitive binding experiment with the dsRNA-binding enzyme, ADAR2, was performed. Our result showed that the crosslinked dsRNA has nearly the same binding affinity as the natural dsRNA, confirming that our crosslinked dsRNA is well recognized by ADAR2. One of the advantages of this dsRNA crosslinking method is that, in principle, crosslink formation can be easily performed with the chemically unmodified natural RNA. The potential to form a less distorted native-like stable dsRNA will find application in biochemical studies for dsRNA-binding enzymes and RNA nanotechnology. For example, the crosslinking would be useful to develop a new inhibitor for dsRNA-binding enzymes since the crosslinking leads to increase the resistance toward nuclease activity. In addition, duplex stabilization could allow for short duplex inhibitors to be used. The less distorted RNA-RNA crosslinking will be also useful to form thermally stable RNA nanostructures as well as previously reported DNA-DNA crosslinked nanostructures.38,39
4. Materials and methods
4.1. General
The 1H-NMR spectra (400 MHz) and 32P-NMR (162 MHz) were recorded by a Bruker 400 spectrometer. The 1H-NMR spectra (600 MHz) and 13C-NMR spectra (150 MHz) were recorded by a Bruker AVANCE III 600 spectrometer. High resolution electrospray mass analysis was performed by a Bruker MicrOTOFQ II. The MALDI-TOF mass analysis was performed by a Bruker autoflex speed mass spectrometer.
4.2. Synthesis of SMe-protected VP CPG and phosphoramidite precursor
4.2.1. Synthesis of 9-(5-O-dimethoxytrityl-D-ribofuranosyl)-6-chloropurine (1)
6-Chloropurine riboside (1.0 g, 3.5 mmol) was co-evaporated with acetonitrile and pyridine, then dissolved in pyridine (10 mL). To the solution was added DMTrCl (2.4 g, 7.0 mmol), and the mixture was stirred at RT for 1 h. The reaction mixture was then diluted with ethyl acetate (50 mL), washed with water (50 mL) and brine (100 mL). The organic layer was dried over Na2SO4, then concentrated under reduced pressure. The crude was purified by column chromatography (Hexane:Ethyl acetate = 1:1→1:3→1:6→Ethyl acetate only containing 1% pyridine) to give 1 as a yellow foam (1.74 g, 85%). 1H-NMR (400 MHz, CDCl3) δ (ppm) 8.69 (1H, s), 8.37 (1H, s), 7.27–26 (3H, m), 7.19–17 (6H, m), 6.74 (4H, dd, J = 1.6, 8.8 Hz), 6.05 (1H, d, J = 5.6), 5.04 (1H, brs), 4.86 (1H, t, J = 5.2 Hz), 4.47 (1H, dd, J = 5.2, 6.0 Hz), 4.39 (1H, dd, J = 3.2, 6.0 Hz), 3.76 (6H, s), 3.45 (1H, dd, J = 10.8, 3.6 Hz) 3.35 (1H, dd, J = 10.8, 3.2 Hz), 3.25 (1H, brs). 13C-NMR (150 MHz, CDCl3) δ (ppm) 158.7, 151.7, 151.6, 151.0, 144.3, 144.0, 135.4, 132.5, 130.1, 130.0, 128.0, 127.1, 113.3, 90.9, 86.9, 85.9, 75.9, 72.4, 63. 5, 55.4. ESI-HRMS (m/z) calcd for C13H29ClN4O6 (M+H) + 589.1848, found 589.1864.
4.2.2. Synthesis of 9-(5-O-dimethoxytrityl-D-ribofuranosyl)-6-vinylpurine (2)
To a solution of 1 (0.77 g, 1.3 mmol) in DMF were added PdCl2(PPh3)2 (92 mg, 0.13 mmol) and tributyl(vinyl)tin (0.57 mL, 2.0 mmol). The reaction mixture was stirred at 85° C for 40 min and then allowed to cool at room temperature. The reaction mixture was diluted with CH2Cl2 (70 mL) and washed with saturated aqueous NaHCO3 (70 mL) and brine (100 mL). The organic layer was dried over Na2SO4, concentrated under reduced pressure and purified by column chromatography (CH2Cl2:MeOH = CH2Cl2 only→ 49:1→ 29:1→ 9:1 containing 1% pyridine) to give 2 as a brown foam (0.53 g). The product was immediately used for next reaction.
4.2.3. Synthesis of 9-(5-O-dimethoxytrityl-D-ribofuranosyl)-6-(2-methylthioethyl)purine (3)
To a solution of 2 (0.53 g, 0.91 mmol) in acetonitrile (4 mL) was added CH3SNa (0.32 g, 0.22 mmol) which was dissolved in water (0.4 mL), and the mixture was stirred at RT for 3 h. The reaction mixture was diluted with CH2Cl2 (15 mL) and washed with water (70 mL) and brine (100 mL). The organic layer was dried over Na2SO4, concentrated under reduced pressure and purified by column chromatography (CHCl3:MeOH = CHCl3 only→ 49:1→ 29:1→ 9:1 containing 1% pyridine) to give 3 as an orange foam (0.48 g, 60% in two steps). 1H-NMR (600 MHz, CDCl3) δ (ppm) 8.85 (1H, s), 8.32 (1H, s), 7.27–26 (2H, m), 7.20–16 (7H, m), 6.74 (4H, dd, J = 9.0, 2.4 Hz), 6.04 (1H, d, J = 5.4), 5.71 (1H, d, J = 2.4), 4.85 (1H, td, J = 6.0, 3.6 Hz), 4.45 (1H, m), 4.43 (1H, dd, J = 6.0, 3.0 Hz), 3.77 (6H, s), 3.52 (2H, td, J = 7.2 4.8 Hz), 3.46 (1H, dd, J = 10.8, 3.6 Hz), 3.29 (1H, dd, J = 10.8, 3.6 Hz), 3.27 (1H, d, J = 2.4), 3.10 (2H, t, J = 7.2 Hz), 2.18 (3H, s). 13C-NMR (150 MHz, CDCl3) δ (ppm) 161.4, 158.7, 151.8, 150.1, 149.7, 144.4, 142.6, 135.5, 135.4, 133.8, 130.1, 128.0, 127.1, 113.3, 90.8, 86.7, 86.1, 76.0, 72.7, 63.5, 55.3, 32.9, 32.1, 15.6. ESI-HRMS (m/z) calcd for C34H36N4O6S (M+H)+ 629.2428, found 629.2428.
4.2.4. Synthesis of 9-(5-O-dimethoxytrityl-2/3-O-succinylated-LCAA-CPG-D-ribofuranosyl)-6-(2-methylthioethyl)purine (4 and 5)
Preparation of succinylated LCAA-CPG
LCAA-CPG (0.5 g, 76 μmol) in a vial was slowly stirred in a solution of 3% trichloroacetic acid in CH2Cl2 (4.5 mL) at room temperature for 4 h. The LCAA-CPG filtered off and washed first with 9:1 triethylamine:diisopropylethylamine (50 mL), then with CH2Cl2 and ether. The activated support was dried under vacuum for 2 h. Into the vial containing the acid-activated LCAA-CPG was added succinic anhydride (0.1 g, 1.0 mmol) and 4-dimethyl aminopyridine (4-DMAP, 20mg, 0.16 mmol). Anhydrous pyridine (3 mL) was added via a syringe, and the vial was shaken at room temperature for 20 h. The CPG was filtered off, washed successively with pyridine, CH2Cl2 and ether. The succinylated CPG was dried under vacuum for 3 h.
Nucleoside succinylated LCAA-CPG coupling
The succinylated CPG (0.1 g, ~16 μmol) in a vial was co-evaporated with acetonitrile three times, then dried overnight under vacuum. To the vial were added EDC (0.62 g, 400 μmol), 4-DMAP (2.4 mg, 20 μmol), and pyridine (0.6 mL). To this solution, precursor 3 that was coevaporated with ACN, vacuumed overnight and diluted in pyridine (0.6 mL) and N,N-diisopropylethylamine (DIPEA, 20.9 μL) were added and the solution was shaken at room temperature for 48 h. Pentachlorophenol (26.6 mg, 0.1 mmol) was added and the mixture was shaken for an additional period of 19 h. The CPG was washed using pyridine, CH2Cl2, and ACN in a small amount and dried under vacuum for 1 h. Piperidine (2.5 mL) was added and the mixture was shaken for 1 h, then washed with a small amount of pyridine, CH2Cl2, and ACN dried under vacuum for 3h. Cap A (0.5 mL) and cap B (0.5 mL) were added and the mixture was shaken for 3 h at room temperature, washed with a small amount of pyridine three times, CH2Cl2 three times, THF and ether, then it was dried under vacuum overnight to give 4 and 5 as a dark orange powder (86 μmol/g loading, 56%).
4.2.5. Synthesis of 9-(5-O-dimethoxytrityl-2-O-tert-butyldimethylsilyl)-6-(2-methylthioethyl)purine (6)
Compound 3 (0.58 g, 0.92 mmol) was co-evaporated with acetonitrile, and then the dried residue was dissolved in THF (4 mL). To the solution were added pyridine (371 μL, 4.6 mmol), AgNO3 (240 mg, 1.4 mmol) and TBDMSCl (249 mg, 1.65 mmol), and the mixture was stirred for 3 h at room temperature. The reaction mixture was diluted with CH2Cl2 (50 mL), filtered through celite, and washed with NaHCO3 (50mL) and brine (50 mL). The organic layer was dried over Na2SO4, concentrated under reduced pressure and purified by column chromatography (Hexane/Ethyl acetate = 9:1→ 6:1→ 4:1→ 3:1→ 1:1→ Ethyl acetate only, containing 1% pyridine) to give 6 as an orange foam (412 mg, 61%). 1H-NMR (600MHz, CDCl3) δ (ppm) 8.81 (1H, s), 8.27 (1H, s), 7.45 (2H, d, J = 9.0 Hz), 7.34 (4H, dd, J = 9.0, 0.6 Hz), 7.27–26 (3H, m), 6.81 (4H, dd, J = 11.4, 2.4 Hz), 6.11 (1H, d, J = 5.4), 5.01 (1H, t, J = 5.4 Hz), 4.36 (1H, dd, J = 4.2, 9.0 Hz), 4.29 (1H, dd, J = 9.0, 7.2 Hz), 3.79 (6H, s), 3.55–51 (3H, m), 3.39 (1H, dd, J = 13.6, 4.2 Hz), 3.10 (2H, t, J = 5.4 Hz), 2.71 (d, J = 4.2 Hz), 2.18 (3H, s), 0.83 (9H, s), −0.02 (3H, s), −0.17 (3H, s). 13C-NMR (150 MHz, CDCl3) δ (ppm) 160.8, 158.7, 152.6, 150.8, 144.7, 142.8, 135.7, 133.5, 130.2, 128.2, 128.1, 127.1, 113.3, 88.3, 86.8, 84.3, 75.8, 71.6, 63.4, 55.4, 32.9, 32.2, 25.7, 18.0, 15.6, −4.9, −5.1. ESI-HRMS (m/z) calcd for C40H50N4O6SSi (M+H)+ 743.3293, found 743.3293.
4.2.6. Synthesis of 9-(5-O-dimethoxytrityl-3-O-(N,N-diisopropyl-2-cyanoethylphosphoramidyl)-2-O-tert-butyldimethylsilyl-D-ribofuranosyl)-6-(2-methylthioethyl)purine (7)
Compound 6 (120.0 mg, 0.162 mmol) was co-evaporated with acetonitrile and dissolved in CH2Cl2 (1 mL). To the dry residue were added DIPEA (170 μL, 0.98 mmol) and phosphoramidite (90 μL, 0.40 mmol), and the mixture was stirred at RT for 1 h. The reaction mixture was then quenched with NaHCO3 (20 mL), and extracted three times with EtOAc (20 mL). The combined organic layers were washed with brine (20 mL), dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (Hexane/Ethyl acetate = 4:1 →3:1 →2:1 →1:1, containing 1% pyridine) to give 7 as a pale orange foam (141 mg, 92%). 31P NMR (162 MHz, CDCl3): δ (ppm) 151.0, 149.0. ESI-HRMS (m/z) calcd for C49H68N6O7PSSi (M+H)+ 943.4372, found 943.4372.
4.3. Synthesis and purification of SMe-protected VP-containing oligonucleotides
The ORNs were synthesized following standard protocols on a DNA/RNA synthesizer. Cleavage from the solid support and deprotection were accomplished under a mild condition with NH4OH:EtOH for 12 h at 30°C, followed by TBS deprotection by incubation with trimethylamine trihydrofluoride (TEA-3HF) (125 μL) in DMSO (100 μL) for 2.5 h at 65°C. The reaction mixture was desalted by an NAP-10 column (GE Healthcare). The ORNs were then purified by gel electrophoresis (20% polyacrylamide) in 1× TBE buffer. The extract was filtered and desalted by a Sep-pak C-18 Cartridge (Waters Co.). Further purification was carried out by HPLC. The purified ORNs were treated with Zip-tip (Merck-Millipore), and then MALDI-TOF mass spectroscopy was measured. The concentration of the ORNs was determined by UV absorption at 260 nm.
4.4. General procedure for crosslinking reaction
30 μM of ORN-B (5 μL, 150 pmol) and acetic acid (5 μL) were mixed, and the mixture was incubated at 37°C for 3h. The mixture was evaporated to dryness, and then water (10 μL) was added. 15 μM of RNA-D having a vinyl group (1 μL, 15 pmol, final 2 μM) was combined with 10 μM of the RNA target (0.75 μL, 7.5 pmol, final 1 μM) and MES buffer (pH 7.0 or 5.0, final 50 mM) containing NaCl (final 100 mM). After incubation, a part of the reaction mixture was collected in a small tube and quenched with loading buffer (80% formamide, 10 mM EDTA). The collected reaction mixture was analyzed by denaturing 16% polyacrylamide gel electrophoresis containing 20% formamide, run at 300V × 45 mins to 1 hour in 1× TBE buffer. The FAM-labelled RNAs were visualized and quantified with FLA-5100 (Fujifilm Co., Tokyo, Japan).
4.5. Alkali mediated foot-printing
To the duplex RNA (0.13 μM) was added K2CO3 diluted in MeOH with 5% H2O (45 mM). The reaction was carried out at 30°C for 4 h. The reaction solution was neutralized by the addition of 0.5M of TEAA (20 μL) and 1M of HCl (0.5 μL) and then analyzed by denaturing 16% polyacrylamide gel electrophoresis containing 20% formamide, run at 300V × 45 mins to 1 hour in 1× TBE buffer. The FAM-labelled RNAs were visualized with FLA-5100 (Fujifilm Co., Tokyo, Japan).
4.6. Melting temperature (Tm) measurement
The duplex RNA (0.2 μM, natural or crosslinked) in 50 mM of MES buffer (pH 7.0) containing 100 mM NaCl was transferred to a microquartz cell with a 1-cm path length. The melting temperature was then measured under UV absorption at 260 nm from 25°C to 95°C at the rate of 1°C/min. The measurements were carried out 3 times per each sample, and averaged for obtaining the final value. The melting temperature measurement was performed by a DU-800 (BECKMAN-COULTER) equipped with a temperature controller.
4.7. Circular Dichroism (CD) measurement
The CD spectra were measured using 1 μM duplex RNA (natural or crosslinked) in 20 mM phosphate buffer (pH 7.0) containing 100 mM NaCl at 25°C by a J-720WI (JASCO Co., Hachioji, Japan) equipped with a Peltier temperature controller.
4.8. Competitive binding assay with ADAR2
ORN14 (30 pmol) was 5′-end-labeled by incubating the oligonucleotide with T4 polynucleotide kinase and [γ-32P] ATP (6000 Ci mmol−1) for 37 °C for 1 h. Sample was filtered through a G25 column and purified on a 19% (w/v) denaturing polyacrylamide gel. The labeled RNA was visualized by storage phosphor autoradioradiography, excised, crushed, and soaked overnight at 4°C using a solution containing 500 nM NH4OAC and 0.1 mM EDTA. The sample was ethanol precipitated, washed with 70% (v/v) ethanol, lyophilized to dryness, and dissolved in nuclease-free water. ORN6(X=A) and ORN14 duplex was hybridized in a 1:1 ratio in 1× TE buffer and 0.1 M NaCl by heating to 95 °C for 5 minutes and slow cooling to 30 °C.
Samples containing 63 nM hADAR2, 2.8 nM 32P-labeled, ORN6(X=A) and ORN14 duplex and different concentrations of unlabeled natural or crosslinked ORN6-ORN14 duplex (1000, 500, 250, 125, 62.5, 31.25, 15.6, 7.81, and 0 nM) were equilibrated in 20-mM Tris-HCl, pH 7.0, 6% glycerol, 0.5 mM DTT, 60 mM KCl, 20 mM NaCl, 0.1 mM 2- mercaptoethanol, 1.5 mM EDTA, 0.003% NP-40, 160-units/ml RNasin, 100-μg/ml BSA and 1.0-μg/ml yeast tRNA for 30 min at 30 °C. Samples were run on a 6% 80:1 native polyacrylamide gel at 16V/cm for 45 min, dried at 80 °C for 1 hr and imaged on a Typhoon Trio Variable Mode Image from GE Healthcare. Bands were quantified by volume integration using ImageQuant software such that all radioactive signal not found in the free RNA band was quantified as bound RNA. The data were fitted to the following equation: m4+((m1−m4)/(1+(m0/m3)^m2)) where m0 = concentration of competing RNA, m1 = curve maximum, m2 = slope, m3 = IC50 and m4 = curve minimum. IC50 assays were carried out in triplicate. Reported IC50 values are average of individual assays and error reported is standard deviation of the individual IC50 values.
Supplementary Material
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas (“Middle Molecular Strategy.” JP15H05838) and a Grant-in-Aid for Scientific Research (B) [25288073] from the Japan Society for the Promotion of Science (JSPS). This work was also supported in part by the research program of “Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials”.
Footnotes
Supplementary data associated with this article can be found, in the online version.
References and notes
- 1.Lunde BM, Moore C, Varani G. Nat Rev Mol Cell Biol. 2007;8:479. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beal PA. ChemBioChem. 2005;6:257. doi: 10.1002/cbic.200400303. [DOI] [PubMed] [Google Scholar]
- 3.Bass BL. Annu Rev Biochem. 2002;71:817. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nishikura K. Annu Rev Biochem. 2010;79:321. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tomaselli S, Galeano F, Alon S, Raho S, Galardi S, Polito VA, Presutti C, Vincenti S, Eisenberg E, Locatelli F, Gallo A. Genome Biol. 2015;16:5. doi: 10.1186/s13059-014-0575-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paz N, Levanon EY, Amariglio N, Heimberger AB, Ram Z, Constantini S, Barbash ZS, Adamsky K, Safran M, Hirschberg A, Krupsky M, Ben-Dov I, Cazacu S, Mikkelsen T, Brodie C, Eisenberg E, Rechavi G. Genome Res. 2007;17:1586. doi: 10.1101/gr.6493107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, Battini R, Bertini E, Brogan PA, Brueton LA, Carpanelli M, De Laet C, de Lonlay P, del Toro M, Desguerre I, Fazzi E, Garcia-Cazorla A, Heiberg A, Kawaguchi M, Kumar R, Lin JP, Lourenco CM, Male AM, Marques W, Jr, Mignot C, Olivieri I, Orcesi S, Prabhakar P, Rasmussen M, Robinson RA, Rozenberg F, Schmidt JL, Steindl K, Tan TY, van der Merwe WG, Vanderver A, Vassallo G, Wakeling EL, Wassmer E, Whittaker E, Livingston JH, Lebon P, Suzuki T, McLaughlin PJ, Keegan LP, O’Connell MA, Lovell SC, Crow YJ. Nat Genet. 2012;44:1243. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiden MD, Hoshino S, Levy DN, Li Y, Kumar R, Burke SA, Dawson R, Hioe CE, Borkowsky W, Rom WN, Hoshino Y. PLoS One. 2014;9:e108476. doi: 10.1371/journal.pone.0108476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meister G, Tuschl T. Nature. 2004;431:343. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- 10.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Nature. 2001;409:363. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 11.Mozaffari-Jovin S, Wandersleben T, Santos KF, Will CL, Luhrmann R, Wahl MC. RNA Biol. 2014;11:298. doi: 10.4161/rna.28353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo P. Nat Nanotechnol. 2010;5:833. doi: 10.1038/nnano.2010.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grabow WW, Jaeger L. Acc Chem Res. 2014;47:1871. doi: 10.1021/ar500076k. [DOI] [PubMed] [Google Scholar]
- 14.Patel MR, Kozuch SD, Cultrara CN, Yadav R, Huang S, Samuni U, Koren J, III, Chiosis G, Sabatino D. Nano Lett. 2016;16:6099. doi: 10.1021/acs.nanolett.6b02274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sloane JL, Greenberg MM. J Org Chem. 2014;79:9792. doi: 10.1021/jo501982r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christian EL, McPheeters DS, Harris ME. Biochemistry. 1998;37:17618. doi: 10.1021/bi982050a. [DOI] [PubMed] [Google Scholar]
- 17.Hiley SL, Sood VD, Fan J, Collins RA. EMBO J. 2002;21:4691. doi: 10.1093/emboj/cdf462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamiya Y, Iishiba K, Doi T, Tsuda K, Kashida H, Asanuma H. Biomater Sci. 2015;3:1534. doi: 10.1039/c5bm00231a. [DOI] [PubMed] [Google Scholar]
- 19.Nagatsugi F, Imoto S. Org Biomol Chem. 2011;9:2579. doi: 10.1039/c0ob00819b. [DOI] [PubMed] [Google Scholar]
- 20.Kocalka P, El-Sagheer AH, Brown T. ChemBioChem. 2008;9:1280. doi: 10.1002/cbic.200800006. [DOI] [PubMed] [Google Scholar]
- 21.Xiong H, Seela F. Bioconjugate Chem. 2012;23:1230. doi: 10.1021/bc300074k. [DOI] [PubMed] [Google Scholar]
- 22.Sakamoto T, Ooe M, Fujimoto K. Bioconjugate Chem. 2015;26:1475. doi: 10.1021/acs.bioconjchem.5b00352. [DOI] [PubMed] [Google Scholar]
- 23.Ichikawa K, Kojima N, Hirano Y, Takebayashi T, Kowata K, Komatsu Y. Chem Commun. 2012;48:2143. doi: 10.1039/c2cc16785a. [DOI] [PubMed] [Google Scholar]
- 24.Ghosh S, Greenberg MM. J Org Chem. 2014;79:5948. doi: 10.1021/jo500944g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fakhari F, Rokita SE. Nat Commun. 2014;5:5591. doi: 10.1038/ncomms6591. [DOI] [PubMed] [Google Scholar]
- 26.Sugihara Y, Nakata Y, Yamayoshi A, Murakami A, Kobori A. J Org Chem. 2016;81:981. doi: 10.1021/acs.joc.5b02573. [DOI] [PubMed] [Google Scholar]
- 27.Op de Beeck M, Madder A. J Am Chem Soc. 2011;133:796. doi: 10.1021/ja1048169. [DOI] [PubMed] [Google Scholar]
- 28.Nagatsugi F, Kawasaki T, Usui D, Maeda M, Sasaki S. J Am Chem Soc. 1999;121:6753. [Google Scholar]
- 29.Kawasaki T, Nagatsugi F, Ali MM, Maeda M, Sugiyama K, Hori K, Sasaki S. J Org Chem. 2005;70:14. doi: 10.1021/jo048298p. [DOI] [PubMed] [Google Scholar]
- 30.Imoto S, Hori T, Hagihara S, Taniguchi Y, Sasaki S, Nagatsugi F. Bioorg Med Chem Lett. 2010;20:6121. doi: 10.1016/j.bmcl.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 31.Imoto S, Chikuni T, Kansui H, Kunieda T, Nagatsugi F. Nucleos Nucleot Nucl. 2012;31:752. doi: 10.1080/15257770.2012.726756. [DOI] [PubMed] [Google Scholar]
- 32.Hagihara S, Kusano S, Lin WC, Chao XG, Hori T, Imoto S, Nagatsugi F. Bioorg Med Chem Lett. 2012;22:3870. doi: 10.1016/j.bmcl.2012.04.123. [DOI] [PubMed] [Google Scholar]
- 33.Damha MJ, Giannaris PA, Zabarylo SV. Nucleic Acids Res. 1990;18:3813. doi: 10.1093/nar/18.13.3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Phelps KJ, Tran K, Eifler T, Erickson AI, Fisher AJ, Beal PA. Nucleic Acids Res. 2015;43:1123. doi: 10.1093/nar/gku1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matthews MM, Thomas JM, Zheng Y, Tran K, Phelps KJ, Scott AI, Havel J, Fisher AJ, Beal PA. Nat Struct Mol Biol. 2016;23:426. doi: 10.1038/nsmb.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kusano S, Ishiyama S, Lam SL, Mashima T, Katahira M, Miyamoto K, Aida M, Nagatsugi F. Nucleic Acids Res. 2015;43:7717. doi: 10.1093/nar/gkv797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lesnik EA, Freier SM. Biochemistry. 1995;34:10807. doi: 10.1021/bi00034a013. [DOI] [PubMed] [Google Scholar]
- 38.Rajendran A, Endo M, Katsuda Y, Hidaka K, Sugiyama H. J Am Chem Soc. 2011;133:14488. doi: 10.1021/ja204546h. [DOI] [PubMed] [Google Scholar]
- 39.Tagawa M, Shohda K, Fujimoto K, Suyama A. Soft Matter. 2011;7:10931. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.