

Abstract
Centrioles are conserved microtubule-based organelles that form the core of the centrosome and act as templates for the formation of cilia and flagella. Centrioles have important roles in most microtubule related processes, including motility, cell division and cell signaling. To coordinate these diverse cellular processes, centriole number must be tightly controlled. In cycling cells, one new centriole is formed next to each preexisting centriole in every cell cycle. Advances in imaging, proteomics, structural biology and genome editing have revealed new insights into centriole biogenesis, how centriole numbers are controlled and how alterations in these structures contribute to diseases such as cancer and neurodevelopmental disorders. Moreover, recent work has uncovered the existence of surveillance pathways that limit proliferation of cells with numerical centriole aberrations. Here we discuss recent progress in this field with a focus on signaling pathways and molecular mechanisms.
Introduction
Centrosomes function in animal cells as microtubule-organizing centers to influence cell shape, polarity and motility, as well as spindle formation, chromosome segregation and cytokinesis1–4. Each centrosome typically comprises a pair of centrioles, which assemble a protein matrix, the pericentriolar material (PCM). The PCM harbors not only proteins important for microtubule nucleation5, but also regulators of the cell cycle and its checkpoints, in line with important roles for centrosomes in intracellular signaling6. Fully mature centrioles can also dock at the plasma membrane where they function as basal bodies for the formation of cilia and flagella7, and dysfunction of the basal body-ciliary apparatus gives rise to ciliopathies8. In recent years, much progress has been made towards understanding how centriole duplication and centrosome assembly are controlled, and how deregulation of these processes can contribute to human disease1,9,10. Here we summarize our current understanding of the mechanisms underlying the regulation of centriole duplication, and we discuss how centrosome aberrations contribute to human diseases such as cancer and neurodevelopmental disorders. We will focus primarily on vertebrate centrosomes, but incorporate data from other organisms where appropriate. To provide a guide to nomenclature, the names of prominent orthologous proteins in different species are presented in Table 1.
Table 1.
A brief guide to nomenclature
| H. sapiens | D. melanogaster | C. elegans | Chlamydomonas |
|---|---|---|---|
|
| |||
| PLK4 | Plk4 (SAK) | zyg-1 | |
| SAS-6 | DSas-6 | sas-6 | BLD12 |
| STIL | Ana2 | sas-5 | |
| CPAP (CENPJ) | DSas-4 | sas-4 | |
| CEP135 | DCep135 | BLD10 | |
| CEP152 | Asl | ||
| CEP192 | DSpd-2 | spd-2 | |
| CEP215 (CDK5RAP2) | Cnn | spd-5 | |
| CEP295 | Ana1 | ||
Footnotes: Plk4 = Polo-like kinase 4; SAS-6 = Spindle Assembly Abnormal 6; STIL = SCL/TAL1 Interrupting Locus; CPAP = Centrosomal P4.1-Associated Protein/CENPJ = Centromere Protein J; CEP135= Centrosomal Protein 135; CEP152 = Centrosomal Protein 152; CEP192 = Centrosomal Protein 192; CDK5RAP2/CEP216 = CDK5 Regulatory Subunit Associated Protein 2/Centrosomal Protein 216; CEP295 = Centrosomal Protein 295; Ana2 = Anastral Spindle 2; SAS-4 = Spindle Assembly Abnormal 4; Asl = Asterless; Dspd-2 = Spindle Defective 2; Cnn = centrosomin; Ana1 = Anastral Spindle 1; zyg-1 = Zygote defective 1; sas-5 = Spindle Assembly Abnormal 5; spd-5 = Spindle Defective 5; BLD12 = Bald 12; BLD10 = Bald 10.
Centrosome structure and assembly
Centriole duplication and centrosome assembly are complex processes that need to be tightly regulated during proliferation and development. Key components involved in these processes have recently been identified, setting the stage for mechanistic analyses of centriole biogenesis and PCM assembly.
Establishing centriole structure
Centrioles are cylindrical structures characterized by an evolutionarily conserved radial 9-fold symmetry11,12 (Figure 1A). In vertebrates, the walls of centrioles are composed of 9 triplet microtubule blades that are arranged circumferentially. The wall of a fully mature centriole carries two sets of appendages: subdistal appendages, which are required for anchoring of cytoskeletal microtubules, and distal appendages, which are needed for membrane docking during ciliogenesis. Several appendage markers have been identified, but much remains to be learned about the assembly and function of these structures13,14. The proximal part of the procentriole [G] lumen harbors a scaffolding structure known as the ‘cartwheel’ (Figure 1A)15, whose assembly represents the first step in the construction of a new procentriole and onto which microtubules are added to form the centriolar wall. In some organisms, cartwheels are permanent features of centrioles, but in human cells they act as transient scaffolding structures and are disassembled as cells exit mitosis. At the center of the cartwheel is a ring-shaped hub, from which nine spokes emanate to connect to the A-tubules of the microtubule triplets. In side views, the cartwheel appears as a stack of rings, whose height varies depending on species and cell cycle stage (Figure 1A)11,16–20.
Figure 1. Centriole architecture and the centrosome duplication-segregation cycle.

(A) (a) Schematic showing fully mature parent centriole (upright) and tightly associated procentriole. Prominent markers representative for the different structures are indicated to the right. (b) Micrograph shows lattice of in vitro reconstituted cartwheel hub and spoke structures visualized by cryo-electron microscopy. Adapted with permission from23. (c) Image derived from cryotomogram sections of Chlamydomonas procentriole emphasizes cartwheel and triplet microtubules. Adapted with permission from19. (d) Transmission electron microscopy shows longitudinal section (top) and cross sections at proximal (lower left) and distal parts (lower right) of Paramecium basal body (Anne-Marie Tassin, unpublished). (B) Shared pathways ensure coordination of centrosome duplication-segregation and chromosome replication-segregation cycles. At the G1/S transition both centriole duplication and DNA replication depend on CDK2 as well as phosphorylation of the retinoblastoma protein pRb and liberation of E2F transcription factors203. Similarly, overlapping sets of enzymes, including the kinases CDK1 and PLK1 and the protease Separase govern entry into mitosis, chromosome segregation, and licensing of DNA and centrioles for a new round of duplication. Lastly, several proteins with well-established functions in DNA transactions have been proposed to play additional roles in the centrosome cycle, but indirect effects on centrosomes remain difficult to exclude204. Centrioles are depicted in different shades of grey to indicate different states of maturity. A procentriole (light grey) is a newly created centriole that is not yet duplication competent. A procentriole converts into an immature parent centriole (middle grey) following disengagement in mitosis. An immature parent centriole becomes a mature parent centriole (dark grey) following the acquisition of appendages. Appendage structures undergo a transient modification/disassembly during mitosis. Cartwheels are shown in red; loose tethers connecting parent centrioles in dashed green lines; tight linkers connecting procentrioles to their parents in dark blue; subdistal and distal appendages are shown in light and dark blue respectively.
Structural studies and cell free reconstitution experiments have revealed that each cartwheel ring is comprised of nine homodimers of SAS-6 proteins. In vitro, SAS-6 can oligomerize into structures closely resembling the cartwheel hub, suggesting that SAS-6 may impart the typical nine-fold symmetry to centrioles21–23. However, the assembly of stable cartwheels in vivo likely requires additional proteins, interactions with the microtubule wall and/or preexisting centrioles24,25. The conserved centriole duplication factor STIL (Ana2 in Drosophila), interacts with SAS-6 and plays a central role in promoting SAS-6 recruitment and/or assembly26–32. In Chlamydomonas, cartwheel formation requires the protein Bld10p19,33, which interacts with SAS-6 to relieve an inhibitory action of the SAS-6 C-terminus on cartwheel assembly23. In human cells, the putative Bld10p homolog CEP135, also interacts with hSAS-634, but most CEP135 localizes to the parent centriole and not the procentriole35,36, suggesting additional roles in centriole biogenesis and PCM assembly. The exact role of CEP135 in cartwheel formation in vertebrates therefore remains unclear and no homolog of Bld10p has been identified in Caenorhabditis elegans. Finally, the deposition of microtubules onto the cartwheel clearly requires CPAP (Centrosomal P4.1-Associated Protein)/hSAS-4)37,38,39.
Centriole length control
Human centrioles display a length of 450–500 nm and a diameter of 200–250 nm11. The dimensions of centrioles are remarkably constant in most cells of any given organism, but occasional striking deviations can be seen in specific cell types40. In principle, organelle size can be governed by a variety of mechanisms, including molecular rulers [G] or the regulation of kinetics of subunit assembly and disassembly41. For centriole length, polymerization and depolymerization of centriolar microtubules is likely to be critical. The most direct evidence for this notion stems from the demonstration that the Drosophila kinesin-13 Klp10A acts as a microtubule depolymerase to control centriole length42. Mammalian Kif24, another member of the kinesin-13 subfamily, has similarly been shown to localize to centrioles, but although Kif24 is required for normal cilia assembly, it does not influence centriole length43. Interestingly, both Klp10A and Kif24 interact with CP110, a protein previously implicated in centriole length control. While the precise functions of CP110 may differ between species44, in humans it caps the distal tips of centrioles and its depletion causes the extension of overly long centriolar microtubules36,45. Given that the removal of CP110 is required to extend the centriolar microtubules and form the axoneme [G] during ciliogenesis43,45,46, it is not surprising that CP110 levels are regulated by multiple mechanisms47–50.
Consistent with structural studies that showed CPAP controls the speed of microtubule growth during centriole assembly37–39, overexpression of CPAP or its interaction partners, CEP120 and SPICE1 (Spindle and Centriole Associated Protein 1), triggers the assembly of excessively long centrioles45,51–54. Centriole length can also be modulated by deregulation of proteins implicated in building the distal halves of centrioles, including the WD40 protein POC1 (Proteome Of Centriole protein 1)55, the centrin-binding protein hPOC556, or the microtubule binding protein CEP295 (Ana1 in Drosophila)57,58. Interestingly, depletion of CEP295 not only impairs the recruitment of hPOC5 and POC1, but also blocks the acetylation and glutamylation of centriolar microtubules57. In vertebrates, these tubulin modifications accumulate on centrioles as well as cilia, and polyglutamylation is required for long-term stability of centriolar microtubules59. It may be rewarding to explore whether enzymes implicated in post-translational microtubule modifications contribute to centriole length control60.
PCM assembly
Human centrosomes comprise ~200–300 proteins, many of which harbor coiled-coil domains61,62. However, centrosome composition is not static and some proteins rapidly exchange through trafficking on microtubules whose minus ends are anchored within the centrosome63. Others assemble on centrosomes through transient incorporation into highly dynamic cytoplasmic granules, termed centriolar satellites13,64. Satellites have been implicated in the delivery of proteins for centrosome assembly as well as ciliogenesis, and they form and dissolve rapidly in response to a variety of internal and external cues. Although numerous satellite components have recently been identified, satellites do not seem to occur in all cell types and their exact physiological roles remain to be fully understood.
Centrosomes are not surrounded by membranes, raising the question of how the PCM assembles and how its boundaries can be defined. Early electron microscopy led to the perception of PCM as an amorphous structure, but super-resolution microscopy has revealed that individual proteins occupy distinct radial “layers” within the PCM65–68. Large PCM proteins may self-assemble into micron-scale structures through multimerization69,70, and this view is strongly supported by recent structural work on the formation of Cnn scaffolds in Drosophila71. An alternative model is centered on the role of phase separation in the formation of non-membrane bounded organelles72,73. Recent work was focused on C. elegans SPD-5, a core PCM component and putative functional homolog of Drosophila Cnn74. Recombinant SPD-5 was shown to assemble in vitro into spherical condensates that concentrate tubulin and other proteins required for microtubule polymerization and stabilization75. In future, it will be interesting to determine to what extent in vivo PCM assembly occurs through a liquid to condensate phase transition, as opposed to high-affinity, well ordered interactions between complementary surfaces on large proteins. The two mechanisms are not necessarily mutually exclusive, as PCM could form by an initial phase separation that concentrates components which then harden into a gel-like or solid structure with ordered protein-protein interactions.
Control of centriole number
Much like DNA replication, centrosome duplication is tightly regulated to ensure that centriole duplication occurs once and only once (cell cycle control) and that only one new centriole is produced per pre-existing centriole (copy number control)76. Furthermore, duplication and segregation of centrosomes must be coordinated with the chromosome duplication-segregation cycle and these processes are co-regulated (Figure 1B). The following discussion will focus on three main stages of the centrosome cycle. First, we will describe the processes that occur around the time of mitosis and endow procentrioles with competence for duplication (Figure 2A). Second, we will summarize the salient features that underpin the biogenesis of new procentrioles at the G1/S transition (Figure 2B). And, third, we will discuss the final steps that result in full maturation of both centrioles and centrosomes at the G2/M transition (Figure 2C).
Figure 2. Key aspects of the centrosome duplication cycle.
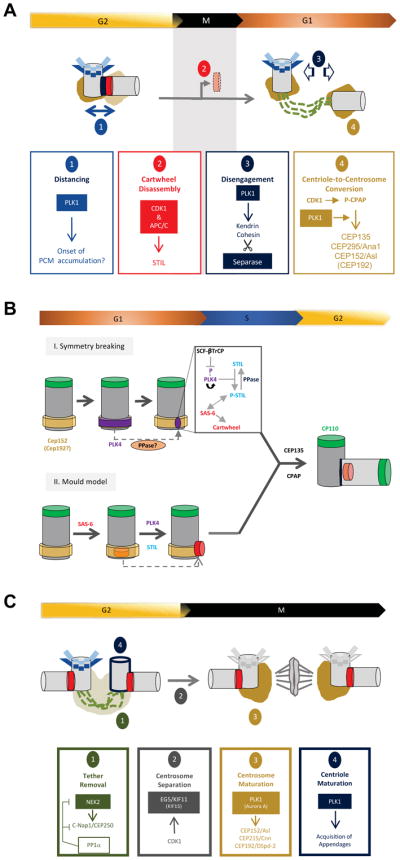
(A) Mitotic events licensing a new round of centriole duplication. Schematic describing four major events that occur during the progression from late G2 through M and into early G1. All four events, distancing, removal of the cartwheel, centriole disengagement and centriole-to-centrosome conversion, are considered necessary for the licensing of centrioles for a new round of duplication. Although the four events are conceptually distinct, they are expected to be integrated at a molecular and structural level.
(B) The birth of a new centriole. The master regulator PLK4 is initially recruited to a ring of CEP152 and CEP192 at the proximal end of the parent centriole. According to one model (I), a symmetry breaking event triggers accumulation of active PLK4 at one single site (dot) on the ring. The mechanism underlying symmetry breaking remains to be understood, but presumably involves self-enforcing feedback loops centered on PLK4, STIL, proteases and yet unidentified phosphatases. An alternative model (II) attributes an important role to the lumen of the parent centriole in assisting SAS-6 self-assembly into a cartwheel structure. PLK4 and STIL subsequently cooperate to remove the pre-formed cartwheel scaffold from the mould and position it laterally on the parent centriole.
(C) Coming of age: centriole and centrosome maturation. A G2 cell typically comprises 2 pairs of centrioles. The two parent centrioles are initially connected by a loose tether and form a single microtubule-organizing center. This tether is removed by a shift in the balance of activities of the NEK2 kinase and an opposing type 1 phosphatase (PP1α) acting on C-Nap1/CEP250 and other substrates118,119,205. Subsequently, the two centrosomes are separated by the microtubule-dependent motor EG5 (and the partially redundant motor KIF15)206, with EG5 being recruited to centrosomes in response to CDK1 phosphorylation207. Entry into mitosis requires expansion of PCM, termed centrosome maturation, in preparation for mitotic spindle formation. This step is triggered by PLK1 and Aurora A and results in the sequential recruitment of CEP152/Asl, CEP215/Cnn and CEP192/DSpd-2. Finally, only one parent centriole is fully mature (i.e. carries appendages) in a G2 cell, but during G2 and/or M phase the second parent centriole matures and acquires appendages in an event triggered by PLK1121. Centrioles are depicted in different shades of grey and PCM in different shades of brown, to indicate different states of maturity. Cartwheels are shown in red; loose tethers connecting parent centrioles as dashed green lines; tight linkers connecting procentrioles to their parents in dark blue; subdistal and distal appendages in light and dark blue respectively.
Licensing centrioles for a new round of duplication
Like DNA replication, which depends on licensing of DNA replication origins, centrioles only acquire the competence for duplication after cells pass through mitosis. In molecular terms, the ‘licensing’ of centrioles is now recognized to depend on two main processes: centriole disengagement which permits the reduplication of the parent centriole and centriole-to-centrosome conversion which is required for the procentriole to acquire competence for duplication.
Centriole engagement, the tight, near-orthogonal connection between each parent centriole and its procentriole, has long been shown to block the reduplication of the parent centriole77–79. Both PLK1 and the protease Separase have been implicated in promoting the loss of this tight connection, a process termed disengagement, prompting searches for the substrates of these enzymes78 (Figure 2A). One likely substrate of Separase is the PCM component PCNT (Pericentrin/Kendrin), which is released from centrosomes following cleavage by Separase in late mitosis80,81. Moreover, cleavage of PCNT is regulated by PLK182 and expression of a non-cleavable PCNT mutant suppressed centriole disengagement80,81. Centriole-associated cohesin has also been reported as a Separase substrate83. However, cohesin cleavage is not sufficient for centriole disengagement in Drosophila embryos and thus, further experiments are needed clarify the role of cohesin in centriole engagement84.
Early electron microscopy showed that a loss of the orthogonal orientation between the parent centriole and procentriole (disengagement) occurs in late M phase/early G185. More recently, correlative live/electron microscopy revealed that PLK1 drives the ‘distancing’ of procentrioles during early prophase, thereby conferring parent centrioles with competence for re-duplication even if the procentriole remains orthogonal to the parent (Figure 2A)86. The PCM is likely to maintain the close association of the centriole pair during mitosis, with the action of Separase contributing to PCM remodeling and the loss of this orthogonal orientation at mitotic exit. While the activity of Plk1 is essential for conferring competence for re-duplication, Separase likely plays a supporting role that ensures disengagement occurs soon after mitotic exit78. Finally, the CDK1-dependent removal of the cartwheel from the procentriole87 has also been shown to be important for relieving the block to reduplication of the parental centriole (Figure 2A)88.
For procentrioles, competence for duplication additionally requires the acquisition of PCM, a process termed ‘centriole to centrosome conversion’89,90, which is also governed by CDK1 and PLK191,92 (Figure 2A). Best described are events in Drosophila, where PLK1 is first recruited to CPAP, through CDK1-dependent phosphorylation of a single docking site92. In both Drosophila and mammalian cells, PLK1 then triggers the sequential assembly of CEP135, CEP295/Ana1 and CEP152/Asl and downstream PCM formation58,89,90. Importantly, recruitment of Asl in Drosophila embryos only occurs after disengagement, indicating that these licensing processes occur sequentially93. In mammalian cells, CEP295 directly binds to CEP192 and contributes to the stabilization of centrioles after the loss of the cartwheel upon mitotic exit89,94. Considering that CEP152 and CEP192 form scaffolds for the recruitment of PLK495–99, the kinase essential for centriole duplication (see below), these results explain why post-mitotic PCM assembly is required to confer duplication competence to procentrioles91. While C.elegans lack an obvious CEP295/Ana1 homolog, recent work has shown that the SAS-7 protein interacts with SPD-2 and is required for procentrioles to acquire competence to duplicate, suggesting SAS-7 may function analogously to CEP295100.
The birth of a new centriole
While cell cycle-coupled mechanisms of centriole licensing ensure that centriole duplication occurs only once per cell cycle, it remains to be explained how cells limit the building of procentrioles to one per pre-existing parent centriole. Whereas PLK1 plays a key role in cell cycle control of centriole duplication, PLK4 takes center stage as the linchpin for copy number control101,102. As indicated by morphological studies, at the G1/S transition one single procentriole begins to assemble perpendicularly to the parent centriole, and this newly formed procentriole then remains closely linked to its parent centriole while it elongates throughout G2 (Figures 1 and 2B).
Consistent with a central role in controlling centriole biogenesis, the levels and activity of PLK4 are tightly regulated. Plk4 exists as a stable homodimer and low steady-state levels arise from PLK4 trans-autophosphorylation within the dimer, which triggers SCF-β-TrCP-mediated proteolytic degradation103–107. Upon binding to STIL, PLK4 undergoes a conformational change and is activated through trans-phosphorylation within the activation segment28,108,109. Activated PLK4 then phosphorylates STIL within the so-called STAN motif, triggering the centriolar recruitment of SAS-6 and cartwheel formation26–29 (Figure 2B). However, in C. elegans, recruitment of a SAS-6/SAS-5 complex was shown to require a direct interaction with the PLK4-related kinase (ZYG-1), independent of catalytic activity110. Further downstream events in centriole biogenesis remain to be fully elucidated, but there is evidence that CEP135 serves to connect SAS-6 to CPAP and outer microtubules of the microtubule triplets34. During centriole elongation CPAP then regulates the growth of centriolar microtubules37–39,52, which are inserted underneath a cap of CP11036. Interestingly, CPAP also interacts with STIL and it will be important to understand how CPAP and STIL modulate each other’s activities32,111–113.
One major question that remains to be answered is how the ‘construction site’ for a new procentriole is chosen on the circumference of the parent centriole (Figure 2B). In mammalian cells, PLK4 is recruited to centrioles through binding to two distinct scaffolding proteins, CEP152 and CEP19295–99. Super-resolution microscopy shows that both CEP152 and CEP192 form rings around parent centrioles and, accordingly, PLK4 can also be seen to form rings in G1 phase. However, PLK4, STIL and SAS-6 then coalesce to a precise region on the circumference of the parent centriole (a dot on the CEP152/CEP195 ring) that marks the site of procentriole assembly26,35,97.A priori, there is no structural limitation to impose the formation of a single procentriole around the circumference of the parental cylinder, as indicated by the near-simultaneous formation of multiple procentrioles in response to overexpression of PLK436,101. So what mechanisms ensure copy-number control? One plausible view invokes a symmetry-breaking event that leads to the stochastic choice of a building site and suppression of all other potential sites (Figure 2B). In one attractive model, STIL is proposed to stabilize PLK4 at the site of procentriole assembly, allowing the remaining PLK4 within the ring to be turned over by self-catalyzed degradation26,109. Such a process would be controlled by both PLK4 kinase activity and counteracting phosphatases and would likely involve multiple feed-back loops, as suggested by theoretical modeling of the role of GTPases in symmetry-breaking during yeast cell polarization114. If correct, this symmetry-breaking model raises the challenge of understanding how PLK4 is regulated in time and space.
According to an alternative model, the lumen of the parent centriole acts as a mould for the assembly of a cartwheel that is subsequently released and used to direct formation of a procentriole (Figure 2B)115. In this case, future work would have to explain how cells limit the use of the mould to once per centriole and cell cycle. It will also be important to better define when and where different complexes involving the centriole duplication factors PLK4, STIL and SAS-6 are formed and stabilized116. Another attractive area ripe for investigation relates to the role of phosphatases in the spatio-temporal control of centriole duplication117.
Coming of age: maturation of centrioles and centrosomes
In a proliferating human cell, both centrioles and centrosomes undergo final maturation during G2 and M phase (Figure 2C). In late G2, each of the two duplicated centrosomes comprises one parental centriole, associated with PCM, and one procentriole, that lacks the ability to recruit PCM. The two parent centrioles are connected by a tether comprising rootletin and other proteins, anchored to C-Nap1/CEP250 118,119. Concomitantly, each procentriole is closely associated with the proximal end of the parent cylinder, through a linkage that remains to be characterized120. Importantly, only one of the two parental centrioles is fully mature and competent to function as a basal body for ciliogenesis, a feature indicated by the presence of subdistal and distal appendages. Mitotic progression is accompanied by transient modification/disassembly of appendage structures and acquisition of appendages by the younger parental centriole requires Plk1 (Figure 2C)121.
At the G2/M transition, the PCM expands significantly in preparation for mitotic spindle formation (Figure 2C). This process, termed ‘centrosome maturation5, has long been known to be governed by PLK1122,123, and a contribution of Aurora A is also well documented124. More recent work, carried out largely in Drosophila and C. elegans embyos, has yielded additional insight into the mechanisms underlying PCM expansion3. The emerging view is that PLK1 triggers the ordered assembly of an initial set of core scaffolding proteins which subsequently recruit all other PCM components. In Drosophila, these core proteins are Asl, Cnn and DSpd-2, corresponding to CEP152, CEP215/CDK5RAP2 and CEP192 in mammalian cells70. According to one model, phosphorylation of Cnn by PLK1 promotes its continuous recruitment around the centrioles, generating a constant outward flux of this scaffolding protein. One attractive feature of this model is that the activity of PLK1 controls the rate of Cnn incorporation into the PCM, offering a plausible mechanism for calibrating the size of PCM associated with each mitotic centrosome3. However, it is not immediately clear how to reconcile this flux model with data from C. elegans, where incorporation of SPD-5 into PCM was found to be isotropic125.
Sensing centriole number
While centriole number is normally tightly maintained at two or four copies per cell in cycling cells, there are several instances where centriole number is altered as part of a normal developmental program. One striking example is in multiciliated epithelial cells that line the airways, ventricles and oviducts of vertebrates. These specialized cells form hundreds of centrioles that serve as basal bodies for formation of multiple cilia126. However, as we will describe in the following sections, in general aberrations to centriole number are not well-tolerated in cycling cells and can contribute to pathologies. The mechanisms by which cells survey centriole number are now starting to emerge.
Centriole loss and the mitotic surveillance pathway
While centrosomes are a major source of spindle microtubules during mitosis, it is clear that chromatin and microtubule-mediated nucleation pathways can support spindle assembly in the absence of centrosomes63. A striking example of the dispensability of centrosomes for cell division are planarians [G], where cell divisions and regeneration occur in the absence of centrosomes, and centrioles are only assembled in terminally differentiated multiciliated cells to allow the formation of cilia used in locomotion127. In Drosophila, centrosomes are required during rapid-syncytial cell divisions in the early stages of embryogenesis, but are dispensable thereafter128. Importantly, flies lacking centrioles from the late stages of development grow to a normal size and are morphologically normal, but perish soon after hatching because of a lack of sensory cilia. These examples support the view that the ancestral role of centrioles was to direct the formation of cilia and flagella and that their association with the poles of the mitotic spindle acted to ensure their equal segregation into the daughter cells129.
Although cell division can proceed in the absence of centrosomes in some circumstances130–132, centrosomes are generally required for sustained proliferation of mammalian cells. Mouse embryos lacking centrioles undergo widespread p53-dependent apoptosis at an earlier developmental stage than mutants that lack cilia133. In cultured mammalian cells, centrosome loss resulted in a robust cell cycle arrest within a few divisions134,135. This arrest could be overcome by removal of p53, explaining why cancer cells often fail to respond to centrosome loss. Therefore, in contrast to planarians and flies, mammalian cells possess mechanisms to “sense” centrosome loss and prevent continued cell proliferation.
Insights into how centrosome depletion was signaled to p53 came from genome-wide knockout screens that led to the identification of a USP28-53BP1-p53-p21 signaling axis referred to as the mitotic surveillance pathway136–138. Deletion of any component of this pathway allowed the continued proliferation of cells in the absence of centrosomes. 53BP1 interacts with p53 and is a pivotal regulator of DNA double-strand break repair, while USP28 is a deubiquitinase that interacts with 53BP1 and has a minor function in DNA damage response signaling139–141. However, the role of 53BP1 in responding to centrosome loss is distinct from its established role in DNA damage repair136–138,142. While much remains to be learned about how the mitotic surveillance pathway functions to survey centrosomes, a plausible model is that in response to centrosome loss, 53BP1 binds to USP28 and p53 to facilitate USP28-dependent deubiquitination and activation of p53, leading to cell cycle arrest137,142 (Figure 3).
Figure 3. Responding to centrosome defects.
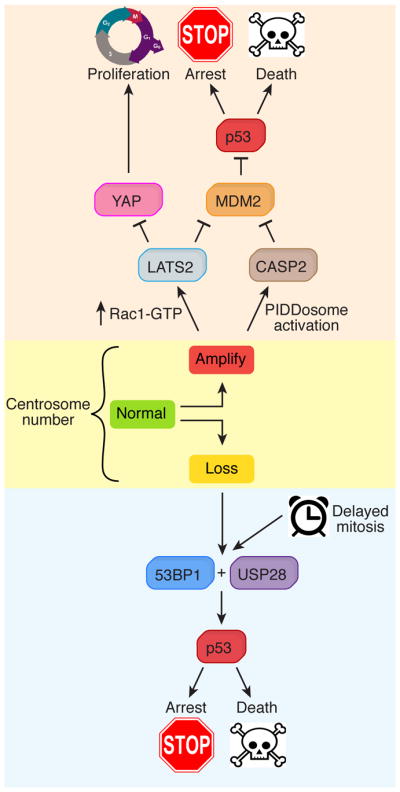
Pathways activated by centrosome loss (bottom) and centrosome amplification (top). Centrosome loss leads to 53BP1 and USP28-dependent stabilization of p53, which in turn promotes either cell death or cell cycle arrest133,136–138. An increased duration of mitosis also activates p53 through the same pathway. Centrosome amplification leads to hyper-activation of Rac1 and a corresponding decline in RhoA-GTP. RhoA-GTP activates the LATS2 kinase, which stabilizes p53 through inhibition of MDM2. In addition, LATS2 phosphorylates and inactivates the transcription factor YAP to inhibit proliferation148. In an alternative pathway, supernumerary centrosomes promote activation of the PIDDosome, which leads to activation of Caspase-2149. Active Caspase-2 cleaves MDM2 and thereby stabilizes p53208.
None of the components of the mitotic surveillance pathway show robust localization to the centrosomes, making it unlikely that they directly monitor centrosome number. How then is centrosome loss ‘sensed’? In the absence of centrosomes, spindle assembly is less efficient and cell division time increased135–138. Remarkably, increasing the duration of mitosis past a specific threshold elicits a durable p53-dependent G1 arrest in human epithelial cells143. This raises the possibility that centrosome loss triggers a cell cycle arrest by delaying mitosis. Consistent with this view, all the components of the mitotic surveillance pathway were also required to arrest the cell cycle following a prolonged mitosis136–138. Moreover, activation of p53 in mouse embryos lacking centrioles was associated with an increase in the duration of mitosis133. Additional evidence comes from the identification of the E3 ligase TRIM37 as a hit in genome wide screens for knockouts that allow proliferation without centrosomes137,138. While TRIM37 is required to arrest the cell cycle after centrosome loss, it is not required to prevent cell proliferation following a delayed mitosis. Loss of TRIM37 enables the formation of extra-centrosomal microtubule organizing centers that speed up spindle assembly in cells lacking centrosomes. TRIM37 deletion may thus “bypass” the arrest caused by centrosome loss by reducing the duration of mitosis in cells lacking centrosomes138.
Surprisingly, USP28 knockout mice are viable and have no clear phenotypes144,145. Nevertheless, there is evidence to suggest that activation of the mitotic surveillance pathway could underlie the growth defects observed in primary microcephaly (see below). Future work will be required to elucidate cell and tissue specific differences in signaling though the mitotic surveillance pathway as well as the impact of activation of this pathway in normal physiology and disease146.
Suppression of cell proliferation following centrosome amplification
Like centrosome loss, increases in centrosome number also suppress the proliferation of cells in culture107,147. This defect can be overcome by removal of p53, but does not depend on USP28 and 53BP1136, suggesting that distinct pathways activate p53 in response to an increase or decrease in centrosome number. Initial insight into how centrosome amplification suppresses cell proliferation came from the discovery that tetraploid cells, which contain twice the normal number of centrosomes, stabilize p53 through the Hippo pathway [G] kinase LATS2148 (Figure 3). Inducing extra centrosomes led to LATS2-dependent p53 stabilization, suggesting that extra centrosomes may, at least in part, be responsible for the activation of LATS2 in tetraploid cells.
Recently, an additional pathway, controlled by the PIDDosome [G], was found to be important in preventing the proliferation of cells with extra centrosomes149. The PIDDosome controls the proximity-induced activation of Caspase-2150 and was required to stabilize p53 after cytokinesis failure (Figure 3). Importantly, some PIDDosome components localize to the older parent centriole, suggesting that PIDDosome activation may be controlled by the presence of additional mature centrioles149. Consistent with this idea, depletion of the appendage protein ODF2, reduced Caspase-2 activation and p53 stabilization in cells overexpressing PLK4149. While ‘counting’ mature parent centrioles offer a method to detect centriole amplification, it remains unclear how excess mature parent centrioles would be detected and in turn, how they would promote activation of the PIDDosome. It will be interesting to test if driving premature maturation of the younger parent centriole with constitutively active Plk1 can promote PIDDosome activation in the absence of centriole amplification121.
Unlike p53 loss, LATS2 or Caspase-2 knockout does not allow the continued proliferation of cells with extra centrosomes136,149. It is therefore likely that additional pathways feed into p53 activation in response to centrosome amplification. Since many tumor cells possess supernumerary centrosomes (see below), overcoming the inhibitory effect of extra centrosomes on cell proliferation will be a key step to allow cells with extra centrosomes to acquire the necessary oncogenic mutations required for tumor development151.
Centrosome defects and cancer
Over a century ago the German cytologist Theodor Boveri postulated that centrosome aberrations could contribute to human cancer. Indeed, centrosome defects are present in a broad array of both solid and hematopoietic human cancers and in some tumors types, centrosome abnormalities have been observed early in disease development and correlate with advanced tumor grade and poor clinical outcome10,151. Centrosome anomalies can be subdivided into either numerical or structural alterations151. While structural alterations are likely to originate from alterations in the levels or activity of centrosome proteins152, numerical alterations reflect increases in centrosome copy number, and arise due to the acquisition of an excessive number of centrioles. While structural and numerical centrosome aberrations are conceptually distinct, they often co-exist in tumors.
Do extra centrosomes promote tumorigenesis?
To test the role of extra centrosomes in cancer, many studies have exploited PLK4 overexpression to increase centrosome number. Pioneering work in Drosophila showed that while centrosome amplification does not promote the development of spontaneous tumors, neuroblast and epithelial cells with extra centrosomes can initiate tumorigenesis when transplanted into host flies153,154. However, how centrosome amplification impacts tumor development in mammals is complex. In the mouse brain, extra centrosomes do not promote tumorigenesis155. Similarly, centrosome amplification in the skin epidermis resulted in spindle orientation defects and aneuploidy, but these abnormalities were not able to initiate spontaneous tumorigenesis or enhance the development of carcinogen-induced skin tumors156. By contrast, centrosome amplification did accelerate tumorigenesis in a p53-deficient skin epidermis157. Moreover, global PLK4 overexpression also accelerated the onset of lymphomas and sarcomas in p53 null mice and promoted hyperproliferation in the skin and pancreas158. Taken together, these studies validate a central role of p53 in restricting the continued proliferation of cells with centrosome amplification in mammals107.
While initial studies failed to observe the development of spontaneous tumors in animals with widespread PLK4 overexpression156,158,159, a more modest increase in PLK4 levels was shown to promote persistent centrosome amplification that promoted the development spontaneous tumors147. Importantly, these tumors exhibited dramatic numerical and structural chromosomal alterations, mirroring the complex karyotype changes frequently observed in human tumors with extra centrosomes147. Some impairment of the p53 pathway is to be expected in tumors that form spontaneously in response to centrosome amplification. Accordingly, spontaneous lymphomas that develop in mice with centrosome amplification show down-regulation of p53 target genes147. In the future, it will be interesting to test if knockout of LATS2 or PIDDosome components accelerate the development of tumors driven by centrosome amplification.
The origin of centrosome defects in tumor cells
Cancer cell lines show wide variation in the penetrance and extent of centrosome amplification. Reversible depletion of centrosomes using a PLK4 kinase inhibitor has shown that tumor cell lines reach an equilibrium of centrosome number distribution that is determined by the rate at which extra centrosomes are accumulated and the rate at which cells harboring them are selected against134. One pathway leading to the acquisition of extra centrosomes is dysregulation of the centriole duplication cycle. While genes encoding centrosome proteins are rarely mutated in human cancers, increased or decreased expression of centrosome proteins is more common (Table 2)1,10,151. In addition, perturbation of cell cycle progression can lead to defects in centriole biogenesis. The clearest example is that of a prolonged arrest in G2 phase, which leads to Plk1 activation, centriole disengagement and premature centriole reduplication160. Consequently, DNA damage can induce centrosome amplification by increasing the time cells spend in G2 phase161,162.
Table 2.
Proteins involved in centriole number control, their functions and links to disease
| Gene symbol | Function | Links with disease |
|---|---|---|
|
| ||
| Centriole genes linked with tumorigenesis | ||
|
| ||
| PLK4 | Centriole duplication | Overexpressed in breast cancer. Controls invasion through regulation of the actin cytoskeleton. PLK4+/− mice are predisposed to liver and lung cancer |
| STIL | Centriole duplication | Promoter fused to TAL1 in T-Cell acute lymphoblastic leukaemia |
| NLP | Microtubule nucleation | Overexpressed in multiple cancers |
|
| ||
| Genes linked to MCPH | ||
|
| ||
| PLK4 | Centriole duplication | Mutations reduce the levels of PLK4 |
| STIL | Centriole duplication | Mutations inhibit the cell-cycle controlled degradation of STIL |
| CPAP | Controls centriole length and centriole duplication | Mutations weaken binding of CPAP to STIL |
| CEP135 | Centriole duplication | |
| CEP152 | Centriole duplication | |
| CEP63 | Centriole duplication | |
| CDK5RAP2/CEP215 | Centriole duplication | |
| WDR62 | Spindle pole organization; Centriole duplication | |
| ASPM | Spindle pole organization; Centriole duplication | |
| TUBGCP6 | Centriole duplication, component of the γTubulin ring complex | |
| TUBGCP4 | Component of the γTubulin ring complex | |
| CDK6 | Centrosome associated in mitosis | Mutations mislocalize CDK6 |
|
| ||
| Centriole genes linked to primordial dwarfism | ||
|
| ||
| PLK4 | Centriole duplication | Mutations reduce the levels of PLK4 |
| CPAP | Centriole duplication | |
| CEP152 | Centriole duplication | |
| CEP63 | Centriole duplication | |
| PCNT | Component of the PCM | Mutations mislocalize PCNT |
|
| ||
| Centriole genes linked with other disorders | ||
|
| ||
| ALMS1 | Functions in ciliogenesis | Alstrom Syndrome results in retinitis pigmentosa, deafness, obesity and diabetes. |
| OFD1 | Controls centriole length | Orofaciodigital syndrome results in malformations of the face, oral cavity and digits |
| C2CD3 | Controls centriole length | Orofaciodigital syndrome results in malformations of the face, oral cavity and digits |
Footnotes: Plk4 = Polo-like kinase 4; STIL = SCL/TAL1 Interrupting Locus; NLP = Ninein-Like Protein; CPAP = Centrosomal P4.1-Associated Protein; CEP135= Centrosomal Protein 135; CEP152 = Centrosomal Protein 152; CEP63 = Centrosomal Protein 63; CDK5RAP2/CEP216 = CDK5 Regulatory Subunit Associated Protein 2/Centrosomal Protein 216; WDR62 = WD Repeat Domain 62; ASPM = Abnormal Spindle Microtubule Assembly; TUBGCP6 = Tubulin Gamma Complex Associated Protein 6; TUBGCP4= Tubulin Gamma Complex Associated Protein 4; CDK6 = Cyclin Dependent Kinase 6; PCNT = Pericentrin/Kendrin; ALSM1= Alstrom Syndrome Protein 1; OFD1= Oral-Facial-Digital Syndrome 1; C2D3 = C2 Calcium Dependent Domain Containing 3; TAL1 = T-Cell Acute Lymphocytic Leukemia 1; PCM = Pericentriolar Material. MCPH = Autosomal recessive primary microcephaly.
A final pathway to generate extra centrosomes is through failed cell division. In addition to the doubling of centrosome number, failing division provides the benefit of doubling the genome to buffer against deleterious mutations or chromosome segregation errors. These properties allow tetraploid cells to sample novel karyotypes, eventually landing upon a rare combination that provides a growth advantage163. Consistent with a pro-tumorigenic role of tetraploid cells, a growing body of evidence suggests that a large fraction of human tumors arise from a tetraploid intermediate164. Although the uncontrolled proliferation of tetraploid cells can drive tumorigenesis165, extra centrosomes in tetraploid cells initially trigger a p53-dependent cell cycle arrest148. As a consequence, repeated cytokinesis failure does not result in the long-term establishment of centrosome amplification in cell culture166. This suggests that further genetic alterations, such as loss of LATS2, Caspase-2 or p53, are required to bypass this fitness disadvantage and generate long-term increases in centrosome number following cytokinesis failure.
Deregulation of oncogenes or tumor suppressor genes have been shown to lead to the formation of supernumerary centrosomes. For example, KLF14 is a transcriptional repressor of PLK4 and knockout of KLF14 leads to PLK4-induced centrosome amplification and tumor formation in mice167. PLK4 is also transcriptionally repressed by the p53 tumor suppressor158,168. Nevertheless, p53 knockout is insufficient to induce centrosome amplification in human cell lines and in tissues of mice135,136,147,155,156,158. Rather than playing a direct role in controlling centrosome number as originally proposed169, loss of p53 is likely to offer a permissive environment for the continued proliferation of cells with centrosome abnormalities, as it allows cells to bypass centrosome number surveillance pathways107,156–158.
Consequences of centrosome defects
Irrespective of how they arise, extra centrosomes are capable of nucleating microtubules that lead to the formation of multi-polar mitotic spindles. If not corrected, this results in a multi-polar division leading to extensive chromosome missegregation and inviable progeny (Figure 4A)170. The primary mechanism by which tumor cells suppress multi-polar divisions is through the coalescence of centrosomes into two groups to form a pseudo-bipolar spindle171. The efficiency of the clustering process is likely to be an important parameter in determining the ability of cells to tolerate centrosome amplification172,173. Centrosome clustering increases the frequency of incorrect merotelic [G] attachments of chromosomes to the mitotic spindle, leading to low rates of chromosome segregation errors that can be compatible with cell viability (Figure 4A)153,170,174. Through this pathway, supernumerary centrosomes can promote the frequent gains and losses of chromosomes during division, providing an explanation for the tight correlation of centrosome amplification and aneuploidy [G] in human cancer10,151.
Figure 4. Mechanisms through which centrosome amplification can contribute to tumorigenesis.

(A) Genome instability. Cells with supernumerary centrosomes form multi-polar mitotic spindles. Multipolar divisions lead to the production of highly aneuploid daughter cells that are typically inviable. To avoid multipolar divisions, cells cluster their centrosomes prior to anaphase. Centrosome clustering enriches for incorrect merotelic attachments of chromosomes to the mitotic spindle, resulting in chromosome segregation errors153,170,174. In addition to creating whole chromosome aneuploidy, mitotic errors caused by extra centrosomes can promote the acquisition of DNA double strand breaks that result in chromosomal rearrangements179–181.
(B) Defective asymmetric divisions. Drosophila neuroblasts undergo asymmetric cell division to self-renew and produce a differentiated Ganglion Mother Cell. Centrosome amplification can lead to a failure to correctly align the spinde resulting in the equal partioning of cell fate determinates (red and green crescents) into the daughter cells. This leads to an expansion of the stem cell pool and tissue overgrowth153. However, centrosome amplification did not produce spindle orientation defects in mouse neuronal cells, indicating this defect is likely to species or cell type specific155.
(C) Invasive behavior. Increased microtubule nucleation promotes Rac1 hyper-activation that drives invasive behavior184.
(D) Reduced ciliary signaling. Ciliary signaling can be disrupted in response to centrosome amplification by either dilution of cilia signaling components or a failure to form cilia158,185.
An additional source of mitotic errors emerges from the improper timing of centrosome separation prior to cell division. Both accelerating and delaying centrosome separation increase the frequency of chromosome misattachments to the mitotic spindle leading to chromosome segregation errors175–178. It will be interesting to investigate if structural or numerical alterations in centrosomes can contribute to defects in the timing of centrosome separation.
Along with whole chromosome aneuploidy, mitotic errors driven by supernumerary centrosomes also promote the formation of DNA double strand breaks that lead to chromosomal rearrangements. Extra centrosomes increase the frequency of chromosomes that lag in the middle of the spindle during anaphase and these chromosomes can be damaged by constriction in the cleavage furrow during cytokinesis179. Moreover, lagging chromosomes are often partitioned into micronuclei [G], which accumulate high levels of DNA damage that promote chromosomal rearrangements180,181. Supernumerary centrosomes can therefore facilitate karyotype evolution by acting as a source of both numerical and structural chromosomal alterations.
While centrosome amplification provides a source of genetic instability, extra centrosomes could also contribute to tumorigenesis through additional mechanisms. Drosophila neural stem cells (neuroblasts) or epithelial cells with extra centrosomes are capable of initiating tumorigenesis when transplanted into host flies153,154. While aneuploidy was observed in transplanted epithelial cells with extra centrosomes, supernumerary centrosomes generated only a modest increase in aneuploidy in neuroblasts, suggesting that genomic instability is unlikely to be the cause of the uncontrolled proliferation of the transplanted brain cells. Instead, neuroblasts with extra centrosomes have spindle alignment defects that result in an increase in symmetric over asymmetric cell divisions (Figure 4B)153. Impaired asymmetric divisions lead to amplification of the neuroblast stem cell pool and subsequent tissue overgrowth182. Examining whether defects in asymmetric cell division contribute to tumorigenesis in vertebrates is an exciting area of future work.
In addition to perturbing cell divisions, numerical and structural centrosome aberrations can also alter the architecture of the interphase microtubule cytoskeleton152,183. Centrosome amplification promotes the formation of invasive protrusions in non-transformed mammary cells grown in a three-dimensional culture system184. Importantly, this invasive behavior was not caused by aneuploidy. Instead, cells with extra centrosomes exhibited increased microtubule nucleation that activated the small GTPase RAC1 (Figure 4C). This provides a possible explanation for the association of centrosome amplification and advanced tumor grade. Further work will be needed to define the impact of centrosome aberrations on cellular invasion and metastasis in vivo.
In addition to their role at the centrosome, centrioles also serve as basal bodies required for primary cilia formation. In cultured human cells, PLK4-induced centriole amplification frequently resulted in the formation of more than one primary cilium185. Surprisingly, cells with additional cilia had reduced levels of ciliary signaling molecules and defective activation of the cilia-regulated Sonic Hedgehog pathway. By contrast, in the mouse epidermis and primary keratinocytes, PLK4 overexpression leads to centriole amplification and the formation of fewer primary cilia158. Centriole amplification can therefore disrupt ciliary signaling, either due to dilution of ciliary signaling components or the loss of cilia themselves (Figure 4D). Since dysregulation of cilia-regulated signaling pathways are known to contribute to tumorigenesis, supernumerary centrioles could impact cell proliferation by perturbing normal ciliary signaling186,187.
Centrosome anomalies in primary microcephaly
Autosomal recessive primary microcephaly (MCPH) is a severe developmental disorder caused by reduced neuronal proliferation during embryonic development and characterized by small brain size and mental retardation. Surprisingly, the major genetic causes of MCPH are mutations in widely expressed genes coding for proteins that function at the centrosome. Currently, mutations in twelve genes encoding centrosome-localized proteins have been shown to cause MCPH and at least eight of these have established roles in centriole duplication (Table 2)188–190. This suggests that defects in centriole biogenesis may be an underlying cause of neurogenesis defects in MCPH191. Consistently, microcephaly causing mutations in PLK4 and CPAP have been shown to impair centriole biogenesis and depletion of proteins required for centriole duplication reduces the brain size of mice32,111–113,192–194. On the other hand, microcephaly mutations in STIL can promote centriole amplification and overexpression of PLK4 in the developing mouse brain resulted in centriole amplification and reduced brain size at birth87,155. Taken together, the evidence supports the idea that either elevated or reduced numbers of centrioles can cause MCPH.
During brain development, neural progenitors undergo symmetric proliferative divisions to self-renew. Since centrosomes play an important role in orienting the mitotic spindle, defects in the centrosome number or structure could impair symmetric divisions and lead to the premature depletion of neural progenitors195. In agreement with this view, spindle orientation defects have been observed in brain organoids [G] and mice with MCPH-causing mutations in CDK5RAP2196,197. While this mechanism is appealing, randomizing spindle orientation in mouse neuroepithelial progenitors does not affect the rate at which neurons are produced198, and defects in mitotic spindle orientation were not observed in the microcephalic brains of some mouse models191.
Cells with abnormal centriole numbers exhibit delayed spindle assembly and an increased duration of mitosis107,134,135,199. Since a mitotic delay is observed in neural progenitors in the brains of some mouse models of microcephaly, it is plausible this delay activates the mitotic surveillance pathway to restrict the proliferation of neural progenitors during embryogenesis192,193,200. In support of this idea, extending mitosis was shown to promote both differentiation and death of neural progenitors in the developing mouse brain200. Moreover, mouse models with reduced levels of centrosomal proteins exhibit microcephaly that is rescued by loss of p53192,193. Importantly, while deletion of p53 rescued brain size, it did not correct defects in tissue architecture caused by abnormal spindle orientation and the incorrect spatial arrangement of neural progenitor cells192. The available data support a new model in which centrosome defects lead to mitotic delays that trigger activation of the mitotic surveillance pathway in the developing brain. Future work should focus on testing whether the mitotic surveillance pathway is activated in neural progenitor cells with centrosome defects and whether deletion of USP28 and 53BP1 can rescue brain size in models of MCPH. Mutations in some non-centrosomal proteins also cause MCPH and it will be interesting to test if these mutations also delay mitosis and activate the mitotic surveillance pathway188–190.
A central unanswered question is why mutations in widely expressed centrosome proteins lead to specific defects in brain development? In fact, mutations in some centrosome proteins cause microcephalic primordial dwarfism, where a reduction in brain size is observed alongside a corresponding reduction in body size (Table 2)188,189. Since MCPH or microcephalic primordial dwarfism can be caused by mutations in the same gene, they may represent a phenotypic spectrum with overlap in the underlying pathological mechanisms. Weak hypomorphic mutations [G] in a gene could result in MCPH, while stronger hypomorphs cause global growth defects leading to microcephalic primordial dwarfism. One explanation for the increased sensitivity of the brain is that cortical development requires extensive proliferation in a brief developmental time window, while other organs might be able to “catch up” if there are minor delays in achieving the required number of cells. An alternative possibility is that neural progenitors have a lower threshold for activation of the mitotic surveillance pathway compared with other cell types.
Perspective
The past decade has witnessed a dramatic increase in our understanding of the molecules and molecular mechanisms that control centriole biogenesis and function. We will continue to benefit from insights provided by structural work on centriole and PCM components and continued research into the role of phosphorylation in controlling centriole assembly. In particular, additional substrates of kinases PLK1, PLK4 and CDK2 are likely to await identification. Moreover, little is presently known about the role of phosphatases in centriole biogenesis and it will be interesting to further explore the role of other posttranslational modifications of centrosome proteins.
An increased understanding of the molecular mechanisms underlying centriole number, structure and function will have important ramifications for the understanding and treatment of diseases linked to centrosome dysfunction and potential therapeutic approaches are now being explored (Box 1). In this regard, the identification of pathways that restrain the cell cycle in response to abnormal centrosome numbers is particularly exciting. However, we lack a comprehensive understanding of how these pathways are triggered and how they function in the context of an organism. In future, animal models that faithfully mimic the phenotypes produced by centrosome dysfunction will play a critical role to elucidate the mechanisms by which centrosome defects contribute to human disease. At present, studies that have examined the effect of centrosome amplification in mammals interfere with PLK4 expression. However, PLK4 also plays a critical role in spindle assembly in the absence of centrioles in the early mouse embyro132, and recent work also suggested PLK4 can control cancer cell migration and invasion through regulation of the actin cytoskeleton201. It will be important, therefore, to further explore these non-canonical functions of PLK4 and extend previous studies by employing alternative means to modify centriole numbers.
Box 1. Centrosomes as therapeutic targets.
PLK4 has emerged as a therapeutic target based on its key role in controlling centrosome duplication and recent evidence that it functions to promote cancer cell migration and invasion101,102,201. CFI-400945 was the first described inhibitor of PLK4 and potently suppresses the growth of human Xenograft tumors in mice202. However, CFI-400945 also inhibits the activity of other kinases including Aurora B, making it unclear whether PLK4 is the only relevant therapeutic target of CFI-400945. The recent development of the highly specific PLK4 inhibitor centrinone provides a precise means to study the effect of inhibiting centrosome biogenesis on tumor growth. Work in cultured cells showed centrinone prevents the proliferation of non-transformed cells, but allows continued proliferation of most transformed cell lines134. This suggests that inhibiting centrosome duplication alone may not be an efficacious anti-cancer strategy. Nevertheless, it may be possible to identify genetic alterations that are synthetically lethal with centrosome loss and PLK4 inhibitors could offer therapeutic value in suppressing functions of PLK4 that promote invasion and metastasis201.
An alternative therapeutic strategy is to exacerbate the challenge of dividing with abnormal centrosome numbers. Since centrosome clustering is not required in cells with normal centrosome numbers, but is required to ensure bipolar spindle assembly in cells with supernumerary centrosomes, one idea is to suppress centrosome clustering and force cancer cells with extra centrosomes into lethal multipolar divisions170,171,174. However, the fact that most cancer cell lines can proliferate in vitro without centrosomes suggests that they do not require supernumerary centrosomes for their survival134. Inhibiting centrosome clustering may therefore reduce the initial tumor burden, but eventually allow the outgrowth of resistant cell populations. An alternative to targeting the centrosome directly is to manipulate proteins that control the response to errors in centrosome duplication. USP28 is an enzymatic component of the mitotic surveillance pathway and in principle can be inhibited. Since USP28 knockout mice lack any clear phenotypes144,145, USP28 inhibition could be used therapeutically in conditions such as microcephaly, where the mitotic surveillance pathway may be pathologically activated.
Key points.
Centrosome duplication is tightly regulated to ensure that centriole duplication occurs only once per cell cycle and that only one new centriole is produced per pre-existing centriole.
PLK1 plays a key role in cell cycle control of centriole duplication, while PLK4 takes center stage in controlling centriole copy number.
Recent work has uncovered the existence of distinct signaling pathways that limit proliferation of cells with an increase or decrease in centrosome number.
Overcoming the inhibitory effect of extra centrosomes on cell proliferation is necessary to allow cells with extra centrosomes to acquire the necessary oncogenic mutations required for tumor development.
Primary microcephaly may be caused by deregulation of centriole numbers and, potentially, by pathological activation of the mitotic surveillance pathway in the developing brain.
Acknowledgments
We thank our lab members for helpful discussions and apologize to colleagues whose work could not be cited due to space limitations. Work in the author’s laboratory was supported by grants from the Swiss National Science Foundation (310030B-149641) to EAN and a R01 research grant from the National Institutes of Health (GM 114119), an American Cancer Society Scholar Grant (129742-RSG-16-156-01-CCG) and a March of Dimes Research Grant (1-FY17-698) to AJH.
Glossary
- Procentriole
A newly constructed centriole that is unable to duplicate
- Molecular rulers
Molecules of defined size that can be used to set distances between other structures.
- Axoneme
The nine-fold symmetrical microtubule based structure at the center of cilia and flagella.
- Planarians
A flatworm used as a model system to study regeneration.
- Hippo pathway
A signaling pathway that controls organ size in animals by restraining cell proliferation and promoting apoptosis.
- PIDDosome
A protein complex comprised of RAIDD and PIDD that is implicated in the activation of Caspase 2.
- Merotelic
A type of attachment where one kinetochore binds microtubules emanating from two centrosomes located on opposite sides of the mitotic spindle.
- Aneuploidy
The presence of an abnormal chromosome number that is not a multiple of the haploid chromosome complement.
- Micronuclei
A small nucleus separate from the daughter nucleus that contains one or a few chromosomes or chromosome fragments.
- Organoids
An in vitro culture system that mimics the micro-anatomy of an organ.
- Hypomorphic mutations
A mutation that causes a partial loss of gene function.
Biographies
Erich Nigg –Erich is Full Professor for Cell Biology and director of the Biozentrum at the University of Basel, Switzerland. His major research interests focus on the regulation of mitotic chromosome segregation and the centrosome duplication cycle.
Andrew Holland – Andrew is an Assistant Professor at Johns Hopkins University School of Medicine. His major research interests are in understanding the molecular mechanisms that control accurate chromosome distribution and the role that mitotic errors play in human health and disease.
References
- 1.Nigg EA, Raff JW. Centrioles, centrosomes, and cilia in health and disease. Cell. 2009;139:663–678. doi: 10.1016/j.cell.2009.10.036. [DOI] [PubMed] [Google Scholar]
- 2.Bornens M. The centrosome in cells and organisms. Science. 2012;335:422–426. doi: 10.1126/science.1209037. [DOI] [PubMed] [Google Scholar]
- 3.Conduit PT, Wainman A, Raff JW. Centrosome function and assembly in animal cells. Nat Rev Mol Cell Biol. 2015;16:611–624. doi: 10.1038/nrm4062. [DOI] [PubMed] [Google Scholar]
- 4.Fu J, Hagan IM, Glover DM. The centrosome and its duplication cycle. Cold Spring Harb Perspect Biol. 2015;7:a015800. doi: 10.1101/cshperspect.a015800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woodruff JB, Wueseke O, Hyman AA. Pericentriolar material structure and dynamics. Philos Trans R Soc Lond B Biol Sci. 2014;369 doi: 10.1098/rstb.2013.0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arquint C, Gabryjonczyk AM, Nigg EA. Centrosomes as signalling centres. Philos Trans R Soc Lond B Biol Sci. 2014;369 doi: 10.1098/rstb.2013.0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanchez I, Dynlacht BD. Cilium assembly and disassembly. Nat Cell Biol. 2016;18:711–717. doi: 10.1038/ncb3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braun DA, Hildebrandt F. Ciliopathies. Cold Spring Harb Perspect Biol. 2017;9 doi: 10.1101/cshperspect.a028191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bettencourt-Dias M, Hildebrandt F, Pellman D, Woods G, Godinho SA. Centrosomes and cilia in human disease. Trends Genet. 2011;27:307–315. doi: 10.1016/j.tig.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonczy P. Centrosomes and cancer: revisiting a long-standing relationship. Nat Rev Cancer. 2015;15:639–652. doi: 10.1038/nrc3995. [DOI] [PubMed] [Google Scholar]
- 11.Azimzadeh J, Marshall WF. Building the centriole. Curr Biol. 2010;20:R816–825. doi: 10.1016/j.cub.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonczy P. Towards a molecular architecture of centriole assembly. Nat Rev Mol Cell Biol. 2012;13:425–435. doi: 10.1038/nrm3373. [DOI] [PubMed] [Google Scholar]
- 13.Gupta GD, et al. A Dynamic Protein Interaction Landscape of the Human Centrosome-Cilium Interface. Cell. 2015;163:1484–1499. doi: 10.1016/j.cell.2015.10.065. A massive proteomics effort to probe protein interactions at the centrosome-cilium interface. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia G, 3rd, Reiter JF. A primer on the mouse basal body. Cilia. 2016;5:17. doi: 10.1186/s13630-016-0038-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirono M. Cartwheel assembly. Philos Trans R Soc Lond B Biol Sci. 2014;369 doi: 10.1098/rstb.2013.0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keller D, et al. Mechanisms of HsSAS-6 assembly promoting centriole formation in human cells. J Cell Biol. 2014;204:697–712. doi: 10.1083/jcb.201307049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guichard P, Chretien D, Marco S, Tassin AM. Procentriole assembly revealed by cryo-electron tomography. Embo J. 2010;29:1565–1572. doi: 10.1038/emboj.2010.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guichard P, et al. Cartwheel architecture of Trichonympha basal body. Science. 2012;337:553. doi: 10.1126/science.1222789. [DOI] [PubMed] [Google Scholar]
- 19.Guichard P, et al. Native architecture of the centriole proximal region reveals features underlying its 9-fold radial symmetry. Curr Biol. 2013;23:1620–1628. doi: 10.1016/j.cub.2013.06.061. [DOI] [PubMed] [Google Scholar]
- 20.Bauer M, Cubizolles F, Schmidt A, Nigg EA. Quantitative analysis of human centrosome architecture by targeted proteomics and fluorescence imaging. Embo J. 2016;35:2152–2166. doi: 10.15252/embj.201694462. An initial effort at obtaining quantitative information about the abundance of centrosomal proteins within cells and isolated organelles. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitagawa D, et al. Structural basis of the 9-fold symmetry of centrioles. Cell. 2011;144:364–375. doi: 10.1016/j.cell.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Breugel M, et al. Structures of SAS-6 suggest its organization in centrioles. Science. 2011;331:1196–1199. doi: 10.1126/science.1199325. [DOI] [PubMed] [Google Scholar]
- 23.Guichard P, et al. Cell-free reconstitution reveals centriole cartwheel assembly mechanisms. Nat Commun. 2017;8:14813. doi: 10.1038/ncomms14813. A pioneering study demonstrating successful in vitro reconstitution of early steps of centriole assembly. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang WJ, et al. De novo centriole formation in human cells is error-prone and does not require SAS-6 self-assembly. Elife. 2015;4 doi: 10.7554/eLife.10586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hilbert M, et al. SAS-6 engineering reveals interdependence between cartwheel and microtubules in determining centriole architecture. Nat Cell Biol. 2016;18:393–403. doi: 10.1038/ncb3329. [DOI] [PubMed] [Google Scholar]
- 26.Ohta M, et al. Direct interaction of Plk4 with STIL ensures formation of a single procentriole per parental centriole. Nat Commun. 2014;5:5267. doi: 10.1038/ncomms6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dzhindzhev NS, et al. Plk4 phosphorylates Ana2 to trigger Sas6 recruitment and procentriole formation. Curr Biol. 2014;24:2526–2532. doi: 10.1016/j.cub.2014.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moyer TC, Clutario KM, Lambrus BG, Daggubati V, Holland AJ. Binding of STIL to Plk4 activates kinase activity to promote centriole assembly. J Cell Biol. 2015;209:863–878. doi: 10.1083/jcb.201502088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kratz AS, Barenz F, Richter KT, Hoffmann I. Plk4-dependent phosphorylation of STIL is required for centriole duplication. Biol Open. 2015;4:370–377. doi: 10.1242/bio.201411023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stevens NR, Dobbelaere J, Brunk K, Franz A, Raff JW. Drosophila Ana2 is a conserved centriole duplication factor. J Cell Biol. 2010;188:313–323. doi: 10.1083/jcb.200910016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arquint C, Sonnen KF, Stierhof YD, Nigg EA. Cell-cycle-regulated expression of STIL controls centriole number in human cells. J Cell Sci. 2012;125:1342–1352. doi: 10.1242/jcs.099887. [DOI] [PubMed] [Google Scholar]
- 32.Tang CJ, et al. The human microcephaly protein STIL interacts with CPAP and is required for procentriole formation. Embo J. 2011;30:4790–4804. doi: 10.1038/emboj.2011.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hiraki M, Nakazawa Y, Kamiya R, Hirono M. Bld10p constitutes the cartwheel-spoke tip and stabilizes the 9-fold symmetry of the centriole. Curr Biol. 2007;17:1778–1783. doi: 10.1016/j.cub.2007.09.021. [DOI] [PubMed] [Google Scholar]
- 34.Lin YC, et al. Human microcephaly protein CEP135 binds to hSAS-6 and CPAP, and is required for centriole assembly. Embo J. 2013;32:1141–1154. doi: 10.1038/emboj.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sonnen KF, Schermelleh L, Leonhardt H, Nigg EA. 3D-structured illumination microscopy provides novel insight into architecture of human centrosomes. Biol Open. 2012;1:965–976. doi: 10.1242/bio.20122337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kleylein-Sohn J, et al. Plk4-induced centriole biogenesis in human cells. Dev Cell. 2007;13:190–202. doi: 10.1016/j.devcel.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 37.Pelletier L, O’Toole E, Schwager A, Hyman AA, Muller-Reichert T. Centriole assembly in Caenorhabditis elegans. Nature. 2006;444:619–623. doi: 10.1038/nature05318. [DOI] [PubMed] [Google Scholar]
- 38.Sharma A, et al. Centriolar CPAP/SAS-4 Imparts Slow Processive Microtubule Growth. Dev Cell. 2016;37:362–376. doi: 10.1016/j.devcel.2016.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng X, et al. Molecular basis for CPAP-tubulin interaction in controlling centriolar and ciliary length. Nat Commun. 2016;7:11874. doi: 10.1038/ncomms11874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galletta BJ, Jacobs KC, Fagerstrom CJ, Rusan NM. Asterless is required for centriole length control and sperm development. J Cell Biol. 2016;213:435–450. doi: 10.1083/jcb.201501120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marshall WF. Cell Geometry: How Cells Count and Measure Size. Annual review of biophysics. 2016;45:49–64. doi: 10.1146/annurev-biophys-062215-010905. [DOI] [PubMed] [Google Scholar]
- 42.Delgehyr N, et al. Klp10A, a microtubule-depolymerizing kinesin-13, cooperates with CP110 to control Drosophila centriole length. Curr Biol. 2012;22:502–509. doi: 10.1016/j.cub.2012.01.046. [DOI] [PubMed] [Google Scholar]
- 43.Kobayashi T, Tsang WY, Li J, Lane W, Dynlacht BD. Centriolar kinesin Kif24 interacts with CP110 to remodel microtubules and regulate ciliogenesis. Cell. 2011;145:914–925. doi: 10.1016/j.cell.2011.04.028. [DOI] [PubMed] [Google Scholar]
- 44.Franz A, Roque H, Saurya S, Dobbelaere J, Raff JW. CP110 exhibits novel regulatory activities during centriole assembly in Drosophila. J Cell Biol. 2013;203:785–799. doi: 10.1083/jcb.201305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmidt TI, et al. Control of centriole length by CPAP and CP110. Curr Biol. 2009;19:1005–1011. doi: 10.1016/j.cub.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 46.Tsang WY, et al. CP110 suppresses primary cilia formation through its interaction with CEP290, a protein deficient in human ciliary disease. Dev Cell. 2008;15:187–197. doi: 10.1016/j.devcel.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Al-Hakim AK, Bashkurov M, Gingras AC, Durocher D, Pelletier L. Interaction proteomics identify NEURL4 and the HECT E3 ligase HERC2 as novel modulators of centrosome architecture. Mol Cell Proteomics. 2012;11:M111.014233. doi: 10.1074/mcp.M111.014233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li J, et al. USP33 regulates centrosome biogenesis via deubiquitination of the centriolar protein CP110. Nature. 2013;495:255–259. doi: 10.1038/nature11941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li J, et al. Neurl4, a novel daughter centriole protein, prevents formation of ectopic microtubule organizing centres. EMBO Rep. 2012;13:547–553. doi: 10.1038/embor.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cao J, et al. miR-129-3p controls cilia assembly by regulating CP110 and actin dynamics. Nat Cell Biol. 2012;14:697–706. doi: 10.1038/ncb2512. [DOI] [PubMed] [Google Scholar]
- 51.Kohlmaier G, et al. Overly long centrioles and defective cell division upon excess of the SAS-4-related protein CPAP. Curr Biol. 2009;19:1012–1018. doi: 10.1016/j.cub.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tang CJ, Fu RH, Wu KS, Hsu WB, Tang TK. CPAP is a cell-cycle regulated protein that controls centriole length. Nat Cell Biol. 2009;11:825–831. doi: 10.1038/ncb1889. [DOI] [PubMed] [Google Scholar]
- 53.Comartin D, et al. CEP120 and SPICE1 cooperate with CPAP in centriole elongation. Curr Biol. 2013;23:1360–1366. doi: 10.1016/j.cub.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 54.Lin YN, et al. CEP120 interacts with CPAP and positively regulates centriole elongation. J Cell Biol. 2013;202:211–219. doi: 10.1083/jcb.201212060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Keller LC, et al. Molecular architecture of the centriole proteome: the conserved WD40 domain protein POC1 is required for centriole duplication and length control. Mol Biol Cell. 2009;20:1150–1166. doi: 10.1091/mbc.E08-06-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Azimzadeh J, et al. hPOC5 is a centrin-binding protein required for assembly of full-length centrioles. J Cell Biol. 2009;185:101–114. doi: 10.1083/jcb.200808082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang CW, Hsu WB, Tsai JJ, Tang CJ, Tang TK. CEP295 interacts with microtubules and is required for centriole elongation. J Cell Sci. 2016;129:2501–2513. doi: 10.1242/jcs.186338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saurya S, et al. Drosophila Ana1 is required for centrosome assembly and centriole elongation. J Cell Sci. 2016;129:2514–2525. doi: 10.1242/jcs.186460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bobinnec Y, et al. Centriole disassembly in vivo and its effect on centrosome structure and function in vertebrate cells. J Cell Biol. 1998;143:1575–1589. doi: 10.1083/jcb.143.6.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Janke C, Bulinski JC. Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol. 2011;12:773–786. doi: 10.1038/nrm3227. [DOI] [PubMed] [Google Scholar]
- 61.Andersen JS, et al. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 2003;426:570–574. doi: 10.1038/nature02166. [DOI] [PubMed] [Google Scholar]
- 62.Jakobsen L, et al. Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. Embo J. 2011;30:1520–1535. doi: 10.1038/emboj.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Prosser SL, Pelletier L. Mitotic spindle assembly in animal cells: a fine balancing act. Nat Rev Mol Cell Biol. 2017;18:187–201. doi: 10.1038/nrm.2016.162. [DOI] [PubMed] [Google Scholar]
- 64.Hori A, Toda T. Regulation of centriolar satellite integrity and its physiology. Cell Mol Life Sci. 2017;74:213–229. doi: 10.1007/s00018-016-2315-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fu J, Glover DM. Structured illumination of the interface between centriole and peri-centriolar material. Open Biol. 2012;2:120104. doi: 10.1098/rsob.120104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lawo S, Hasegan M, Gupta GD, Pelletier L. Subdiffraction imaging of centrosomes reveals higher-order organizational features of pericentriolar material. Nat Cell Biol. 2012;14:1148–1158. doi: 10.1038/ncb2591. [DOI] [PubMed] [Google Scholar]
- 67.Mennella V, et al. Subdiffraction-resolution fluorescence microscopy reveals a domain of the centrosome critical for pericentriolar material organization. Nat Cell Biol. 2012;14:1159–1168. doi: 10.1038/ncb2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sonnen KF, Schermelleh L, Leonhardt H, Nigg EA. 3D-structured illumination microscopy provides novel insight into architecture of human centrosomes. Biol Open. 2012;1:965–976. doi: 10.1242/bio.20122337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Conduit PT, et al. The centrosome-specific phosphorylation of Cnn by Polo/Plk1 drives Cnn scaffold assembly and centrosome maturation. Dev Cell. 2014;28:659–669. doi: 10.1016/j.devcel.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Conduit PT, et al. A molecular mechanism of mitotic centrosome assembly in Drosophila. Elife. 2014;3:e03399. doi: 10.7554/eLife.03399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feng Z, et al. Structural Basis for Mitotic Centrosome Assembly in Flies. Cell. 2017;169:1078–1089.e1013. doi: 10.1016/j.cell.2017.05.030. This structural study, focused on the Drosophila PCM scaffolding protein Cnn, provides insight into molecular interactions required for mitotic centrosome assembly (compare with Woodruff, et al. 2017, ref. 68) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017;18:285–298. doi: 10.1038/nrm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zwicker D, Decker M, Jaensch S, Hyman AA, Julicher F. Centrosomes are autocatalytic droplets of pericentriolar material organized by centrioles. Proc Natl Acad Sci U S A. 2014;111:E2636–2645. doi: 10.1073/pnas.1404855111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Woodruff JB, et al. Centrosomes. Regulated assembly of a supramolecular centrosome scaffold in vitro. Science. 2015;348:808–812. doi: 10.1126/science.aaa3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Woodruff JB, et al. The Centrosome Is a Selective Condensate that Nucleates Microtubules by Concentrating Tubulin. Cell. 2017;169:1066–1077.e1010. doi: 10.1016/j.cell.2017.05.028. This study shows that the C. elegans PCM protein SPD-5 forms a selective condensate able to nucleate microtubules, suggesting that PCM formation in vivo involves a phase separation process (compare with Feng, et al. 2017, ref. 64) [DOI] [PubMed] [Google Scholar]
- 76.Nigg EA. Centrosome duplication: of rules and licenses. Trends Cell Biol. 2007;17:215–221. doi: 10.1016/j.tcb.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 77.Tsou MF, Stearns T. Mechanism limiting centrosome duplication to once per cell cycle. Nature. 2006;442:947–951. doi: 10.1038/nature04985. [DOI] [PubMed] [Google Scholar]
- 78.Tsou MF, et al. Polo kinase and separase regulate the mitotic licensing of centriole duplication in human cells. Dev Cell. 2009;17:344–354. doi: 10.1016/j.devcel.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Loncarek J, Hergert P, Magidson V, Khodjakov A. Control of daughter centriole formation by the pericentriolar material. Nat Cell Biol. 2008;10:322–328. doi: 10.1038/ncb1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Matsuo K, et al. Kendrin is a novel substrate for separase involved in the licensing of centriole duplication. Curr Biol. 2012;22:915–921. doi: 10.1016/j.cub.2012.03.048. This study (along with Kim, et al. 2015, ref. 75) identifies kendrin/pericentrin as a key substrate of separase, important for licensing of centriole duplication. [DOI] [PubMed] [Google Scholar]
- 81.Lee K, Rhee K. Separase-dependent cleavage of pericentrin B is necessary and sufficient for centriole disengagement during mitosis. Cell Cycle. 2012;11:2476–2485. doi: 10.4161/cc.20878. [DOI] [PubMed] [Google Scholar]
- 82.Kim J, Lee K, Rhee K. PLK1 regulation of PCNT cleavage ensures fidelity of centriole separation during mitotic exit. Nat Commun. 2015;6:10076. doi: 10.1038/ncomms10076. This study (along with Matuso, et al. 2012, ref. 73) identifies Pericentrin/Kendrin as a key substrate of separase, important for licensing of centriole duplication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schockel L, Mockel M, Mayer B, Boos D, Stemmann O. Cleavage of cohesin rings coordinates the separation of centrioles and chromatids. Nat Cell Biol. 2011;13:966–972. doi: 10.1038/ncb2280. [DOI] [PubMed] [Google Scholar]
- 84.Oliveira RA, Nasmyth K. Cohesin cleavage is insufficient for centriole disengagement in Drosophila. Curr Biol. 2013;23:R601–603. doi: 10.1016/j.cub.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 85.Kuriyama R, Borisy GG. Centriole cycle in Chinese hamster ovary cells as determined by whole-mount electron microscopy. J Cell Biol. 1981;91:814–821. doi: 10.1083/jcb.91.3.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shukla A, Kong D, Sharma M, Magidson V, Loncarek J. Plk1 relieves centriole block to reduplication by promoting daughter centriole maturation. Nat Commun. 2015;6:8077. doi: 10.1038/ncomms9077. A correlative live/electron microscopy study emphasizing the role of Plk1 in triggering early licensing events important for a new round of centriole duplication (see also Wang, et al. 2011, ref. 83; and Novak, et al. ref. 84) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Arquint C, Nigg EA. STIL microcephaly mutations interfere with APC/C-mediated degradation and cause centriole amplification. Curr Biol. 2014;24:351–360. doi: 10.1016/j.cub.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 88.Kim M, et al. Promotion and Suppression of Centriole Duplication Are Catalytically Coupled through PLK4 to Ensure Centriole Homeostasis. Cell Rep. 2016;16:1195–1203. doi: 10.1016/j.celrep.2016.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Izquierdo D, Wang WJ, Uryu K, Tsou MF. Stabilization of cartwheel-less centrioles for duplication requires CEP295-mediated centriole-to-centrosome conversion. Cell Rep. 2014;8:957–965. doi: 10.1016/j.celrep.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fu J, et al. Conserved molecular interactions in centriole-to-centrosome conversion. Nat Cell Biol. 2016;18:87–99. doi: 10.1038/ncb3274. This study illuminates an evolutionarily conserved mechanism underlying centriole-to-centrosome conversion (see also Novak, et al. ref. 84) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang WJ, Soni RK, Uryu K, Tsou MF. The conversion of centrioles to centrosomes: essential coupling of duplication with segregation. J Cell Biol. 2011;193:727–739. doi: 10.1083/jcb.201101109. This study emphasizes a key role for Plk1 in both centriole disengagement and PCM assembly on procentrioles, two mitotic events critical for enabling centrioles for a next round of duplication (see also Shulka, et al. 2016, Ref. 78; Novak, et al. ref. 84) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Novak ZA, Wainman A, Gartenmann L, Raff JW. Cdk1 Phosphorylates Drosophila Sas-4 to Recruit Polo to Daughter Centrioles and Convert Them to Centrosomes. Dev Cell. 2016;37:545–557. doi: 10.1016/j.devcel.2016.05.022. This study describes an early key step leading to centrosomal recruitment of Plk1, important for centriole-to-centrosome conversion (see also Shulka, et al. 2016, Ref. 78; Fu, et al. 2016, Ref. 82; and Wang, et al. 2011, ref. 83) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Novak ZA, Conduit PT, Wainman A, Raff JW. Asterless licenses daughter centrioles to duplicate for the first time in Drosophila embryos. Curr Biol. 2014;24:1276–1282. doi: 10.1016/j.cub.2014.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tsuchiya Y, Yoshiba S, Gupta A, Watanabe K, Kitagawa D. Cep295 is a conserved scaffold protein required for generation of a bona fide mother centriole. Nat Commun. 2016;7:12567. doi: 10.1038/ncomms12567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hatch EM, Kulukian A, Holland AJ, Cleveland DW, Stearns T. Cep152 interacts with Plk4 and is required for centriole duplication. J Cell Biol. 2010;191:721–729. doi: 10.1083/jcb.201006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cizmecioglu O, et al. Cep152 acts as a scaffold for recruitment of Plk4 and CPAP to the centrosome. J Cell Biol. 2010;191:731–739. doi: 10.1083/jcb.201007107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kim TS, et al. Hierarchical recruitment of Plk4 and regulation of centriole biogenesis by two centrosomal scaffolds, Cep192 and Cep152. Proc Natl Acad Sci U S A. 2013;110:E4849–4857. doi: 10.1073/pnas.1319656110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sonnen KF, Gabryjonczyk AM, Anselm E, Stierhof YD, Nigg EA. Human Cep192 and Cep152 cooperate in Plk4 recruitment and centriole duplication. J Cell Sci. 2013;126:3223–3233. doi: 10.1242/jcs.129502. [DOI] [PubMed] [Google Scholar]
- 99.Park SY, et al. Molecular basis for unidirectional scaffold switching of human Plk4 in centriole biogenesis. Nat Struct Mol Biol. 2014;21:696–703. doi: 10.1038/nsmb.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sugioka K, et al. Centriolar SAS-7 acts upstream of SPD-2 to regulate centriole assembly and pericentriolar material formation. Elife. 2017;6 doi: 10.7554/eLife.20353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA. The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol. 2005;7:1140–1146. doi: 10.1038/ncb1320. [DOI] [PubMed] [Google Scholar]
- 102.Bettencourt-Dias M, et al. SAK/PLK4 is required for centriole duplication and flagella development. Curr Biol. 2005;15:2199–2207. doi: 10.1016/j.cub.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 103.Cunha-Ferreira I, et al. Regulation of autophosphorylation controls PLK4 self-destruction and centriole number. Curr Biol. 2013;23:2245–2254. doi: 10.1016/j.cub.2013.09.037. [DOI] [PubMed] [Google Scholar]
- 104.Holland AJ, Lan W, Niessen S, Hoover H, Cleveland DW. Polo-like kinase 4 kinase activity limits centrosome overduplication by autoregulating its own stability. J Cell Biol. 2010;188:191–198. doi: 10.1083/jcb.200911102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Klebba JE, et al. Polo-like kinase 4 autodestructs by generating its Slimb-binding phosphodegron. Curr Biol. 2013;23:2255–2261. doi: 10.1016/j.cub.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Guderian G, Westendorf J, Uldschmid A, Nigg EA. Plk4 trans-autophosphorylation regulates centriole number by controlling betaTrCP-mediated degradation. J Cell Sci. 2010;123:2163–2169. doi: 10.1242/jcs.068502. [DOI] [PubMed] [Google Scholar]
- 107.Holland AJ, et al. The autoregulated instability of Polo-like kinase 4 limits centrosome duplication to once per cell cycle. Genes Dev. 2012;26:2684–2689. doi: 10.1101/gad.207027.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lopes CA, et al. PLK4 trans-Autoactivation Controls Centriole Biogenesis in Space. Dev Cell. 2015;35:222–235. doi: 10.1016/j.devcel.2015.09.020. [DOI] [PubMed] [Google Scholar]
- 109.Arquint C, et al. STIL binding to Polo-box 3 of PLK4 regulates centriole duplication. Elife. 2015;4 doi: 10.7554/eLife.07888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lettman MM, et al. Direct binding of SAS-6 to ZYG-1 recruits SAS-6 to the mother centriole for cartwheel assembly. Dev Cell. 2013;25:284–298. doi: 10.1016/j.devcel.2013.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cottee MA, et al. Crystal structures of the CPAP/STIL complex reveal its role in centriole assembly and human microcephaly. Elife. 2013;2:e01071. doi: 10.7554/eLife.01071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hatzopoulos GN, et al. Structural analysis of the G-box domain of the microcephaly protein CPAP suggests a role in centriole architecture. Structure. 2013;21:2069–2077. doi: 10.1016/j.str.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zheng X, et al. Conserved TCP domain of Sas-4/CPAP is essential for pericentriolar material tethering during centrosome biogenesis. Proc Natl Acad Sci U S A. 2014;111:E354–363. doi: 10.1073/pnas.1317535111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Goryachev AB, Leda M. Many roads to symmetry breaking: molecular mechanisms and theoretical models of yeast cell polarity. Mol Biol Cell. 2017;28:370–380. doi: 10.1091/mbc.E16-10-0739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fong CS, Kim M, Yang TT, Liao JC, Tsou MF. SAS-6 assembly templated by the lumen of cartwheel-less centrioles precedes centriole duplication. Dev Cell. 2014;30:238–245. doi: 10.1016/j.devcel.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zitouni S, et al. CDK1 Prevents Unscheduled PLK4-STIL Complex Assembly in Centriole Biogenesis. Curr Biol. 2016;26:1127–1137. doi: 10.1016/j.cub.2016.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Peel N, et al. Protein Phosphatase 1 Down Regulates ZYG-1 Levels to Limit Centriole Duplication. PLoS Genet. 2017;13:e1006543. doi: 10.1371/journal.pgen.1006543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Agircan FG, Schiebel E, Mardin BR. Separate to operate: control of centrosome positioning and separation. Philos Trans R Soc Lond B Biol Sci. 2014;369 doi: 10.1098/rstb.2013.0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mayor T, Stierhof YD, Tanaka K, Fry AM, Nigg EA. The centrosomal protein C-Nap1 is required for cell cycle-regulated centrosome cohesion. J Cell Biol. 2000;151:837–846. doi: 10.1083/jcb.151.4.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nigg EA, Stearns T. The centrosome cycle: Centriole biogenesis, duplication and inherent asymmetries. Nat Cell Biol. 2011;13:1154–1160. doi: 10.1038/ncb2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kong D, et al. Centriole maturation requires regulated Plk1 activity during two consecutive cell cycles. J Cell Biol. 2014;206:855–865. doi: 10.1083/jcb.201407087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lane HA, Nigg EA. Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J Cell Biol. 1996;135:1701–1713. doi: 10.1083/jcb.135.6.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lee K, Rhee K. PLK1 phosphorylation of pericentrin initiates centrosome maturation at the onset of mitosis. J Cell Biol. 2011;195:1093–1101. doi: 10.1083/jcb.201106093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Joukov V, Walter JC, De Nicolo A. The Cep192-organized aurora A-Plk1 cascade is essential for centrosome cycle and bipolar spindle assembly. Mol Cell. 2014;55:578–591. doi: 10.1016/j.molcel.2014.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Laos T, Cabral G, Dammermann A. Isotropic incorporation of SPD-5 underlies centrosome assembly in C. elegans. Curr Biol. 2015;25:R648–649. doi: 10.1016/j.cub.2015.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Spassky N, Meunier A. The development and functions of multiciliated epithelia. Nat Rev Mol Cell Biol. 2017;18:423–436. doi: 10.1038/nrm.2017.21. [DOI] [PubMed] [Google Scholar]
- 127.Azimzadeh J, Wong ML, Downhour DM, Sanchez Alvarado A, Marshall WF. Centrosome loss in the evolution of planarians. Science. 2012;335:461–463. doi: 10.1126/science.1214457. By showing that, in planarians, centrioles assemble only in terminally differentiating ciliated cells, but are not required for mitotic cell divisions, this study has important implications for the evolution of the animal centrosome and its role in development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Basto R, et al. Flies without centrioles. Cell. 2006;125:1375–1386. doi: 10.1016/j.cell.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 129.Debec A, Sullivan W, Bettencourt-Dias M. Centrioles: active players or passengers during mitosis? Cell Mol Life Sci. 2010;67:2173–2194. doi: 10.1007/s00018-010-0323-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Szollosi D, Calarco P, Donahue RP. Absence of centrioles in the first and second meiotic spindles of mouse oocytes. J Cell Sci. 1972;11:521–541. doi: 10.1242/jcs.11.2.521. [DOI] [PubMed] [Google Scholar]
- 131.Howe K, FitzHarris G. A non-canonical mode of microtubule organization operates throughout pre-implantation development in mouse. Cell Cycle. 2013;12:1616–1624. doi: 10.4161/cc.24755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Coelho PA, et al. Spindle formation in the mouse embryo requires Plk4 in the absence of centrioles. Dev Cell. 2013;27:586–597. doi: 10.1016/j.devcel.2013.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bazzi H, Anderson KV. Acentriolar mitosis activates a p53-dependent apoptosis pathway in the mouse embryo. Proc Natl Acad Sci U S A. 2014;111:E1491–1500. doi: 10.1073/pnas.1400568111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wong YL, et al. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science. 2015;348:1155–1160. doi: 10.1126/science.aaa5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lambrus BG, et al. p53 protects against genome instability following centriole duplication failure. J Cell Biol. 2015;210:63–77. doi: 10.1083/jcb.201502089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lambrus BG, et al. A USP28-53BP1-p53-p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J Cell Biol. 2016;214:143–153. doi: 10.1083/jcb.201604054. Together with Fong et al. 2016 (Ref. 135) and Meitinger et al. 2016 (Ref. 136), this study uses genome-wide screening to uncover a surveillance pathway, involving 53BP1 and USP28, that acts to activate p53 in response to loss of centrosomes or extended duration of mitosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fong CS, et al. 53BP1 and USP28 mediate p53-dependent cell cycle arrest in response to centrosome loss and prolonged mitosis. Elife. 2016;5 doi: 10.7554/eLife.16270. Together with Lambrus et al. 2016 (Ref. 134) and Meitinger et al. 2016 (Ref. 136), this study uses genome-wide screening to uncover a surveillance pathway, involving 53BP1 and USP28, that acts to activate p53 in response to loss of centrosomes or extended duration of mitosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Meitinger F, et al. 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J Cell Biol. 2016;214:155–166. doi: 10.1083/jcb.201604081. This study presents results in line with Lambrus et al. 2016 (Ref. 134) and Fong et al. 2016 (Ref. 136) and, additionally, shows that loss of the ubiquitin ligase TRIM37 enables the formation of extra-centrosomal microtubule organizing centers, thereby allowing bypass of the arrest caused by centrosome loss. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Knobel PA, et al. USP28 is recruited to sites of DNA damage by the tandem BRCT domains of 53BP1 but plays a minor role in double-strand break metabolism. Mol Cell Biol. 2014;34:2062–2074. doi: 10.1128/MCB.00197-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhang D, Zaugg K, Mak TW, Elledge SJ. A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell. 2006;126:529–542. doi: 10.1016/j.cell.2006.06.039. [DOI] [PubMed] [Google Scholar]
- 141.Zimmermann M, de Lange T. 53BP1: pro choice in DNA repair. Trends Cell Biol. 2014;24:108–117. doi: 10.1016/j.tcb.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cuella-Martin R, et al. 53BP1 Integrates DNA Repair and p53-Dependent Cell Fate Decisions via Distinct Mechanisms. Mol Cell. 2016 doi: 10.1016/j.molcel.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Uetake Y, Sluder G. Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr Biol. 2010;20:1666–1671. doi: 10.1016/j.cub.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Schulein-Volk C, et al. Dual regulation of Fbw7 function and oncogenic transformation by Usp28. Cell Rep. 2014;9:1099–1109. doi: 10.1016/j.celrep.2014.09.057. [DOI] [PubMed] [Google Scholar]
- 145.Diefenbacher ME, et al. The deubiquitinase USP28 controls intestinal homeostasis and promotes colorectal cancer. J Clin Invest. 2014;124:3407–3418. doi: 10.1172/JCI73733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lambrus BG, Holland AJ. A New Mode of Mitotic Surveillance. Trends Cell Biol. 2017 doi: 10.1016/j.tcb.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Levine MS, et al. Centrosome Amplification Is Sufficient to Promote Spontaneous Tumorigenesis in Mammals. Dev Cell. 2017;40:313–322 e315. doi: 10.1016/j.devcel.2016.12.022. Using a mouse model allowing moderately increased expression of PLK4, this study provides long-awaited evidence for the hypothesis that centrosome amplification is sufficient to promote spontaneous tumorigenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ganem NJ, et al. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell. 2014;158:833–848. doi: 10.1016/j.cell.2014.06.029. This study reports that cytokinesis failure and concomitant centrosome amplification result in the activation of the Hippo pathway (see also Fava et al. 2017; Ref. 148) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fava LL, et al. The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev. 2017;31:34–45. doi: 10.1101/gad.289728.116. This study reports that PIDDosome-Caspase 2 activation plays a key role in stabilizing p53 and thereby halt cell proliferation in response to centrosome amplification (see also Ganem et al. 2014; Ref. 146) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tinel A, Tschopp J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304:843–846. doi: 10.1126/science.1095432. [DOI] [PubMed] [Google Scholar]
- 151.Chan JY. A clinical overview of centrosome amplification in human cancers. Int J Biol Sci. 2011;7:1122–1144. doi: 10.7150/ijbs.7.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Schnerch D, Nigg EA. Structural centrosome aberrations favor proliferation by abrogating microtubule-dependent tissue integrity of breast epithelial mammospheres. Oncogene. 2016;35:2711–2722. doi: 10.1038/onc.2015.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Basto R, et al. Centrosome amplification can initiate tumorigenesis in flies. Cell. 2008;133:1032–1042. doi: 10.1016/j.cell.2008.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sabino D, et al. Moesin is a major regulator of centrosome behavior in epithelial cells with extra centrosomes. Curr Biol. 2015;25:879–889. doi: 10.1016/j.cub.2015.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Marthiens V, et al. Centrosome amplification causes microcephaly. Nat Cell Biol. 2013;15:731–740. doi: 10.1038/ncb2746. [DOI] [PubMed] [Google Scholar]
- 156.Vitre B, et al. Chronic centrosome amplification without tumorigenesis. Proc Natl Acad Sci U S A. 2015;112:E6321–6330. doi: 10.1073/pnas.1519388112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sercin O, et al. Transient PLK4 overexpression accelerates tumorigenesis in p53-deficient epidermis. Nat Cell Biol. 2016;18:100–110. doi: 10.1038/ncb3270. [DOI] [PubMed] [Google Scholar]
- 158.Coelho PA, et al. Over-expression of Plk4 induces centrosome amplification, loss of primary cilia and associated tissue hyperplasia in the mouse. Open Biol. 2015;5:150209. doi: 10.1098/rsob.150209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kulukian A, et al. Epidermal development, growth control, and homeostasis in the face of centrosome amplification. Proc Natl Acad Sci U S A. 2015;112:E6311–6320. doi: 10.1073/pnas.1518376112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Loncarek J, Hergert P, Khodjakov A. Centriole reduplication during prolonged interphase requires procentriole maturation governed by Plk1. Curr Biol. 2010;20:1277–1282. doi: 10.1016/j.cub.2010.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Douthwright S, Sluder G. Link between DNA damage and centriole disengagement/reduplication in untransformed human cells. Journal of cellular physiology. 2014;229:1427–1436. doi: 10.1002/jcp.24579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Inanc B, Dodson H, Morrison CG. A centrosome-autonomous signal that involves centriole disengagement permits centrosome duplication in G2 phase after DNA damage. Mol Biol Cell. 2010;21:3866–3877. doi: 10.1091/mbc.E10-02-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev. 2007;17:157–162. doi: 10.1016/j.gde.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 164.Zack TI, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Fujiwara T, et al. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437:1043–1047. doi: 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- 166.Krzywicka-Racka A, Sluder G. Repeated cleavage failure does not establish centrosome amplification in untransformed human cells. J Cell Biol. 2011;194:199–207. doi: 10.1083/jcb.201101073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Fan G, et al. Loss of KLF14 triggers centrosome amplification and tumorigenesis. Nat Commun. 2015;6:8450. doi: 10.1038/ncomms9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Li J, et al. SAK, a new polo-like kinase, is transcriptionally repressed by p53 and induces apoptosis upon RNAi silencing. Neoplasia. 2005;7:312–323. doi: 10.1593/neo.04325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF. Abnormal centrosome amplification in the absence of p53. Science. 1996;271:1744–1747. doi: 10.1126/science.271.5256.1744. [DOI] [PubMed] [Google Scholar]
- 170.Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278–282. doi: 10.1038/nature08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Quintyne NJ, Reing JE, Hoffelder DR, Gollin SM, Saunders WS. Spindle multipolarity is prevented by centrosomal clustering. Science. 2005;307:127–129. doi: 10.1126/science.1104905. [DOI] [PubMed] [Google Scholar]
- 172.Leber B, et al. Proteins required for centrosome clustering in cancer cells. Sci Transl Med. 2:33ra38. doi: 10.1126/scitranslmed.3000915. [DOI] [PubMed] [Google Scholar]
- 173.Kwon M, et al. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008;22:2189–2203. doi: 10.1101/gad.1700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Silkworth WT, Nardi IK, Scholl LM, Cimini D. Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS One. 2009;4:e6564. doi: 10.1371/journal.pone.0006564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhang Y, et al. USP44 regulates centrosome positioning to prevent aneuploidy and suppress tumorigenesis. J Clin Invest. 2012;122:4362–4374. doi: 10.1172/JCI63084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Silkworth WT, Nardi IK, Paul R, Mogilner A, Cimini D. Timing of centrosome separation is important for accurate chromosome segregation. Mol Biol Cell. 2012;23:401–411. doi: 10.1091/mbc.E11-02-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.van Ree JH, Nam HJ, Jeganathan KB, Kanakkanthara A, van Deursen JM. Pten regulates spindle pole movement through Dlg1-mediated recruitment of Eg5 to centrosomes. Nat Cell Biol. 2016;18:814–821. doi: 10.1038/ncb3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Nam HJ, van Deursen JM. Cyclin B2 and p53 control proper timing of centrosome separation. Nat Cell Biol. 2014;16:538–549. doi: 10.1038/ncb2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science. 2011;333:1895–1898. doi: 10.1126/science.1210214. [DOI] [PubMed] [Google Scholar]
- 180.Crasta K, et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482:53–58. doi: 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhang CZ, et al. Chromothripsis from DNA damage in micronuclei. Nature. 2015;522:179–184. doi: 10.1038/nature14493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Castellanos E, Dominguez P, Gonzalez C. Centrosome dysfunction in Drosophila neural stem cells causes tumors that are not due to genome instability. Curr Biol. 2008;18:1209–1214. doi: 10.1016/j.cub.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 183.Godinho SA, Pellman D. Causes and consequences of centrosome abnormalities in cancer. Philos Trans R Soc Lond B Biol Sci. 2014;369 doi: 10.1098/rstb.2013.0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Godinho SA, et al. Oncogene-like induction of cellular invasion from centrosome amplification. Nature. 2014;510:167–171. doi: 10.1038/nature13277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Mahjoub MR, Stearns T. Supernumerary centrosomes nucleate extra cilia and compromise primary cilium signaling. Curr Biol. 2012;22:1628–1634. doi: 10.1016/j.cub.2012.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Han YG, et al. Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med. 2009;15:1062–1065. doi: 10.1038/nm.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wong SY, et al. Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat Med. 2009;15:1055–1061. doi: 10.1038/nm.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Nano M, Basto R. Consequences of Centrosome Dysfunction During Brain Development. Adv Exp Med Biol. 2017;1002:19–45. doi: 10.1007/978-3-319-57127-0_2. [DOI] [PubMed] [Google Scholar]
- 189.Chavali PL, Putz M, Gergely F. Small organelle, big responsibility: the role of centrosomes in development and disease. Philos Trans R Soc Lond B Biol Sci. 2014;369 doi: 10.1098/rstb.2013.0468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Faheem M, et al. Molecular genetics of human primary microcephaly: an overview. BMC Med Genomics. 2015;8(Suppl 1):S4. doi: 10.1186/1755-8794-8-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Jayaraman D, et al. Microcephaly Proteins Wdr62 and Aspm Define a Mother Centriole Complex Regulating Centriole Biogenesis, Apical Complex, and Cell Fate. Neuron. 2016;92:813–828. doi: 10.1016/j.neuron.2016.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Insolera R, Bazzi H, Shao W, Anderson KV, Shi SH. Cortical neurogenesis in the absence of centrioles. Nat Neurosci. 2014;17:1528–1535. doi: 10.1038/nn.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Marjanovic M, et al. CEP63 deficiency promotes p53-dependent microcephaly and reveals a role for the centrosome in meiotic recombination. Nat Commun. 2015;6:7676. doi: 10.1038/ncomms8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Martin CA, et al. Mutations in PLK4, encoding a master regulator of centriole biogenesis, cause microcephaly, growth failure and retinopathy. Nat Genet. 2014;46:1283–1292. doi: 10.1038/ng.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Morin X, Bellaiche Y. Mitotic spindle orientation in asymmetric and symmetric cell divisions during animal development. Dev Cell. 2011;21:102–119. doi: 10.1016/j.devcel.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 196.Lizarraga SB, et al. Cdk5rap2 regulates centrosome function and chromosome segregation in neuronal progenitors. Development. 2010;137:1907–1917. doi: 10.1242/dev.040410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lancaster MA, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Konno D, et al. Neuroepithelial progenitors undergo LGN-dependent planar divisions to maintain self-renewability during mammalian neurogenesis. Nat Cell Biol. 2008;10:93–101. doi: 10.1038/ncb1673. [DOI] [PubMed] [Google Scholar]
- 199.Sir JH, et al. Loss of centrioles causes chromosomal instability in vertebrate somatic cells. J Cell Biol. 2013;203:747–756. doi: 10.1083/jcb.201309038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Pilaz LJ, et al. Prolonged Mitosis of Neural Progenitors Alters Cell Fate in the Developing Brain. Neuron. 2016;89:83–99. doi: 10.1016/j.neuron.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kazazian K, et al. Plk4 Promotes Cancer Invasion and Metastasis through Arp2/3 Complex Regulation of the Actin Cytoskeleton. Cancer Res. 2017;77:434–447. doi: 10.1158/0008-5472.CAN-16-2060. [DOI] [PubMed] [Google Scholar]
- 202.Mason JM, et al. Functional characterization of CFI-400945, a Polo-like kinase 4 inhibitor, as a potential anticancer agent. Cancer Cell. 2014;26:163–176. doi: 10.1016/j.ccr.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 203.Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA. Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nat Cell Biol. 1999;1:88–93. doi: 10.1038/10054. [DOI] [PubMed] [Google Scholar]
- 204.Knockleby J, Lee H. Same partners, different dance: involvement of DNA replication proteins in centrosome regulation. Cell Cycle. 2010;9:4487–4491. doi: 10.4161/cc.9.22.14047. [DOI] [PubMed] [Google Scholar]
- 205.Meraldi P, Nigg EA. Centrosome cohesion is regulated by a balance of kinase and phosphatase activities. J Cell Sci. 2001;114:3749–3757. doi: 10.1242/jcs.114.20.3749. [DOI] [PubMed] [Google Scholar]
- 206.Tanenbaum ME, et al. Kif15 cooperates with eg5 to promote bipolar spindle assembly. Curr Biol. 2009;19:1703–1711. doi: 10.1016/j.cub.2009.08.027. [DOI] [PubMed] [Google Scholar]
- 207.Blangy A, et al. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell. 1995;83:1159–1169. doi: 10.1016/0092-8674(95)90142-6. [DOI] [PubMed] [Google Scholar]
- 208.Oliver TG, et al. Caspase-2-mediated cleavage of Mdm2 creates a p53-induced positive feedback loop. Mol Cell. 2011;43:57–71. doi: 10.1016/j.molcel.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]