

Abstract
The increased production of reactive oxygen species (ROS) has been postulated to play a key role in the progression of nonalcoholic fatty liver disease (NAFLD). However, the source of ROS and mechanisms underlying the development of NAFLD have yet to be established. We observed a significant up-regulation of a minor isoform of NADPH oxidase, NOX1, in the liver of nonalcoholic steatohepatitis (NASH) patients as well as of mice fed a high-fat and high-cholesterol (HFC) diet for 8 weeks. In mice deficient in Nox1 (Nox1KO), increased levels of serum alanine aminotransferase and hepatic cleaved caspase-3 demonstrated in HFC diet-fed wild-type mice (WT) were significantly attenuated. Concomitantly, increased protein nitrotyrosine adducts, a marker of peroxynitrite-induced injury detected in hepatic sinusoids of WT, were significantly suppressed in Nox1KO. The expression of NOX1 mRNA was much higher in the fractions of enriched liver sinusoidal endothelial cells (LSECs) than in those of hepatocytes. In primary cultured LSECs, palmitic acid (PA) up-regulated the mRNA level of NOX1, but not of NOX2 or NOX4. The production of nitric oxide by LSECs was significantly attenuated by PA-treatment in WT but not in Nox1KO. When the in vitro relaxation of TWNT1, a cell line that originated from hepatic stellate cells, was assessed by the gel contraction assay, the relaxation of stellate cells induced by LSECs was attenuated by PA treatment. In contrast, the relaxation effect of LSECs was preserved in cells isolated from Nox1KO. Taken together, the up-regulation of NOX1 in LSECs may elicit peroxynitrite-mediated cellular injury and impaired hepatic microcirculation through the reduced bioavailability of nitric oxide. ROS derived from NOX1 may therefore constitute a critical component in the progression of NAFLD.
Keywords: Nonalcoholic fatty liver disease (NAFLD), Reactive oxygen species (ROS), Liver sinusoidal endothelial cells (LSECs), Nitrotyrosine, Nitric oxide (NO)
1. Introduction
The involvement of reactive oxygen species (ROS) in the development of non-alcoholic fatty liver diseases (NAFLD) has been documented [1]. As a source of ROS in the liver, cytochrome P450 2E1 (CYP2E1) that metabolizes a variety of small xenobiotic substrates, produces superoxide as a byproduct and promotes high-fat diet-induced nonalcoholic steatohepatitis (NASH) [2]. Another source of ROS in the liver is NADPH oxidase, a multi-subunit enzyme comprised of the catalytic subunit NOX, p22phox, and several cytosolic subunits. Over the last decade, several isoforms of NOX have been identified [3], among which NOX1, NOX2, and NOX4 were demonstrated in the liver [4,5]. While the role of NOX2 in the NASH pathology has been variable in different animal models [6,7], the involvement of NOX4/NADPH oxidase was demonstrated in a fast-food diet-induced NASH model using hepatocyte-specific Nox4 knockout mice [8].
NOX1 is a non-phagocytic homolog of NOX2 (gp91phox), an isoform characterized in chronic granulomatous disease. We previously reported that NOX1 promotes the proliferation of hepatic stellate cells (HSCs) to accelerate the development of liver fibrosis induced by bile duct ligation [9]. ROS derived from NOX1 inactivate the phosphatase and tensin homolog (PTEN) and enhance the proliferative PI3K/Akt signaling in activated HSCs. On the other hand, the involvement of NOX1 in fatty liver diseases has not been examined. Therefore, this study was undertaken to clarify the role of NOX1 in a diet-induced fatty liver model using Nox1-deficient mice (Nox1KO). In this study, we fed mice with a high-fat and high-cholesterol (HFC) diet. It has been documented that supplementation of cholesterol aggravates liver steatosis and ensuing steatohepatitis in a rodent model of NAFLD [10,11]. We here demonstrate the critical role of NOX1 in the formation of protein nitrotyrosine adducts and reduction of nitric oxide bioavailability in hepatic sinusoids. Our findings suggest a novel mechanism for the progression from steatosis to early-stage NASH through peroxynitrite-mediated hepatocellular injury and impairment of the hepatic microcirculation.
2. Materials and methods
Additional Material and Methods are included in the Supplemental Information.
2.1. Liver biopsy samples
Formalin-fixed and paraffin-embedded (FFPE) liver tissue blocks from NASH patients and healthy controls were obtained from the University of California Davis Cancer Center Biorepository (Sacramento, CA) funded by the National Cancer Institute (Bethesda, MD). Following the manufacturer's instructions, mRNA was extracted from the FFPE liver tissue using the RecoverALL Total Nucleic Acid Isolation kit (Life Technologies, Grand Island, NY). Reverse transcription was performed using a kit from Bio-Rad (Hercules, CA). For the study using liver biopsy samples (Supplemental Fig. 1), all patients had given written informed consent for the analysis of genes and liver biopsies before the study. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the Ethics Committee of the Kyoto Prefectural University of Medicine.
2.2. Animal model
All animals were treated in compliance with the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health. All experimental procedures received approval from the Committee for Animal Research at Kyoto Prefectural University of Medicine. Nox1-deficient (Nox1KO) and control littermate (WT) mice were maintained on a 12-hr light/dark cycle and fed ad libitum [12]. Male WT and Nox1KO were fed either control diet (CE2 diet; containing 4.6% fat; 12% calories from fat; Japan CLEA, Tokyo, Japan) or HFC diet (modified atherogenic diet F2HFD1 with 1.5% cholesterol, 8% lard, 0% cholic acid; containing 20.9% fat; 43.7% calories from fat; OrientalBioService, Kyoto, Japan) for 8 weeks starting at 8–10 weeks of age.
2.3. Biochemical measurements
The analyses of serum alanine aminotransferase (ALT), cholesterol (T-CHO), non-esterified free fatty acids (NEFAs), and hepatic triglycerides were carried out by enzymatic assay (Nagahama Life Science Laboratory, Nagahama, Japan). The activity of caspase-3 was determined using the Apo-ONE® Homogeneous Caspase-3/7 assay purchased from Promega (Madison, WI).
2.4. Statistics
Results are expressed as the mean ± standard error of the mean (SEM). Statistical analyses were performed by Student's t-test for the comparison of two unpaired groups, or analysis of variance (ANOVA) followed by the post-hoc Tukey-Kramer's test for the comparison of multiple groups.
3. Results
3.1. NOX1 mRNA expression was increased in NASH patients and mice fed HFC diet
The up-regulation of NOX4 mRNA in human liver biopsy samples from NASH patients has been reported [8]. When the expression of NOX1 mRNA was examined, a significant increase in NOX1 mRNA was observed in NASH patients compared with those from healthy controls in liver samples obtained from the University of California Davis Cancer Center Biorepository (Fig. 1A). In a separate study with biopsy samples obtained from Kyoto Prefectural University of Medicine, the level of NOX1 tended to be higher in NASH patients, although there was no significant difference between patients diagnosed with nonalcoholic fatty liver (NAFL) and those with NASH (Supplemental Fig. 1).
Fig. 1. NOX1 mRNA expression was increased in NASH patients and mice fed HFC diet.

(A) NOX1 mRNA expression in the liver of normal subjects (NL) and NASH patients (NASH). N=5–6 per group. **p < 0.01 versus NL. (B) Levels of NOX1, NOX2, NOX4, and CYP2E1 mRNAs in the liver of mice fed a control diet (control) or a high-fat and high-cholesterol (HFC) diet for 8 weeks. N=4–5 per group. *p < 0.05, ** p < 0.01 versus control.
When the expression of NOX1 mRNA was determined in a mouse model of NAFLD fed HFC diet for 8 weeks, the level of NOX1 in the liver was significantly increased compared with that in mice fed a control diet. In addition, the expression of NOX2 was slightly but significantly increased in HFC diet-fed mice (Fig. 1B). The mRNA expression of NOX3 was not detected (data not shown). While the up-regulation of NOX4 and CYP2E1 was previously reported in NASH models [2,8,13], there was no difference in mRNA levels between our experimental groups (Fig. 1B). The detection of NOX1 protein in the liver was unsuccessful with the usage of the established polyclonal antibody raised against mouse NOX1 [14], possibly due to its detection limit.
3.2. Liver steatosis and inflammation induced by HFC diet were unaffected by Nox1 deficiency
When WT and Nox1KO were fed control or HFC diet for 8 weeks, a time-dependent increase in body weight was demonstrated in WT as well as Nox1KO (Fig. 2A). There was no difference in the elevated serum cholesterol (T-CHO) or non-esterified free fatty acids (NEFAs) level between the genotypes (Fig. 2B, C). The hepatic triglyceride content was significantly increased after 8 weeks of HFC diet feeding, but no difference was observed between the genotypes (Fig. 2D). Oil Red O staining also showed comparable levels of liver steatosis in the two genotypes (Supplemental Fig. 2). Levels of inflammatory cytokines, interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α), were un-affected by HFC diet (Fig. 2E, F). In addition, the activation of hepatic stellate cells was not apparent in this model, because αSMA mRNA expression was equivalent in mice fed control or HFC diet (Supplemental Fig. 3). Thus, this HFC diet model shows low inflammatory phenotype and possibly corresponds to the early-stage of human NAFLD.
Fig. 2.

Liver steatosis and inflammation induced by HFC diet were unaffected by Nox1 deficiency. (A) Body weight, (B) serum level of total cholesterol (T-CHO), and (C) serum levels of non-esterified fatty acids (NEFAs) and hepatic triglycerides (D) of WT littermates and Nox1KO fed control or HFC diet for 8 weeks. (E, F) The expression of IL-1β (E) and TNF-α (F) mRNAs in the liver of WT and Nox1KO fed HFC diet for 8 weeks. N=6–9 per group. *p < 0.05, **p < 0.01 versus corresponding control.
3.3. Liver injury and cellular apoptosis induced by HFC diet were ameliorated in Nox1KO
On the other hand, a significant increase in the serum level of ALT was observed in WT on HFC diet for 8 weeks, reflecting hepatocellular damage (Fig. 3A). This increase was partially but significantly attenuated in Nox1KO. Although some TUNEL-positive cells were observed in the liver of HFC diet-fed mice (Fig. 3B), it was too few to assess the significance of differences among the experimental groups (data not shown). On the other hand, an increase in the level of hepatic cleaved-caspase 3 in HFC diet-fed WT was significantly blunted in Nox1KO (Fig. 3C). Thus, Nox1 deficiency protected against HFC diet-induced liver damage, possibly by reducing cellular apoptosis.
Fig. 3. Liver injury and cellular apoptosis induced by HFC diet were ameliorated in Nox1KO.

(A) Levels of serum ALT in mice fed HFC diet for 8 weeks. N=4 per group. Results are representative of two separate experiments. (B) Representative photographs of TUNEL staining of sections obtained from the liver. Nuclei of apoptotic cells were stained brown (DAB) and all nuclei were counterstained with hematoxylin (purple). Arrows indicate apoptotic cells. Scale bar=50 μm. (C) Representative immunoblots and quantitative densitometric analysis of cleaved-caspase 3 in the liver. N=6–8 per group. **p < 0.01 versus corresponding control, #p < 0.05.
3.4. Protein nitrotyrosine adducts in the hepatic sinusoids were decreased in Nox1KO
NOX1/NADPH oxidase generates superoxide, which reacts with nitric oxide (NO) to form a highly-reactive molecule, peroxynitrite [15]. While this reaction reduces the bioavailability of NO [12], peoxynitrite causes protein tyrosine nitration to generate protein nitrotyrosine adducts. When immunohistochemical staining was carried out, protein nitrotyrosine adducts were mainly deposited in the hepatic sinusoids radially expanding from the central vein (Fig. 4A). Compared with control diet-fed mice, the intensity was higher in the liver of HFC diet-fed WT, but not of Nox1KO. To verify these findings, a competitive ELISA assay was carried out. As demonstrated in Fig. 4B, the increased levels of protein nitrotyrosine adducts in HFC diet-fed WT were significantly suppressed in Nox1KO.
Fig. 4. Protein nitrotyrosine adducts were decreased in Nox1KO.

(A) Representative photographs of immunostaining with anti-nitrotyrosine antibody (DAB) in the liver of mice fed HFC diet for 8 weeks. Nuclei were counterstained with hematoxylin (purple). Scale bar=100 μm. (B) Quantitative analysis of protein nitrotyrosine adducts by ELISA. N=4–6 per group. *p < 0.05 versus corresponding control, #p < 0.05.
To elucidate the potential involvement of altered expression of nitric oxide synthase (NOS) in protein nitrotyrosine adduct formation, the levels of NOS mRNAs in the liver were examined. Neither the iNOS nor eNOS mRNA level was altered following HFC diet feeding (Supplemental Fig. 4A, B). The increased level of protein nitrotyrosine adducts in HFC diet-fed mice was therefore attributed to up-regulation of NOX1 in the liver.
As shown in Fig. 5A, the hepatic sinusoids that make up the liver microvasculature were clearly stained by red fluorescent-labeled tomato lectin. When protein nitrotyrosine adducts were co-stained with tomato lectin, they were partially co-localized with tomato lectin or observed contiguous to tomato lectin. Such a distribution pattern of protein nitrotyrosine adducts was similar among experimental groups (Fig. 5B). These results suggest that the hepatic sinusoids are the main locus of NOX1-mediated protein tyrosine nitration.
Fig. 5. Protein nitrotyrosine adducts were observed in the hepatic sinusoids.

(A) Immunohistochemistry in the liver of WT mice fed HFC diet for 8 weeks. Arrows indicate nitrotyrosine contiguous to tomato lectin-positive sinusoidal endothelium. (B) Representative photographs of immunostaining for nitrotyrosine (green) and tomato lectin (red) in WT or Nox1KO fed control or HFC diet. Scale bar=30 μm.
3.5. NOX1 was up-regulated by palmitic acid in cultured liver sinusoidal endothelial cells
Consistent with a previous report [16], the expression of NOX1 mRNA was abundant in freshly isolated CD146-positive liver sinusoidal endothelial cells (LSECs) compared with that in hepatocytes. NOX2 mRNA was also preferentially expressed in LSECs, whereas NOX4 mRNA was less abundant in LSECs than in hepatocytes (Fig. 6A).
Fig. 6. NOX1 was up-regulated by palmitic acid in cultured liver sinusoidal endothelial cells.

(A) Expression of NOX isoform mRNAs in freshly isolated liver sinusoidal endothelial cells (LSECs) relative to enriched hepatocytes (left). N=3 per group. Representative photographs of hepatocytes and LSECs cultured for 24 h (right). (B, C) Time- and dose-dependent expression of NOX1 mRNA in primary cultured LSECs. Cells were incubated with palmitic acid (PA) for 24 h (B), or with 400 μM PA (C). (D) Expression of NOX1 mRNA in the presence of 400 μM PA or 400 μM oleic acid (OA) for 24 h. LSECs were pretreated with 10 μM TAK242 before PA treatment. (E) Expression of NOX2 and NOX4 mRNAs in LSECs treated with PA for 24 h. N=3–12 per group. *p < 0.05, **p < 0.01, #p < 0.01.
The primary isolated LSECs were treated with palmitic acid (PA), the most abundant saturated FFA in plasma. As shown in Fig. 6B and C, NOX1 mRNA expression was significantly increased by PA treatment in time- and dose-dependent manners. On the other hand, the level of NOX1 mRNA was not affected by oleic acid (OA), an unsaturated FFA. Pre-treatment of LSECs with TAK-242, an inhibitor for Toll-like receptor 4 (TLR4), significantly suppressed the PA-induced increase in NOX1 expression (Fig. 6D), suggesting that the up-regulation of NOX1 by PA was mediated by TLR4. The levels of NOX2 and NOX4 mRNA were not increased by PA treatment (Fig. 6E). These results suggest that saturated FFA-mediated TLR4 activation elicits up-regulation of NOX1 in the liver of HFC diet-fed mice. In fact, a significant increase in the serm level of NEFAs was demonstrated in HFC diet-fed mice (Fig. 2C).
3.6. NOX1 is required for PA-mediated regulation of NO bioavailability
When the cellular toxicity of PA was first examined by a cleaved caspase 3/7 assay, high concentrations of PA (> 800 μM) induced the lipoapoptosis of LSECs isolated from Nox1KO as well as WT (Supplemental Fig. 6). Thus, it was suggested that the lipoapoptosis induced by PA was not mediated by NOX1.
LSECs play an important role in the regulation of hepatic vascular tone by releasing vasoactive molecules. Endothelial cell-derived NO is a predominant vasodilator, which can be quenched and converted to peroxynitrite upon direct reaction with superoxide [15]. We therefore examined the production of NO in LSECs using the fluorescent NO probe 4,5-diaminofluorescein (DAF-2). For the detection of low levels of NO, a low concentration of DAF2 (0.1 μM) was used to limit the autofluorescence [17]. As shown in Fig. 7A, equivalent levels of NO were detected in LSECs from WT and Nox1KO in the absence of PA. However, treatment with 400 μM PA for 24 h significantly impaired NO production in WT, but not Nox1KO. These data suggest that ROS derived from NOX1 in PA-tretated LSECs may reduce the bioavailability of NO to form peroxynitrite, resulting in loss of the vasodilatory function of LSECs and increased peroxinitrite-mediated toxicity.
Fig. 7. Reduced bioavailability of NO induced by palmitic acid was abolished by Nox1 deficiency.

(A) Levels of nitric oxide (NO) released from LSECs treated with 200 or 400 μM PA for 24 h. N=6 per group. (B) Representative photographs and quantitative analysis of collagen gel contraction of TWNT1 cells. TWNT1, an immortalized hepatic stellate cell line, was seeded onto the collagen gel in the absence (−) or presence of LSECs isolated from WT or Nox1KO. Cells were cultured with serum-free medium containing 400 μM PA for 24 h. The gel area was measured 24 h after FBS stimulation. N=7 per group. *p < 0.05, **p < 0.01 versus vehicle (Veh), #p < 0.05. n.s.: not significant.
The dysfunction of LSECs due to the reduced bioavailability of NO may impair the hepatic microcirculation [18]. To evaluate LSEC dysfunction induced by PA, a collagen gel contraction assay was performed, in which LSECs were co-cultured with hepatic stellate cells. Upon 10% FBS stimulation, TWNT1, an immortalized human hepatic stellate cell line, showed a contractile response to release collagen lattices from their substrata. The co-culture of TWNT1 with WT- or Nox1KO-derived LSECs significantly suppressed the contraction, indicating that LSECs induced the relaxation of HSCs. PA treatment itself did not affect the gel contraction of TWNT1 when incubated without LSECs (Fig. 7B). On the other hand, the relaxation was attenuated in TWNT1 co-cultured with WT-LSECs in the presence of PA, but not with Nox1KO-LSECs (Fig. 7B). These findings suggest that NOX1-derived ROS impaired the LSEC-induced relaxation of HSCs, possibly through the reduction of NO bioavailability. The dysfunction of LSECs may subsequently impair hepatic microcirculation to accelerate hepatic cell death and the ensuing development of NASH (Fig. 8).
Fig. 8. A schematic model proposed for the role of NOX1 in HFC diet-induced liver injury.
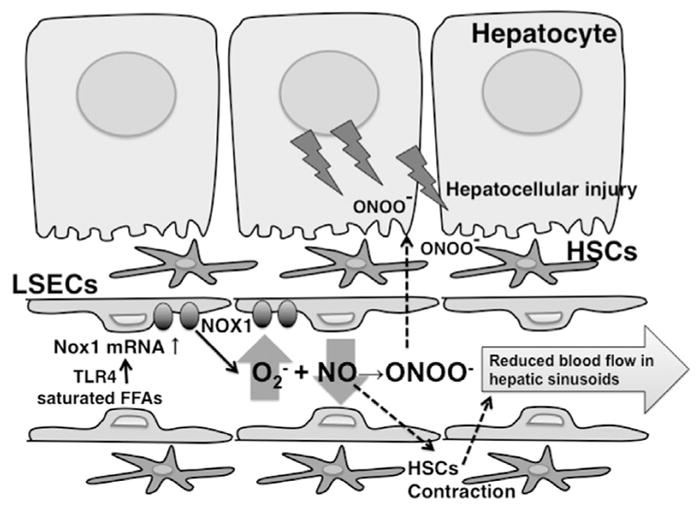
Saturated FFAs up-regulate NOX1 in LSECs though TLR4. The generation of peroxynitrite by ROS derived from NOX1 may directly cause hepatocellular injury. At the same time, NOX1-derived ROS may impair the hepatic microcirculation through the reduced bioactivity of NO, which will further enhance peroxynitrite-mediated hepatocellular injury under hypoxic conditions.
4. Discussion
In this study we demonstrated that ROS derived from NOX1/ NADPH oxidase play a critical role in HFC diet-induced liver injury. We herein showed that: (1) Expression of NOX1 was elevated in the liver of NASH patients and HFC diet-fed mice. (2) Nox1 deficiency ameliorated not only HFC diet-induced liver damage but also the production of protein nitrotyrosine adducts in hepatic sinusoids. (3) NOX1 mRNA was mainly expressed in LSECs and up-regulated by PA through TLR4 activation. (4) PA reduced the bioavailability of NO in WT-LSECs but not in Nox1KO-LSECs. (5) PA-treatment attenuated the relaxation of HSCs induced by WT-LSECs but not by Nox1KO-LSECs. Taken together, elevated levels of saturated FFAs in NASH patients or in HFC diet-fed animals may up-regulate NOX1 and increase oxidative stress in hepatic sinusoids, which impairs the hepatic microcirculation through the reduced bioactivity of NO. In addition, the suppressed formation of protein nitrotyrosine adducts in Nox1KO suggests that the generation of cell-toxic peroxynitrites in the liver is mediated by NOX1.
A significant increase in NOX1 mRNA was observed in the liver of NASH patients as well as of mice fed HFC diet for 8 weeks. In a separate study on biopsy samples obtained from a different cohort, the level of NOX1 mRNA in NASH patients was compared with that in patients with NAFL. However, no significant difference was demonstrated between the groups (Supplemental Fig. 1). Because healthy control samples were unavailable in this cohort, we could not compare the level of NOX1 with the normal liver. There is a possibility that NOX1 may be up-regulated in the liver of patients who already developed hepatic steatosis. In the normal mouse liver, the expression of NOX1 mRNA was relatively low compared with other NOX isoforms. Considering the fact that hepatocytes express a very low level of NOX1 mRNA vs. LSECs or HSCs [9], the limited expression of NOX1 mRNA detected in the whole liver may be due to the predominance of hepatocytes in the isolated tissue.
The involvement of TLR4 in the up-regulation of NOX1 has been documented [19,20]. Indeed, the up-regulation of NOX1 by PA in isolated LSECs was attenuated by TLR4 inhibitor. On the other hand, OA, an unsaturated FFA, did not affect NOX1 expression, suggesting that the up-regulation of NOX1 is not due to the nonspecific surfactant effects of lipids. Recently, it was reported that fetuin-A, a liver secretory glycoprotein and a major carrier protein of FFAs, acts as an endogenous ligand of TLR4 [21]. Another line of study demonstrated that saturated FFAs, but not polyunsaturated FFAs, induce the dimerization of TLR2/4 to cause receptor activation in a ligand-independent manner [22]. The concentrations of FFAs used for cultured LSECs were almost equivalent to those in mouse serum following HFC diet feeding, in which the serum NEFA level was approximately 500 μEq/L, comparable to 500 μM of monovalent FFAs. Accordingly, it can be speculated that increased levels of circulating saturated FFAs may induce NOX1 expression through TLR4 activation not only in LSECs of HFC diet-fed mice, but also of obese individuals with hyperlipidemia. It is also conceivable that gut-derived endotoxin in circulating plasma following the intake of a high-fat diet [23] may take part in the up-regulation of NOX1. In this context, the activation of TLR4 may be critical for the up-regulation of NOX1.
Nox1 deficiency significantly ameliorated liver injury induced by HFC diet feeding, but it did not affect the increase in body weight, serum lipids, or hepatic steatosis. We previously reported that liver injury and fibrosis induced by bile duct ligation (BDL) were significantly attenuated in Nox1KO. While NOX1 was shown to promote the proliferation of HSCs in the BDL model [9], the activation of HSCs was not apparent in mice fed HFC diet (Supplemental Fig. 2), suggesting that this HFC diet model represents the early phase of NASH development. Accordingly, the molecular mechanism underlying HFC diet-induced liver injury may be distinct from that of BDL-induced injury. In mice fed HFC diet, a crosstalk between NOX1 and NO was demonstrated. Namely, ROS derived from NOX1 increased the formation of protein nitrotyrosine adducts. A clear correlation exists between the relatively high expression of NOX1 in LSECs and the localization of protein nitrotyrosine adducts along hepatic sinusoids. Given that protein nitrotyrosine adducts indirectly suggest the accelerated formation of peroxynitrite, which is highly toxic to cells [24,25], the NOX1-dependent generation of peroxynitrite in hepatic sinusoids may have promoted hepatocellular injury.
The reduced bioavailability of NO by reaction with superoxide generated by NOX1 in LSECs could impair intrahepatic microcirculation. LSECs produce NO as an important vasodilator to maintain the intrahepatic circulation. When we examined the effect of LSEC-derived NO on stellate cells with a collagen gel contraction assay, primary cultured LSECs significantly attenuated the contraction of stellate cells induced by FBS-stimulation. The relaxation of stellate cells induced by LSECs was impaired by PA-treatment of WT but not of Nox1KO LSECs, suggesting that NO was inactivated by increased superoxide generated by NOX1. This is similar to our previous findings obtained in the an-giotensin II-infused hypertension model, in which ROS derived from NOX1 perturbed endothelium-dependent relaxation of aortic strips, leading to the development of systemic hypertension [12]. The dysfunction of LSECs is known to augment intrahepatic resistance to impair microcirculation in the liver [18,26]. In fact, increased intrahepatic resistance has been demonstrated in an isolated-perfused rat liver obtained from a cafeteria diet-fed model of NAFLD [27], a methionine-choline-deficient model of NASH [28], and from a model of LPS-induced endotoxemia [29]. While the assessment of intrahepatic micro-circulation is technically limited in mice [30], impaired blood flow in the liver may accelerate hepatocyte cell death in hypoxic micro-environments.
The protective roles of NO against liver disease were demonstrated using mice deficient in NOS [31–33]. Considering the fact that eNOS is endogenously expressed in LSECs [34], NO derived from eNOS may take part in the impaired relaxation of sinusoids induced by NOX1. In contrast with the significant up-regulation of NOX1, the expression of eNOS was unaltered following HFC diet feeding. The lack of a significant induction of iNOS in our animal model also suggests the predominant contribution of eNOS to the development of endothelial dysfunction in liver sinusoids.
Recently, NOX4 was demonstrated to play a critical role in the development of NASH [8]. Given that levels of NOX1 mRNA expressed in freshly-isolated fractions of Kupffer cells and HSCs were almost equivalent to that in LSECs (data not shown), NOX1 may be mainly expressed in non-parenchymal cells, not in parenchymal hepatocytes. In contrast, NOX4 was predominantly expressed in hepatocytes and the hepatocyte-specific deletion of NOX4 attenuated liver injury, fibrosis, and hepatic insulin resistance induced by a fast-food diet [8]. While up-regulation of NOX4 mRNA was demonstrated in the study, we did not observe alteration in NOX4 expression in mice fed HFC diet. This discrepancy may be due to the difference in dietary fat composition and feeding period. The NOX1-mediated pathway may constitute an early component of NASH pathogenesis in patients with hepatic steatosis. Considering that each NOX isoform expressed in a specific cell lineage plays a different role in the disease pathogenesis, the inhibition of both NOX1 and NOX4 may be a potential treatment strategy to suppress the development of NASH.
5. Conclusions
ROS derived from NOX1 may not only reduce the bioavailability of NO to cause an impaired hepatic microcirculation, but also enhance peroxynitrite-induced hepatocellular injury. The present study provides insight into the potential significance of NOX1/NADPH oxidase in the progression of NAFLD.
Supplementary Material
Acknowledgments
Financial support
This work was partly supported by JSPS KAKENHI Grant numbers JP24390063 (to C.Y.-N.) and JP15K18993 (to M.M.).
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.freeradbiomed.2017.12.019.
Footnotes
Conflicts of interest
C.Y.-N. is a cofounder of a startup company developing NOX inhibitors.
References
- 1.Leung TM, Nieto N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J Hepatol. 2013;58:395–398. doi: 10.1016/j.jhep.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 2.Abdelmegeed MA, Banerjee A, Yoo SH, Jang S, Gonzalez FJ, Song BJ. Critical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitis. J Hepatol. 2012;57:860–866. doi: 10.1016/j.jhep.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 4.Jiang JX, Venugopal S, Serizawa N, Chen X, Scott F, Li Y, et al. Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology. 2010;139:1375–1384. doi: 10.1053/j.gastro.2010.05.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Mochel NS, Seronello S, Wang SH, Ito C, Zheng JX, Liang TJ, et al. Hepatocyte NAD(P)H oxidases as an endogenous source of reactive oxygen species during hepatitis C virus infection. Hepatology. 2010;52:47–59. doi: 10.1002/hep.23671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.dela Pena A, Leclercq IA, Williams J, Farrell GC. NADPH oxidase is not an essential mediator of oxidative stress or liver injury in murine MCD diet-induced steatohepatitis. J Hepatol. 2007;46:304–313. doi: 10.1016/j.jhep.2006.08.025. [DOI] [PubMed] [Google Scholar]
- 7.Jiang JX, Chen X, Fukada H, Serizawa N, Devaraj S, Torok NJ. Advanced glycation endproducts induce fibrogenic activity in nonalcoholic steatohepatitis by modulating TNF-alpha-converting enzyme activity in mice. Hepatology. 2013;58:1339–1348. doi: 10.1002/hep.26491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bettaieb A, Jiang JX, Sasaki Y, Chao TI, Kiss Z, Chen X, et al. Hepatocyte nicotinamide adenine dinucleotide phosphate reduced oxidase 4 regulates stress signaling, fibrosis, and insulin sensitivity during development of steatohepatitis in mice. Gastroenterology. 2015;149:468–480. e410. doi: 10.1053/j.gastro.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui W, Matsuno K, Iwata K, Ibi M, Matsumoto M, Zhang J, et al. NOX1/nicotinamide adenine dinucleotide phosphate, reduced form (NADPH) oxidase promotes proliferation of stellate cells and aggravates liver fibrosis induced by bile duct ligation. Hepatology. 2011;54:949–958. doi: 10.1002/hep.24465. [DOI] [PubMed] [Google Scholar]
- 10.Ichimura M, Kawase M, Masuzumi M, Sakaki M, Nagata Y, Tanaka K, et al. High-fat and high-cholesterol diet rapidly induces non-alcoholic steatohepatitis with advanced fibrosis in Sprague-Dawley rats. Hepatol Res: Off J Jpn Soc Hepatol. 2015;45:458–469. doi: 10.1111/hepr.12358. [DOI] [PubMed] [Google Scholar]
- 11.Matsuzawa N, Takamura T, Kurita S, Misu H, Ota T, Ando H, et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology. 2007;46:1392–1403. doi: 10.1002/hep.21874. [DOI] [PubMed] [Google Scholar]
- 12.Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M, et al. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation. 2005;112:2677–2685. doi: 10.1161/CIRCULATIONAHA.105.573709. [DOI] [PubMed] [Google Scholar]
- 13.Leclercq IA, Farrell GC, Field J, Bell DR, Gonzalez FJ, Robertson GR. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J Clin Investig. 2000;105:1067–1075. doi: 10.1172/JCI8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsumoto M, Katsuyama M, Iwata K, Ibi M, Zhang J, Zhu K, et al. Characterization of N-glycosylation sites on the extracellular domain of NOX1/ NADPH oxidase. Free Radic Biol Med. 2014;68:196–204. doi: 10.1016/j.freeradbiomed.2013.12.013. [DOI] [PubMed] [Google Scholar]
- 15.Nguyen Dinh Cat A, Montezano AC, Burger D, Touyz RM. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid Redox Signal. 2013;19:1110–1120. doi: 10.1089/ars.2012.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paik YH, Iwaisako K, Seki E, Inokuchi S, Schnabl B, Osterreicher CH, et al. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology. 2011;53:1730–1741. doi: 10.1002/hep.24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rathel TR, Leikert JJ, Vollmar AM, Dirsch VM. Application of 4,5-diamino-fluorescein to reliably measure nitric oxide released from endothelial cells in vitro. Biol Proced Online. 2003;5:136–142. doi: 10.1251/bpo55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwakiri Y. Endothelial dysfunction in the regulation of cirrhosis and portal hypertension. Liver Int: Off J Int Assoc Study Liver. 2012;32:199–213. doi: 10.1111/j.1478-3231.2011.02579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maitra U, Singh N, Gan L, Ringwood L, Li L. IRAK-1 contributes to lipopolysaccharide-induced reactive oxygen species generation in macrophages by inducing NOX-1 transcription and Rac1 activation and suppressing the expression of anti-oxidative enzymes. J Biol Chem. 2009;284:35403–35411. doi: 10.1074/jbc.M109.059501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsuno K, Iwata K, Matsumoto M, Katsuyama M, Cui W, Murata A, et al. NOX1/NADPH oxidase is involved in endotoxin-induced cardiomyocyte apoptosis. Free Radic Biol Med. 2012;53:1718–1728. doi: 10.1016/j.freeradbiomed.2012.08.590. [DOI] [PubMed] [Google Scholar]
- 21.Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, et al. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med. 2012;18:1279–1285. doi: 10.1038/nm.2851. [DOI] [PubMed] [Google Scholar]
- 22.Hwang DH, Kim JA, Lee JY. Mechanisms for the activation of Toll-like receptor 2/4 by saturated fatty acids and inhibition by docosahexaenoic acid. Eur J Pharmacol. 2016;785:24–35. doi: 10.1016/j.ejphar.2016.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–1846. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- 24.Shacka JJ, Sahawneh MA, Gonzalez JD, Ye YZ, D'Alessandro TL, Estevez AG. Two distinct signaling pathways regulate peroxynitrite-induced apoptosis in PC12 cells. Cell Death Differ. 2006;13:1506–1514. doi: 10.1038/sj.cdd.4401831. [DOI] [PubMed] [Google Scholar]
- 25.Szabo C, Virag L, Cuzzocrea S, Scott GS, Hake P, O'Connor MP, et al. Protection against peroxynitrite-induced fibroblast injury and arthritis development by inhibition of poly(ADP-ribose) synthase. Proc Natl Acad Sci USA. 1998;95:3867–3872. doi: 10.1073/pnas.95.7.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwakiri Y, Kim MY. Nitric oxide in liver diseases. Trends Pharmacol Sci. 2015;36:524–536. doi: 10.1016/j.tips.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pasarin M, La Mura V, Gracia-Sancho J, Garcia-Caldero H, Rodriguez-Vilarrupla A, Garcia-Pagan JC, et al. Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PloS One. 2012;7:e32785. doi: 10.1371/journal.pone.0032785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Francque S, Laleman W, Verbeke L, Van Steenkiste C, Casteleyn C, Kwanten W, et al. Increased intrahepatic resistance in severe steatosis: endothelial dysfunction, vasoconstrictor overproduction and altered microvascular architecture. Lab Investig: J Tech Methods Pathol. 2012;92:1428–1439. doi: 10.1038/labinvest.2012.103. [DOI] [PubMed] [Google Scholar]
- 29.La Mura V, Pasarin M, Meireles CZ, Miquel R, Rodriguez-Vilarrupla A, Hide D, et al. Effects of simvastatin administration on rodents with lipopolysaccharide-induced liver microvascular dysfunction. Hepatology. 2013;57:1172–1181. doi: 10.1002/hep.26127. [DOI] [PubMed] [Google Scholar]
- 30.Zhao ZS, Shibamoto T, Tsutsumi M, Cui S, Zhang WM, Takano H, et al. Mouse hepatic portal venoconstrictive response to vasoconstrictors is much weaker than that in rat. J Cardiovasc Pharmacol. 2009;54:421–426. doi: 10.1097/FJC.0b013e3181bad2a6. [DOI] [PubMed] [Google Scholar]
- 31.Chen Y, Hozawa S, Sawamura S, Sato S, Fukuyama N, Tsuji C, et al. Deficiency of inducible nitric oxide synthase exacerbates hepatic fibrosis in mice fed high-fat diet. Biochem Biophys Res Commun. 2005;326:45–51. doi: 10.1016/j.bbrc.2004.10.202. [DOI] [PubMed] [Google Scholar]
- 32.Nozaki Y, Fujita K, Wada K, Yoneda M, Kessoku T, Shinohara Y, et al. Deficiency of iNOS-derived NO accelerates lipid accumulation-independent liver fibrosis in non-alcoholic steatohepatitis mouse model. BMC Gastroenterol. 2015;15:42. doi: 10.1186/s12876-015-0269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tateya S, Rizzo NO, Handa P, Cheng AM, Morgan-Stevenson V, Daum G, et al. Endothelial NO/cGMP/VASP signaling attenuates Kupffer cell activation and hepatic insulin resistance induced by high-fat feeding. Diabetes. 2011;60:2792–2801. doi: 10.2337/db11-0255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shah V, Haddad FG, Garcia-Cardena G, Frangos JA, Mennone A, Groszmann RJ, et al. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of resistance in the hepatic sinusoids. J Clin Investig. 1997;100:2923–2930. doi: 10.1172/JCI119842. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.