

Abstract
The spleen is an important site for generating protective immune responses against pathogens. After infection, immune cells undergo rapid reorganization to initiate and maintain localized inflammatory responses; however, the mechanisms governing this spatial and temporal cellular reorganization remain unclear. We show that the strategic position of splenic marginal zone CD169+ macrophages is vital for rapid initiation of antibacterial responses. In addition to controlling initial bacterial growth, CD169+ macrophages orchestrate a second phase of innate protection by mediating the transport of bacteria to splenic T cell zones. This compartmentalization of bacteria within the spleen was essential for driving the reorganization of innate immune cells into hierarchical clusters and for local interferon-γ production near sites of bacterial replication foci. Our results show that both phases of the antimicrobial innate immune response were dependent on CD169+ macrophages, and, in their absence, the series of events needed for pathogen clearance and subsequent survival of the host was disrupted. Our study provides insight into how lymphoid organ structure and function are related at a fundamental level.
INTRODUCTION
The importance of the spleen for resistance against infection is well established (1). Innate immune cells in the spleen are strategically positioned to rapidly detect invading pathogens. After an infection, innate immune cells in the spleen undergo reorganization into “hierarchical clusters” that allow for the initiation and progression of an effective immune response against infections (2–4); however, it remains unclear how this process is regulated in lymphoid tissues. Moreover, the dynamics and functional consequences of immune cell remodeling after bacterial infection are still not well understood. CD169+ macrophages are a subpopulation of tissue-resident macrophages positioned in the splenic marginal zone (MZ) that are among the first cell types to encounter invading pathogens (2, 5–8). Analogous to this, in the lymph nodes (LNs), CD169+ macrophages reside in the subcapsular sinus and have been shown to play a protective role against viral infections by capturing LNs draining viral particles (9), as well as for initiating humoral and adaptive immune responses against other infections (7, 10, 11) and tumors (12). However, little is known about the functional outcome and downstream consequences of pathogen uptake by splenic MZ CD169+ macrophages after infections.
Spatial remodeling of cells in the spleen is necessary for mediating protection against bacterial infection; nevertheless, it remains unclear how reorganization of innate immune cells is regulated in secondary lymphoid tissues. Organized “hierarchical clustering” of neutrophils, monocytes, and natural killer (NK) cells at sites of Listeria monocytogenes (Lm) infection enables focal innate immune cell activation and inflammatory cytokine production in Lm-infected T cell zones (3). Before the formation of hierarchical clusters, bacteria are actively transported from the MZs to the T cell zones, where they continue to replicate (13, 14). The current paradigm is based on previous work that demonstrated that splenic CD8αα+ dendritic cells (DCs) provide a requisite permissive bacterial replication niche for Lm, and thus, it has been proposed that CD8αα+ DCs are required for the establishment of splenic infection (13, 15). In the absence of splenic CD8αα+ DCs (Batf3−/− mice), Lm invasion, as well as the subsequent transport and propagation of the bacteria in the T cell zones, is markedly impaired (13, 15, 16). Thus, it is believed that CD8αα+ DCs are necessary not only for establishing a productive infection in the spleens of mice but also for transporting Lm to the splenic T cell zones. Although the consequences of CD8αα+ DC infection are well established, MZ CD169+ macrophages have also been demonstrated to harbor Lm early after infection (8). The strategic anatomical positioning of CD169+ macrophages in the spleen makes it likely that these cells are the first to interact with an invading pathogen. Hence, we reasoned that the CD169+ macrophages play a central role in mediating bacterial clearance and translocation and sought to reconcile how these two cellular niches regulated susceptibility to Lm.
Here, we have examined the role of splenic CD169+ macrophages after systemic infection with Lm. We demonstrate how the localization of a pathogen within the spleen is controlled by MZ CD169+ macrophages and how important the location of a pathogen within a secondary lymphoid organ is for generating a protective innate immune response.
RESULTS
CD169+ macrophages capture Lm and control Lm replication
To understand the role of CD169+ macrophages during bacterial infections, we first determined whether these cells play host to Lm after infection (17). The spleens isolated from infected wild-type (WT) animals were sectioned, stained for various cell surface markers, and analyzed using confocal microscopy. As early as 3 hours postinfection (hpi), Lm was captured by CD169+ macrophages in WT mice (Fig. 1A and movies S1 and S2). Quantification of intracellular Lm at 3 hpi showed that most of the Lm were localized within CD169+ macrophages (Fig. 1B). Over the course of 9 hours, the frequency of Lm- infected CD169+ macrophages decreased, indicating that CD169+ macrophages were the primary host of Lm early after infection, but as the infection progressed, the bacterial niche shifted to other cells in the spleen. The direct uptake of Lm by CD169+ macrophages was further validated by purifying CD169+ macrophages from the spleens of WT mice and quantifying viable bacteria. These studies showed that CD169+ macrophages rapidly trapped Lm as early as 1 hpi (Fig. 1C) and served as primary cellular host to Lm early after infection.
Fig. 1. Splenic CD169+ macrophages trap and prevent early Lm replication and spread.
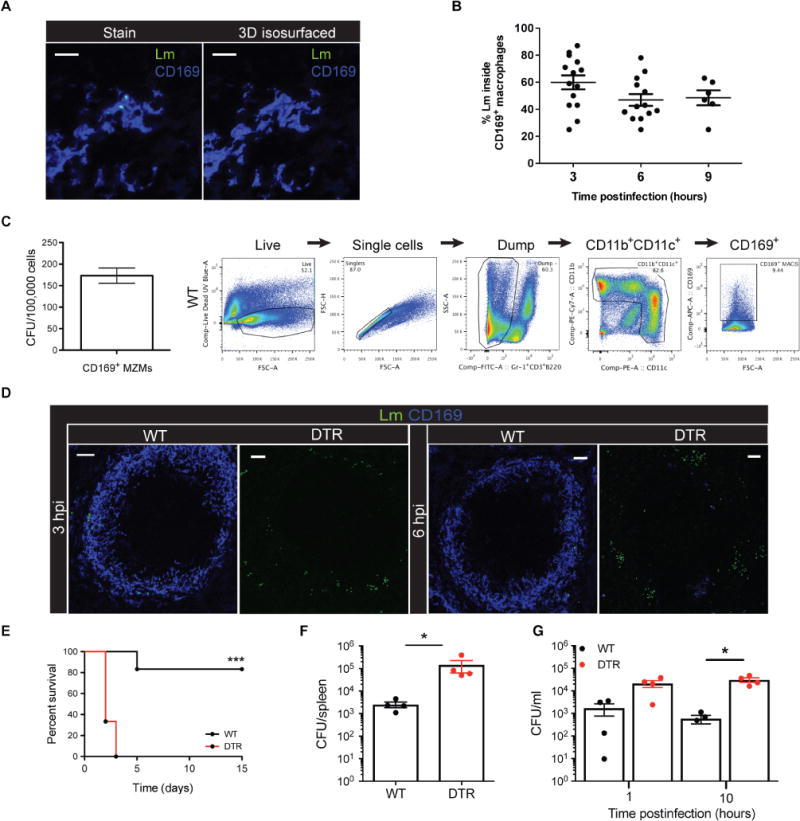
(A) Confocal image (left) and isosurfaced image (right) of intracellular Lm located within a CD169+ macrophage in the MZ. Scale bars, 10 μm. 3D, three-dimensional. (B) Quantification of intracellular Lm within CD169+ macrophages in the spleens of WT mice. Each symbol represents a distinct region of the spleen. Data are shown as means ± SEM. (C) Quantification of viable Lm from sorted WT CD169+ macrophages at 1 hpi. Data are shown as means ± SEM. Representative example of gating strategy used to sort CD169+ macrophages from the spleen. CFU, colony-forming units; MZMs, MZ macrophages; UV, ultraviolet; FSC-A, forward scatter–area; FSC-H, forward scatter–height; SSC-A, side scatter–area; PE, phycoerythrin. (D) Confocal images of spleen sections from WT or CD169-DTR mice. Lm are rendered with spots. Scale bars, 50 μm. (E) Survival of WT or CD169-DTR mice (***P < 0.0005, Mantel-Cox log-rank test). (F) Quantification of Lm numbers from the spleens of WT or CD169-DTR mice at 10 hpi. Each symbol represents an individual mouse, shown as means ± SEM (*P < 0.05, Mann-Whitney test). (G) Quantification of Lm in the blood of WT or CD169-DTR mice. Each symbol represents an individual mouse, shown as mean ± SEM (*P < 0.05, Mann-Whitney test). All data are representative of two to three separate experiments with three to six mice per group.
To determine whether CD169+ macrophages mediate innate immune protection after infection, we selectively depleted CD169+ macrophages using heterozygous transgenic mice expressing the knocked-in human diphtheria toxin (DT) receptor (DTR) under the control of CD169 gene transcription [referred to as CD169-DTR; (12, 18)]. We found that, unlike in WT mice where most of the Lm were confined to CD169+ macrophages in the MZ, at very early time points after infection, CD169-DTR mice exhibited markedly increased Lm replication in the MZ and red pulp (Fig. 1D). Depletion of CD169+ macrophages resulted in notable susceptibility to Lm, with all CD169-DTR mice succumbing to sublethal infection by 3 days after infection (Fig. 1E). Bacterial quantification from the spleens of WT and CD169-DTR mice showed that CD169-DTR mice were unable to control early Lm growth and had significantly greater bacterial burdens at 10 hpi (Fig. 1F). This impaired ability to control splenic Lm replication coincided with significantly more Lm circulating in the blood at 10 hpi in CD169-DTR mice (Fig. 1G).
We characterized CD169-DTR mice to verify that susceptibility to infection was not due to off-target effects of DT administration (fig. S1). We found that MZ vasculature and microarchitecture remained normal after DT treatment and CD169+ macrophage depletion, as judged by mucosal vascular addressin cell adhesion molecule–1 (MAdCAM-1) staining (fig. S1A). A recent publication showed that CD169+ Kupffer cells in the liver can also be depleted in CD169-DTR mice after several days of DT treatment (19). To prevent depletion of CD169+ macrophages in the liver, we used a single dose of DT, which was sufficient to deplete splenic MZ CD169+ macrophages but not CD169+ Kupffer cells in the liver (fig. S1B). Untreated or DT-treated naïve WT or CD169-DTR mice had comparable innate immune cell frequencies (dot plots showing gating strategy; fig. S1, C and D), indicating that the increased susceptibility to Lm observed in CD169-DTR mice was specifically due to the absence of CD169+ macrophages and not due to the nonspecific depletion of other important cells, such as red pulp macrophages, DCs, and granulocytes in the spleen. These data indicate that CD169+ macrophages are vital for protection in the context of Lm infection.
Systemic infections are the result of pathogen breaching the original site of infection and hijacking the body’s vascular system. Orally ingested Listeria was previously shown to breach the intestinal barrier 2 to 3 days after infection and was transported to the spleen (20–22). We examined the physiological role of splenic CD169+ macrophages after intestinal Lm infection. WT or CD169-DTR mice were with DT and then infected orally with Lm. We quantified bacterial burdens in the small intestines, mesenteric LNs (MLNs), and the spleen. We found that gastrointestinal infection also resulted in significantly greater Lm burdens in the spleen (fig. S2A). Impaired control of bacterial replication was specific to the spleen because CD169-DTR mice controlled Lm infection comparably with WT mice in the small intestines and MLNs (fig. S2, B and C). Last, we sought to test whether the protective role of CD169+ macrophages extended beyond intracellular Gram-positive bacterium. We infected WT and CD169-DTR mice intravenously with Escherichia coli, an extracellular Gram-negative bacterium. Bacterial quantification in the spleen at 24 hpi showed that CD169+ macrophages were also required for controlling E. coli growth in the spleen because CD169-DTR mice had significantly more E. coli (fig. S2D). Another clinically relevant blood-borne pathogen is cytomegalovirus (CMV). WT and CD169-DTR mice were infected intravenously with murine CMV (MCMV) expressing green fluorescent protein (GFP), and we used confocal microscopy to determine viral replication in the spleen. We observed that CD169-DTR mice had noticeably more MCMV present in the MZ compared with WT mice (fig. S2E). These findings demonstrate that CD169+ macrophages are the primary line of defense against a wide range of systemic pathogens.
CD169+ macrophages express an inflammatory transcriptional profile after infection
To obtain some insight into the function of CD169+ macrophages, we sorted CD169+ macrophages from the spleen and carried out RNA sequencing. CD11b+ and CD11c+ cells were negatively enriched, and naïve red pulp macrophages (F4/80+), CD169+ macrophages (from uninfected mice), or CD169+ macrophages from infected mice were sorted (fig. S3A). Extracted RNA integrity was validated (fig. S3B), and reproducibility between biological replicates was confirmed by Pearson correlation coefficients (fig. S3C). Two-dimensional principal components analysis (PCA) and cluster analysis showed that naïve CD169+ macrophages expressed a distinct gene expression profile when compared with red pulp macrophages (labeled F4/80+ naïve) under steady-state conditions (Fig. 2A). We identified more than 709 differentially expressed genes between red pulp macrophages and CD169+ macrophages from naïve mice (Fig. 2B). Previous studies characterizing the transcriptional profiles of tissue-resident macrophages identified a distinct genetic signature for red pulp macrophages (23–25). As part of this core gene signature, Spi-C, Axl, F4/80, Cd68, Vcam1, Pecam1, and Ccr3 were exclusively highly expressed in red pulp macrophages compared with other tissue-resident macrophages. Similarly, our data show that these genes were also significantly differentially expressed in red pulp macrophages compared with CD169+ macrophages (Fig. 2C). In line with known functions of red pulp macrophages in red blood cell clearance and heme recycling (26), we found that ferroportin (Slc40a1) and heme oxygenase expression was significantly up-regulated in red pulp macrophages compared with CD169+ macrophages (Fig. 2C). Of the 709 differentially expressed genes between red pulp macrophages and CD169+ macrophages, 365 differentially expressed genes were up-regulated, and 344 were down-regulated. Among the top 15 most highly up-regulated genes in CD169+ macrophages relative to red pulp macrophages were genes associated with antimicrobial activity. Gene ontology (GO) enrichment analysis indicated that genes related to inflammatory response, defense response, and cell activation were overrepresented in the up-regulated genes at steady state (table S1).
Fig. 2. Splenic CD169+ macrophages express a unique gene profile.
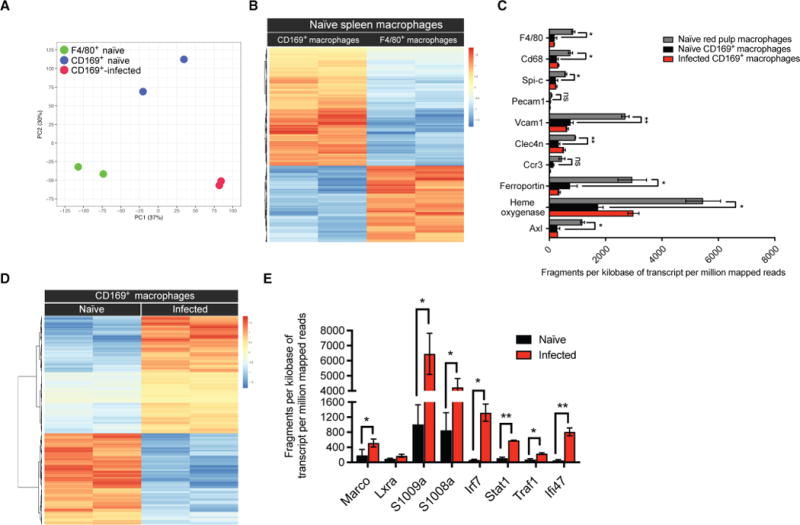
(A) PCA of 17,336 genes using each sample’s log2[fragments per kilobase of transcript per million mapped reads (FPKM) + 1]. (B) Heat map using z scores for 709 differentially expressed genes between naïve CD169+ macrophages and red pulp macrophages (F4/80+). (C) Transcript expression (FPKM) of paired-end sample sequencing represented as normalized values based on transcript length and depth of coverage. Genes represent transcripts that were previously identified to distinguish red pulp macrophages from other subsets of tissue macrophages. Data are shown as means ± SEM (*P < 0.05 and **P < 0.005, Student’s t test). ns, not significant. (D) Heat map using z scores for the identified 795 differentially expressed genes comparing CD169+ macrophages from naïve and Lm-infected mice. (E) Transcript expression (FPKM) of genes exclusively up-regulated in CD169+ macrophages 12 hours after Lm infection. Data are shown as means ± SEM (*P < 0.05 and **P < 0.005, Student’s t test).
The gene expression profile of activated CD169+ macrophages isolated from infected animals was changed when compared with CD169+ macrophages from naïve mice. After Lm infection, CD169+ macrophages adopted an inflammatory macrophage (M1) signature. Our analysis identified 795 differentially expressed genes in CD169+ macrophages after infection when compared with cells from uninfected animals (Fig. 2D). We found that inflammatory cytokine transcripts, as well as accessory molecules of inflammatory cytokine receptors, were up-regulated in CD169+ macrophages after infection (Fig. 2E).
GO enrichment analysis of differentially expressed genes revealed that genes related to immune system response, defense response, inflammatory response, innate response, and response to bacterium were significantly overrepresented in the up-regulated genes in CD169+ macrophages after infection (P values ranging from 10−22 to 10−17; table S2). Furthermore, the GO enrichment analysis on down-regulated genes among activated CD169+ macrophages versus macrophages isolated from naïve mice showed no enrichment of immune- and proinflammatory-related processes (table S3). Analysis of enriched transcription factor (TF) sites in the promoters of the up-regulated genes between activated and naïve CD169+ macrophages identified 58 TFs highly expressed only in CD169+ macrophages from infected animals (table S4). A GO enrichment analysis indicated that genes related to immune response were significantly overrepresented in the identified TFs (P values ranging from 10−17 to 10−4). In table S5, we show the 18 enriched GO terms and the corresponding TFs associated with them.
CD169+ macrophages regulate splenic localization of Lm
Previous studies have shown that Lm is actively transported from the MZs to the T cell zones (14, 27), and this transport is mediated by CD8αα+ DCs (13). Once localized in the T cell zones, Lm growth is controlled by innate immune cell clusters that form and locally release inflammatory cytokines (3). Given our results showing the rapid capture of Lm by CD169+ macrophages, we reasoned that CD169+ macrophages may also play a role in mediating Lm localization within the spleen. Between 9 and 12 hpi, bacteria were observed transiting from the MZs to the T cell zones in WT mice (Fig. 3A, white arrowheads). However, in the absence of CD169+ macrophages, Lm was largely excluded from the white pulp. In CD169-DTR mice, Lm was observed replicating in the splenic red pulp and MZ (Fig. 3A, red arrowheads), with fewer than 10% of Lm seen moving into the white pulp by 12 hpi (Fig. 3B). By 24 hpi, when more than 80% of Lm was localized in the T cell zones of WT mice, CD169-DTR mice still had significantly less Lm confined to the T cell zones, despite more bacteria available for transport (Fig. 3, C and D). These results show how pathogens that disseminate systemically are captured and controlled by CD169+ macrophages in the spleen that are ideally positioned to serve as gatekeepers for invading pathogens. F4/80+ red pulp macrophages failed to guard the red pulp compartment from bacterial replication in the absence of CD169+ MZ macrophages, despite the fact that the number of red pulp macrophages in WT mice was comparable (or slightly increased) with CD169-DTR mice after infection (Fig. 3, E and F).
Fig. 3. CD169+ macrophages mediate Lm translocation to the splenic T cell zones.
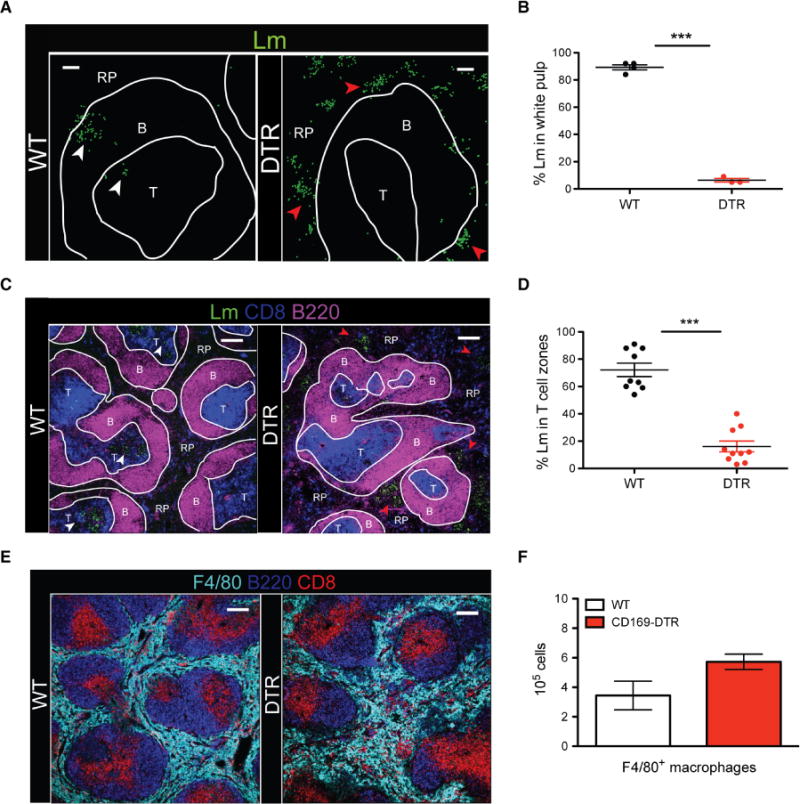
(A) Confocal images of spleen sections from WT and CD169-DTR mice at 12 hpi. Lm are rendered with spots in green, B cell zones (B), T cell zones (T), and red pulp (RP). White and red arrowheads indicate Lm inside and outside the white pulp, respectively. Scale bars, 50 μm. (B) Frequency of Lm located in splenic T cell zones of WT or CD169-DTR mice at 12 hpi. Each symbol represents a distinct region of splenic tissue. Data are shown as means ± SEM (***P < 0.0005, Student’s t test). (C) Confocal image of spleen section from WT or CD169-DTR mice at 24 hpi. Lm are rendered with spots in green, B cell zones, T cell zones, and red pulp. White and red arrowheads indicate Lm inside and outside the white pulp, respectively. Scale bars, 150 μm. (D) Frequency of Lm found in splenic T cell zones of WT or CD169-DTR mice at 24 hpi. Each symbol represents a distinct region of splenic tissue. Data are shown as means ± SEM (***P < 0.0005, Student’s t test). (E) Confocal image of spleen section from WT and CD169-DTR mice at 24 hpi stained for F4/80 (light blue), B220 (dark blue), and CD8α+ DCs (red). Scale bars, 150 μm. (F) FACS quantification of F4/80+ macrophages in the spleens of WT or CD169-DTR mice at 48 hpi. Data are shown as means ± SEM. All data are representative of three separate experiments with two to four mice per group.
CD169+ macrophages mediate a second wave of innate immune cell protection
Bacterial transport to the T cell zones is followed by the formation of organized clusters consisting of neutrophils, monocytes, and NK cells (3). These dense clusters of CD11b+ cells and NK cells provide a localized inflammatory response at sites of bacterial replication, which is essential for preventing protracted replication and spread of the bacteria (3). Given the disruption of Lm transport and unchecked Lm replication at and beyond 24 hpi in CD169-DTR mice (Fig. 4A), despite having equal or greater recruitment of innate immune cells (fig. S4A), we next assessed the formation of granulomas and hierarchical clustering of innate immune cells in the absence of CD169+ macrophages. Unlike WT mice where neutrophils and monocytes formed large, dense clusters around the bacteria in the T cell zones, myeloid cell clusters were smaller and dispersed throughout the splenic red pulp of CD169-DTR mice (Fig. 4B). Moreover, several Lm replication foci were completely devoid of neutrophils (Fig. 4B, white arrowheads).
Fig. 4. CD169+ macrophages play a critical role in mediating innate immune cell reorganization.
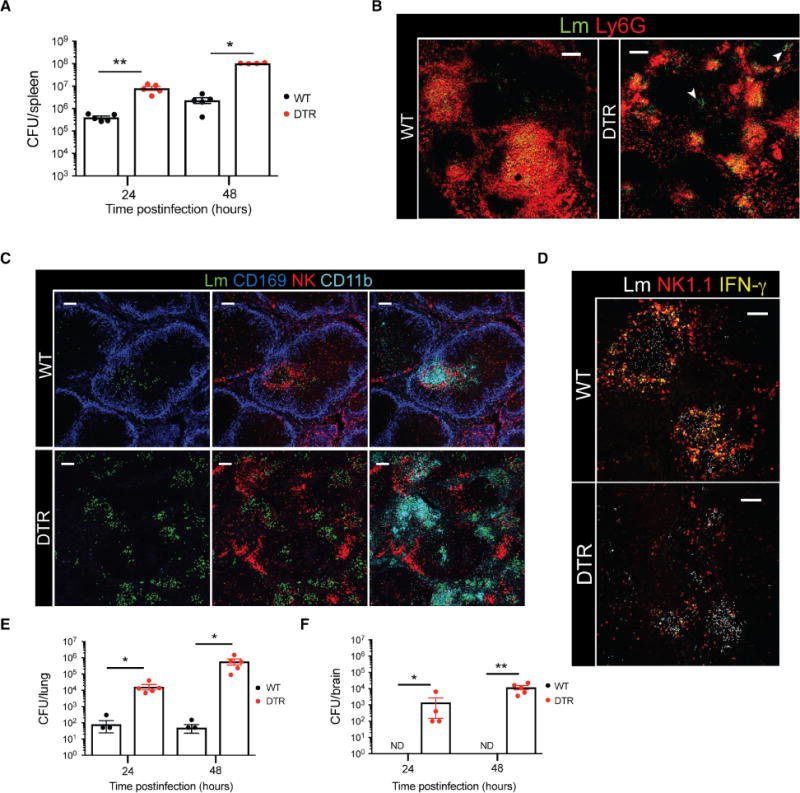
(A) Splenic Lm quantification in WT or CD169-DTR mice at 24 and 48 hpi. Each symbol represents an individual mouse. Data are shown as means ± SEM (*P < 0.05 and **P < 0.005, Mann-Whitney test). (B) Confocal images of WT or CD169-DTR mice at 24 hpi. Spleen sections stained for Lm (green) and Ly6G (red). Lm are rendered using spots. White arrowheads indicate Lm foci devoid of neutrophil clusters. Scale bars, 100 μm. (C) Confocal images of spleen sections from WT or CD169-DTR mice at 24 hpi showing organized clustering versus disrupted hierarchical clustering of innate immune cells. Lm are rendered with spots in green. CD169+ macrophages are shown in blue, NK cells are shown in red, and CD11b+ cells are shown in turquoise. Scale bars, 100 μm. (D) Representative images of spleen sections showing IFN-γ (yellow), NK1.1 (red), and Lm (white). Lm are rendered with spots. Scale bars, 80 μm. (E and F) Lm numbers quantified from (E) lungs and (F) brains of WT or CD169-DTR mice. ND, not detected. Each symbol represents an individual mouse. Data are shown as means ± SEM (*P < 0.05 and **P < 0.005, Mann-Whitney test). All data are representative of two to four separate experiments with two to four mice per group.
In WT mice, NK cells closely associated with myeloid cells, forming a distinct border around clusters of CD11b+ cells. However, in the absence of CD169+ macrophages, hierarchical clustering between NK cells and CD11b+ cells was abolished (Fig. 4C). Specifically, in CD169-DTR mice, NK cells were dissociated from CD11b+ cell clusters and Lm replication foci (fig. S4B, yellow arrows). These data show that hierarchical clustering was primarily driven by pathogen localization to the T cell zones, and organized granulomatous clusters failed to form adequately in the red pulp. We next examined the functional consequences of displaced innate immune cells in the absence of CD169+ macrophages. In accordance with a previous report (3), confocal immunofluorescence analysis of interferon-γ (IFN-γ) showed that cytokine production was concentrated at the periphery of Lm replication foci in splenic T cell zones of WT mice (Fig. 4D). In contrast, IFN-γ was widely dispersed throughout the red pulp of CD169-DTR mice and not entirely confined to Lm replication foci (Fig. 4D). In the absence of proper innate cell cluster formation, Lm spread to the lungs and brains of CD169-DTR mice, whereas bacterial central nervous system invasion in WT mice was negligible (Fig. 4, E and F). Dissociated clustering of CD11b+ cells and NK cells did not impair the production of proinflammatory cytokines or chemokines in the spleen (fig. S4, C and D). These findings indicated that, in addition to rapid capture of Lm, CD169+ macrophages played an important role in mediating the efficient transport of bacteria to the T cell zones.
CD169+ macrophages mediate bacterial transport to the T cell zones by trans-infecting CD8α+ DCs
Given that CD8αα+ DCs have previously been implicated in transporting Lm to T cell zones (13, 14), we considered the possibility that CD169+ macrophages were interacting with CD8αα+ DCs early after infection to enable Lm translocation to the T cell zone. As compared with CD169-DTR mice, confocal imaging analysis of splenic sections revealed the formation of prominent CD8αα+ DC clusters in the MZ, where Lm- infected CD169+ macrophages were present in WT mice (Fig. 5, A and B, white arrowheads). CD8αα+ DC clusters failed to form in mice depleted of CD169+ macrophages; instead, areas where Lm replication foci were observed were devoid of prominent CD8αα+ DC clusters (Fig. 5, A and B). Moreover, most of the Lm found transiting from the MZs to the T cell zones in WT mice were located within CD11c+ DCs (fig. S5A, white arrows). Even at later time points after infection (15 hpi), most of the Lm in WT mice were associated with CD8αα+ DCs; conversely, in CD169-DTR mice, very few CD8αα+ DCs were observed near Lm replication sites in the red pulp (fig. S5B). Higher magnification revealed that Lm-infected CD169+ macrophages interacted closely with CD8αα+ DCs, and bacteria were present at junctions between CD169+ macrophages and CD8αα+ DCs (Fig. 5C and movie S3).
Fig. 5. CD169+ macrophages mediate the transport of bacteria to T cell zones by trans-infecting CD8α + DCs.
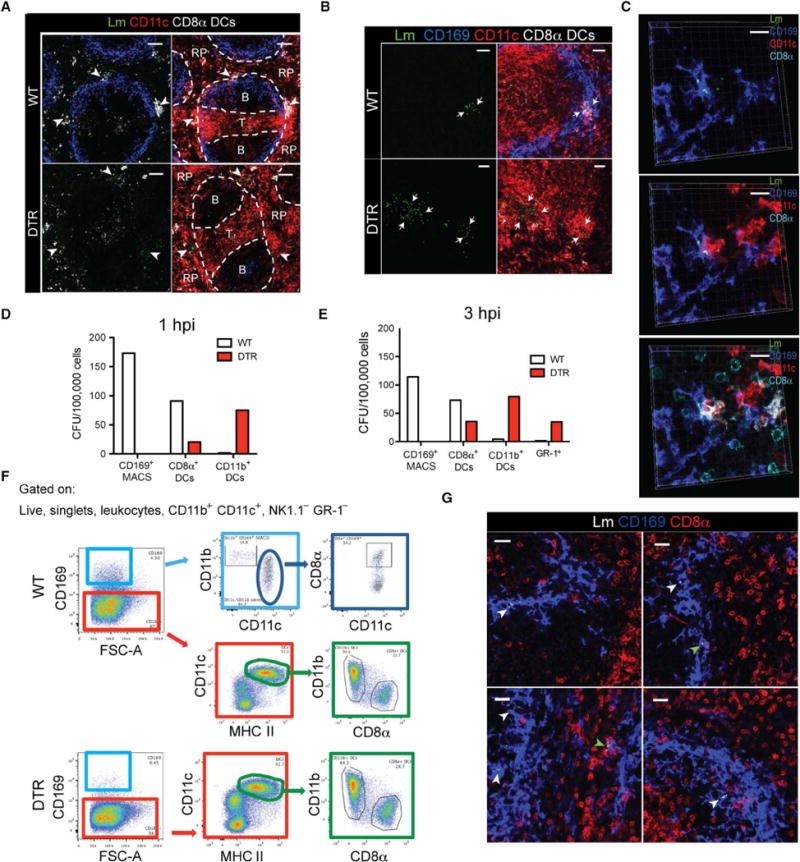
(A) Confocal images of spleen sections from WT or CD169-DTR mice at 6 hpi. Colocalization of CD11c and CD8α is shown in white. Lm are rendered with spots in green. White arrowheads indicate Lm in the MZ. Scale bars, 70 to 80 μm. (B) Confocal images of spleen sections from WT or CD169-DTR mice at 9 hpi. Spleen sections stained for Lm (green), CD169 (blue), and CD11c (red). Colocalization of CD11c and CD8α is shown in white. Lm are rendered with spots. White arrows indicate areas of Lm. Scale bars, 30 μm. (C) High-magnification confocal image showing Lm-infected CD169+ macrophages and CD8α+ DCs in close contact at 3 hpi. Spleen sections stained for Lm (green), CD169 (blue), CD11c (red), and CD8α (light blue). Scale bars, 20 μm. (D and E) Quantification of viable Lm recovered from FACS-sorted myeloid cells from pooled WT or CD169-DTR mice at (D) 1 hpi and (E) 3 hpi (n = 2 to 4). (F) General gating strategy used for CD169+ macrophages and DC subset identification from the spleen. (G) Representative confocal image of WT mice at 3 hpi. Spleen sections stained for Lm (white), CD169 (blue), and CD11c (red). Lm are rendered with spots. White and green arrowheads indicate Lm in CD169+ cells and CD169+CD11c+ cells, respectively. Scale bars, 20 μm. All data are representative of three to four separate experiments with two to four mice per group.
We hypothesized that infected CD169+ macrophages mediated the recruitment and clustering of CD8αα+ DCs at the sites of infection and facilitated the trans-infection of CD8αα+ DCs, enabling efficient Lm transport to T cell zones. We sorted CD8αα+ DCs and quantified the number of live Lm from these cells in the presence and absence of CD169+ macrophages, as performed previously (16, 28). These studies revealed that, in the absence of CD169+ macrophages, the number of live Lm harbored by CD8αα+ DCs was markedly reduced compared with WT mice (Fig. 5, D and E). This was not due to inherent differences in the number or spatial positioning of CD8αα+ DCs in WT and CD169-DTR mice at later time points of infection (fig. S6, A and B). As shown previously for the LNs (28), we find that a subset of splenic CD169+ macrophages also expresses CD8αα (Fig. 5F). Given that CD8αα+CD169+ macrophages were also depleted after DT treatment, it was possible that CD8αα+CD169+CD11c+ cells were trafficking Lm to the T cell zones, and in their absence, Lm translocation was disrupted. However, our confocal imaging analysis showed that most of the Lm were harbored by CD8αα−CD169+ macrophages early after infection (Fig. 5G, white arrowheads). Although in some instances, CD8αα+CD169+ cells were infected (green arrowheads), these cells appeared to be CD8αα+ DCs clustering with infected CD169+ macrophages. Throughout this study, we used CD8αα and CD11c costaining to identify CD8αα+ DCs because it is widely used in flow cytometry analysis. To ensure that the costaining method identifies these DC subsets with high fidelity, we stained spleen sections with XCR1 antibody (a prominent chemokine receptor preferentially expressed on CD8αα+ DCs). As shown in fig. S6C, all CD8αα+CD11c+ costained DCs also stained for XCR1. Furthermore, we costained spleen sections for CD8αα, CD11c, and T cell receptor β (TCRβ) and observed that CD8αα+CD11c+ cells did not costain with TCRβ (fig. S6D), supporting our conclusions that CD8αα+ DCs alone and not CD8αα+ T cells were clustering in the spleen after infection. Together, these data show that, despite greater bacterial burdens, CD8αα+ DCs from CD169-DTR mice harbored fewer Lm, with the cellular tropism of Lm significantly skewed toward CD11b+ DCs (Fig. 5D) and GR-1+ cells (Fig. 5E).
Batf3−/− mice are susceptible to Lm infection in the absence of CD169+ macrophages
Previous reports have shown that splenic CD8αα+ DCs are required for a productive Lm infection in the spleen because they provide a permissive replication niche for bacteria (13, 15, 16). Our findings indicated that CD169+ macrophages play a primary host to Lm, so we sought to reconcile our results with the current paradigm. We hypothesized that if CD8αα+ DCs are solely required for a productive Lm infection in the spleen, mice lacking CD8αα+ DCs should not exhibit impaired bacterial clearance in the absence of CD169+ macrophages. We crossed CD169-DTR mice with Batf3−/− mice, generated CD169-DTR–Batf3−/− mice (DTR-Batf3−/−) that lacked splenic CD8αα+ DCs, and determined their ability to control Lm infection when CD169+ macrophages were depleted. In accordance with a previous report (21), Batf3−/− mice had significantly reduced splenic Lm burdens compared with WT mice (Fig. 6, A and B). However, CD169-DTR–Batf3−/− mice harbored significantly greater Lm burdens at 12 and 24 hpi (Fig. 6, A and B). These results indicated that the protection provided by CD169+ macrophages precedes the pathogen-favorable CD8αα+ DC niche. Furthermore, CD169-DTR–Batf3−/− mice susceptibility was not due to deficiencies in innate immune cell recruitment because the infiltration of innate immune cells in the spleens of CD169-DTR–Batf3−/− mice recapitulated the cellular differentials seen in CD169-DTR mice (fig. S7A). We did not observe BATF3-independent generation of CD8αα+ DCs after infection in Batf3−/− or CD169-DTR–Batf3−/− mice (fig. S7B). CD169-DTR–Batf3−/− mice also exhibited impaired Lm translocation to a similar extent as observed in CD169-DTR mice (Fig. 6, C and D). Quantification of splenic bacterial burdens at 1 hpi showed that WT and Batf3−/− mice harbored equal numbers of Lm (Fig. 6E), indicating that the early deposition and control of Lm in the spleen were unaltered between WT and Batf3−/− mice. These findings suggested that resistance to Lm infection in Batf3−/− mice was likely mediated by a combination of rapid capture and clearance by CD169+ macrophages, as well as by a lack of the replication-favorable CD8αα+ DC niche onto which CD169+ macrophages transfer Lm. Our confocal microscopy analysis of Batf3−/− mice early after infection showed that most of the bacteria in Batf3−/− mice at 3 hpi were located in CD169+ macrophages (Fig. 6F).
Fig. 6. CD8α+ DC-deficient mice are highly susceptible to Lm infection in the absence of CD169+ macrophages.
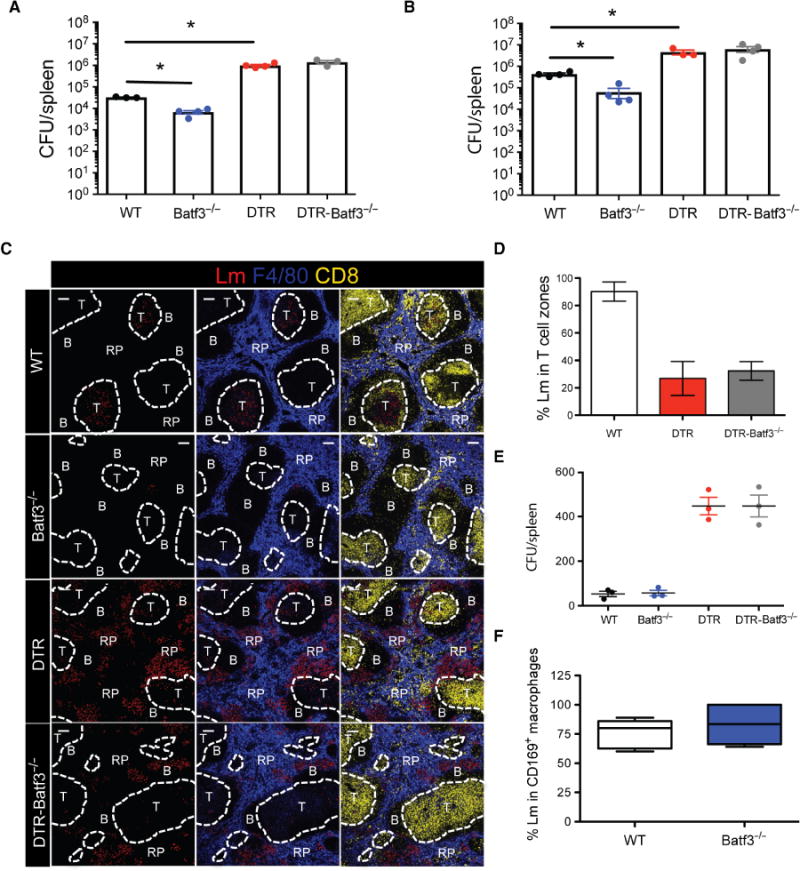
(A and B) Splenic Lm quantification of WT, Batf3−/−, CD169-DTR, or CD169-DTR–Batf3−/− mice at (A) 12 hpi and (B) 24 hpi. Each symbol represents individual mouse. Data are shown as means ± SEM (*P < 0.05, Mann-Whitney test). (C) Confocal images of spleen sections from WT, Batf3−/−, CD169-DTR, or CD169-DTR–Batf3−/− mice at 24 hpi. Spleen sections stained for Lm (red), F4/80 (blue), and CD8 (yellow). Lm are rendered with spots. Scale bars, 100 μm. (D) Frequency of Lm found in splenic T cell zones at 24 hpi in WT, CD169-DTR, or CD169-DTR–Batf3−/− mice. Data are shown as means ± SEM. (E) Lm quantification in the spleens of WT, Batf3−/−, CD169-DTR, or CD169-DTR–Batf3−/− mice at 1 hpi. Each symbol represents an individual mouse. Data are shown as means ± SEM (**P < 0.005, Mann-Whitney test). (F) Frequency of intracellular Lm located within CD169+ macrophages in WT or Batf3−/− mice at 3 hpi. All data are representative of two to four separate experiments.
CD8α DC recruitment to infected CD169+ macrophages requires phagosomal escape of Lm
We sought to further explore the mechanisms driving CD8αα+ DC recruitment to Lm-infected CD169+ macrophages. First, we determined whether Lm-encoded actin assembly–induced protein (ActA) was required for CD8αα+ DC trans-infection. Using an ActA-deficient Lm strain, which is unable to efficiently polymerize the host cells’ actin filaments for cell-to-cell motility but able to escape the phagolyso-some (19), we observed that, similar to WT Lm, ActA-deficient Lm infection also resulted in the clustering of CD8αα+ DCs in the MZ at sites of infection (Fig. 7A) and the subsequent trans-infection of the DC population, indicating that ActA mediating cell-to-cell spread was not important for DC trans-infection. Another Lm-encoded virulent factor listeriolysin O (LLO) is a pore-forming hemolysin protein that allows the bacteria to escape the phagosomes and replicate in the cytosol. In sharp contrast to WT and ActA-deficient Lm infection, after LLO-deficient Lm infection, CD8αα+ DCs did not cluster with Lm-infected macrophages (Fig. 7A). These findings indicate that CD169+ macrophage–mediated recruitment of CD8αα+ DCs to sites of infection is mediated by bacterial cytosol invasion. Quantification of intracellular Lm at 12 hpi with WT Lm, ActA-deficient Lm, or LLO-deficient Lm showed that, in the case of WT and ActA-deficient Lm infections, because the bacteria were being transferred to CD8αα+ DCs continuously, we observed a greater spread of data points with regard to percentage of Lm in CD169+ macrophages (Fig. 7B). However, LLO-deficient Lm was almost exclusively found in CD169+ macro phages, suggesting that when unable to escape the phagolysosome, the cellular niche of Lm was entirely confined to these cells.
Fig. 7. Cytosolic entry of Lm required for CD8α + DC recruitment.
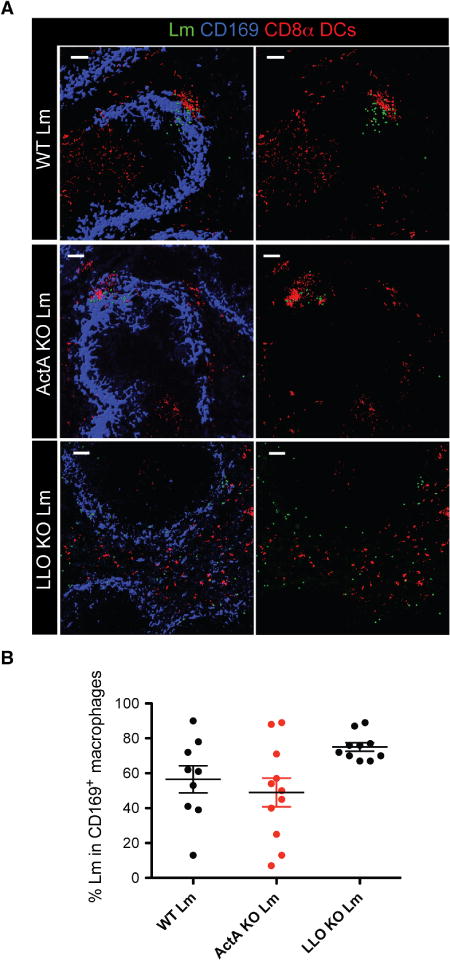
(A) Confocal microscopy of spleen sections from WT mice at 12 hpi with WT Lm (top), ActA knockout (KO) Lm (middle), and LLO KO Lm (bottom). Lm are rendered with spots in green. CD169 is shown in blue, and colocalization of CD11c and CD8α is shown in red. Scale bars, 50 μm. (B) Quantification of intracellular Lm within CD169+ macrophages at 12 hpi. Each symbol represents a different confocal image quantified. All data are representative of two separate experiments with two to three mice per group.
DISCUSSION
Here, we examined the role of splenic CD169+ macrophages during systemic infections. Using several blood-borne pathogens, we demonstrate the essential role of splenic CD169+ macrophages in host protection. A previous study (5) used clodronate liposomes to broadly deplete all phagocytic cells to show that mice were highly susceptible to Lm. Here, using a CD169-DTR system to exclusively deplete MZ macrophages, we demonstrate that the local trafficking of Lm within the spleen was also controlled by CD169+ macrophages. We found that CD169+ macrophages play host to Lm early after infection, and their absence resulted in protracted bacterial growth and spread, which ultimately lead to death of the infected animals. The bacteria were not controlled despite CD169-DTR mice mounting equal or greater inflammatory responses during infection. This led us to further investigate how CD169+ macrophages were contributing to innate immune protection during acute bacterial infection. In addition to their essential role in controlling bacterial growth, our study revealed that CD169+ macrophages play a critical role in mediating the localization of Lm, which ultimately drives the effective spatial reorganization of innate immune cells, vital for providing protracted innate immune protection.
Currently, it is understood that, after initial deposition in the spleen, Lm is transported from the MZs to the T cell zones by CD8αα+ DCs, where Lm further replicates (14, 29). The redistribution of Lm to the T cell zones is followed by a second wave of innate immune responses characterized by the subsequent reorganization of monocytes, neutrophils, and NK cells into hierarchical clusters at sites of Lm replication foci (3). We document here that, in the absence of CD169+ macrophages, transport of Lm to the T cell zones was impaired, and reorganization of innate immune cells into organized hierarchical clusters was abrogated. Thus, the localization of Lm in the T cell zones was critical for the subsequent reorganization of monocytes, neutrophils, and NK cells into hierarchical clusters, allowing for the appropriate geographical production of cytokines such as IFN-γ and preventing the spread and unchecked growth of Lm (fig. S8). Our results showed that the environment in the splenic red pulp was not conducive to forming organized innate immune cell granulomas, which was likely due to several factors, including altered chemokine expression and increased sheer forces as a result of fast-moving blood that prevent the homotypic and heterotypic adhesions required between innate immune cells (30, 31) for forming large organized granulomatous structures. Although the red pulp is densely populated by F4/80+ macrophages, these macrophages were unable to control Lm replication and spread.
A role for CD169+ macrophages mediating pathogen localization in the spleen has not been previously described. Thus, we sought to reconcile our results showing that CD169+ macrophages were the primary cellular host for Lm early after infection with previous studies that have shown that Lm transport to the T cell zones occurs via migration of Lm-infected CD8αα+ DCs to T cell zones (13, 14). We considered that CD169+ macrophages could potentially contribute to pathogen transport. We observed that after infection, CD8αα+ DCs clustered closely with Lm-infected CD169+ macrophages, which did not occur in CD169-DTR mice. This clustering of CD8αα+ DCs resulted in apparent trans-infection of CD8αα+ DCs by CD169+ macrophages, which was in line with previous reports that have demonstrated the ability of CD169+ macrophages to directly transfer viral particles to B cells (10) and lymphocytes (32), as well as transfer antigen to CD8αα+ DCs for cross-presentation (33). Although a small percentage of CD169+ cells in the spleen can express CD8αα, our results showed that most of the Lm were harbored by CD169+CD8αα− macrophages early after infection. Moreover, most of the Lm being transported to the T cell zones (between 12 and 20 hpi) were only in CD11c+ cells and not in CD169+ cells. Although collectively our data strongly indicate that CD169+ macrophages trans-infect CD8αα+ DCs that then transport the bacteria to the T cell zones, we cannot exclude the possibility that a portion of the initially deposited Lm in the spleen is directly transported to the splenic T cell zones by infected CD169+CD8αα+CD11c+ cells that may be a subset of DCs rather than macrophages. Moreover, CD169+CD8αα+CD11c+ cells are among the cells that are deleted after DT treatment in CD169-DTR mice.
The establishment of a productive splenic Lm infection was previously shown to require CD8αα+ DCs because they provide a permissive replication niche for bacteria (13, 15, 16). Unexpectedly, DTR-Batf3−/− mice that were deficient in both CD8αα+ DCs and CD169+ macrophages were unable to control Lm infection. This result demonstrated that the protective role of CD169+ macrophages predominates the pathogen-favorable CD8αα+ DC niche. Differences in bacterial burdens between WT and Batf3−/− mice were not due to altered initial deposition of bacteria into the spleen because both WT and Batf3−/− mice harbored equal numbers of Lm at early time points after infection. Our imaging studies showed that CD169+ macrophages harbored most of the Lm in Batf3−/− mice after infection, indicating that the resistance to Lm infection in these mice was due, in part, to rapid uptake and clearance of Lm by CD169+ macrophages and the inability to subsequently trans-infect CD8αα+ DCs, allowing further bacterial replication in the T cell zones. Thus, our findings will necessitate the reassessment of the current paradigms related to the requirement of CD8αα+ DCs for establishing a productive infection and bacterial localization in the spleen after Lm infection.
We were intrigued by the fact that CD8αα+ DCs only clustered and formed close contacts with infected CD169+ macrophages. We reasoned that the infection was essential for inducing the signals in the macrophages that resulted in the recruitment of CD8αα+ DCs. Initially, we explored the possibility that ActA-dependent cell-to-cell spread may be required for the recruitment and trans-infection of CD8αα+ DCs. However, unexpectedly, ActA-mediated cell-to-cell spread was not important for the recruitment, clustering, and trans-infection of CD8αα+ DCs. Conversely, LLO-mediated phagosomal escape in the Lm-infected macrophages was required for proper recruitment and clustering of CD8αα+ DCs. Thus, the induction of signals that promote CD8αα+ DC trans-infection likely requires cytosolic bacterial replication, which suggests that DNA sensing in the cytoplasm of the macrophages may promote activation of genes that are important for regulating this process. This result explains why LLO-deficient Lm-based vaccine vectors are poor at inducing a potent CD8 T cell response. Because ActA- assisted cell-to-cell spread was not required for the trans-infection of Lm from CD169+ macrophages to CD8αα DCs, other processes such as trogocytosis (34) likely play a more important role.
Together, our findings shed new light on the role of CD169+ MZ macrophages in mediating protection against pathogens. The multi-step progression of the antimicrobial innate immune response was heavily dependent on the CD169+ macrophages, and without their presence, the entire hierarchy of events needed for the clearance of the pathogen and the subsequent survival of the host was disrupted. Our report provides fundamental insights into how cellular communication of innate immune cells is critical for regulating the anatomical positioning of the pathogen in the context of protective innate immune responses in secondary lymphoid organs.
MATERIALS AND METHODS
Mice
Female C57BL/6 mice were purchased from Charles River, and B6.129S(C)-Batf3tm1Kmm (Batf3−/−) mice were purchased from the Jackson Laboratory. All mice, including Siglec1DTR/+ (CD169-DTR) mice, were bred and genotyped at the University of Connecticut (UConn) Health Center. CD169-DTR mice were crossed with Batf3−/− mice. All mice were maintained in an Association for Assessment and Accreditation of Laboratory Animal Care–accredited facility. All procedures were performed in accordance with approved University of Connecticut Health Center Institutional Animal Care and Use Committee animal protocols.
Infections
Mice were treated with DT (40 ng/g body weight intravenously) (Sigma- Aldrich) 2 days before infection. Mice were infected with WT Lm (10403S) or ovalbumin-expressing Lm (Lm-OVA; 10403S) intravenously. For early infection studies (1 to 12 hpi), mice were infected with 105 to 106 WT Lm. For time points beyond 12 hpi, mice were infected with 103 to 104 WT Lm. Mice were infected with 107 LLO- deficient GFP Lm (NF-L519) and 105 ActA-deficient Lm (10403S) intravenously. For oral infection, mice were infected with 1010 internalin A mutant Lm (10403S) by gavage (35). For E. coli studies, mice were infected with 1.66 × 108 colony-forming units of E. coli (strain BL21). For MCMV studies, mice were infected with 3 × 105 plaque-forming units of MCMV expressing GFP (Smith strain).
Cell sorting and purification for intracellular bacterial quantification
Quantification of live bacteria within different sorted splenic cellular populations was performed as previously described (16, 28). Briefly, for cell sorting, splenic tissue was incubated in RPMI 1640 containing 10% fetal calf serum, collagenase IV (100 U/ml), gentamicin (5 μg/ml), 1 mM Hepes, 5 mM glutamine, 1 mM CaCl2, 1 mM MgCl2, and deoxy-ribonuclease I (1 μg/ml) for 30 min at 37°C. After incubation, splenic tissue was dissociated into a single-cell suspension and treated with tris-buffered ammonium chloride, and the remaining leukocytes were positively enriched for CD11b and CD11c expression using MACS magnetic beads (Miltenyi Biotec). Enriched fractions were stained for a live-dead marker, CD11b, CD11c, major histocompatibility complex II, CD8αα, CD169, B220, NK1.1, and GR-1 (table S6). CD169+ cells, CD11b+ DCs, and CD8αα+ DCs were sorted on a Becton Dickinson FACSAria II.
Supplementary Material
Movie S1. Listeria is captured by CD169+ macrophages early after infection.
Movie S2. Listeria is captured by CD169+ macrophages early after infection.
Movie S3. CD169+ macrophages transfer bacteria to CD8αα+ DCs.
Fig. S1. Characterization of WT and CD169-DTR mice.
Fig. S2. Splenic CD169+ macrophages are essential for protection against various blood-borne pathogens.
Fig. S3. Sorting strategy and quality control for RNA sequencing.
Fig. S4. CD169-DTR mice have elevated but displaced cytokine production.
Fig. S5. CD169+ macrophages recruit CD8αα+ DCs to early sites of infection.
Fig. S6. Characterization of CD8αα+ DCs in CD169-DTR mice.
Fig. S7. Myeloid cell differentials comparing Batf3−/− knockout and CD169-DTR–Batf3−/− knockout mice.
Fig. S8. Model depicting the role of CD169+ macrophages after Lm infection.
Table S1. Top 15 GO enriched biological processes up-regulated in CD169+ macrophages versus red pulp macrophages at steady state.
Table S2. Top 15 GO enriched biological processes up-regulated in CD169+ macrophages during infection versus steady state.
Table S3. Top 15 GO enriched biological processes down-regulated in CD169+ macrophages during infection versus steady state.
Table S4. List of identified TF candidates for up-regulated genes in CD169+ macrophages isolated from infected mice versus uninfected mice.
Table S5. Enriched immune-related GO terms for corresponding TFs in CD169+ macrophages during infection versus steady state.
Table S6. Antibodies used for fluorescence-activated cell sorting (FACS) and confocal microscopy.
Acknowledgments
We thank X. Pham for technical assistance, E. Jellison for help with flow cytometry, B. Reese for help with RNA sequencing, the Center for Cell Analysis and Modeling at UConn Health for help with confocal imaging, and J. C. Brunson for help with RNA sequencing data analysis. We thank L. Puddington and V. Rathinam for critically reading the manuscript.
Funding: This work was supported by NIH grant AI097375 (to K.M.K.).
Footnotes
SUPPLEMENTARY MATERIALS
immunology.sciencemag.org/cgi/content/full/2/16/eaah5520/DC1 Materials and Methods
Source data (Excel file)
References (36–38)
Author contributions: K.M.K. directed the study. O.A.P. and K.M.K. designed the study. O.A.P., S.T.Y., and L.M. performed the experiments. P.A.R., B.B.U., and M.T. provided reagents and analytical tools. P.V.-L. and T.S. provided help with bioinformatics. O.A.P. and K.M.K. analyzed the data and wrote the paper.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: Sequencing data have been deposited in the National Center for Biotechnology Information Sequence Read Archive (SRA#SUB2908162).
REFERENCES AND NOTES
- 1.Taniguchi LU, Correia MDT, Zampieri FG. Overwhelming post-splenectomy infection: Narrative review of the literature. Surg Infect (Larchmt) 2014;15:686–693. doi: 10.1089/sur.2013.051. [DOI] [PubMed] [Google Scholar]
- 2.Mebius RE, Kraal G. Structure and function of the spleen. Nat Rev Immunol. 2005;5:606–616. doi: 10.1038/nri1669. [DOI] [PubMed] [Google Scholar]
- 3.Kang SJ, Liang HE, Reizis B, Locksley RM. Regulation of hierarchical clustering and activation of innate immune cells by dendritic cells. Immunity. 2008;29:819–833. doi: 10.1016/j.immuni.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bronte V, Pittet MJ. The spleen in local and systemic regulation of immunity. Immunity. 2013;39:806–818. doi: 10.1016/j.immuni.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aichele P, Zinke J, Grode L, Schwendener RA, Kaufmann SHE, Seiler P. Macrophages of the splenic marginal zone are essential for trapping of blood-borne particulate antigen but dispensable for induction of specific T cell responses. J Immunol. 2003;171:1148–1155. doi: 10.4049/jimmunol.171.3.1148. [DOI] [PubMed] [Google Scholar]
- 6.Mebius RE, Nolte MA, Kraal G. Development and function of the splenic marginal zone. Crit Rev Immunol. 2004;24:449–464. doi: 10.1615/critrevimmunol.v24.i6.40. [DOI] [PubMed] [Google Scholar]
- 7.Honke N, Shaabani N, Cadeddu G, Sorg UR, Zhang DE, Trilling M, Klingel K, Sauter M, Kandolf R, Gailus N, van Rooijen N, Burkart C, Baldus SE, Grusdat M, Löhning M, Hengel H, Pfeffer K, Tanaka M, Häussinger D, Recher M, Lang PA, Lang KS. Enforced viral replication activates adaptive immunity and is essential for the control of a cytopathic virus. Nat Immunol. 2012;13:51–57. doi: 10.1038/ni.2169. [DOI] [PubMed] [Google Scholar]
- 8.Aoshi T, Carrero JA, Konjufca V, Koide Y, Unanue ER, Miller MJ. The cellular niche of Listeria monocytogenes infection changes rapidly in the spleen. Eur J Immunol. 2009;39:417–425. doi: 10.1002/eji.200838718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iannacone M, Moseman EA, Tonti E, Bosurgi L, Junt T, Henrickson SE, Whelan SP, Guidotti LG, von Andrian UH. Subcapsular sinus macrophages prevent CNS invasion on peripheral infection with a neurotropic virus. Nature. 2010;465:1079–1083. doi: 10.1038/nature09118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Junt T, Moseman EA, Iannacone M, Massberg S, Lang PA, Boes M, Fink K, Henrickson SE, Shayakhmetov DM, Di Paolo NC, van Rooijen N, Mempel TR, Whelan SP, von Andrian UH. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature. 2007;450:110–114. doi: 10.1038/nature06287. [DOI] [PubMed] [Google Scholar]
- 11.Gaya M, Castello A, Montaner B, Rogers N, Reis e Sousa C, Bruckbauer A, Batista FD. Inflammation-induced disruption of SCS macrophages impairs B cell responses to secondary infection. Science. 2015;347:667–672. doi: 10.1126/science.aaa1300. [DOI] [PubMed] [Google Scholar]
- 12.Asano K, Nabeyama A, Miyake Y, Qiu C-H, Kurita A, Tomura M, Kanagawa O, Fujii S-i, Tanaka M. CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity. 2011;34:85–95. doi: 10.1016/j.immuni.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 13.Edelson BT, Bradstreet TR, Hildner K, Carrero JA, Frederick KE, KC W, Belizaire R, Aoshi T, Schreiber RD, Miller MJ, Murphy TL, Unanue ER, Murphy KM. CD8αα+ dendritic cells are an obligate cellular entry point for productive infection by. Listeria monocytogenes Immunity. 2011;35:236–248. doi: 10.1016/j.immuni.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aoshi T, Zinselmeyer BH, Konjufca V, Lynch JN, Zhang X, Koide Y, Miller MJ. Bacterial entry to the splenic white pulp initiates antigen presentation to CD8+ T cells. Immunity. 2008;29:476–486. doi: 10.1016/j.immuni.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 15.Neuenhahn M, Kerksiek KM, Nauerth M, Suhre MH, Schiemann M, Gebhardt FE, Stemberger C, Panthel K, Schröder S, Chakraborty T, Jung S, Hochrein H, Rüssmann H, Brocker T, Busch DH. CD8αα+ dendritic cells are required for efficient entry of Listeria monocytogenes into the spleen. Immunity. 2006;25:619–630. doi: 10.1016/j.immuni.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 16.Verschoor A, Neuenhahn M, Navarini AA, Graef P, Plaumann A, Seidlmeier A, Nieswandt B, Massberg S, Zinkernagel RM, Hengartner H, Busch DH. A platelet-mediated system for shuttling blood-borne bacteria to CD8αα+ dendritic cells depends on glycoprotein GPIb and complement C3. Nat Immunol. 2011;12:1194–1201. doi: 10.1038/ni.2140. [DOI] [PubMed] [Google Scholar]
- 17.Fettucciari K, Quotadamo F, Noce R, Palumbo C, Modesti A, Rosati E, Mannucci R, Bartoli A, Marconi P. Group B Streptococcus (GBS) disrupts by calpain activation the actin and microtubule cytoskeleton of macrophages. Cell Microbiol. 2011;13:859–884. doi: 10.1111/j.1462-5822.2011.01584.x. [DOI] [PubMed] [Google Scholar]
- 18.Miyake Y, Asano K, Kaise H, Uemura M, Nakayama M, Tanaka M. Critical role of macrophages in the marginal zone in the suppression of immune responses to apoptotic cell–associated antigens. J Clin Invest. 2007;117:2268–2278. doi: 10.1172/JCI31990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, Cossart P. L. monocytogenes -induced actin assembly requires the actA gene product, a surface protein. Cell. 1992;68:521–531. doi: 10.1016/0092-8674(92)90188-i. [DOI] [PubMed] [Google Scholar]
- 20.Disson O, Grayo S, Huillet E, Nikitas G, Langa-Vives F, Dussurget O, Ragon M, Le Monnier A, Babinet C, Cossart P, Lecuit M. Conjugated action of two species-specific invasion proteins for fetoplacental listeriosis. Nature. 2008;455:1114–1118. doi: 10.1038/nature07303. [DOI] [PubMed] [Google Scholar]
- 21.Nikitas G, Deschamps C, Disson O, Niault T, Cossart P, Lecuit M. Transcytosis of Listeria monocytogenes across the intestinal barrier upon specific targeting of goblet cell accessible E-cadherin. J Exp Med. 2011;208:2263–2277. doi: 10.1084/jem.20110560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bou Ghanem EN, Jones GS, Myers-Morales T, Patil PD, Hidayatullah AN, D’Orazio SEF. InlA promotes dissemination of Listeria monocytogenes to the mesenteric lymph nodes during food borne infection of mice. PLOS Pathog. 2012;8:e1003015. doi: 10.1371/journal.ppat.1003015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lavin Y, Mortha A, Rahman A, Merad M. Regulation of macrophage development and function in peripheral tissues. Nat Rev Immunol. 2015;15:731–744. doi: 10.1038/nri3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma’ayan A, Chua WJ, Hansen TH, Turley SJ, Merad M, Randolph GJ. Immunological Genome Consortium, Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell. 2014;157:832–844. doi: 10.1016/j.cell.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurotaki D, Uede T, Tamura T. Functions and development of red pulp macrophages. Microbiol Immunol. 2015;59:55–62. doi: 10.1111/1348-0421.12228. [DOI] [PubMed] [Google Scholar]
- 27.Conlan JW. Early pathogenesis of Listeria monocytogenes infection in the mouse spleen. J Med Microbiol. 1996;44:295–302. doi: 10.1099/00222615-44-4-295. [DOI] [PubMed] [Google Scholar]
- 28.Neuenhahn M, Schiemann M, Busch DH. DCs in mouse models of intracellular bacterial infection. Methods Mol Biol. 2010;595:319–329. doi: 10.1007/978-1-60761-421-0_21. [DOI] [PubMed] [Google Scholar]
- 29.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM. Batf3 deficiency reveals a critical role for CD8αα+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neelamegham S, Taylor AD, Shankaran H, Smith CW, Simon SI. Shear and time-dependent changes in Mac-1, LFA-1, and ICAM-3 binding regulate neutrophil homotypic adhesion. J Immunol. 2000;164:3798–3805. doi: 10.4049/jimmunol.164.7.3798. [DOI] [PubMed] [Google Scholar]
- 31.Simon SI, Neelamegham S, Taylor A, Smith CW. The multistep process of homotypic neutrophil aggregation: A review of the molecules and effects of hydrodynamics. Cell Adhes Commun. 1998;6:263–276. doi: 10.3109/15419069809004482. [DOI] [PubMed] [Google Scholar]
- 32.Sewald X, Ladinsky MS, Uchil PD, Beloor J, Pi R, Herrmann C, Motamedi N, Murooka TT, Brehm MA, Greiner DL, Shultz LD, Mempel TR, Bjorkman PJ, Kumar P, Mothes W. Retroviruses use CD169-mediated trans-infection of permissive lymphocytes to establish infection. Science. 2015;350:563–567. doi: 10.1126/science.aab2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Backer R, Schwandt T, Greuter M, Oosting M, Jüngerkes F, Tüting T, Boon L, O’Toole T, Kraal G, Limmer A, den Haan JMM. Effective collaboration between marginal metallophilic macrophages and CD8+ dendritic cells in the generation of cytotoxic T cells. Proc Natl Acad Sci USA. 2010;107:216–221. doi: 10.1073/pnas.0909541107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steele S, Radlinski L, Taft-Benz S, Brunton J, Kawula TH. Trogocytosis-associated cell to cell spread of intracellular bacterial pathogens. eLife. 2016;5:e10625. doi: 10.7554/eLife.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheridan BS, Pham QM, Lee YT, Cauley LS, Puddington L, Lefrançois L. Oral infection drives a distinct population of intestinal resident memory CD8+ T cells with enhanced protective function. Immunity. 2014;40:747–757. doi: 10.1016/j.immuni.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, Voss N, Stegmaier P, Lewicki-Potapov B, Saxel H, Kel AE, Wingender E. TRANSFAC® and its module TRANSCompel®: Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–D110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kel AE, Gößling E, Reuter I, Cheremushkin E, Kel-Margoulis OV, Wingender E. MATCH™: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003;31:3576–3579. doi: 10.1093/nar/gkg585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kel A, Voss N, Jauregui R, Kel-Margoulis O, Wingender E. Beyond microarrays: Finding key transcription factors controlling signal transduction pathways. BMC Bioinformatics. 2006;7(suppl. 2):S13. doi: 10.1186/1471-2105-7-S2-S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Listeria is captured by CD169+ macrophages early after infection.
Movie S2. Listeria is captured by CD169+ macrophages early after infection.
Movie S3. CD169+ macrophages transfer bacteria to CD8αα+ DCs.
Fig. S1. Characterization of WT and CD169-DTR mice.
Fig. S2. Splenic CD169+ macrophages are essential for protection against various blood-borne pathogens.
Fig. S3. Sorting strategy and quality control for RNA sequencing.
Fig. S4. CD169-DTR mice have elevated but displaced cytokine production.
Fig. S5. CD169+ macrophages recruit CD8αα+ DCs to early sites of infection.
Fig. S6. Characterization of CD8αα+ DCs in CD169-DTR mice.
Fig. S7. Myeloid cell differentials comparing Batf3−/− knockout and CD169-DTR–Batf3−/− knockout mice.
Fig. S8. Model depicting the role of CD169+ macrophages after Lm infection.
Table S1. Top 15 GO enriched biological processes up-regulated in CD169+ macrophages versus red pulp macrophages at steady state.
Table S2. Top 15 GO enriched biological processes up-regulated in CD169+ macrophages during infection versus steady state.
Table S3. Top 15 GO enriched biological processes down-regulated in CD169+ macrophages during infection versus steady state.
Table S4. List of identified TF candidates for up-regulated genes in CD169+ macrophages isolated from infected mice versus uninfected mice.
Table S5. Enriched immune-related GO terms for corresponding TFs in CD169+ macrophages during infection versus steady state.
Table S6. Antibodies used for fluorescence-activated cell sorting (FACS) and confocal microscopy.