

Abstract
Introduction
Translocation of phosphatidylserine from the inner leaflet to the outer leaflet of the endothelial membrane via phospholipid scramblase-1 (PLSCR1) is an apoptotic signal responsible for the loss of endothelial barrier integrity after ischemia-reperfusion injury. We hypothesized that inhibiting phosphatidylserine expression on endothelial cells would attenuate ischemia-reperfusion (IRI) induced increases in hydraulic permeability (Lp).
Methods
Mesenteric hydraulic permeability (Lp) was measured in rat post-capillary mesenteric venules subjected to IRI via superior mesenteric artery (SMA) occlusion (45 minutes) and release (300 minutes) in conjunction with several inhibitors of phosphatidylserine exposure as follows: 1) inhibition of PLSCR1 translocation (DTE - dithioerythritol, n=3), 2) inhibition of PLSCR1 membrane trafficking (2-BP - 2-bromopalmitate, n=3), and 3) inhibition of ion exchange necessary for PLSCR1 function (DIDS - 4,4′-Diisothiocyano-2,2′-stilbenedisulfonic acid, n=3). Under the same IRI conditions, rats were also administered targeted inhibitors of phosphatidylserine exposure including knockdown of phospholipid scramblase-1 (n=3) using RNA interference (RNAi), and as a potential therapeutic tool Diannexin, a selective phosphatidylserine blocker (n=3).
Results
During IRI net hydraulic permeability (Lp) increased by 80% (p<0.01). Net reductions of Lp were accomplished by 2-BP (46% reduction, p=0.005), combined DET+2-BP+DIDS (32% reduction, p=0.04), RNAi (55% reduction, p=0.002), Diannexin administered pre-SMA artery occlusion (73% reduction, p=0.001) and post-SMA occlusion (70% reduction, p=0.002).
Conclusion
Phosphatidylserine exposure is a key event in the pathogenesis of microvascular dysfunction during ischemia-reperfusion injury. Clinically, inhibition of phosphatidylserine exposure is a promising strategy that may one day be used to mitigate the effects of ischemia-reperfusion injury.
Study Type and Level of Evidence
Therapeutic, Level III
Keywords: Scramblase, Ischemia-Reperfusion, Endothelium, Hydraulic Conductivity, Microvascular Permeability
BACKGROUND
Post-injury endothelial dysfunction is linked to molecular changes that occur at the lipid membrane. Phosphatidylserine (PS) is a normally-appearing lipid substrate that is relegated to the inner cytosolic leaflet of lipid bilayer. This lipid asymmetry in the cell wall bilayer is maintained by an ATP-dependent aminophospholipid translocator that returns exteriorized PS against its concentration gradient to the inner cytosolic leaflet in healthy cells. It is postulated that PS is exteriorized to the outer leaflet during pro-inflammatory states [1] and that PS exposure is thought to act as an apoptotic signal that prompts phagocytes to engulf senescent cells. Phosphatidylserine exteriorization occurs partially via membrane “scramblases,” most notably phospholipid scramblase isoform-1 (PLSCR1) and under ATP-depleted states, increased PLSCR1 activity is observed [2]. Additionally, when ATP is depleted, the ATP-dependent aminophospholipid translocator activity becomes less efficient with the end result of increased levels of PS exposed on the outer leaflet of the cell membrane. A proposed mechanism of increased trans-endothelial flux is exteriorization of PS by PLSCR1, leading to scavenging of damaged endothelial cells, loss of the endothelial barrier and increased efflux of extracellular fluid. Endothelial dysfunction promoting extravascular fluid losses can predispose severely injured patients to multiple organ system failure and sepsis.
Phosphatidylserine exposure has been linked to multiple pathologies, such as HIV infection [3], sepsis [4, 5], immunologic function and axon degeneration in the CNS [6, 7], mast cell degranulation [8], autoimmune disease [9], coagulopathy [10] and malignancy [11]. Phosphatidylserine has been studied extensively at the red blood cell (RBC) level, specifically in models of sickle cell anemia [12, 13]. In endothelial cells, PS exposure occurs during the systemic capillary leak syndrome [14] and (LPS)-induced myocardial damage in cardiac endothelium [15]. In models of hypoxia-reoxygenation, PS exposure is linked to IgM binding and promulgation of the inflammatory response [16]. In intestinal ischemia-reperfusion injury, PS exposure promotes intestinal hyperpermeability [17]. Transmembrane exposure of PS causes complement activation in vitro after hypoxia-reoxygenation, producing further inflammation and vascular injury [18].
Because apoptosis of endothelial cells is thought to be one mechanism of microvascular dysfunction after ischemia reperfusion injury [19, 20], our hypothesis was that inhibiting phosphatidylserine expression on endothelial cells would attenuate IRI induced increases in hydraulic permeability (Lp). Hydraulic permeability (Lp) is a measure that describes the ease by which water moves across the endothelial barrier, which can be dramatically increased during times of physiologic stress. It is derived from the Starling equation which takes into account factors that impact on trans-endothelial water movement such as hydrostatic pressure and oncotic pressure. Our specific aims were to describe changes in hydraulic permeability after IRI using 1) inhibitors of PS exposure on endothelial cells in vivo (dithioerythritol (DTE), 2-bromopalmitate (2-BP), and 4,4′-Diisothiocyano-2,2′-stilbenedisulfonic acid (DIDS)), and 2) targeted inhibition of PS exposure (RNAi and Diannexin).
METHODS
All studies were approved by our Institutional Committee for the use of animals in research and complied with animal research protocols.
Animal Experiments
Animal and Solution Preparations
Red blood cells that are used as flow markers were harvested from female Sprague-Dawley rats (250-310 g; Hilltop Lab Animals, Scottsdale, PA). The blood was centrifuged to remove the plasma and buffy coat, and then washed three times in 15 ml of Ringer’s solution. The Ringer’s solution was prepared daily in distilled de-ionized water and contained 135 mM NaCl; 4.6 mM KCl; 2.0 mM CaCl; 2.46 mM MgS04; 5.0 mM NaHCO3; 5.5 mM Dextrose; 9.03 mM Hepes Salt (Research Organics; Cleveland, OH); 11.04 mM Hepes Acid (Research Organics). A 1% bovine serum albumin (fraction V, fatty acid free - Pierce Biotechnology, Rockford IL) Ringer’s solution was prepared before each experiment by adding the appropriate amount of BSA to the Ringer’s. This was used as the perfusate.
Adult female Sprague-Dawley rats were anesthetized with subcutaneous sodium pentobarbital (60 mg/kg body weight). A midline laparotomy incision was performed. The bowel mesentery was gently exposed and positioned on an inverted microscope stage (Diaphot, Nikon; Melville, NY). The superior mesenteric artery was identified at the base of the mesentery and a prolene monofilament suture was placed about it circumferentially for occlusion. Throughout this process the animal’s body temperature was maintained at 37° C and the mesentery was continuously bathed in Ringer’s solution.
In vivo measurement of hydraulic permeability (Lp)
Postcapillary venules, 20 to 30μm in diameter and at least 400μm in length, were identified based on flow patterns. Vessels with no evidence of leukocyte adherence or side branches were chosen. The vessels were cannulated with micropipettes attached to a water manometer to control hydrostatic perfusion pressure.
Single vessel hydraulic permeability (Lp) was determined using the modified Landis micro-occlusion technique [21]. The assumptions and limitations of this technique have been previously described [22]. Briefly, initial cell velocity (dl/dt) was obtained by recording marker cell position as a function of time. Transmural water flux per unit area (Jv/S) was calculated by the equation: Jv/S = (dl/dt) × (r/21), where r is the capillary radius and 1 is the initial distance between the marker cell and the occluded site. Hydraulic permeability (Lp) was determined by using a modified version of Starling’s equation of fluid filtration: Lp = Jv/S[(Pc−Pi) − σΔπ], where Pc is the capillary hydrostatic pressure, Pi is the interstitial hydrostatic pressure, σ is the osmotic reflection coefficient, and Δπ is the osmotic pressure difference. Assuming Pi was near zero and σΔπ (units=cm-H2O) remained constant at 3.78 for 1% BSA Ringer’s solution, Lp was calculated from the slope of the regression of Jv/S on P, where P = (Pc − 3.78). This was derived from several occlusions at three different perfusate pressures. Each n signifies that a single vessel was cannulated and the Lp serially measured in a single rat, such that an n=3 means that 3 different vessels were studied in 3 different rats. Control studies that document the stability of this model over time and after multiple recannulations of the vessels have been previously reported [23].
Ischemia-Reperfusion Protocol
The protocol was adapted from previous work [23, 24]. In brief, study microvessels were perfused with 1% Ringer/BSA solution for 10 min before baseline Lp was assessed. After this assessment, a prolene suture snare was placed around the superior mesenteric artery (SMA) and tightened for 45 min, constituting the gut ischemia phase of the protocol. Cessation of blood flow through the study vessel was confirmed by intra-vital microscopy. During this time, the bowel was covered with gauze and bathed with a warm Ringer solution. After 45 min of ischemia, the prolene suture snare was released and reperfusion of the venule was confirmed and continued for an additional 270 minutes, constituting the reperfusion phase of the protocol. Subsequently, Lp was measured at the following time intervals post SMA occlusion – 105, 165, 195, 225, 255, 285, and 315 minutes. Data points were obtained in triplicate at each time interval. For the hydraulic conductivity studies, the reperfusion component of the study lasted 5h. For control animals (sham animals), all steps were carried out in the same manner as the appropriate comparison experimental group, except the prolene suture snare around the SMA was not tightened, thereby eliminating the ischemic and reperfusion components of the study. Measures of Lp was obtained in the same manner as the other study group animals and serve as time controls.
RNA interference studies
Cloning of rat PLSCR1 cDNA
PLSCR1-specific primers were designed from the nucleotide sequences deposited in the Entrez data bank. Blast search confirmed that the region of PLSCR1 to be amplified did not share any appreciable identity with any known genes in the non-redundant or EST database. Total RNA (1µg) was reverse transcribed using random hexamers in a 20µl reaction volume and 5µl of RT mix, used in a 50µl PCR reaction using PLSCR1-specific primers. The resulting product was analyzed by agarose gel electrophoresis and cloned into a dual-promoter plasmid (pTopoII, Invitrogen Corp., Carlsbad, CA). The cloned product was sequenced to confirm its identity and used to generate dsRNA for the RNAi studies.
Synthesis of dsRNA
Sense and antisense RNAs was synthesized in vitro using the mega-riboprobe kit (Ambion, TX) from PLSCR1 and control cDNAs cloned in plasmid vectors as described [25]. Equimolar concentrations of sense and antisense RNA was mixed, phenol-extracted and precipitated. The RNA pellet was dissolved in annealing buffer, boiled for one minute in a boiling water bath and allowed to cool to 22°C over 16 hours to form double-stranded RNA. For in vivo studies, dsRNA (20µg) was mixed with 1.5µl of lipofectAMINE (Invitrogen) and injected directly into the mesentery.
Synthesis of long double-stranded RNA (dsRNA) and RT-PCR
RT-PCR was used to amplify PLSCR1 fragment from rat colon RNA using PLSCR1-specific primers (forward primer: 5′-CGCGTCATCCAGAGTGAG-3′, reverse primer: 5′-TATAATCACCCAG-3′) at an annealing temperature of 58° C. The PLSCR1 PCR product was cloned into pTOPO4 vector (Invitrogen) and sequenced to confirm identity. This cloned cDNA was used as template to transcribe sense and antisense RNAs in vitro using the Megascript RNA kit (Ambion, Austin, TX) according to the manufacturer’s specifications and as described previously [26]. A nonspecific 300-bp segment of pBluescript (Stratagene, La Jolla, CA) was similarly transcribed for use as control dsRNA. dsRNAs (20μg) for either pBluescript (control RNAi) or PLSCR1 (PLSCR1 RNAi) were mixed with 1.5μl of Lipofectamine 2000 (Invitrogen) and brought to a volume of 250μl before being injected into the ileal mesentery [27].
RNAi protocol
After anesthesia was induced using 2% isoflurane and 2% O2, a midline incision was made, and the small bowel mesentery was gently exposed as described in the animal preparation section. The dsRNA solutions described above were applied onto the thin mesenteric tissue over a mesenteric arcade containing appropriate venule so as not to disrupt the vascular arcades or mesenteric tissue itself. In addition, the dsRNA solution was injected into the mesenteric fat tissue underneath the same arcade by raising a wheal in that area. The ileal wall underneath the injected arcade was marked with a 6-0 Prolene blue monofilament suture to identify the region with knockdown. The laparotomy incision was then sutured closed in two layers, and the rats were allowed to recover from the anesthesia as described previously [23-25]. Three days after dsRNA injections, anesthesia was again induced for ischemia-reperfusion studies below. The midline laparotomy incision was reopened, and the previously marked ileal loop was exposed again. A suitable postcapillary mesenteric venule in the area in which the dsRNA was previously administered was cannulated and measured for hydraulic permeability (Lp) at regular intervals (ischemia-reperfusion protocol described above).
Determination of PS inhibition on Lp after IRI
To determine the effect of PS inhibition on Lp after IRI, we used several inhibitors of PS exposure (DTE, 2-BP, DIDS). Dithioerythritol (DTE) is an inhibitor of the cysteine residue that is essential for PLSCR1 translocation; 2-bromopalmitate (2-BP) is an inhibitor of PLSCR1 trafficking to the plasma membrane, which renders PLSCR-1 nonfunctional; and 4,4′-diisothiocyano-stilbene-2,2′-disulfonic acid (DIDS) is an inhibitor of the anion exchange mechanism necessary for PLSCR1 function. First we measured Lp at baseline (t=0) and during ischemia (t=45 minutes). Serial measurements of Lp were then obtained in triplicate during reperfusion at the following time intervals: 105, 165, 195, 225, 255, 285, and 315 minutes. The measurements for Lp were then compared at baseline, during IRI, and during IRI with pre-ischemia administration of DTE (0.5nM, n=3), 2-BP (100µM, n=3), DIDS (5mg/kg, n=3), DTE+2-BP+DIDS.
To determine the effect of targeted PS inhibition on Lp after IRI, we used RNAi to specifically knockdown PLSCR-1. Three days after the injection of dsRNA for PLSCR1, rats underwent the IRI protocol as described above (n=3). Next, serial measurements of Lp were obtained in triplicate at baseline (t=0), during ischemia (t=45 minutes), and during reperfusion at the following time intervals: 105, 165, 195, 225, 255, 285, and 315 minutes. The measurements for Lp were then compared at baseline, during IRI and during IRI after PLSCR1 RNAi.
Finally, we wanted to investigate a potential therapeutic tool, Diannexin, that blocks exposed PS from interacting with proteins that are attracted to the negatively charged moieties of PS, and as such, it is a selective phosphatidylserine blocker. Rats underwent the IRI protocol as described above. Diannexin (400µg/kg) was administered prior to SMA occlusion (n=3), and to simulate a more clinically relevant scenario after SMA occlusion was released (n=3). Next, serial measurements of Lp were obtained in triplicate at baseline (t=0), during ischemia (t=45 minutes), and during reperfusion at the following time intervals: 105, 165, 195, 225, 255, 285, and 315 minutes.
Statistical Analysis
Comparisons between control and sample means for in vivo modified Landis calculations were made with single-factor analysis-of-variance. Lp test groups differences were compared using the area for the area-under-the-curve (AUC) analysis, which was calculated using the trapezium rule under the Landis curve where the area for each trapezoid = 0.5 × (b1 + b2) × height, where b1 = Lp at t1 and b2 = Lp at t2 and height = (t2) − (t1). Differences between measures of Lp were evaluated with a paired Student’s t-test or repeated measures two-way ANOVA as appropriate. Tukey’s HSD post hoc tests were calculated to verify differences among group comparisons. Percentages are expressed as mean ± SEM based on a normal distribution. P values < 0.05 were considered statistically significant. Values for Lp are expressed as mean ± SEM × 10−7 cm·s −1·cmH2O−1, except when fold increases or percentage change are reported.
RESULTS
Ischemia-Reperfusion Studies
The effect of PS inhibition on Lp after IRI is displayed in Figure 1. Inhibition by DTE attenuated the IRI-induced increase in Lp at all time points during reperfusion except for the 45, 165, and 195 minute time points (p < 0.004). Inhibition by 2-BP attenuated the IRI-induced increase in Lp at all time points during reperfusion except for the 165 and 195 minute time points (p < 0.01). Similarly, inhibition by DIDS attenuated the IRI-induced increase in Lp at all time points during reperfusion except for the 165 and 196 minute time points (p < 0.02). Combined inhibition by DTE + DIDS + 2-BP reduced the IRI-induced increase in Lp at all time points during reperfusion except for the 45, 165, and 196 minute time points (p < 0.002).
Figure 1. Impact of Phosphatidylserine Inhibition on Hydraulic Permeability in Rat Mesenteric Venules During Ischemia/Reperfusion.
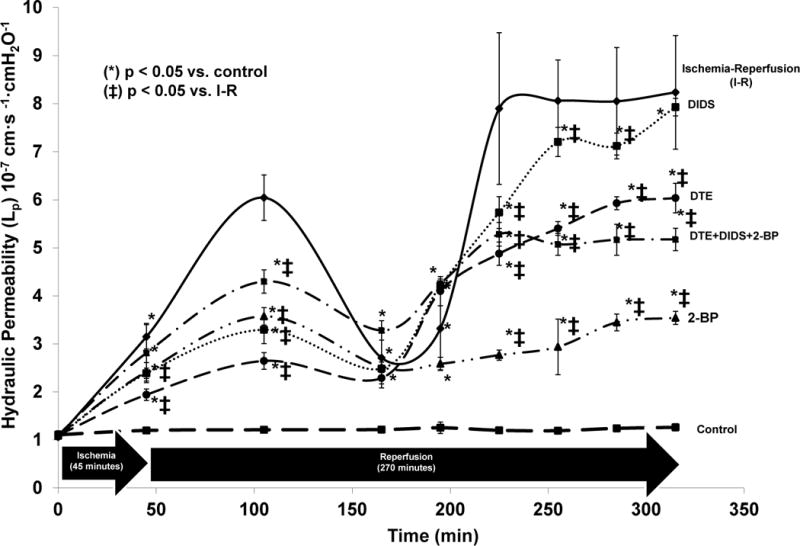
The modified Landis technique with SMA occlusion (45 minutes) and reperfusion (270 minutes) was used to assess for changes in hydraulic permeability after IRI. Potential therapy targeting the trafficking mechanism of PLSCR1 to the endothelial membrane (2-BP) was particularly effective in attenuating the increased hydraulic permeability associated with IRI. Abbreviations = Dithioerythritol (DTE), 2-Bromopalmitate (2-BP), 4,4′-Diisothiocyano-2,2′-stilbenedisulfonic (DIDS), Phospholipid Scramblase-1 (PLSCR1). Units for Lp are expressed as mean ± SEM × 10−7 cm·s −1·cmH2O−1. (*) denotes p < 0.05 vs. control, (ǂ) denotes p < 0.05 vs. ischemia-reperfusion (I-R).
Targeted inhibition by RNA interference and its effect on Lp after IRI is displayed in Figure 2. RNAi had a very robust effect on reducing the IRI-induced increase in Lp after IRI. RNAi attenuated the IRI-induced increase in Lp at all time points during reperfusion (p < 0.02). This effect held even for the 45, 165, and 195 minute time points, that weren’t significantly different in the experiments on inhibition with DTE, 2-BP or DIDS.
Figure 2. Impact of Targeted Phosphatidylserine Inhibition in Rat Mesenteric Venules via RNA Interference.
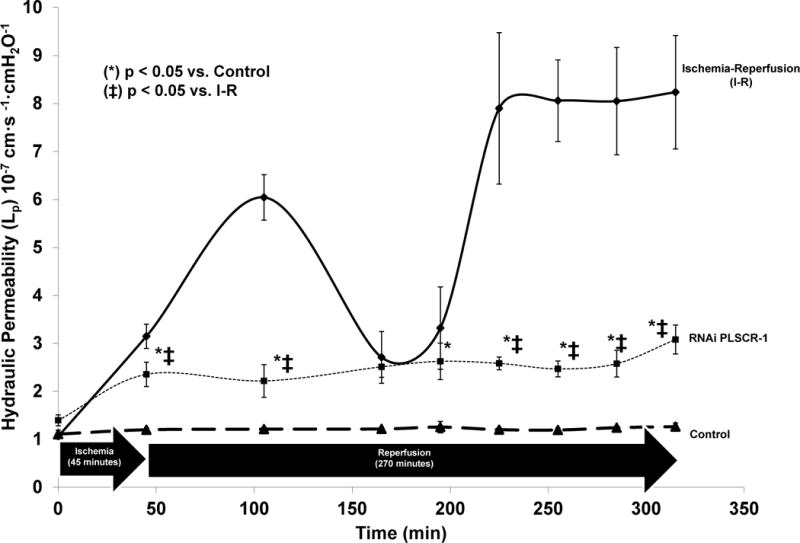
RNAi had a very robust effect on reducing the IRI-induced increase in Lp after IRI. RNAi attenuated the IRI-induced increase in Lp at all time points during reperfusion. Abbreviations = RNA interference (RNAi), Units for Lp are Lp are expressed as mean ± SEM × 10−7 cm·s −1·cmH2O−1. (*) denotes p < 0.05 vs. control, (ǂ) denotes p < 0.05 vs. ischemia-reperfusion (I-R).
Targeted inhibition with Diannexin had the most prominent effect on Lp after IRI, and this is displayed in Figure 3. Both pre- and post-ischemia Diannexin attenuated the IRI-induced increase in Lp at all time points during reperfusion (p < 0.02). This effect held even for the 45, 165, and 195 minute time points that weren’t significantly different in the experiments of inhibition with DTE, 2-BP or DIDS. Diannexin administered pre-ischemia had a slightly more robust effect than Diannexin administered post-ischemia, as pre-ischemia Diannexin returned the IRI-induced increase in Lp to baseline levels at the 45 and 105 minute time periods (p < 0.05).
Figure 3. Impact of Targeted Phosphatidylserine Inhibition in Rat Mesenteric Venules During Ischemia/Reperfusion.
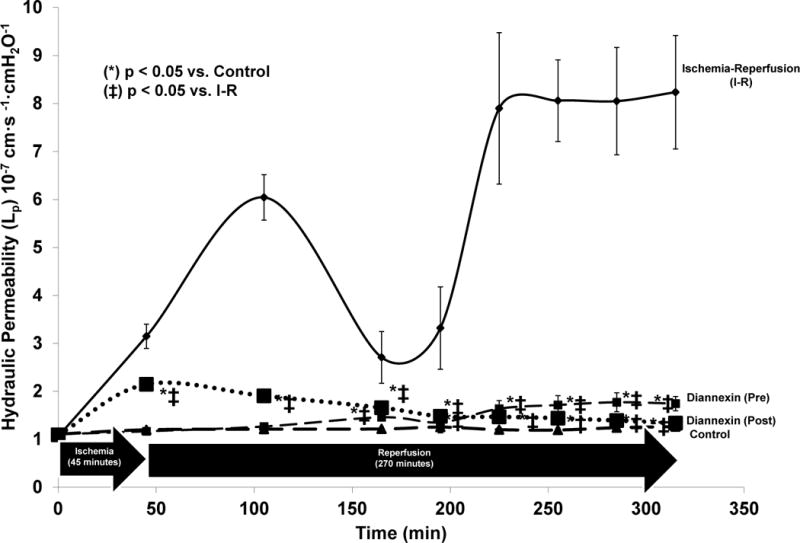
Both pre- and post-ischemia administered Diannexin attenuated the IRI-induced increase in Lp at all time points during reperfusion. Diannexin administered pre-ischemia had a slightly more robust effect than Diannexin administered post-ischemia, as pre-ischemia Diannexin returned the IRI-induced increase in Lp to baseline levels at the 45 and 105 minute time periods. Abbreviations = Phospholipid Scramblase-1 (PLSCR1). Units for Lp are Lp are expressed as mean ± SEM × 10−7 cm·s −1·cmH2O−1. (*) denotes p < 0.05 vs. control, (ǂ) denotes p < 0.05 vs. ischemia-reperfusion (I-R).
An area under the curve analysis was performed for all inhibitors during IRI. With the exception of DTE and DIDS, all agents demonstrated significant attenuation of the IRI-induced increase in Lp (p < 0.001). This is shown in Figure 4.
Figure 4. Area Under the Curve (AUC) Analysis: Phosphatidylserine Inhibition During Ischemia/Reperfusion.
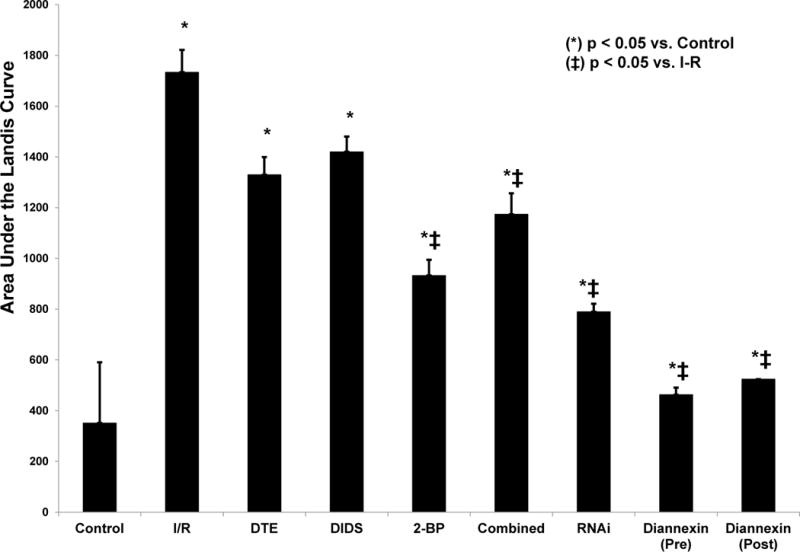
The cumulative area under the Landis curve was calculated for each inhibitor of PS. During IRI net hydraulic permeability (Lp) increased by 80% (p<0.01). Net reductions of Lp were accomplished by 2-BP (46% reduction, p=0.005), combined DTE+2-BP+DIDS (32% reduction, p=0.04), RNAi (55% reduction, p=0.002), Diannexin pre-SMA occlusion (73% reduction, p=0.001) and post-SMA occlusion (70% reduction, p=0.002). As shown, the most effective inhibition (as represented by reduction of the cumulative area) was accomplished by targeted knockdown of PLSCR1 with RNAi and by targeted blockade of PS with Diannexin (p < 0.01 for both). (*) denotes p < 0.05 vs. control, (ǂ) denotes p < 0.05 vs. ischemia-reperfusion (I-R).
DISCUSSION
Endothelial dysfunction promoting extravascular fluid losses can predispose severely injured patients to multiple organ system failure and sepsis. Extravascular fluid loss may be due to loss of the endothelial barrier, in part mediated by PS-exposing cells that serve as an extrinsic signal for phagocytosis. Our hypothesis was that inhibiting phosphatidylserine expression on endothelial cells would attenuate IRI induced increases in hydraulic permeability (Lp). In our first specific aim we sought to determine whether inhibitors of PS exposure on endothelial cells in vivo (DTE, 2-BP, and DIDS) would attenuate the increase in hydraulic permeability observed during IRI. In our second specific aim, we sought to determine whether the increase in hydraulic permeability could be attenuated pharmacologically during IRI using targeted inhibition of PS exposure (RNAi and Diannexin). We found that DTE, 2-BP, and DIDS were modestly effective at decreasing hydraulic permeability and that targeted inhibition of PS exposure via RNAi and Diannexin were very effective at reducing hydraulic permeability during IRI. These results add further support that phosphatidylserine exposure is a key event in the pathogenesis of microvascular dysfunction during ischemia-reperfusion injury and suggests that inhibition of phosphatidylserine exposure is a promising strategy that may one day be used to mitigate the effects of ischemia-reperfusion injury.
Our in vivo studies interrogated the production transport, insertion, modification, flipping, and antagonist properties of PLSCR1 (and resultant PS exposure). The compound 2-BP inhibited the transport and insertion of PLSCR1 into the cell membrane, leading to decreased hydraulic permeability. Using the known enzymatic structure and function of PLSCR1 we used thiol modification of the cysteine residues by DTE to interrupt the enzymatic conformational change required for PLSCR1 function, leading to decreased hydraulic permeability. The combined inhibitors approach (2-BP+DTE+DIDS) was inferior to 2-BP alone, possibly due to DTE and DIDS interfering with 2-BP efficacy (decreased uptake or diminished function). The biodegradation of 2-BP by DTE and DIDS, both with some reducing properties, is one hypothetical mechanism of diminished function. Another plausible mechanism would be DTE competing with 2-bromopalmitate for cysteine residues, preferentially undergoing reduction instead of palmitoylation inhibition, exhibiting a weaker effect than 2-BP alone [28]. Another potential mechanism involves modulation of downstream inflammatory mediator production that contributes to endothelial permeability. For example, DIDS has been shown to inhibit serine palmitoyltransferase activity and downstream sphingolipid synthesis [29], and nuclear sphingolipids propagate genes that are involved in inflammation [30]. As opposed to 2-BP, DIDS may exhibit a dominant effect on palmitoyltransferase activity and downstream sphingolipid production, rendering less of a protective effect compared to 2-BP alone.
With targeted RNAi we were able to significantly attenuate hydraulic permeability due to the decreased production of the PLSCR1 enzyme. Finally, targeted PS inhibition via the specific blocker, Diannexin, significantly decreased the hydraulic permeability observed after IRI when given either before, or after, the initiation of ischemia. Diannexin was more effective than RNAi, possibly due to the completeness of the PS blockade with Diannexin compared to knockdown of PLSCR1 via RNAi or from cells potentially increasing production of PLSCR1 mRNA during progressive IRI. Moreover, the administration of Diannexin before ischemia was more effective compared to Diannexin administration after ischemia, which makes intuitive sense. In the setting of traumatic injury, Diannexin post-ischemia would more closely resemble real-life therapy during the initial injury phase, however, theoretically Diannexin could be administered prophylactically after the initial resuscitation but before an additional systemic insult occurs (such as in the damage control surgery setting before repeated operations) and potentially prevent the sequelae of the second hit phenomenon.
Phosphatidylserine exposure during hypoxia-reoxygenation is associated with microvascular dysfunction. In vitro, hypoxia (2h) and reoxygenation (1h) in murine endothelial cells is associated with increased extracellular beta-2 glycoprotein binding to endothelium (beta-2 glycoprotein has high affinity for anionic phospholipids such as PS) as demonstrated by increased Annexin-V immunofluorescence [16]. In that study, PLSCR1 mRNA was upregulated in hypoxia-treated cells, and hypoxia with subsequent reoxygenation increased protein expression of PLSCR1. In HUVECs, hypoxia and reoxygenation in cells exposing PS by Annexin-V demonstrated increased C3/C5 production and activation. Complement activation increases microvascular dysfunction during reperfusion injury through increased binding and activation of neutrophils [18]. In systemic capillary leak syndrome, endothelial cells exposed to patient sera demonstrated increased PS exposure, DNA fragmentation and increased bax/bcl-2 ratios [14], suggesting that PS exposure and resultant microvascular leak is associated with endothelial loss via apoptosis. During sepsis, lipopolysaccharide (LPS)-induced microparticles (small 1µm vesicles that bud from damaged endothelial cells) exposing PS can cause microvascular dysregulation via endothelial apoptosis, possibly by a nitric oxide intermediate [31]. Microvascular dysfunction and increased endothelial permeability in vivo is associated with PS-exposure [32]. In a murine model of pulmonary sepsis, endothelial barrier dysfunction and increased permeability to albumin was associated with increased PS exposure [33]. In terms of quantifying the deleterious nature of PS production and translocation, this study demonstrated a 10-fold increase in exposure of PS, which corresponded to a 6-fold increase in apoptosis, a 10-fold increase in pulmonary endothelial cell death and a 3-fold increase in microvascular fluid leak.
In our current study, hydraulic permeability during IRI was reduced significantly by inhibiting PLSCR1 and PS exposure, suggesting that PS may act through an apoptotic mechanism to produce vascular dysfunction. A limitation of our in vivo studies was an inability to quantify endothelial apoptosis and cite this as the cause for increased hydraulic permeability during IRI in our experiments.
Phosphatidylserine exposure has deleterious effects beyond endothelial apoptosis. Exteriorization of PS is associated with prothrombinase complex formation and recruitment of cells that participate in further inflammation [34]. As thrombosis and permeability are intrinsically linked, shielding PS-rich surfaces decreases catalytic efficiency of tenase and prothrombinase complexes by 200- and 1000-fold [35] and pharmacologic compounds that act on PS production, trafficking, exteriorization or that modulate PLSCR1 activity may be beneficial. In preclinical models, PS antagonism has been shown to be effective. In a mouse model of bilateral renal IRI, Diannexin pretreatment decreased proximal tubule damage (diminished tubular cast formation), decreased leukocyte influx (decreased granulocyte staining on immunohistochemistry), decreased neutrophil eliciting factors (reduced mRNA production of KIM-1 and NGAL). Specifically, PS inhibition reduced mRNA markers of acute kidney injury by 62-67% and protein expression by 71-74%, correlating to mildly damage renal tubules (approximately 1% scattered casts in tubules) in contrast to untreated controls (25% casts in tubules), which improved renal recovery (normalized urine flow and urinary glucose levels) [36]. In a model of hepatic ischemia-reperfusion, PS inhibition reduced levels of liver transaminases (80% reduction), and decreased necrosis of liver tissue on H&E staining [37]. The authors of both studies concluded that PS inhibition via Diannexin blocked leukocyte activation and recruitment, limiting inflammation in the renal and hepatic microcirculation, preserving parenchymal function.
In conclusion, microvascular dysfunction after ischemia-reperfusion injury is in part linked to endothelial cells with increased levels of PS exposed on their cell surfaces which promotes inflammation and injury. Plausible therapeutic targets exist for targeting both intracellular and extracellular mechanisms of inhibiting PS exposure. Further study on the intracellular mechanistic aspects of PS exposure in endothelial cells and the impact on microvascular dysfunction are warranted.
Acknowledgments
Diannexin was given as a gift by Anthony Allison, Ph.D., Alavita Pharmaceuticals, Mountain View, CA.
Footnotes
Presented at the 76th annual meeting of the American Association for the Surgery of Trauma, September 15, 2017, Baltimore, MD
The authors have no conflicts of interest to report.
AUTHOR CONTRIBUTIONS: A.S., A.B., and G.P.V. all participated in study design, data acquisition and analysis and composition of the manuscript.
References
- 1.Segawa K, Nagata S. An Apoptotic ‘Eat Me’ Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015 Nov;25(11):639–50. doi: 10.1016/j.tcb.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 2.Gleiss B, Gogvadze V, Orrenius S, Fadeel B. Fas-triggered phosphatidylserine exposure is modulated by intracellular ATP. FEBS Lett. 2002 May 22;519(1–3):153–8. doi: 10.1016/s0014-5793(02)02743-6. [DOI] [PubMed] [Google Scholar]
- 3.Zaitseva E, Zaitsev E, Melikov K, Arakelyan A, Marin M, Villasmil R, Margolis LB, Melikyan GB, Chernomordik LV. Fusion Stage of HIV-1 Entry Depends on Virus-Induced Cell Surface Exposure of Phosphatidylserine. Cell Host Microbe. 2017 Jul 12;22(1):99–110.e7. doi: 10.1016/j.chom.2017.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McLean KC, Oppenheimer KH, Sweet LM, Phillippe M. Phospholipid scramblase expression in the pregnant mouse uterus in LPS-induced preterm delivery. Reprod Sci. 2012 Nov;19(11):1211–8. doi: 10.1177/1933719112446078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu B, Sims PJ, Wiedmer T, Moser AH, Shigenaga JK, Grunfeld C, Feingold KR. Expression of the phospholipid scramblase (PLSCR) gene family during the acute phase response. Biochim Biophys Acta. 2007 Sep;1771(9):1177–85. doi: 10.1016/j.bbalip.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 6.Tufail Y, Cook D, Fourgeaud L, Powers CJ, Merten K, Clark CL, Hoffman E, Ngo A, Sekiguchi KJ, O’Shea CC, et al. Phosphatidylserine Exposure Controls Viral Innate Immune Responses by Microglia. Neuron. 2017 Feb 8;93(3):574–586.e8. doi: 10.1016/j.neuron.2016.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wakatsuki S, Araki T. Specific phospholipid scramblases are involved in exposure of phosphatidylserine, an “eat-me” signal for phagocytes, on degenerating axons. Commun Integr Biol. 2017 Feb 17;10(2):e1296615. doi: 10.1080/19420889.2017.1296615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kassas-Guediri A, Coudrat J, Pacreau E, Launay P, Monteiro RC, Blank U, Charles N, Benhamou M. Phospholipid scramblase 1 amplifies anaphylactic reactions in vivo. PLoS One. 2017 Mar 10;12(3):e0173815. doi: 10.1371/journal.pone.0173815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki E, Amengual O, Atsumi T, Oku K, Hashimoto T, Kataoka H, Horita T, Yasuda S, Ieko M, Fukushima K, et al. Increased expression of phospholipid scramblase 1 in monocytes from patients with systemic lupus erythematosus. J Rheumatol. 2010 Aug 1;37(8):1639–45. doi: 10.3899/jrheum.091420. [DOI] [PubMed] [Google Scholar]
- 10.Hidai C, Fujiwara Y, Kokubun S, Kitano H. EGF domain of coagulation factor IX is conducive to exposure of phosphatidylserine. Cell Biol Int. 2017 Apr;41(4):374–383. doi: 10.1002/cbin.10733. [DOI] [PubMed] [Google Scholar]
- 11.Chen CY, Chen JS, Chou YP, Kuo YB, Fan CW, Chan EC. Antibody against N-terminal domain of phospholipid scramblase 1 induces apoptosis in colorectal cancer cells through the intrinsic apoptotic pathway. Chem Biol Drug Des. 2014 Jul;84(1):36–43. doi: 10.1111/cbdd.12347. [DOI] [PubMed] [Google Scholar]
- 12.Neidlinger NM, Larkin SK, Bhagat A, Victorino GP, Kuypers FA. Hydrolysis of phosphatidylserine-exposing red blood cells by secretory phospholipase A2 generates lysophosphatidic acid and results in vascular dysfunction. J Biol Chem. 2006 Jan 13;281(2):775–81. doi: 10.1074/jbc.M505790200. [DOI] [PubMed] [Google Scholar]
- 13.Barber LA, Palascak MB, Joiner CH, Franco RS. Aminophospholipid translocase and phospholipid scramblase activities in sickle erythrocyte subpopulations. Br J Haematol. 2009 Aug;146(4):447–55. doi: 10.1111/j.1365-2141.2009.07760.x. [DOI] [PubMed] [Google Scholar]
- 14.Assaly R, Olson D, Hammersley J, Fan PS, Liu J, Shapiro JI, Kahaleh MB. Initial evidence of endothelial cell apoptosis as a mechanism of systemic capillary leak syndrome. Chest. 2001 Oct;120(4):1301–8. doi: 10.1378/chest.120.4.1301. [DOI] [PubMed] [Google Scholar]
- 15.Pétillot P, Lahorte C, Bonanno E, Signore A, Lancel S, Marchetti P, Vallet B, Slegers G, Neviere R. Annexin V detection of lipopolysaccharide-induced cardiac apoptosis. Shock. 2007 Jan;27(1):69–74. doi: 10.1097/01.shk.0000235085.56100.38. [DOI] [PubMed] [Google Scholar]
- 16.Slone EA, Pope MR, Fleming SD. Phospholipid scramblase 1 is required for β2-glycoprotein I binding in hypoxia and reoxygenation-induced endothelial inflammation. J Leukoc Biol. 2015 Nov;98(5):791–804. doi: 10.1189/jlb.3A1014-480R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Slone EA, Fleming SD. Membrane lipid interactions in intestinal ischemia/reperfusion-induced Injury. Clin Immunol. 2014 Jul;153(1):228–40. doi: 10.1016/j.clim.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mold C, Morris CA. Complement activation by apoptotic endothelial cells following hypoxia/reoxygenation. Immunology. 2001 Mar;102(3):359–64. doi: 10.1046/j.1365-2567.2001.01192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shou X, Wang B, Zhou R, Wang L, Ren A, Xin S, Zhu L. Baicalin Suppresses Hypoxia-Reoxygenation-Induced Arterial Endothelial Cell Apoptosis via Suppressing PKCδ/p53 Signaling. Med Sci Monit. 2017 Dec 22;23:6057–6063. doi: 10.12659/MSM.907989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hashimoto K, Kim H, Oishi H, Chen M, Iskender I, Sakamoto J, Ohsumi A, Guan Z, Hwang D, Waddell TK, Cypel M, Liu M, Keshavjee S. Annexin V homodimer protects against ischemia reperfusion-induced acute lung injury in lung transplantation. J Thorac Cardiovasc Surg. 2016 Mar;151(3):861–869. doi: 10.1016/j.jtcvs.2015.10.112. [DOI] [PubMed] [Google Scholar]
- 21.Landis E. M: Microinjection studies of capillary permeability. II. The relation between capillary pressure and the rate at which fluid passes through the walls of single capillaries. Am J Physiol. 1927;82:217–238. [Google Scholar]
- 22.Curry F, Huxley V, Sarelius I. Cardiovascular Physiology Techniques in the Life Sciences. New York: Elsevier; 1983. pp. 1–34. [Google Scholar]
- 23.Victorino GP, Chong TJ, Curran B. Albumin impacts the effects of tonicity on microvascular hydraulic permeability. J Surg Res. 2004 Dec;122(2):167–72. doi: 10.1016/j.jss.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 24.Victorino GP, Ramirez RM, Chong TJ, Curran B, Sadjadi J. Ischemia-reperfusion injury in rats affects hydraulic conductivity in two phases that are temporally and mechanistically separate. Am J Physiol Heart Circ Physiol. 2008 Nov;295(5):H2164–71. doi: 10.1152/ajpheart.00419.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cureton EL, Ereso AQ, Victorino GP, Curran B, Poole DP, Liao M, Harken AH, Bhargava A. Local secretion of urocortin 1 promotes microvascular permeability during lipopolysaccharide-induced inflammation. Endocrinology. 2009 Dec;150(12):5428–37. doi: 10.1210/en.2009-0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gavrila AM, Robinson B, Hoy J, Stewart J, Bhargava A, Amir S. Double-stranded RNA-mediated suppression of Period2 expression in the suprachiasmatic nucleus disrupts circadian locomotor activity in rats. Neuroscience. 2008 Jun 23;154(2):409–14. doi: 10.1016/j.neuroscience.2008.04.032. [DOI] [PubMed] [Google Scholar]
- 27.la Fleur SE, Wick EC, Idumalla PS, Grady EF, Bhargava A. Role of peripheral corticotropin-releasing factor and urocortin II in intestinal inflammation and motility in terminal ileum. Proc Natl Acad Sci USA. 2005 May 24;102(21):7647–52. doi: 10.1073/pnas.0408531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wiedmer T, Zhao J, Nanjundan M, Sims PJ. Palmitoylation of phospholipid scramblase 1 controls its distribution between nucleus and plasma membrane. Biochemistry. 2003 Feb 11;42(5):1227–33. doi: 10.1021/bi026679w. [DOI] [PubMed] [Google Scholar]
- 29.Mandon EC, Ehses I, Rother J, van Echten G, Sandhoff K. Subcellular localization and membrane topology of serine palmitoyltransferase, 3-dehydrosphinganine reductase, and sphinganine N-acyltransferase in mouse liver. J Biol Chem. 1992 Jun 5;267(16):11144–8. [PubMed] [Google Scholar]
- 30.Fu P, Ebenezer DL, Ha AW, Suryadevara V, Harijith A, Natarajan V. Nuclear Lipid Mediators: Role of Nuclear Sphingolipids and Sphinosine-1-Phosphate Signaling in Epigenetic Regulation of Inflammation and Gene Expression. J Cell Biochem. 2018 Jan 27; doi: 10.1002/jcb.26707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gambim MH, do Carmo Ade O, Marti L, Veríssimo-Filho S, Lopes LR, Janiszewski M. Platelet-derived exosomes induce endothelial cell apoptosis through peroxynitrite generation: experimental evidence for a novel mechanism of septic vascular dysfunction. Crit Care. 2007;11(5):R107. doi: 10.1186/cc6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Molski M, Groth A, Allison AC, Hendrickson M, Siemionow M. Diannexin treatment decreases ischemia-reperfusion injury at the endothelial cell level of the microvascular bed in muscle flaps. Ann Plast Surg. 2009 Nov;63(5):564–71. doi: 10.1097/SAP.0b013e3181935a4e. [DOI] [PubMed] [Google Scholar]
- 33.Gill SE, Rohan M, Mehta S. Role of pulmonary microvascular endothelial cell apoptosis in murine sepsis-induced lung injury in vivo. Respir Res. 2015 Sep 16;16:109. doi: 10.1186/s12931-015-0266-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hugel B, Martínez MC, Kunzelmann C, Freyssinet JM. Membrane microparticles: two sides of the coin. Physiology (Bethesda) 2005 Feb;20:22–7. doi: 10.1152/physiol.00029.2004. [DOI] [PubMed] [Google Scholar]
- 35.del Conde I, Nabi F, Tonda R, Thiagarajan P, López JA, Kleiman NS. Effect of P-selectin on phosphatidylserine exposure and surface-dependent thrombin generation on monocytes. Arterioscler Thromb Vasc Biol. 2005 May;25(5):1065–70. doi: 10.1161/01.ATV.0000159094.17235.9b. [DOI] [PubMed] [Google Scholar]
- 36.Wever KE, Wagener FA, Frielink C, Boerman OC, Scheffer GJ, Allison A, Masereeuw R, Rongen GA. Diannexin protects against renal ischemia reperfusion injury and targets phosphatidylserines in ischemic tissue. PLoS One. 2011;6(8):e24276. doi: 10.1371/journal.pone.0024276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teoh NC, Ito Y, Field J, Bethea NW, Amr D, McCuskey MK, McCuskey RS, Farrell GC, Allison AC. Diannexin, a novel annexin V homodimer, provides prolonged protection against hepatic ischemia-reperfusion injury in mice. Gastroenterology. 2007 Aug;133(2):632–46. doi: 10.1053/j.gastro.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 38.Radhakrishnan RS, Shah SK, Lance SH, Radhakrishnan HR, Xue H, Radhakrishnan GL, Ramaswamy US, Walker PA, Uray KS, Laine GA, et al. Hypertonic saline alters hydraulic conductivity and up-regulates mucosal/submucosal aquaporin 4 in resuscitation-induced intestinal edema. Crit Care Med. 2009 Nov;37(11):2946–52. doi: 10.1097/CCM.0b013e3181ab878b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reynoso R, Perrin RM, Breslin JW, Daines DA, Watson KD, Watterson DM, Wu MH, Yuan S. A role for long chain myosin light chain kinase (MLCK-210) in microvascular hyperpermeability during severe burns. Shock. 2007 Nov;28(5):589–95. doi: 10.1097/SHK.0b013e31804d415f. [DOI] [PubMed] [Google Scholar]
- 40.Mazzuca E, Aliverti A, Miserocchi G. Computational micro-scale model of control of extravascular water and capillary perfusion in the air blood barrier. J Theor Biol. 2016 Jul 7;400:42–51. doi: 10.1016/j.jtbi.2016.03.036. [DOI] [PubMed] [Google Scholar]