

Abstract
The use of imatinib, second and third generation ABL tyrosine kinase inhibitors (TKI) (i.e. dasatinib, nilotinib, bosutinib and ponatinib) made CML a clinically manageable and, in a small percentage of cases, a cured disease. TKI therapy also turned CML blastic transformation into a rare event; however, disease progression still occurs in those patients who are refractory, not compliant with TKI therapy or develop resistance to multiple TKIs. In the past few years, it became clear that the BCR-ABL1 oncogene does not operate alone to drive disease emergence, maintenance and progression. Indeed, it seems that bone marrow (BM) microenvironment-generated signals and cell autonomous BCR-ABL1 kinase-independent genetic and epigenetic alterations all contribute to: i. persistence of a quiescent leukemic stem cell (LSC) reservoir, ii. innate or acquired resistance to TKIs, and iii. progression into the fatal blast crisis stage. Herein, we review the intricate leukemic network in which aberrant, but finely tuned, survival, mitogenic and self-renewal signals are generated by leukemic progenitors, stromal cells, immune cells and metabolic microenvironmental conditions (e.g. hypoxia) to promote LSC maintenance and blastic transformation.
Keywords: Stem cells, microenvironment, chronic myeloid leukemia
1. INTRODUCTION
Exposure to harmful environmental conditions (e.g. ionizing radiation) and/or genetic predisposition seems to be the cause of chronic myeloid leukemia (CML), a biphasic hematopoietic stem cell (HSCs)-derived but progenitor-driven myeloproliferative disorder characterized by the accumulation of apparently normal myeloid cells [1, 2]. The hallmark of CML is the Philadelphia (Ph+) chromosome that originates from the reciprocal translocation t(9;22)(q34;q11). This encodes the constitutively active BCR/ABL1 oncogenic tyrosine kinase whose expression is essential for the onset, maintenance and progression of the disease and its response to TKI therapy although results from a BCR-ABL1 knock-in animal model argue against the plethora of data supporting a role for BCR-ABL1 as the oncogene sufficient to induce CML [1-3].
The initial chronic phase (CML-CP), if left untreated naturally evolves into the fatal blast crisis stage (CML-BC) [1, 2]; however, this became a rare event after the advent of ABL TKIs [4, 5]. Three TKIs (imatinib, nilotinib, and dasatinib) are currently used as first-line treatment for patients with newly diagnosed CML-CP [6, 7]. Although the majority of CML-CP patients show a strong and persistent response to imatinib (IM), ~40% of IM-treated CML-CP patients develop BCR-ABL1 mutation-induced drug resistance [4-6, 8]. Likewise, ~35-50% of patients become resistant to second generation TKIs [6, 8]. For patients failing the frontline therapy, other second or third generation TKIs are available and their use depends on each patient’s clinical characteristics and on the type of BCR-ABL1 mutations present (e.g. ponatinib for patients with BCR-ABL1 gatekeeper T315I mutation); however, only ~50% TKI-resistant CML-CP and ~35% -BC patients show a significant response [6, 8-10]. Furthermore, ponatinib induces severe adverse effects (i.e. vascular occlusion, heart failure and hepatotoxicity) in some patients [5, 8, 11, 12] and, as reported for first and second generation TKIs, CML-BC patients do not show long-term response to ponatinib treatment [11, 13]. Similarly, the selection of clones with BCR-ABL1 compound mutations in multiple TKI-treated patients may also confer resistance to ponatinib [14]. For those patients resistant to multiple TKIs, allogeneic BM transplantation remains the only therapeutic option [7, 15]. Other experimental drugs (e.g. omacetaxine, KPT-330) are also available but their impact on disease-free survival is not clear yet [13, 16, 17]. Furthermore, TKI therapy discontinuation of IM-treated CML-CP patients in complete cytogenetic (CCyR) and molecular (CMR) response for more than 2 years usually results in disease relapse [10, 18], whereas treatment-free remission (TFR) seems to be more durable and frequent in patients who achieved deeper molecular responses (≥4-log reductions in BCR-ABL1 levels) upon treatment with imatinib or second generation TKIs [8, 18, 19]. Notably, sustained MMR, but not CMR, after TKI discontinuation was significantly associated with high (600 – 800 mg per day) dose imatinib or second-generation TKI treatment [20]. By contrast, prior history of suboptimal response or IM resistance correlated with disease relapse after second-generation TKI discontinuation [21].
Based on these considerations and on the evidence showing that disease relapse is the most common event after TKI discontinuation [8] and that BCR-ABL1 kinase-independent TKI resistance is commonly observed in CML patients in advanced phases (accelerated or blastic) or in CML-CP patients presenting additional cytogenetic alterations (~10% of CML) [22, 23], it is becoming clear that cell autonomous genetic or epigenetic changes and/or microenvironmental factors might be responsible for the persistence of Ph+ cells with stem cell behavior. These LSCs survive and self-renew regardless of BCR-ABL1 activity, present innate or acquired TKI resistance, and are capable of reinitiating CML-CP and/or promoting and maintaining CML-BC [1, 23].
Blastic transformation likely results from acquired increase in BCR-ABL1 expression and kinase activity [24-26], which leads to altered mRNA metabolism [27], genetic instability [28] and alteration of pathways regulating survival, proliferation and differentiation of myeloid- or lymphoid-committed CD34+ progenitors [1] (Fig. 1). Thus, BCR-ABL1 may represent the key factor responsible for the genetic heterogeneity of CML-BC. The molecular events leading to enhanced BCR-ABL1 expression/activity in CML-BC progenitors still remain to be fully understood, although BCR-ABL1 gene amplification [29, 30], increased BCR promoter activity [31], impaired protein phosphatase 2A (PP2A) activity [32], and inhibition of SHP1 phosphatase [32, 33] might occur alone or in cooperation to increase BCR-ABL1 expression and activity [25]. Among these events, loss of PP2A activity [32] and reactive oxygen species (ROS)-induced genomic instability [34-36] occur in a BCR-ABL1 dose- and kinase-dependent and kinase-independent manner in leukemic progenitors and LSCs, respectively, and significantly contribute to the epigenetic and genetic heterogeneity that characterizes CML-BC [1]. Thus, disease progression may result from the accumulation of a critical number or combination of different mutations [1, 2, 37-42] occurring at the stem and/or progenitor level, and from the pleiotropic effect of enhanced BCR-ABL1 activity, which starts to increase in accelerated phase [1, 38].
Fig. 1. Mechanisms of CML blastic transformation.
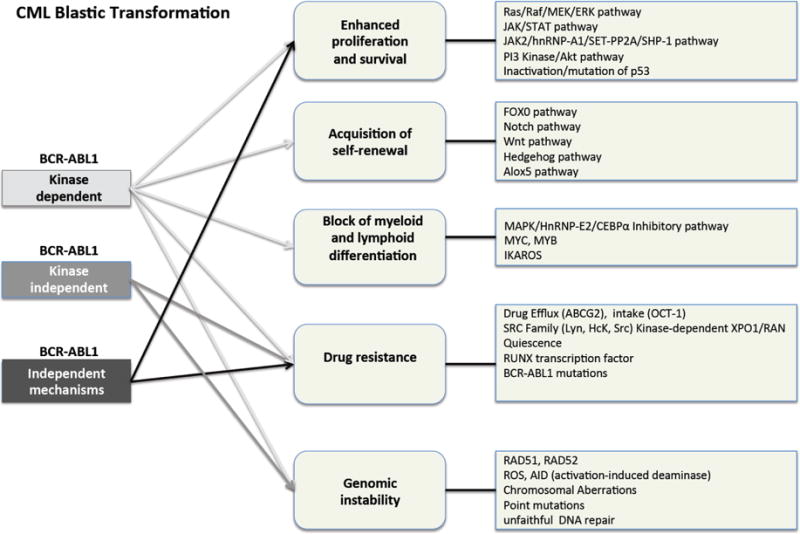
Schematic representation of the main BCR-ABL1 kinase-dependent, kinase-independent and BCR-ABL1 independent molecular pathways that may account for enhanced survival/proliferation, acquired self-renewal, differentiation arrest, TKI resistance and genomic instability of leukemic CD34+ progenitors.
Hereafter, we will review the role of cell autonomous and microenvironmental events controlling TKI resistance, survival, proliferation/differentiation and/or self-renewal of CML-CP and BC LSCs.
2. LSC ORIGIN AND REGULATION IN CML-CP AND CML-BC
Genomic instability seems to be a key feature not only of Ph+ leukemic progenitors, which rely on BCR-ABL1 kinase activity for survival and proliferation [1, 23, 43, 44], but also of CML (CP and BC) LSCs [1, 23, 45] in which BCR-ABL1 kinase but not expression seems to be dispensable for survival and self-renewal [46-48]. Accordingly, a discrepancy between BCR-ABL1 levels and activity has been reported in TKI-resistant quiescent LSCs compared to proliferating CD34+ leukemic progenitors [48]. Indeed, BCR-BL1 expression per se is sufficient to recruit and activate JAK2/β-catenin-regulated self-renewal/survival signals in CML-CP and –BC quiescent LSCs [48]. In LSCs and progenitors from both disease stages, BCR-ABL1 also induces genomic instability in a dose-dependent manner by increasing DNA oxidative damage and unfaithful DNA repair [1, 45, 49]. TKI treatment of BCR-ABL1+ cells revealed that BCR-ABL1 kinase mutations and other genetic aberrations associated with disease progression specifically occur in LSCs but not in leukemic progenitors [43]. This supports the idea that the origin and maintenance of Ph+ LSCs requires BCR-ABL1 expression but not its kinase activity, and that other preexisting or BCR-ABL1 irreversibly-induced genetic or epigenetic changes contribute to LSC survival, self-renewal and innate resistance to TKIs.
Quiescent LSCs are not only resistant to TKIs but also increase upon TKI treatment [23, 50], suggesting that signals generated by TKIs can promote LSC survival. Furthermore, BCR-ABL1 kinase-dependent and –independent genetic (e.g. increased ROS, alterations of GSK3β and IKZF1) and epigenetic (e.g. C/EBPα downmodulation, SET and β-catenin overexpression) events contribute to the acquisition of LIC characteristics and TKI resistance by the CML-BC granulocytic-macrophage progenitors (GMPs) [1, 23, 51, 52]. Notably, in vitro and xenotransplant-based assays aimed at assessing the presence of LICs in different stem and progenitor cell subpopulations from CML-BC patients revealed that acquisition of self-renewal is not restricted to GMPs but can occur in HSCs and different progenitor cell fractions with variability between patients [53]. Thus, a better phenotypic characterization of LSCs in CML-CP and –BC, and a more in-depth analysis of the molecular pathways similarly altered in TKI-resistant quiescent LSCs and progenitors with LSC capability are necessary for eradicating CML (CP and BC) at leukemia-initiating cell (LIC) levels.
2.1. Phenotypic Characterization of LSCs
Targeting of leukemic HSCs in TKI-responsive CML-CP patients and in TKI-resistant CP and BC CML patients requires the correct identification of the leukemic cell pool harboring the Ph+ LIC population capable of colony-forming cell (CFC), LTC-IC and NOD SCID-γ (NSG) repopulating activities. It is well accepted that the TKI-resistant LSCs in both CML-CP and –BC patients include lineage-negative CD34+/CD38−/CD90+/CD45RA−/CD71−/HLA-DRlow cells with innate drug-resistance but also CD34+ cells that have been forced into the G0 phase of the cell cycle by ABL TKI treatment [46, 50, 54-58]. In CML-BC, lineage-negative CD34+/CD38+/CD45RA+/CD123+ GMPs and, perhaps, other progenitors with acquired self-renewal serve as the LICs [51, 53]. Such a simplistic definition does not take into consideration that markers distinguishing between normal and leukemic HSCs are not well established yet. In this regard, CD123 has recently been proposed as a CML LSC marker because of its overexpression in the CML (CP and BC) stem cell-enriched CD34+/CD38− cell fraction [59]. Similarly, Evi1 levels are increased in CD34+/CD38−/CD90+ CML LSCs and correlate with enhanced self-renewal [60]. Interestingly, recent reports show that CD25 and CD26 expression appears to be restricted to leukemic CD34+ cells [61-63]. In addition, their expression is higher in leukemic CD34+/CD38− than CD34+/CD38+ cells and increases during blastic transformation (CD26 only in myeloid BC), suggesting the potential for targeting these surface markers in CML eradication and CML-BC clinical trials [61-63]. IL-1 receptor accessory protein (IL-1RAP) expression seems restricted to CML LSCs [58, 64] although it is controversial whether IL-1RAP antibody-mediated targeting may efficiently eradicate CML because its expression in LSCs might be downregulated by TKIs as shown for leukemic progenitors [58].
3. ROLE OF MICROENVIRONMENT IN LSC MAINTENANCE
The inability to find markers to selectively purge LSCs while sparing normal cells suggests that leukemic stem and progenitor cells promote morphological (e.g. BM fibrosis) and functional (e.g. cytokine/chemokine production; altered stromal-hematopoietic cell interaction) changes in the BM microenvironment that, in turn, provide a protective niche for LSCs thereby contributing to their quiescence, self-renewal and TKI-resistance [65-68] (Fig. 2). Additionally, TKIs (i.e. imatinib) may promote survival of quiescent CML cells by enhancing the expression of CXCR4 that, in turn, favors LSC retention in the BM and their interaction with the mesenchymal stroma [69, 70]. Conversely, G-CSF production by leukemic progenitors decreases CXCL12 expression thereby resulting in decreased LSC homing and BM retention [67]. However, the importance of CXCR4 in LSC maintenance is still unclear as TKI treatment combined with inhibition of CXCR4 mobilized CXCR4+ cells without exerting an effect on leukemia burden [71]. Nonetheless, there is strong evidence indicating that the direct interaction of stromal cells with LSCs plays a pivotal role in LSC survival and TKI resistance. Indeed, N-cadherin-mediated LSC and progenitor cell adhesion to mesenchymal stromal cells enhances LSC self-renewal and survival and protects CML stem/progenitor cells from TKI-induced apoptosis through stabilization/activation of β-catenin signaling [68]. In addition, microenvironment-generated signals also increase ROS-dependent genomic instability in CML-BC GMPs [72-75].
Fig. 2. Cell autonomous and microenvironmental signals regulating LSC behavior.
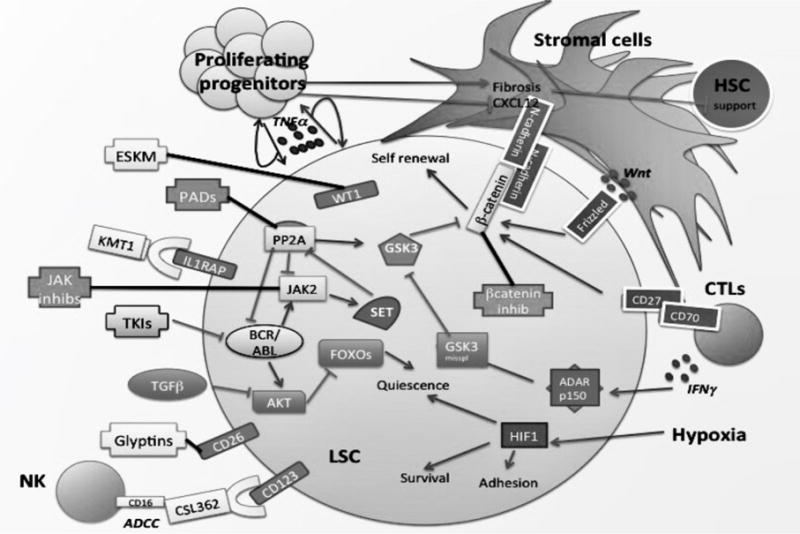
LSCs display both cell autonomous and BM microenvironmentally-driven aberrant signals allowing them to survive, self-renew and escape TKI-induced killing. Potential therapeutic approaches targeting LSC markers (e.g. IL1-RAP, CD26)) and BCR-ABL1 kinase-independent pathways (e.g. SET/PP2A, β-Catenin, Jak2). New therapeutic avenues may also arise from the modulation of cell survival mechanisms triggered by hypoxia and the microenvironmental crosstalk between LSCs and stromal (N-cadherin-mediated) or immune cells (e.g. NK cells).
CML stem/progenitor cells remodel the BM niche to skew hematopoiesis towards the expansion of leukemic over normal cells [67, 76]. This is achieved through the aberrant reprogramming of mesenchymal stromal cells that are conditioned to produce cytokine and chemokine (e.g. MIP1α, MIP1β, IL-1β, IL-4, IL-6, TNFα, CXCL12, LIF, BMP2/4) [67, 76, 77]. Of note, BMP2/BMP4 proteins control BM myelofibrosis, promote BMP4-dependent amplification and maintenance of LSCs, and induce the BMP2-dependent expansion of committed myeloid progenitors [77]. Furthermore, the presence of IL-2 in the microenvironment increases CD25 levels in LSK mouse cells, consistent with the high CD25 expression observed in Ph+ CD34+/CD38− cells [62]. In addition, proliferating leukemic progenitors secrete CCL3 and TPO that, upon inducing BM myelofibrosis through an expansion of osteoblastic progenitors, confer a selective growth advantage to LSCs over normal HSCs [76], an event likely essential for the pathogenesis of CML [78]. Interestingly, specific activation of the pituitary hormone (PTH) receptor increases TGFβ production by the remodeled osteoblast compartment resulting in TGFβ-induced inhibition of LSC engraftment and survival in immunocompromised mice [65]. Unexpectedly, TGFβ, by inhibiting Akt, induces FOXO3a nuclear localization with consequent increase in the LIC population [79].
The BM niche exerts its LSC protective role not only by releasing soluble factors but also through the presence of an oxygen gradient, which progressively decreases from 6% to 1% in the sinusoidal cavity and endosteal niche, respectively [80]. Culturing leukemic CD34+/CD38− cells in hypoxic conditions protects cells from TKIs [81] and increases their survival and self-renewal in a BCR-ABL1 kinase-independent manner [81], suggesting that hypoxia may contribute to LSC quiescence [81, 82]. This likely occurs through modulation of hypoxia-inducible factor-1α (HIF1α). In fact, loss of HIF1α causes cell cycle arrest, impaired engraftment, increased LSC apoptosis, and results in a less severe and non-transplantable CML-BC-like phenotype in immunocompromised mice [83]. Hypoxia also seems to be involved in the modulation of E-cadherin and CXCR4 thus also contributing to LSC homing and retention in the BM niche [84]. Finally, hypoxia increases ROS levels thus contributing to genomic instability and therefore clonal evolution and, perhaps, blastic transformation [85]. Accordingly, hypoxia enhances the capacity of CD34+ CML progenitors to behave as LSCs in a BCR-ABL1 kinase-independent manner [81, 82]. These HSC-like CML progenitors cultured in hypoxic conditions have a gene expression profile similar to normal CD34+ cells [81] while in normoxic conditions, quiescent CML HSCs are more similar to dividing early progenitors than quiescent normal HSCs are to normal progenitors [86, 87].
The BM also functions as a primary site for immune response towards leukemic stem/progenitor cells. Considering the immunogenic nature of CML and the effects of interferon alpha (IFNα) therapy [88], it is not surprising that IFNα treatment prior to TKI discontinuation may reduce relapse and maintain CCyR [89]. As IFNα is capable of activating natural killer (NK) cells, its protective effect may depend on NK cytotoxic activity [90]. Accordingly, it has been reported that BCR-ABL1 also promotes dendritic cell-mediated NK cell activation [91, 92]. Activated NK cells (IFNγ+/CD3−/CD56+) are higher in patients with durable TFR and in IM-treated patients in CMR compared to fluctuating CMR patients and healthy controls, suggesting a role for these immune cells in the maintenance of CMR [93]. Similarly, dasatinib induces an expansion of large granular lymphocytes, T and NK cells, and such an effect seems to correlate with long-lasting response to TKI therapy [94, 95]. However, the lack of NK cell differentiation in the presence of CML progenitor-produced IL-15 [96], the reduced NK cell number, and their functional impairment in CML [97] argue against an immune surveillance role for NK cells in CML even if autologous human NK cells prevent CML development [98] and trigger antibody dependent cell cytotoxity (ADCC) against CD34+ CML progenitors in response to anti-IL-3R (CD123) antibody therapy [59]. CML patients carrying BCR-ABL1 mutations display immunogenic T-cell epitopes [99], suggesting that cytotoxic T lymphocytes (CTL) have the potential to target CML stem/progenitor cells. Nevertheless, CTLs induce proliferation of CML LSCs by secreting IFNγ that, in turn, activates the ADAR1 p150-mediated β-catenin survival/self-renewal pathway [100, 101]. Likewise, CTLs augment β-catenin-mediated CML stem/progenitor cell survival also by CD70-mediated triggering of CD27 [102].
4. CELL AUTONOMOUS EVENTS REGULATING LSC MAINTENANCE IN CML (CP AND BC) AND THEIR ROLE AS THERAPEUTIC TARGETS
The modulation of survival and self-renewal signals by the BM microenvironment would not promote the expansion of the leukemic cell clone, drug resistance, and maintenance of the leukemic stem cell pool if those signals were not aberrantly exacerbated through cell autonomous events occurring in leukemic but not normal HSCs (Fig. 2). However, the correct analysis of the cell autonomous events occurring in TKI-resistant quiescent LSCs is polluted by a series of data generated in proliferating stem cell-enriched CD34+/CD38− BM cells, and in CD34+ CML-CP progenitors which show sensitivity to TKIs and are BCR-ABL1 oncogene-addicted [1, 23]. However, some signaling pathways are similarly activated in both quiescent LSCs and leukemic progenitors with the only difference that in the latter, oncogenic signals mostly rely on BCR-ABL1 kinase activity [1, 23]. While BCR-ABL1 expression but not its activity represents a key factor allowing the acquisition and maintenance of the leukemic CD34+/CD38− LSC phenotype [46-48], it is unclear whether BCR-ABL1 kinase activity is dispensable for the generation of the lineage-negative CD34+/CD38+/CD123+/CD45RA+ GMP subpopulation with self-renewal ability that exits cell cycle and becomes dormant [103, 104]. The use of a kinase-deficient BCR-ABL1 in a CML animal model argues against the BCR-ABL1 kinase-independence of GMP expansion and acquisition of self-renewal [105], unless homing of these cells in the hypoxic BM niche negatively influences BCR-ABL1 expression and kinase activity as reported for CML-BC cells shifted from normoxic to hypoxic culture and vice versa [106, 107]. Thus, downregulation of BCR-ABL1 kinase activity in quiescent LSCs residing in the hypoxic BM niche seems necessary for their self-renewal and survival. This is consistent with the reported ability of BCR-ABL1 to induce differentiation of mouse leukemic LSCs [108].
In the past few years, genetic and epigenetic cell autonomous events leading to the activation of oncoproteins or inhibition of tumor suppressors have been found to be necessary for CML (CP and BC) LSC survival/self-renewal and, likely, disease progression [1, 23, 35, 45, 109] (Fig. 1). Indeed, LSC behavior depends on aberrant, albeit finely tuned, signals that alter, for example, kinase/phosphatase balance, mRNA metabolism (including mRNA processing and miRNA expression), nucleocytoplasmic trafficking, self-renewal, proliferation/differentiation, and response to apoptotic stimuli (e.g. TKI-induced) [109].
4.1. Role of JAK2, PP2A and β-catenin
Among the signaling pathways operational in both CML-CP and –BC, the BCR-ABL1/JAK2/SET-PP2A/GSK3β/β-catenin network has an essential role in the regulation of LSC survival and self-renewal [1, 23, 48, 51, 109]. Indeed, the regulation of β-catenin represents a key factor controlling survival and self-renewal of CML LSCs [23, 54, 103, 110-113]. In quiescent CD34+/CD38− LSCs from both CML-CP and –BC patients, β-catenin-dependent regulation of survival and self-renewal signals requires BCR-ABL1 expression but not its activity [48, 114]. Conversely, in CML-BC GMPs, β-catenin activation is not solely dependent on BCR-ABL1 kinase activity [51, 105] but relies on the cooperation of both BCR-ABL1-independent and –dependent mechanisms [51, 115, 116]. Several lines of evidence support a key role for β-catenin in the regulation of the CML-BC LSCs, including: BCR-ABL1-dependent and imatinib-sensitive β-catenin tyrosine phosphorylation inhibits β-catenin binding to the destruction complex axin/GSK3β/PP2A, thereby activating TCF4-dependent transcription [115]; axin overexpression prevents in vitro replating of CML-BC progenitors; genetic loss of β-catenin impairs self-renewal of normal HSCs and BCR-ABL1-expressing GMPs [112]; GSK3β downregulation increases CML progenitor self-renewal by activating β-catenin and elevating levels of sonic hedgehog pathway mediators such as GLI 1/2 [116, 117]; activation of PP2A impairs self-renewal and survival of leukemic HSCs and CMPs/GMPs in part by inducing BCR-ABL1-independent, JAK2- and GSK3β-dependent degradation of β-catenin [48]. This pathway is further dysregulated in CML-BC as alternative splicing of GSK3β occurs, resulting in a dominant negative form that allows for increased β-catenin activity and GMP self-renewal [116]. This is consistent with previous studies demonstrating that loss of β-catenin prevents LSC self-renewal [112, 114].
Regulation of GSK3β-dependent β-catenin activation in CML (CP and BC) LICs is mediated by induction/activation of JAK2 and SET-dependent inhibition of the PP2A tumor suppressor [32, 48, 118-121]. Although activation of JAK2 in CML-BC progenitors depends on BCR-ABL1 activity [122-124], there is evidence that BCR-ABL1, regardless of its kinase activity, is able to recruit/activate JAK2 that, in turn, leads to PP2A inhibition/β-catenin activation in mouse LSKBCR−ABL1+ and/or quiescent LSCs from CML patients [48]. Furthermore, the notion that JAK2 signaling (e.g. STAT3 and its downstream targets Bcl-XL and Mcl-1) can be activated by cytokines (e.g., IL-6, G-CSF, and GM-CSF) in the microenvironment and/or other extrinsic factors in TKI-treated CML cells [125, 126] suggests that JAK2 can be activated independently of BCR-ABL1 activity. Supporting a critical role for JAK2 in LSC survival it has been reported that combination of BCR-ABL1 TKIs with either pharmacologic suppression of JAK2 or activation of PP2A eliminates both BCR-ABL1-dependent and –independent survival signals, thereby resulting in efficient killing of CML (CP and BC) LSCs and progenitors [48, 127-129]. This led to the development of clinical trials (e.g. NCT01751425 NCT01914484; NCT01702064) with nilotinib and JAK2 inhibitors (e.g. ruxolitinib). However, drawing any conclusion on whether JAK2 inhibition either improves the therapeutic effects of nilotinib or leads to eradication of CML stem cells is still premature.
Inhibition of PP2A also has a major role in the regulation of LSC survival and self-renewal. Indeed, BCR-ABL1 recruits JAK2 that, in turn, stabilizes BCR-ABL1 through direct phosphorylation and induces/activates SET that, in turn, binds and inhibits PP2A in CML (CP and BC)-derived LSCs and progenitors [32, 48, 121-124, 130, 131]. Notably, SET-dependent suppression of PP2A activity requires BCR-ABL1 activity in oncogene-addicted CML progenitors [32, 120] but not TKI-resistant quiescent LSCs, in which BCR-ABL1 expression alone is necessary to recruit JAK2 [48]. This might represent an essential step for survival and self-renewal of CML LICs. Indeed, PP2A directly activates GSK3β and inactivates β-catenin, and indirectly induces inactivation/degradation of BCR-ABL1 and JAK2, likely through SHP1 recruitment/activation [32, 122, 132-134]. The important role played by PP2A and its inhibitors SET and CIP2A is also elegantly supported by a) clinical correlative data showing that levels of inactive PP2A in BM of CML patients at diagnosis directly correlate with blastic transformation [135], and b) preclinical data showing that enhanced expression of SET binding protein 1 (SETBP1), a SET stabilizing factor frequently mutated in hematologic malignancies, confers self-renewal to CML-BC-like mouse progenitors [136].
JAK2 activation might also occur in CML-BC progenitors independently of perturbation of the SET/PP2A interplay [32, 48]. Abelson helper integration site 1 (AHI-1) has been suggested to have a role in the BCR-ABL1 and JAK2 interplay [137]. AHI-1 interacts with JAK2 and BCR-ABL1 to confer TKI resistance [129]. Similar conclusions were drawn using JAK2 and β-catenin inhibitors [128, 138-141], although a recent study with JAK2 null mice [142] argues against JAK2 requirement for BCR-ABL1 leukemogenesis.
4.2. Other Therapeutic Targets
Sonic hedgehog (Shh) signaling also provides survival signals through activation of β-catenin in CML [143]. In fact, levels of the Shh signaling protein Smoothened (Smo1) seem critical for development of TKI resistance and self-renewal of LSCs and CML-BC progenitors [144, 145] but dispensable for normal HSC function [146]. However, it is still unclear whether Shh inhibition leads to CML eradication without harming normal HSC survival and self-renewal [147-152]. Several strategies to target CML LSCs and/or CML-BC GMPs with HDAC [153], Bcl-xL/BCL2/MCL1 [104, 154, 155], mTOR [156], PML [157, 158], Alox-5 [159, 160], autophagy and proteasome inhibitors [161, 162] showed promising results in preclinical settings and are currently under clinical investigation [23]. Moreover, the regulation of LSC behavior seems to also involve other druggable factors (e.g. TGFβ, AKT, FOXO3a, BCL6 and Rac2) that can be tested for their efficacy against CML (CP and BC) LSCs [47, 79, 163-167]. Altogether, these studies have also provided insight into the mechanisms of BCR-ABL1 kinase-independent LSC self-renewal/survival. In some cases, targeting LSC-related pathways does not induce killing but restores TKI sensitivity in LSCs [112, 144, 153, 157, 161, 162, 167-169]. Conversely, the therapeutic efficacy of PP2A-activating drugs [PADs; e.g. FTY720 (Fingolimod; Gilenya), OP449] is still underestimated [121, 170]. PADs are a novel and powerful class of anti-leukemic agents that, upon SET sequestration, reactivate PP2A which, in turn, inhibits BCR-ABL1 and JAK2 and their kinase-dependent and –independent oncogenic signals, suppress in vitro and in vivo BCR-ABL1-driven leukemia regardless of BCR-ABL1 mutation status, and potentiate the pro-apoptotic effect of TKIs [48, 120, 121, 170]. Importantly, PADs target quiescent TKI-resistant CML (CP and BC) LSCs without harming normal hematopoiesis [48]. Finally, targeting the RAS-related nuclear protein RAN and the karyopherin β family member XPO1 (exportin-1, CRM1), two interacting proteins with key functions in nucleocytoplasmic transport may represents a novel strategy to overcome BCR-ABL1 kinase-independent TKI resistance [171], induce apoptosis of CML-BC progenitors [17] and, perhaps, impair their self-renewal activity. In fact, treatment of leukemic progenitors with the clinically relevant XPO1 inhibitor KPT-330 leads to apoptosis and suppression of their clonogenic potential, significantly increased survival of leukemic mice, and restored sensitivity to TKI-induced apoptosis [17, 171]. Interestingly, altered XPO1-dependent subcellular localization of hnRNP-A1 and SET and, subsequently, reactivation of PP2A and inhibition of BCR-ABL1, significantly contribute to KPT-330-induced apoptosis in CML-BC [17].
4.3. Altered mRNA Metabolism in CML (CP and BC)
While there is substantial evidence that BCR-ABL1 kinase-dependent deregulation of mRNA metabolism significantly contributes to the phenotype of CML-BC progenitors through i. differential pre-mRNA splicing, editing, nuclear export and translation; ii. increased mTOR activity and autophagy; iii. altered RNA binding protein (e.g. hnRNPs, La, Msi2) expression; and iv. deregulated miRNA expression (reviewed in [27]), much less is known about the role of BCR-ABL1 kinase-independent mechanisms regulating mRNA metabolism in LSCs. In this regard, altered mRNA splicing and editing, which result in splice variants of BCL2 gene family and misspliced GSK3β, respectively, appear involved in the acquisition of LSC phenotype by CML-BC myeloid progenitors [100, 104, 116]. Likewise, induction of the KPT-330-sensitive [17] hnRNP-A1, which controls JAK2-dependent SET expression [32], occurs in mouse LSKBCR−ABL1+ cells and may contribute to LSC survival/self-renewal [172]. Expression of a series of miRNAs was found to be dysregulated in CML (Fig. 3). miR-486-5p and miR-300 regulation in CML occurs through BCR-ABL1 kinase-dependent and –independent mechanisms and is potentially involved in acquisition of LSC self-renewal/survival and TKI resistance [173, 174]. In fact, miR-486-5p expression increases in CML progenitors and its inhibition potentiates TKI-induced apoptosis likely through Akt signaling (PTEN and FoxO1) modulation, whereas miR-300 appears involved in the regulation of the JAK2/SET/β-catenin self-renewal/survival pathway [173, 174]. In addition, miR-326 is down-regulated in CML-BC progenitors and its restoration results in apoptosis of CD34+ CML cells through suppression of Smo expression [175] whereas TKI-induced downregulation of miR-30a promotes autophagy [176], suggesting a potential role for these miRNAs in β-catenin-dependent regulation of LSC survival and TKI resistance. Other microRNAs (e.g. miR-130a/b, miR-138, miR30e, miR30a, miR29a, miR29b and miR23a), long non-coding RNA (lncRNA)-BGL3, and miRNA-regulating factors (e.g. Lin28/28b) may determine the CML-BC LSC phenotype including acquisition of TKI resistance [177-185] and regulation of BCR-ABL1 levels [186-192].
Fig. 3. MicroRNAs with potential roles in CML.
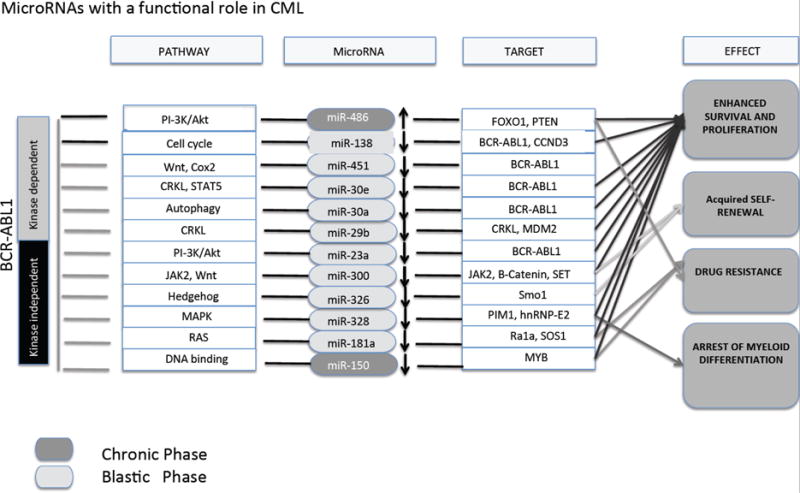
BCR-ABL1 kinase-dependent and -independent pathways modulating miRNAs expression in chronic (dark gray) and blastic phase CML (light grey). These miRNAs control proliferation/survival enhancement, drug resistance, myeloid differentiation and self-renewal capability through modulation of the expression of specific mRNA targets.
Finally, acquisition of self-renewal ability by GMPs requires suppression of differentiation. In most myeloid CML-BC cases, suppression of granulocytic differentiation is a BCR-ABL1 dose- and kinase-dependent effect that relies on inhibition of miR-328 expression [193-195]. Restoration of miR-328 expression in CD34+ CML-BC progenitors negatively regulates PIM1-dependent survival, and exhibits decoy activity by interacting with the RNA binding protein hnRNP-E2, thereby preventing the inhibitory effects of hnRNP-E2 on the translation of C/EBPα [195], a transcription factor essential for normal and leukemic myeloid differentiation [194, 196, 197]. Likewise, downregulation of miR-150 in CML-BC myeloid progenitors appears to be important for Myb expression and differentiation arrest [198, 199].
CONCLUSION
The use of first, second and third generation TKIs has made CML a clinically manageable disease. Despite this major improvement in the standard of care for CML, the complexity of CML-BC physiopathology, together with the failure of TKIs as therapeutic agents in CML-BC and their inability to eradicate CML at the stem cell level, fully justify the ongoing CML research. However, the complexity of such BCR-ABL1 kinase-dependent and/or -independent molecular networks controlling LSC phenotype in both disease stages makes it safe to conclude that the key to a successful CML eradication rests on understanding the bidirectional network of signals between leukemic HSC/progenitors and BM microenvironment. Thus, a better understanding of the events governing LSC behavior might lead to the “biological” cure of CML and effective treatment of CML-BC and, perhaps, other acute leukemias originating at the HSC level.
Acknowledgments
This work was supported in part by NCI CA163800 (Danilo Perrotti).
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
References
- 1.Perrotti D, Jamieson C, Goldman J, Skorski T. Chronic myeloid leukemia: mechanisms of blastic transformation. J Clin Invest. 2010;120(7):2254–64. doi: 10.1172/JCI41246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quintás-Cardama A, Cortes J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood. 2009;113(8):1619–30. doi: 10.1182/blood-2008-03-144790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Foley SB, Hildenbrand ZL, Soyombo AA, et al. Expression of BCR/ABL p210 from a knockin allele enhances bone marrow engraftment without inducing neoplasia. Cell Reports. 2013;5(1):51–60. doi: 10.1016/j.celrep.2013.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldman JM. Initial treatment for patients with CML. Hematology Am Soc Hematol Educ Program. 2009:453–60. doi: 10.1182/asheducation-2009.1.453. [DOI] [PubMed] [Google Scholar]
- 5.Hughes TP, Branford S. Monitoring disease response to tyrosine kinase inhibitor therapy in CML. Hematology Am Soc Hematol Educ Program. 2009:477–87. doi: 10.1182/asheducation-2009.1.477. [DOI] [PubMed] [Google Scholar]
- 6.Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2014 update on diagnosis, monitoring, and management. Am J Hematol. 2014;89(5):547–56. doi: 10.1002/ajh.23691. [DOI] [PubMed] [Google Scholar]
- 7.Apperley JF. Chronic myeloid leukaemia. Lancet. 2015;385(9976):1447–59. doi: 10.1016/S0140-6736(13)62120-0. [DOI] [PubMed] [Google Scholar]
- 8.Ross DM, Hughes TP. How I determine if and when to recommend stopping tyrosine kinase inhibitor treatment for chronic myeloid leukaemia. Br J Haematol. 2014;166(1):3–11. doi: 10.1111/bjh.12892. [DOI] [PubMed] [Google Scholar]
- 9.Apperley JF. Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007;8(11):1018–29. doi: 10.1016/S1470-2045(07)70342-X. [DOI] [PubMed] [Google Scholar]
- 10.Wei G, Rafiyath S, Liu D. First-line treatment for chronic myeloid leukemia: dasatinib, nilotinib, or imatinib. J Hematol Oncol. 2010;3:47. doi: 10.1186/1756-8722-3-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cortes JE, Kim DW, Pinilla-Ibarz J, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369(19):1783–96. doi: 10.1056/NEJMoa1306494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wehrle J, Pahl HL, von Bubnoff N. Ponatinib: a third-generation inhibitor for the treatment of CML. Recent Results Cancer Res. 2014;201:99–107. doi: 10.1007/978-3-642-54490-3_5. [DOI] [PubMed] [Google Scholar]
- 13.Hehlmann R. How I treat CML blast crisis. Blood. 2012;120(4):737–47. doi: 10.1182/blood-2012-03-380147. [DOI] [PubMed] [Google Scholar]
- 14.Khorashad JS, Kelley TW, Szankasi P, et al. BCR-ABL1 compound mutations in tyrosine kinase inhibitor-resistant CML: frequency and clonal relationships. Blood. 2013;121(3):489–98. doi: 10.1182/blood-2012-05-431379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Innes AJ, Apperley JF. Chronic myeloid leukemia-transplantation in the tyrosine kinase era. Hematol Oncol Clin North Am. 2014;28(6):1037–53. doi: 10.1016/j.hoc.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 16.Cortes JE, Kantarjian HM, Rea D, et al. Final analysis of the efficacy and safety of omacetaxine mepesuccinate in patients with chronic- or accelerated-phase chronic myeloid leukemia: Results with 24 months of follow-up. Cancer. 2015;121(10):1637–44. doi: 10.1002/cncr.29240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walker CJ, Oaks JJ, Santhanam R, et al. Preclinical and clinical efficacy of XPO1/CRM1 inhibition by the karyopherin inhibitor KPT-330 in Ph+ leukemias. Blood. 2013;122(17):3034–44. doi: 10.1182/blood-2013-04-495374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahon FX, Réa D, Guilhot J, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11(11):1029–35. doi: 10.1016/S1470-2045(10)70233-3. [DOI] [PubMed] [Google Scholar]
- 19.Rousselot P, Charbonnier A, Cony-Makhoul P, et al. Loss of major molecular response as a trigger for restarting tyrosine kinase inhibitor therapy in patients with chronic-phase chronic myelogenous leukemia who have stopped imatinib after durable undetectable disease. J Clin Oncol. 2014;32(5):424–30. doi: 10.1200/JCO.2012.48.5797. [DOI] [PubMed] [Google Scholar]
- 20.Benjamini O, Kantarjian H, Rios MB, et al. Patient-driven discontinuation of tyrosine kinase inhibitors: single institution experience. Leuk Lymphoma. 2014;55(12):2879–86. doi: 10.3109/10428194.2013.831092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rea D, Nicolini FE, Tulliez M, et al. Dasatinib or nilotinib discontinuation in chronic phase (CP)-chronic myeloid leukemia (CML) patients (pts) with durably undetectable BCR-ABL transcripts: Interim analysis of the STOP 2G-TKI study with a minimum follow-up of 12 months - on behalf of the french CML group filmc. 56th ASH meeting. Blood. 2014;124 Abstract 811. [Google Scholar]
- 22.Balabanov S, Braig M, Brümmendorf TH. Current aspects in resistance against tyrosine kinase inhibitors in chronic myelogenous leukemia. Drug Discov Today Technol. 2014;11:89–99. doi: 10.1016/j.ddtec.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Holyoake TL, Helgason GV. Do we need more drugs for chronic myeloid leukemia? Immunol Rev. 2015;263(1):106–23. doi: 10.1111/imr.12234. [DOI] [PubMed] [Google Scholar]
- 24.Barnes DJ, Palaiologou D, Panousopoulou E, et al. Bcr-Abl expression levels determine the rate of development of resistance to imatinib mesylate in chronic myeloid leukemia. Cancer Res. 2005;65(19):8912–9. doi: 10.1158/0008-5472.CAN-05-0076. [DOI] [PubMed] [Google Scholar]
- 25.Barnes DJ, Schultheis B, Adedeji S, Melo JV. Dose-dependent effects of Bcr-Abl in cell line models of different stages of chronic myeloid leukemia. Oncogene. 2005;24(42):6432–40. doi: 10.1038/sj.onc.1208796. [DOI] [PubMed] [Google Scholar]
- 26.Modi H, McDonald T, Chu S, Yee JK, Forman SJ, Bhatia R. Role of BCR/ABL gene-expression levels in determining the phenotype and imatinib sensitivity of transformed human hematopoietic cells. Blood. 2007;109(12):5411–21. doi: 10.1182/blood-2006-06-032490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perrotti D, Harb JG. BCR-ABL1 kinase-dependent alteration of mRNA metabolism: potential alternatives for therapeutic intervention. Leuk Lymphoma. 2011;52(Suppl 1):30–44. doi: 10.3109/10428194.2010.546914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skorski T. Oncogenic tyrosine kinases and the DNA-damage response. Nat Rev Cancer. 2002;2(5):351–60. doi: 10.1038/nrc799. [DOI] [PubMed] [Google Scholar]
- 29.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293(5531):876–80. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 30.le Coutre P, Tassi E, Varella-Garcia M, et al. Induction of resistance to the Abelson inhibitor STI571 in human leukemic cells through gene amplification. Blood. 2000;95(5):1758–66. [PubMed] [Google Scholar]
- 31.Marega M, Piazza RG, Pirola A, et al. BCR and BCR-ABL regulation during myeloid differentiation in healthy donors and in chronic phase/blast crisis CML patients. Leukemia. 2010;24(8):1445–9. doi: 10.1038/leu.2010.101. [DOI] [PubMed] [Google Scholar]
- 32.Neviani P, Santhanam R, Trotta R, et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell. 2005;8(5):355–68. doi: 10.1016/j.ccr.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 33.Amin HM, Hoshino K, Yang H, Lin Q, Lai R, Garcia-Manero G. Decreased expression level of SH2 domain-containing protein tyrosine phosphatase-1 (Shp1) is associated with progression of chronic myeloid leukaemia. J Pathol. 2007;212(4):402–10. doi: 10.1002/path.2178. [DOI] [PubMed] [Google Scholar]
- 34.Koptyra M, Cramer K, Slupianek A, Richardson C, Skorski T. BCR/ABL promotes accumulation of chromosomal aberrations induced by oxidative and genotoxic stress. Leukemia. 2008;22(10):1969–72. doi: 10.1038/leu.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skorski T. Chronic myeloid leukemia cells refractory/resistant to tyrosine kinase inhibitors are genetically unstable and may cause relapse and malignant progression to the terminal disease state. Leuk Lymphoma. 2011;52(Suppl 1):23–9. doi: 10.3109/10428194.2010.546912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stoklosa T, Poplawski T, Koptyra M, et al. BCR/ABL inhibits mismatch repair to protect from apoptosis and induce point mutations. Cancer Res. 2008;68(8):2576–80. doi: 10.1158/0008-5472.CAN-07-6858. [DOI] [PubMed] [Google Scholar]
- 37.Dierov J, Sanchez PV, Burke BA, et al. BCR/ABL induces chromosomal instability after genotoxic stress and alters the cell death threshold. Leukemia. 2009;23(2):279–86. doi: 10.1038/leu.2008.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. 2004;103(11):4010–22. doi: 10.1182/blood-2003-12-4111. [DOI] [PubMed] [Google Scholar]
- 39.Feinstein E, Cimino G, Gale RP, et al. p53 in chronic myelogenous leukemia in acute phase. Proc Natl Acad Sci USA. 1991;88(14):6293–7. doi: 10.1073/pnas.88.14.6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roche-Lestienne C, Deluche L, Corm S, et al. RUNX1 DNA-binding mutations and RUNX1-PRDM16 cryptic fusions in BCR-ABL+ leukemias are frequently associated with secondary trisomy 21 and may contribute to clonal evolution and imatinib resistance. Blood. 2008;111(7):3735–41. doi: 10.1182/blood-2007-07-102533. [DOI] [PubMed] [Google Scholar]
- 41.Zhang SJ, Shi JY, Li JY. GATA-2 L359 V mutation is exclusively associated with CML progression but not other hematological malignancies and GATA-2 P250A is a novel single nucleotide polymorphism. Leuk Res. 2009;33(8):1141–3. doi: 10.1016/j.leukres.2009.02.025. [DOI] [PubMed] [Google Scholar]
- 42.Beer PA, Knapp DJ, Miller PH, et al. Disruption of IKAROS activity in primitive chronic-phase CML cells mimics myeloid disease progression. Blood. 2015;125(3):504–15. doi: 10.1182/blood-2014-06-581173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bolton-Gillespie E, Schemionek M, Klein HU, et al. Genomic instability may originate from imatinib-refractory chronic myeloid leukemia stem cells. Blood. 2013;121(20):4175–83. doi: 10.1182/blood-2012-11-466938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muvarak N, Nagaria P, Rassool FV. Genomic instability in chronic myeloid leukemia: targets for therapy? Curr Hematol Malig Rep. 2012;7(2):94–102. doi: 10.1007/s11899-012-0119-0. [DOI] [PubMed] [Google Scholar]
- 45.Skorski T. Genetic mechanisms of chronic myeloid leukemia blastic transformation. Curr Hematol Malig Rep. 2012;7(2):87–93. doi: 10.1007/s11899-012-0114-5. [DOI] [PubMed] [Google Scholar]
- 46.Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121(1):396–409. doi: 10.1172/JCI35721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hamilton A, Helgason GV, Schemionek M, et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood. 2012;119(6):1501–10. doi: 10.1182/blood-2010-12-326843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neviani P, Harb JG, Oaks JJ, et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J Clin Invest. 2013;123(10):4144–57. doi: 10.1172/JCI68951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cramer K, Nieborowska-Skorska M, Koptyra M, et al. BCR/ABL and other kinases from chronic myeloproliferative disorders stimulate single-strand annealing, an unfaithful DNA double-strand break repair. Cancer Res. 2008;68(17):6884–8. doi: 10.1158/0008-5472.CAN-08-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Copland M, Hamilton A, Elrick LJ, et al. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood. 2006;107(11):4532–9. doi: 10.1182/blood-2005-07-2947. [DOI] [PubMed] [Google Scholar]
- 51.Jamieson CH, Ailles LE, Dylla SJ, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351(7):657–67. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 52.Mughal TI, Barbui T, Abdel-Wahab O, et al. Novel insights into the biology and treatment of chronic myeloproliferative neoplasms. Leuk Lymphoma. 2015;56(7):1938–48. doi: 10.3109/10428194.2014.974594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kinstrie R, Karamitros D, Goardon N, et al. Leukemia stem cell potential of different progenitor sub-populations in myeloid blast phase CML. 56th ASH meeting. Blood. 2014;124(21):Abstract 3489. [Google Scholar]
- 54.Chu S, McDonald T, Lin A, et al. Persistence of leukemia stem cells in chronic myelogenous leukemia patients in prolonged remission with imatinib treatment. Blood. 2011;118(20):5565–72. doi: 10.1182/blood-2010-12-327437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Holyoake T, Jiang X, Eaves C, Eaves A. Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood. 1999;94(6):2056–64. [PubMed] [Google Scholar]
- 56.Holyoake TL, Jiang X, Jorgensen HG, et al. Primitive quiescent leukemic cells from patients with chronic myeloid leukemia spontaneously initiate factor-independent growth in vitro in association with up-regulation of expression of interleukin-3. Blood. 2001;97(3):720–8. doi: 10.1182/blood.v97.3.720. [DOI] [PubMed] [Google Scholar]
- 57.Janssen JJ, Deenik W, Smolders KG, et al. Residual normal stem cells can be detected in newly diagnosed chronic myeloid leukemia patients by a new flow cytometric approach and predict for optimal response to imatinib. Leukemia. 2012;26(5):977–84. doi: 10.1038/leu.2011.347. [DOI] [PubMed] [Google Scholar]
- 58.Järås M, Johnels P, Hansen N, et al. Isolation and killing of candidate chronic myeloid leukemia stem cells by antibody targeting of IL-1 receptor accessory protein. Proc Natl Acad Sci USA. 2010;107(37):16280–5. doi: 10.1073/pnas.1004408107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nievergall E, Ramshaw HS, Yong AS, et al. Monoclonal antibody targeting of IL-3 receptor α with CSL362 effectively depletes CML progenitor and stem cells. Blood. 2014;123(8):1218–28. doi: 10.1182/blood-2012-12-475194. [DOI] [PubMed] [Google Scholar]
- 60.Sato T, Goyama S, Kataoka K, et al. Evi1 defines leukemia-initiating capacity and tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Oncogene. 2014;33(42):5028–38. doi: 10.1038/onc.2014.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Herrmann H, Sadovnik I, Cerny-Reiterer S, et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood. 2014;123(25):3951–62. doi: 10.1182/blood-2013-10-536078. [DOI] [PubMed] [Google Scholar]
- 62.Kobayashi CI, Takubo K, Kobayashi H, et al. The IL-2/CD25 axis maintains distinct subsets of chronic myeloid leukemia-initiating cells. Blood. 2014;123(16):2540–9. doi: 10.1182/blood-2013-07-517847. [DOI] [PubMed] [Google Scholar]
- 63.Valent P, Sadovnik I, Ráčil Z, et al. DPPIV (CD26) as a novel stem cell marker in Ph+ chronic myeloid leukaemia. Eur J Clin Invest. 2014;44(12):1239–45. doi: 10.1111/eci.12368. [DOI] [PubMed] [Google Scholar]
- 64.Zhang B, Ho Y, McDonald T, et al. Role of enhanced microenvironmental interleukin-1 (IL-1) expression and increased IL-1 responsiveness in persistence of leukemia stem cells in TKI treated CML patients. 56th ASH meeting. Blood. 2014;124(21) Abstract 4357. [Google Scholar]
- 65.Krause DS, Fulzele K, Catic A, et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat Med. 2013;19(11):1513–7. doi: 10.1038/nm.3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505(7483):327–34. doi: 10.1038/nature12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang B, Ho YW, Huang Q, et al. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell. 2012;21(4):577–92. doi: 10.1016/j.ccr.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang B, Li M, McDonald T, et al. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-β-catenin signaling. Blood. 2013;121(10):1824–38. doi: 10.1182/blood-2012-02-412890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jin L, Tabe Y, Konoplev S, et al. CXCR4 up-regulation by imatinib induces chronic myelogenous leukemia (CML) cell migration to bone marrow stroma and promotes survival of quiescent CML cells. Mol Cancer Ther. 2008;7(1):48–58. doi: 10.1158/1535-7163.MCT-07-0042. [DOI] [PubMed] [Google Scholar]
- 70.Konopleva MY, Jordan CT. Leukemia stem cells and microenvironment: biology and therapeutic targeting. J Clin Oncol. 2011;29(5):591–9. doi: 10.1200/JCO.2010.31.0904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Agarwal A, Fleischman AG, Petersen CL, et al. Effects of plerixafor in combination with BCR-ABL kinase inhibition in a murine model of CML. Blood. 2012;120(13):2658–68. doi: 10.1182/blood-2011-05-355396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jamieson CH, Barroga CF, Vainchenker WP. Miscreant myeloproliferative disorder stem cells. Leukemia. 2008;22(11):2011–9. doi: 10.1038/leu.2008.290. [DOI] [PubMed] [Google Scholar]
- 73.Schemionek M, Spieker T, Kerstiens L, et al. Leukemic spleen cells are more potent than bone marrow-derived cells in a transgenic mouse model of CML. Leukemia. 2012;26(5):1030–7. doi: 10.1038/leu.2011.366. [DOI] [PubMed] [Google Scholar]
- 74.Sengupta A, Arnett J, Dunn S, Williams DA, Cancelas JA. Rac2 GTPase deficiency depletes BCR-ABL+ leukemic stem cells and progenitors in vivo. Blood. 2010;116(1):81–4. doi: 10.1182/blood-2009-10-247437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tabe Y, Jin L, Iwabuchi K, et al. Role of stromal microenvironment in nonpharmacological resistance of CML to imatinib through Lyn/CXCR4 interactions in lipid rafts. Leukemia. 2012;26(5):883–92. doi: 10.1038/leu.2011.291. [DOI] [PubMed] [Google Scholar]
- 76.Schepers K, Pietras EM, Reynaud D, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell. 2013;13(3):285–99. doi: 10.1016/j.stem.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Laperrousaz B, Jeanpierre S, Sagorny K, et al. Primitive CML cell expansion relies on abnormal levels of BMPs provided by the niche and on BMPRIb overexpression. Blood. 2013;122(23):3767–77. doi: 10.1182/blood-2013-05-501460. [DOI] [PubMed] [Google Scholar]
- 78.Baba T, Naka K, Morishita S, Komatsu N, Hirao A, Mukaida N. MIP-1α/CCL3-mediated maintenance of leukemia-initiating cells in the initiation process of chronic myeloid leukemia. J Exp Med. 2013;210(12):2661–73. doi: 10.1084/jem.20130112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Naka K, Hoshii T, Muraguchi T, et al. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010;463(7281):676–80. doi: 10.1038/nature08734. [DOI] [PubMed] [Google Scholar]
- 80.Spencer JA, Ferraro F, Roussakis E, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature. 2014;508(7495):269–73. doi: 10.1038/nature13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ng KP, Manjeri A, Lee KL, et al. Physiologic hypoxia promotes maintenance of CML stem cells despite effective BCR-ABL1 inhibition. Blood. 2014;123(21):3316–26. doi: 10.1182/blood-2013-07-511907. [DOI] [PubMed] [Google Scholar]
- 82.Kida A, Kahn M. Hypoxia selects for a quiescent, CML stem/leukemia initiating-like population dependent on CBP/catenin transcription. Curr Mol Pharmacol. 2013;6(3):204–10. doi: 10.2174/1874467207666140219121219. [DOI] [PubMed] [Google Scholar]
- 83.Zhang H, Li H, Xi HS, Li S. HIF1α is required for survival maintenance of chronic myeloid leukemia stem cells. Blood. 2012;119(11):2595–607. doi: 10.1182/blood-2011-10-387381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Azab AK, Weisberg E, Sahin I, et al. The influence of hypoxia on CML trafficking through modulation of CXCR4 and E-cadherin expression. Leukemia. 2013;27(4):961–4. doi: 10.1038/leu.2012.353. [DOI] [PubMed] [Google Scholar]
- 85.Slupianek A, Falinski R, Znojek P, et al. BCR-ABL1 kinase inhibits uracil DNA glycosylase UNG2 to enhance oxidative DNA damage and stimulate genomic instability. Leukemia. 2013;27(3):629–34. doi: 10.1038/leu.2012.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bruns I, Czibere A, Fischer JC, et al. The hematopoietic stem cell in chronic phase CML is characterized by a transcriptional profile resembling normal myeloid progenitor cells and reflecting loss of quiescence. Leukemia. 2009;23(5):892–9. doi: 10.1038/leu.2008.392. [DOI] [PubMed] [Google Scholar]
- 87.Graham SM, Vass JK, Holyoake TL, Graham GJ. Transcriptional analysis of quiescent and proliferating CD34+ human hemopoietic cells from normal and chronic myeloid leukemia sources. Stem Cells. 2007;25(12):3111–20. doi: 10.1634/stemcells.2007-0250. [DOI] [PubMed] [Google Scholar]
- 88.Brauer KM, Werth D, von Schwarzenberg K, et al. BCR-ABL activity is critical for the immunogenicity of chronic myelogenous leukemia cells. Cancer Res. 2007;67(11):5489–97. doi: 10.1158/0008-5472.CAN-07-0302. [DOI] [PubMed] [Google Scholar]
- 89.Malagola M, Breccia M, Skert C, et al. Long term outcome of Ph+ CML patients achieving complete cytogenetic remission with interferon based therapy moving from interferon to imatinib era. Am J Hematol. 2014;89(2):119–24. doi: 10.1002/ajh.23593. [DOI] [PubMed] [Google Scholar]
- 90.Ilander M, Kreutzman A, Mustjoki S. IFNα induces prolonged remissions modeling curative immunologic responses in chronic myeloid leukemia. OncoImmunology. 2014;3:e28781. doi: 10.4161/onci.28781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Qiao Y, Liu B, Li Z. Activation of NK cells by extracellular heat shock protein 70 through induction of NKG2D ligands on dendritic cells. Cancer Immun. 2008;8:12. [PMC free article] [PubMed] [Google Scholar]
- 92.Terme M, Borg C, Guilhot F, et al. BCR/ABL promotes dendritic cell-mediated natural killer cell activation. Cancer Res. 2005;65(14):6409–17. doi: 10.1158/0008-5472.CAN-04-2675. [DOI] [PubMed] [Google Scholar]
- 93.Mizoguchi I, Yoshimoto T, Katagiri S, et al. Sustained upregulation of effector natural killer cells in chronic myeloid leukemia after discontinuation of imatinib. Cancer Sci. 2013;104(9):1146–53. doi: 10.1111/cas.12216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Qiu ZY, Xu W, Li JY. Large granular lymphocytosis during dasatinib therapy. Cancer Biol Ther. 2014;15(3):247–55. doi: 10.4161/cbt.27310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kreutzman A, Juvonen V, Kairisto V, et al. Mono/oligoclonal T and NK cells are common in chronic myeloid leukemia patients at diagnosis and expand during dasatinib therapy. Blood. 2010;116(5):772–82. doi: 10.1182/blood-2009-12-256800. [DOI] [PubMed] [Google Scholar]
- 96.Carayol G, Giron-Michel J, Azzarone B, et al. Altered natural killer cell differentiation in CD34+ progenitors from chronic myeloid leukemia patients. Oncogene. 2000;19(23):2758–66. doi: 10.1038/sj.onc.1203584. [DOI] [PubMed] [Google Scholar]
- 97.Mellqvist UH, Hansson M, Brune M, Dahlgren C, Hermodsson S, Hellstrand K. Natural killer cell dysfunction and apoptosis induced by chronic myelogenous leukemia cells: role of reactive oxygen species and regulation by histamine. Blood. 2000;96(5):1961–8. [PubMed] [Google Scholar]
- 98.Kijima M, Gardiol N, Held W. Natural killer cell mediated missing-self recognition can protect mice from primary chronic myeloid leukemia in vivo. PLoS One. 2011;6(11):e27639. doi: 10.1371/journal.pone.0027639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cai A, Keskin DB, DeLuca DS, et al. Mutated BCR-ABL generates immunogenic T-cell epitopes in CML patients. Clin Cancer Res. 2012;18(20):5761–72. doi: 10.1158/1078-0432.CCR-12-1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jiang Q, Crews LA, Barrett CL, et al. ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc Natl Acad Sci USA. 2013;110(3):1041–6. doi: 10.1073/pnas.1213021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schürch CM, Riether C, Ochsenbein AF. Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell. 2014;14(4):460–72. doi: 10.1016/j.stem.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 102.Schürch C, Riether C, Matter MS, Tzankov A, Ochsenbein AF. CD27 signaling on chronic myelogenous leukemia stem cells activates Wnt target genes and promotes disease progression. J Clin Invest. 2012;122(2):624–38. doi: 10.1172/JCI45977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Crews LA, Jamieson CH. Chronic myeloid leukemia stem cell biology. Curr Hematol Malig Rep. 2012;7(2):125–32. doi: 10.1007/s11899-012-0121-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Goff DJ, Court Recart A, Sadarangani A, et al. A Pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. Cell Stem Cell. 2013;12(3):316–28. doi: 10.1016/j.stem.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Minami Y, Stuart SA, Ikawa T, et al. BCR-ABL-transformed GMP as myeloid leukemic stem cells. Proc Natl Acad Sci USA. 2008;105(46):17967–72. doi: 10.1073/pnas.0808303105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Giuntoli S, Rovida E, Barbetti V, Cipolleschi MG, Olivotto M, Dello Sbarba P. Hypoxia suppresses BCR/Abl and selects imatinib-insensitive progenitors within clonal CML populations. Leukemia. 2006;20(7):1291–3. doi: 10.1038/sj.leu.2404224. [DOI] [PubMed] [Google Scholar]
- 107.Rovida E, Peppicelli S, Bono S, et al. The metabolically-modulated stem cell niche: a dynamic scenario regulating cancer cell phenotype and resistance to therapy. Cell Cycle. 2014;13(20):3169–75. doi: 10.4161/15384101.2014.964107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schemionek M, Elling C, Steidl U, et al. BCR-ABL enhances differentiation of long-term repopulating hematopoietic stem cells. Blood. 2010;115(16):3185–95. doi: 10.1182/blood-2009-04-215376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Van Etten RA, Mauro M, Radich JP, et al. Advances in the biology and therapy of chronic myeloid leukemia: proceedings from the 6th post-ASH international chronic myeloid leukemia and myeloproliferative neoplasms workshop. Leuk Lymphoma. 2013;54(6):1151–8. doi: 10.3109/10428194.2012.745524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Benoit YD, Guezguez B, Boyd AL, Bhatia M. Molecular pathways: epigenetic modulation of Wnt-glycogen synthase kinase-3 signaling to target human cancer stem cells. Clin Cancer Res. 2014;20(21):5372–8. doi: 10.1158/1078-0432.CCR-13-2491. [DOI] [PubMed] [Google Scholar]
- 111.Lim S, Saw TY, Zhang M, et al. Targeting of the MNK-eIF4E axis in blast crisis chronic myeloid leukemia inhibits leukemia stem cell function. Proc Natl Acad Sci USA. 2013;110(25):E2298–307. doi: 10.1073/pnas.1301838110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhao C, Blum J, Chen A, et al. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. 2007;12(6):528–41. doi: 10.1016/j.ccr.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li L, Bhatia R. Stem cell quiescence. Clin Cancer Res. 2011;17(15):4936–1. doi: 10.1158/1078-0432.CCR-10-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hu Y, Chen Y, Douglas L, Li S. beta-Catenin is essential for survival of leukemic stem cells insensitive to kinase inhibition in mice with BCR-ABL-induced chronic myeloid leukemia. Leukemia. 2009;23(1):109–16. doi: 10.1038/leu.2008.262. [DOI] [PubMed] [Google Scholar]
- 115.Coluccia AM, Vacca A, Duñach M, et al. Bcr-Abl stabilizes beta-catenin in chronic myeloid leukemia through its tyrosine phosphorylation. EMBO J. 2007;26(5):1456–66. doi: 10.1038/sj.emboj.7601485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Abrahamsson AE, Geron I, Gotlib J, et al. Glycogen synthase kinase 3beta missplicing contributes to leukemia stem cell generation. Proc Natl Acad Sci USA. 2009;106(10):3925–9. doi: 10.1073/pnas.0900189106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Radich JP, Dai H, Mao M, et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc Natl Acad Sci USA. 2006;103(8):2794–9. doi: 10.1073/pnas.0510423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liu YC, Lai WC, Chuang KA, et al. Blockade of JAK2 activity suppressed accumulation of β-catenin in leukemic cells. J Cell Biochem. 2010;111(2):402–11. doi: 10.1002/jcb.22714. [DOI] [PubMed] [Google Scholar]
- 119.Samanta AK, Lin H, Sun T, Kantarjian H, Arlinghaus RB. Janus kinase 2: a critical target in chronic myelogenous leukemia. Cancer Res. 2006;66(13):6468–72. doi: 10.1158/0008-5472.CAN-06-0025. [DOI] [PubMed] [Google Scholar]
- 120.Neviani P, Santhanam R, Oaks JJ, et al. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J Clin Invest. 2007;117(9):2408–21. doi: 10.1172/JCI31095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Perrotti D, Neviani P. Protein phosphatase 2A: a target for anticancer therapy. Lancet Oncol. 2013;14(6):e229–38. doi: 10.1016/S1470-2045(12)70558-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Samanta AK, Chakraborty SN, Wang Y, et al. Jak2 inhibition deactivates Lyn kinase through the SET-PP2A-SHP1 pathway, causing apoptosis in drug-resistant cells from chronic myelogenous leukemia patients. Oncogene. 2009;28(14):1669–81. doi: 10.1038/onc.2009.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Samanta AK, Chakraborty SN, Wang Y, Schlette E, Reddy EP, Arlinghaus RB. Destabilization of Bcr-Abl/Jak2 network by a Jak2/Abl kinase inhibitor ON044580 overcomes drug resistance in blast crisis chronic myelogenous leukemia (CML) Genes Cancer. 2010;1(4):346–59. doi: 10.1177/1947601910372232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Samanta A, Perazzona B, Chakraborty S, et al. Janus kinase 2 regulates Bcr-Abl signaling in chronic myeloid leukemia. Leukemia. 2011;25(3):463–72. doi: 10.1038/leu.2010.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Traer E, MacKenzie R, Snead J, et al. Blockade of JAK2-mediated extrinsic survival signals restores sensitivity of CML cells to ABL inhibitors. Leukemia. 2012;26(5):1140–3. doi: 10.1038/leu.2011.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bewry NN, Nair RR, Emmons MF, Boulware D, Pinilla-Ibarz J, Hazlehurst LA. Stat3 contributes to resistance toward BCR-ABL inhibitors in a bone marrow microenvironment model of drug resistance. Mol Cancer Ther. 2008;7(10):3169–75. doi: 10.1158/1535-7163.MCT-08-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gallipoli P, Cook A, Rhodes S, et al. JAK2/STAT5 inhibition by nilotinib with ruxolitinib contributes to the elimination of CML CD34+ cells in vitro and in vivo. Blood. 2014;124(9):1492–501. doi: 10.1182/blood-2013-12-545640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lin H, Chen M, Rothe K, Lorenzi MV, Woolfson A, Jiang X. Selective JAK2/ABL dual inhibition therapy effectively eliminates TKI-insensitive CML stem/progenitor cells. Oncotarget. 2014;5(18):8637–50. doi: 10.18632/oncotarget.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chen M, Gallipoli P, DeGeer D, et al. Targeting primitive chronic myeloid leukemia cells by effective inhibition of a new AHI-1-BCR-ABL-JAK2 complex. J Natl Cancer Inst. 2013;105(6):405–23. doi: 10.1093/jnci/djt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yokoyama N, Reich NC, Miller WT. Determinants for the interaction between Janus kinase 2 and protein phosphatase 2A. Arch Biochem Biophys. 2003;417(1):87–95. doi: 10.1016/s0003-9861(03)00333-3. [DOI] [PubMed] [Google Scholar]
- 131.Perrotti D, Neviani P. Protein phosphatase 2A (PP2A), a drugable tumor suppressor in Ph1(+) leukemias. Cancer Metastasis Rev. 2008;27(2):159–68. doi: 10.1007/s10555-008-9119-x. [DOI] [PubMed] [Google Scholar]
- 132.Patturajan M, Nomoto S, Sommer M, et al. DeltaNp63 induces beta-catenin nuclear accumulation and signaling. Cancer Cell. 2002;1(4):369–79. doi: 10.1016/s1535-6108(02)00057-0. [DOI] [PubMed] [Google Scholar]
- 133.Seeling JM, Miller JR, Gil R, Moon RT, White R, Virshup DM. Regulation of beta-catenin signaling by the B56 subunit of protein phosphatase 2A. Science. 1999;283(5410):2089–91. doi: 10.1126/science.283.5410.2089. [DOI] [PubMed] [Google Scholar]
- 134.Zhang W, Yang J, Liu Y, et al. PR55 alpha, a regulatory subunit of PP2A, specifically regulates PP2A-mediated beta-catenin dephosphorylation. J Biol Chem. 2009;284(34):22649–56. doi: 10.1074/jbc.M109.013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lucas CM, Harris RJ, Giannoudis A, Copland M, Slupsky JR, Clark RE. Cancerous inhibitor of PP2A (CIP2A) at diagnosis of chronic myeloid leukemia is a critical determinant of disease progression. Blood. 2011;117(24):6660–8. doi: 10.1182/blood-2010-08-304477. [DOI] [PubMed] [Google Scholar]
- 136.Oakley K, Han Y, Vishwakarma BA, et al. Setbp1 promotes the self-renewal of murine myeloid progenitors via activation of Hoxa9 and Hoxa10. Blood. 2012;119(25):6099–108. doi: 10.1182/blood-2011-10-388710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhou LL, Zhao Y, Ringrose A, et al. AHI-1 interacts with BCR-ABL and modulates BCR-ABL transforming activity and imatinib response of CML stem/progenitor cells. J Exp Med. 2008;205(11):2657–71. doi: 10.1084/jem.20072316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Court Recart AC, Sadarangani A, Goff D, Shih AY, Wall R, Leu H, et al. Combination Targeted Therapy to Impair Self-Renewal Capacity of Human Blast Crisis Leukemia Stem Cells. 53rd ASH meeting. Blood. 2011;118 Abstract 1693. [Google Scholar]
- 139.Heidel FH, Bullinger L, Feng Z, et al. Genetic and pharmacologic inhibition of β-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell. 2012;10(4):412–24. doi: 10.1016/j.stem.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fiskus W, Sharma S, Saha S, et al. Pre-clinical efficacy of combined therapy with novel β-catenin antagonist BC2059 and histone deacetylase inhibitor against AML cells. Leukemia. 2015;29(6):1267–78. doi: 10.1038/leu.2014.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ringel F, Kaeda J, Schwarz M, et al. Effects of Jak2 type 1 inhibitors NVP-BSK805 and NVP-BVB808 on Jak2 mutation-positive and Bcr-Abl-positive cell lines. Acta Haematol. 2014;132(1):75–86. doi: 10.1159/000356784. [DOI] [PubMed] [Google Scholar]
- 142.Hantschel O, Warsch W, Eckelhart E, et al. BCR-ABL uncouples canonical JAK2-STAT5 signaling in chronic myeloid leukemia. Nat Chem Biol. 2012;8(3):285–93. doi: 10.1038/nchembio.775. [DOI] [PubMed] [Google Scholar]
- 143.Su W, Meng F, Huang L, Zheng M, Liu W, Sun H. Sonic hedgehog maintains survival and growth of chronic myeloid leukemia progenitor cells through β-catenin signaling. Exp Hematol. 2012;40(5):418–27. doi: 10.1016/j.exphem.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 144.Dierks C, Beigi R, Guo GR, et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell. 2008;14(3):238–49. doi: 10.1016/j.ccr.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 145.Zhao C, Chen A, Jamieson CH, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458(7239):776–9. doi: 10.1038/nature07737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hofmann I, Stover EH, Cullen DE, et al. Hedgehog signaling is dispensable for adult murine hematopoietic stem cell function and hematopoiesis. Cell Stem Cell. 2009;4(6):559–67. doi: 10.1016/j.stem.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bhardwaj G, Murdoch B, Wu D, et al. Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nat Immunol. 2001;2(2):172–80. doi: 10.1038/84282. [DOI] [PubMed] [Google Scholar]
- 148.Detmer K, Walker AN, Jenkins TM, Steele TA, Dannawi H. Erythroid differentiation in vitro is blocked by cyclopamine, an inhibitor of hedgehog signaling. Blood Cells Mol Dis. 2000;26(4):360–72. doi: 10.1006/bcmd.2000.0318. [DOI] [PubMed] [Google Scholar]
- 149.Mar BG, Amakye D, Aifantis I, Buonamici S. The controversial role of the Hedgehog pathway in normal and malignant hematopoiesis. Leukemia. 2011;25(11):1665–73. doi: 10.1038/leu.2011.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Merchant A, Joseph G, Wang Q, Brennan S, Matsui W. Gli1 regulates the proliferation and differentiation of HSCs and myeloid progenitors. Blood. 2010;115(12):2391–6. doi: 10.1182/blood-2009-09-241703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Plaisant M, Giorgetti-Peraldi S, Gabrielson M, Loubat A, Dani C, Peraldi P. Inhibition of hedgehog signaling decreases proliferation and clonogenicity of human mesenchymal stem cells. PLoS One. 2011;6(2):e16798. doi: 10.1371/journal.pone.0016798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Irvine DA, Copland M. Targeting hedgehog in hematologic malignancy. Blood. 2012;119(10):2196–204. doi: 10.1182/blood-2011-10-383752. [DOI] [PubMed] [Google Scholar]
- 153.Zhang B, Strauss AC, Chu S, et al. Effective targeting of quiescent chronic myelogenous leukemia stem cells by histone deacetylase inhibitors in combination with imatinib mesylate. Cancer Cell. 2010;17(5):427–42. doi: 10.1016/j.ccr.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Papayannidis C, Guadagnuolo V, Iacobucci I, et al. PF-04449913 reverts multi drug resistance (MDR) by a strong down-regulation of ABCA2 and BCL2 on leukemia stem cells in phase I acute myeloid leukemia and chronic myeloid leukemia treated patients. 53rd ASH meeting. Blood. 2011;118 Abstract 1429. [Google Scholar]
- 155.Harb JG, Neviani P, Chyla BJ, et al. Bcl-xL anti-apoptotic network is dispensable for development and maintenance of CML but is required for disease progression where it represents a new therapeutic target. Leukemia. 2013;27(10):1996–2005. doi: 10.1038/leu.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Airiau K, Mahon FX, Josselin M, Jeanneteau M, Belloc F. PI3K/mTOR pathway inhibitors sensitize chronic myeloid leukemia stem cells to nilotinib and restore the response of progenitors to nilotinib in the presence of stem cell factor. Cell Death Dis. 2013;4:e827. doi: 10.1038/cddis.2013.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ito K, Bernardi R, Morotti A, et al. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 2008;453(7198):1072–8. doi: 10.1038/nature07016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.El Eit RM, Iskandarani AN, Saliba JL, et al. Effective targeting of chronic myeloid leukemia initiating activity with the combination of arsenic trioxide and interferon alpha. Int J Cancer. 2014;134(4):988–6. doi: 10.1002/ijc.28427. [DOI] [PubMed] [Google Scholar]
- 159.Chen Y, Hu Y, Zhang H, Peng C, Li S. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat Genet. 2009;41(7):783–92. doi: 10.1038/ng.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chen Y, Li D, Li S. The Alox5 gene is a novel therapeutic target in cancer stem cells of chronic myeloid leukemia. Cell Cycle. 2009;8(21):3488–92. doi: 10.4161/cc.8.21.9852. [DOI] [PubMed] [Google Scholar]
- 161.Bellodi C, Lidonnici MR, Hamilton A, et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest. 2009;119(5):1109–23. doi: 10.1172/JCI35660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Heaney NB, Pellicano F, Zhang B, et al. Bortezomib induces apoptosis in primitive chronic myeloid leukemia cells including LTC-IC and NOD/SCID repopulating cells. Blood. 2010;115(11):2241–50. doi: 10.1182/blood-2008-06-164582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Miyazono K. Tumour promoting functions of TGF-β in CML-initiating cells. J Biochem. 2012;152(5):383–5. doi: 10.1093/jb/mvs106. [DOI] [PubMed] [Google Scholar]
- 164.Hui RC, Gomes AR, Constantinidou D, et al. The forkhead transcription factor FOXO3a increases phosphoinositide-3 kinase/Akt activity in drug-resistant leukemic cells through induction of PIK3CA expression. Mol Cell Biol. 2008;28(19):5886–98. doi: 10.1128/MCB.01265-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Duy C, Hurtz C, Shojaee S, et al. BCL6 enables Ph+ acute lymphoblastic leukaemia cells to survive BCR-ABL1 kinase inhibition. Nature. 2011;473(7347):384–8. doi: 10.1038/nature09883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Saito M, Gao J, Basso K, et al. A signaling pathway mediating downregulation of BCL6 in germinal center B cells is blocked by BCL6 gene alterations in B cell lymphoma. Cancer Cell. 2007;12(3):280–92. doi: 10.1016/j.ccr.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 167.Nieborowska-Skorska M, Kopinski PK, Ray R, et al. Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors. Blood. 2012;119(18):4253–63. doi: 10.1182/blood-2011-10-385658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pellicano F, Copland M, Jorgensen HG, Mountford J, Leber B, Holyoake TL. BMS-214662 induces mitochondrial apoptosis in chronic myeloid leukemia (CML) stem/progenitor cells, including CD34+38- cells, through activation of protein kinase Cbeta. Blood. 2009;114(19):4186–96. doi: 10.1182/blood-2009-05-219550. [DOI] [PubMed] [Google Scholar]
- 169.Li L, Wang L, Li L, et al. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell. 2012;21(2):266–81. doi: 10.1016/j.ccr.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Neviani P, Perrotti D. SETting OP449 into the PP2A-activating drug family. Clin Cancer Res. 2014;20(8):2026–8. doi: 10.1158/1078-0432.CCR-14-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Khorashad JS, Eiring AM, Mason CC, et al. shRNA library screening identifies nucleocytoplasmic transport as a mediator of BCR-ABL1 kinase-independent resistance. Blood. 2015;125(11):1772–81. doi: 10.1182/blood-2014-08-588855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Harb J, Neviani P, Huettner C, Marcucci G, Perrotti D. BCR/ABL dosage hierarchically and temporally influences hnRNP A1, hnRNP K and hnRNP E2 expression in hematopoietic stem and progenitor cells. 51st ASH meeting. Blood. 2009;114(22) Abstract 191. [Google Scholar]
- 173.Wang LS, Li L, Li L, et al. MicroRNA-486 regulates normal erythropoiesis and enhances growth and modulates drug response in CML progenitors. Blood. 2015;125(8):1302–13. doi: 10.1182/blood-2014-06-581926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Silvestri G, Ellis J, Stramucci L, et al. MiR-300 acts as a tumor supressor in Ph+ progenitors by modulating the JAK2-SET/PP2A/β-catenin interplay. 56th ASH meeting. Blood. 2014;124 Abstract 4529. [Google Scholar]
- 175.Babashah S, Sadeghizadeh M, Hajifathali A, et al. Targeting of the signal transducer Smo links microRNA-326 to the oncogenic Hedgehog pathway in CD34+ CML stem/progenitor cells. Int J Cancer. 2013;133(3):579–89. doi: 10.1002/ijc.28043. [DOI] [PubMed] [Google Scholar]
- 176.Yu Y, Cao L, Yang L, Kang R, Lotze M, Tang D. microRNA 30A promotes autophagy in response to cancer therapy. Autophagy. 2012;8(5):853–5. doi: 10.4161/auto.20053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gao SM, Yang J, Chen C, et al. miR-15a/161 enhances retinoic acid-mediated differentiation of leukemic cells and is up-regulated by retinoic acid. Leuk Lymphoma. 2011;52(12):2365–71. doi: 10.3109/10428194.2011.601476. [DOI] [PubMed] [Google Scholar]
- 178.Bueno MJ, Pérez de Castro I, Gómez de Cedrón M, et al. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell. 2008;13(6):496–506. doi: 10.1016/j.ccr.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 179.Suresh S, McCallum L, Lu W, Lazar N, Perbal B, Irvine AE. Mi-croRNAs 130a/b are regulated by BCR-ABL and downregulate expression of CCN3 in CML. J Cell Commun Signal. 2011;5(3):183–91. doi: 10.1007/s12079-011-0139-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chim CS, Wong KY, Leung CY, et al. Epigenetic inactivation of the hsa-miR-203 in haematological malignancies. J Cell Mol Med. 2011;15(12):2760–7. doi: 10.1111/j.1582-4934.2011.01274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Venturini L, Battmer K, Castoldi M, et al. Expression of the miR-1792 polycistron in chronic myeloid leukemia (CML) CD34+ cells. Blood. 2007;109(10):4399–405. doi: 10.1182/blood-2006-09-045104. [DOI] [PubMed] [Google Scholar]
- 182.Lopotová T, Záčková M, Klamová H, Moravcová J. MicroRNA-451 in chronic myeloid leukemia: miR-451-BCR-ABL regulatory loop? Leuk Res. 2011;35(7):974–7. doi: 10.1016/j.leukres.2011.03.029. [DOI] [PubMed] [Google Scholar]
- 183.McCallum L, Price S, Planque N, et al. A novel mechanism for BCR-ABL action: stimulated secretion of CCN3 is involved in growth and differentiation regulation. Blood. 2006;108(5):1716–23. doi: 10.1182/blood-2006-04-016113. [DOI] [PubMed] [Google Scholar]
- 184.McCallum L, Lu W, Price S, Lazar N, Perbal B, Irvine AE. CCN3 suppresses mitogenic signalling and reinstates growth control mechanisms in Chronic Myeloid Leukaemia. J Cell Commun Signal. 2012;6(1):27–35. doi: 10.1007/s12079-011-0142-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Viswanathan SR, Powers JT, Einhorn W, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet. 2009;41(7):843–8. doi: 10.1038/ng.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hershkovitz-Rokah O, Modai S, Pasmanik-Chor M, et al. MiR-30e induces apoptosis and sensitizes K562 cells to imatinib treatment via regulation of the BCR-ABL protein. Cancer Lett. 2015;356(2 Pt B):597–605. doi: 10.1016/j.canlet.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 187.Liu Y, Song Y, Ma W, Zheng W, Yin H. Decreased microRNA-30a levels are associated with enhanced ABL1 and BCR-ABL1 expression in chronic myeloid leukemia. Leuk Res. 2013;37(3):349–56. doi: 10.1016/j.leukres.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 188.Xu L, Xu Y, Jing Z, et al. Altered expression pattern of miR-29a, miR-29b and the target genes in myeloid leukemia. Exp Hematol Oncol. 2014;3:17. doi: 10.1186/2162-3619-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Li Y, Wang H, Tao K, et al. miR-29b suppresses CML cell proliferation and induces apoptosis via regulation of BCR/ABL1 protein. Exp Cell Res. 2013;319(8):1094–101. doi: 10.1016/j.yexcr.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 190.Zhu X, Lin Z, Du J, Zhou X, Yang L, Liu G. Studies on microR-NAs that are correlated with the cancer stem cells in chronic mye-loid leukemia. Mol Cell Biochem. 2014;390(1–2):75–84. doi: 10.1007/s11010-013-1958-2. [DOI] [PubMed] [Google Scholar]
- 191.Guo G, Kang Q, Zhu X, et al. A long noncoding RNA critically regulates Bcr-Abl-mediated cellular transformation by acting as a competitive endogenous RNA. Oncogene. 2015;34(14):1768–79. doi: 10.1038/onc.2014.131. [DOI] [PubMed] [Google Scholar]
- 192.Xu C, Fu H, Gao L, et al. BCR-ABL/GATA1/miR-138 mini circuitry contributes to the leukemogenesis of chronic myeloid leukemia. Oncogene. 2014;33(1):44–54. doi: 10.1038/onc.2012.557. [DOI] [PubMed] [Google Scholar]
- 193.Chang JS, Santhanam R, Trotta R, et al. High levels of the BCR/ABL oncoprotein are required for the MAPK-hnRNP-E2 dependent suppression of C/EBPalpha-driven myeloid differentiation. Blood. 2007;110(3):994–1003. doi: 10.1182/blood-2007-03-078303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Eiring AM, Harb JG, Neviani P, et al. miR-328 functions as an RNA decoy to modulate hnRNP E2 regulation of mRNA translation in leukemic blasts. Cell. 2010;140(5):652–65. doi: 10.1016/j.cell.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Perrotti D, Cesi V, Trotta R, et al. BCR-ABL suppresses C/EBPalpha expression through inhibitory action of hnRNP E2. Nat Genet. 2002;30(1):48–58. doi: 10.1038/ng791. [DOI] [PubMed] [Google Scholar]
- 196.Tenen DG, Hromas R, Licht JD, Zhang DE. Transcription factors, normal myeloid development, and leukemia. Blood. 1997;90(2):489–519. [PubMed] [Google Scholar]
- 197.Tenen DG. Disruption of differentiation in human cancer: AML shows the way. Nat Rev Cancer. 2003;3(2):89–101. doi: 10.1038/nrc989. [DOI] [PubMed] [Google Scholar]
- 198.Machová Poláková K, Lopotová T, Klamová H, et al. Expression patterns of microRNAs associated with CML phases and their disease related targets. Mol Cancer. 2011;10:41. doi: 10.1186/1476-4598-10-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Morris VA, Zhang A, Yang T, et al. MicroRNA-150 expression induces myeloid differentiation of human acute leukemia cells and normal hematopoietic progenitors. PLoS One. 2013;8(9):e75815. doi: 10.1371/journal.pone.0075815. [DOI] [PMC free article] [PubMed] [Google Scholar]