

Abstract
We have previously reported benzimidazole-based compounds to be potent inhibitors of FabI for Francisella tularensis (FtFabI), making them promising antimicrobial hits. Optically active enantiomers exhibit markedly differing affinities toward FtFabI. The IC50 of benzimidazole (-)-1 is ∼100× lower than the (+)-enantiomer, with similar results for the 2 enantiomers. Determining the absolute configuration for these optical compounds and elucidating their binding modes is important for further design. Electronic circular dichroism (ECD) quantum calculations have become important in determining absolute configurations of optical compounds. We determined the absolute configuration of (-)/(+)-1 and (-)/(+)-2 by comparing experimental spectra and theoretical density functional theory (DFT) simulations of ECD spectra at the B3LYP/6-311+G(2d, p) level using Gaussian09. Comparison of experimental and calculated ECD spectra indicates that the S configuration corresponds to the (-)-rotation for both compounds 1 and 2, while the R configuration corresponds to the (+)-rotation. Further, molecular dynamics simulations and MM-GBSA binding energy calculations for these two pairs of enantiomers with FtFabI show much tighter binding MM-GBSA free energies for S-1 and S-2 than for their enantiomers, R-1 and R-2, consistent with the S configuration being the more active one, and with the ECD determination of the S configuration corresponding to (-) and the R configuration corresponding to (+). Thus, our computational studies allow us to assign (-) to (S)- and (+) to (R)- for compounds 1 and 2, and to further evaluate structural changes to improve efficacy.
Keywords: absolute configuration, Gaussian, electronic circular dichroism (ECD), molecular dynamics (MD), Molecular Mechanics/Generalized Born Surface Area (MM-GBSA)
The need for new antimicrobial agents is increasing consequent to the spread of antibiotic-resistant bacteria, the emergence of new infections, and the potential of using serious pathogenic bacteria in bioweapons.1-3 Hence, it is important to develop new, highly potent antibiotics with novel mechanisms for treatment of bacterial infections. In this regard, the enoyl-acyl carrier protein reductase, FabI, is a promising target for antibacterial drug development.4 FabI is an NAD(P)H-dependent oxidoreductase that acts to reduce enoyl-ACP substrates in the final and rate-controlling step of the bacterial fatty acid biosynthesis pathway (FAS II).5 The FAS II pathway is the result of a series of discrete enzymes at separate steps and is essential in the biosynthesis of the bacterial fatty acid components of bacterial lipid membranes and energy stores.6-8 The mammalian counterpart (FAS I) uses a single, large, multifunctional enzyme. Because of the differing structure and mechanism, FAS-II is an attractive and potentially safe target for antibacterial agent development. Although there are multiple isozymes (including FabK, FabL, and FabV)9-11 that can be present in addition to, or in place of FabI in various bacterial species, FabI is recognized as a target for specific, narrow-spectrum antibacterial agents, for treating species that express FabI as the sole or dominant enoyl reductase enzyme in the FAS II pathway. Francisella tularensis (F. tularensis) is a bacterial species for which FabI is an essential enzyme in fatty acid synthesis.12, 13 F. tularensis is a Gram-negative bacterium responsible for tularemia and is a potential bioweapon due to its ease of cultivation and spread by aerosol as well as its high virulence.14 Although streptomycin, ciprofloxacin and tetracycline have been used for treatment of tularemia15, each has limitations, such as ototoxicity of streptomycin, teeth staining by tetracyclines or even the emergence of ciprofloxacin-resistant strains of F tularensis.16 Therefore, development of novel agents with improved efficacy and different mechanisms of action against F tularensis is an important priority.
In prior work, we identified benzimidazole compounds as novel and potent FabI inhibitors, with antibacterial activities against both F tularensis and Staphylococcus aureus (S. aureus).17-21 Our current work demonstrates that enantiomers of optical benzimidazole compounds possess markedly differing inhibitory efficacies toward FtFabI. The IC50 of benzimidazole (-)-1 is about one hundred times lower than its (+)-enantiomer, with similar results for enantiomers of 2 (Fig. 1). This motivated us to determine the absolute configuration (AC) for the optical benzimidazole compounds to correlate the differing efficacies with their distinctive binding modes to FtFabI.
Fig. 1.
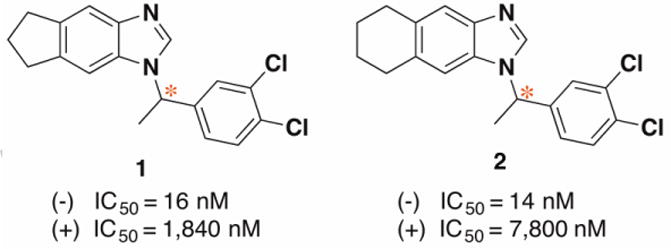
Structures and FtFabI IC50 inhibitory activities of enantiomers of compounds 1 and 2.
However, neither the crystal structures for the optical compounds nor the crystal complexes with FtFabI have been determined, which could directly determine the AC for these chiroptical compounds. We thus used two strategies to determine the AC of chiral benzimidazole compounds by comparing experimental and theoretical results. The first strategy focused on the optically active benzimidazoles themselves by comparing the experimental and calculated spectra for the chiroptical compounds. That benefits from the dramatic growth in the applications of chiroptical spectroscopy for molecular structure determination, and the development of computational resources with faster processor speeds for quantum chemical spectral predictions in recent years.22 The use of electronic circular dichroism (ECD) can provide reliable compound structure determination.23, 24 We thus used ECD to determine the AC for both optical benzimidazole compounds 1 and 2. We have also previously used molecular mechanics -Poisson Boltzmann /generalized Born surface area (MM-PB/GBSA) methods to evaluate the detailed correlation between experimental and predicted results for benzimidazoles binding to FtFabI.25 Herein, we report similar computations to predict the binding affinity of the enantiomers of compounds 1 and 2 to FtFabI. Entropy calculations were combined in this study with MM-GBSA to improve predicted binding affinity. The resulting MM-GBSA calculations, in conjunction with experimental IC50 measurements, are highly consistent with the ECD absolute configuration calculations, allowing us to unambiguously correlate optical enantiomers with inhibitory activity.
Due to the advances in quantum mechanics and the availability of faster computers, determining the 3D structure for optical compounds with confidence has been successfully achieved in recent years.22-24, 26, 27 In the present study, the calculated and experimental ECD spectra of S-1, R-1, S-2, and R-2 were recorded in MeOH from 200-260 nm (see Supporting information). The predicted ECD spectrum of S-1 shows a negative Cotton effect (CE) at 210 nm, which compares well with the (-)-1 experimental ECD with negative CE at 209 nm (Fig. 2A). Similar results were observed between calculated S-2 and experimental (-)-2 in Fig. 2C. Likewise, the intense positive band in the predicted ECD spectrum of R-1 at 210 nm compares well with the (+)-1 experimental ECD which exhibits a positive band at 209nm (Fig. 2B). Similar results were achieved between calculated R-2 and experimental (+)-2 (Fig. 2D).
Fig. 2.
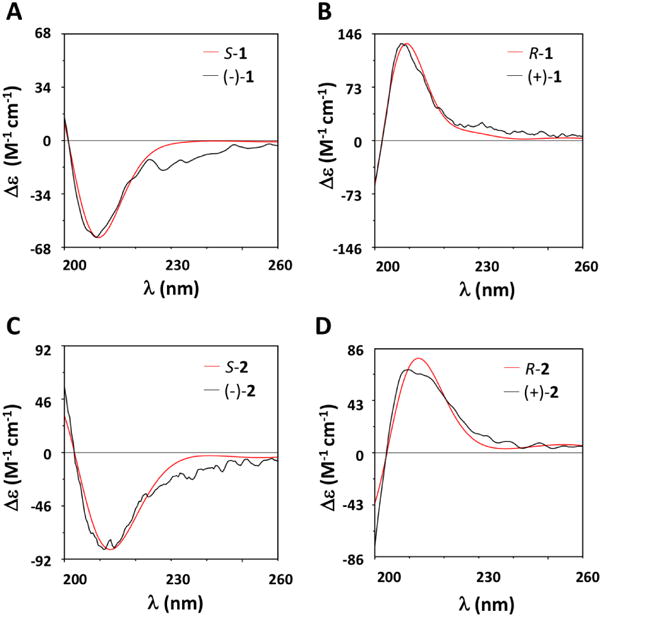
Comparison of experimental (black) and calculated (red) ECD spectra for (A) S-1 and (-)-1; (B) R-1 and (+)-1; (C) S-2 and (-)-2; and (D) R-2 and (+)-2. To facilitate the comparison between experimental and computational spectra, which is sometimes hampered by misalignment of Cotton Effects (CE), we have blue shifted the computational spectrum by 1 nm.
The good agreement in position and intensity of the ECD CEs with experimental spectra supports the accuracy in the theoretical reproduction of conformer populations and our deduction that the absolute configuration of the (-)-sample is determined to be (S), while (+) is (R).
Moreover, the agreement between experimental and calculated ECD spectra of S-1, R-1, S-2, and R-2, was quantitatively tested by the maximum similarity values for ECD (SimECD) spectra with SpecDis.31 SimECD similarity is 0.96 for S-1 with (-)-1, 0.99 for R-1 with (+)-1, 0.98 for S-2 with (-)-2, 0.98 for R-2 with (+)-2, respectively. These high similarity values provide high confidence in the AC assignment of S corresponding to (-) and R corresponding to (+).
MD simulations and binding free energy calculations are efficient tools to investigate many thermodynamic properties of proteins and protein-inhibitor interactions.32-35 In this work, the enantiomers of compounds 1 and 2 were chosen to probe their different activity toward FtFabI at the atomic level. To achieve this, 50 ns-MD simulations and the MM-GBSA were combined to perform a systematic comparative study (see Supporting information for methods). To model the water environment around the protein, MM-GBSA strips the explicit water molecules and uses a parametrized implicit water model (GB). The entropy (-TΔS) contribution to the binding affinity was estimated by a computationally simple and efficient approach recently proposed by Duan et al. based on the fluctuations of protein-ligand interaction energies36 that is commonly used to improve MM-PB/GBSA prediction.37-39
RMSD calculations for the atoms of compounds 1 and 2 enantiomers showed that the simulations were stable for all four systems during the 50-ns MD simulations (Fig. 3).
Fig. 3.
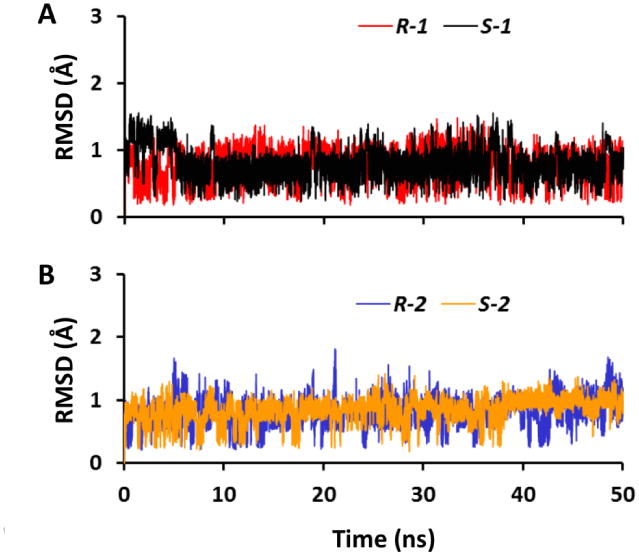
RMSD of the ligands within FtFabI with respect to their initial conformations in MD simulations. (A) RMSD for enantiomers of compound 1; (B) RMSD for enantiomers of compound 2.
The MM-GBSA binding energies were calculated for these four systems, and the results reveal the impact of the chiral carbons on the binding affinities of 1 and 2 to FtFabI (Table 1). Our calculations reveal that compounds with chiral carbons in the S-configuration exhibit significantly tighter binding toward FtFabI compared to the R-configuration (ΔΔGbind = -2.28 kcal mol1 and -1.55 kcal mol1 for compounds 1 and 2, respectively). The predicted activity difference between the S and R configurations correlates well with the experimental activity difference between (-) and (+) rotations, with ΔΔGexp = -2.25 kcal mol-1 and -2.97 kcal mol-1 for compounds 1 and 2, respectively. Further, the MM-GBSA analysis also provides individual contributions to the binding affinity. A general feature is that the contributions are quite similar for all compounds, with the van der Waals interactions being the most prominent, followed by the electrostatic contributions and the nonpolar contributions to solvation. As the most prominent interactions, van der Waals interactions show similar results between S-1 and R-1, as well as between S-2 and R-2. That results from FtFabI having a very hydrophobic binding pocket, with 1 and 2 being lipophilic compounds that also show similar binding modes. The electrostatic portion contributes the more obvious differences between the enantiomers of 1 and 2, which appears to result from differences in H-bonding interactions. The final predicted binding energy shows S-1 binding tighter than its enantiomer R-1, with same behavior of S-2 binding rather tighter than its enantiomer R-2. Comparing with the experimental results of (-)-rotation exhibiting a much lower IC50 than (+)-rotation similarly leads to the conclusion that the S-configuration corresponds to (-) and the R -configuration corresponds to (+). That conclusion is consistent with that derived from our above ECD calculation, which also concludes that the S-configuration corresponds to the (-)-rotation and the R-configuration corresponds to the (+)-rotation.
Table 1. Energetic analysis for S-1, R-1, S-2, and R-2 with FtFabI complexes as obtained by MM-GBSA.
| Energy (kcal mol-1) | S-1 | R-1 | S-2 | R-2 |
|---|---|---|---|---|
| AEvdw | -45.61 ± 0.02 a | -45.91 ± 0.02 | -47.95 ± 0.02 | -47.16 ± 0.02 |
| ΔEelec | -12.08 ± 0.02 | -8.92 ± 0.02 | -12.86 ± 0.02 | -9.95 ± 0.02 |
| ΔEMMb | -57.68 ± 0.02 | -54.84 ± 0.03 | -60.81 ± 0.03 | -57.11 ± 0.03 |
| ΔGnonpol | -5.40 ± 0.01 | -4.84 ± 0.01 | -5.38 ± 0.01 | -5.20 ± 0.01 |
| ΔGpol | 20.07 ± 0.01 | 18.03 ± 0.02 | 21.87 ± 0.02 | 19.05 ± 0.02 |
| ΔGsolvc | 14.68 ± 0.01 | 13.19 ± 0.02 | 16.48 ± 0.02 | 13.86 ± 0.02 |
| ΔH(MM+solv) | -43.01 ± 0.02 | -41.65 ± 0.02 | -44.32 ± 0.02 | -43.25 ± 0.02 |
| -TΔS | 5.91 | 6.83 | 7.89 | 8.37 |
| ΔGpredict | -37.10 ± 0.02 | -34.82 ± 0.02 | -36.43 ± 0.02 | -34.88 ± 0.02 |
| ΔΔGpredict | -2.28 ± 0.02 | -1.55 ± 0.02 | ||
| ΔGexpd | -10.25 | -8.00 | -10.11 | -7.14 |
| ΔΔGexp | -2.25 | -2.97 | ||
Values are average ± standard error of the mean.
ΔEMM = ΔEvdw + ΔEelec.
ΔGsolv = ΔGnonpol + ΔGpol.
ΔGexp is the experimental binding energy as calculated by ΔGexp = RT lnKi, where R is the ideal gas constant (1.9872 × 10-3 kcal K-1 mol-1) and T is 300 K.
The effect of the differing enantiomers on the binding affinity with FtFabI can be further elucidated by calculating the enthalpy (ΔH) contributions of individual amino acids. A comparison among binding compounds S-1, R-1, S-2, and R-2 is presented in Fig. 4. The graph shows the enthalpy contributions of important residues. The NADH and residue Y156 show the most important enthalpy contributions with ΔH more than -2.0 kcal mol-1. That is consistent with our experience for most FabI inhibitors, that those two residues are stable and indispensable H-bond donors for inhibitor binding,40, 41 and consistent with the benzimidazole compounds interactions with FtFabI from the crystal complex.18, 19 Y146 could also form a π stacking interaction with the chloride substituted phenyl ring (Fig. 5), which shows an obvious contribution to the enthalpy. A196, A197, I200, F203 as well as NADH are the surrounding residues for the chiral carbon atoms. A92, F93, A94, L99 and M159 are surrounding the five or six-member ring and build the larger tunnel-like pocket for benzimidazoles (Fig. 5).
Fig. 4.
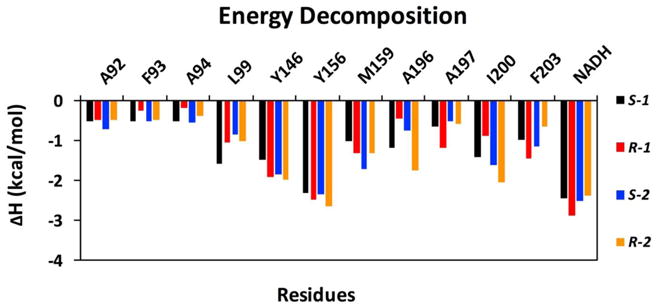
Per r sidu contributions to ΔH for FtFabI with S-1, R-1, S-2, and R-2 complexes. Individual contributions for active site residues are displayed.
Fig. 5.
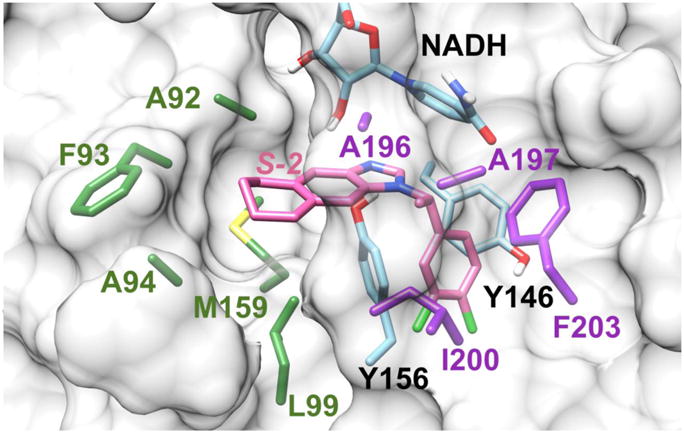
Binding mode of S-2 and FtFabI obtained by modifying the atoms of the ligand from the crystal structure of the FtFabI-benzimidazole complex (PDB IDs: 4J4T).19 The residues of A196, A197, I200, and F203 surrounding the chiral methyl are colored in purple. The residues (A92, F93, A94, L99, and M159) surrounding the six-member ring, which also build the larger tunnel-like pocket for benzimidazoles, are colored in green. Y146, Y156 and NADH are colored in cyan, and S-2 is shown in pink. The FtFabI enzyme is shown as a grey surface with only important residue sidechains shown, and with NADH partly shown to save space. The picture was prepared with Chimera1.10.2.42
The MM-GBSA calculations imply that the different electronic interactions among the enantiomers contribute strongly to differing binding affinities toward FtFabI. To explore this further, HBond analysis was performed on the MD trajectory. The occupancies of HBonds are listed in Table 2, which reveals that differing configurations of the benzimidazole compounds produces an obvious impact on the HBond formation among the benzimidazole-N and Y156 and NADH in FtFabI. Both of the HBonds formed between the benzimidazole-N and OH of Y156 and OH of 2′-ribose of NADH show the same trends, with more frequent HBond formation between compounds with the S-configuration and FtFabI. The occupancy of the HBond between the N of S-1 and Y156 is 67%, slightly higher than the 64% in R-1. However, the 34% HBond occupancy between the N of S-1 and NADH is significantly higher than that of 4% in R-1. A similar result occurred for the enantiomers of compound 2, which reaffirms that the S-configuration is the more stable binding one. The HBond formed with Y156 is more frequent than that formed with NADH, but the larger difference of HBond occupancies with cofactor NADH provides a more prominent contribution toward the different electronic interactions between enantiomers.
Table 2. Hydrogen bonds in FtFabI-benzimidazole complexes.
| Interactions | Occupancy (%) | Interactions | Occupancy (%) | ||
|---|---|---|---|---|---|
| Acceptor | Donor | Acceptor | Donor | ||
| S-1 Na | Y156 OH | 67 | S-1 N | NADH OHb | 34 |
| R-1 N | Y156 OH | 64 | R-1 N | NADH OH | 4 |
| s-2 n | Y156 OH | 54 | S-2 N | NADH OH | 29 |
| R-2 N | Y156 OH | 45 | R-2 N | NADH OH | 3 |
N is the nitrogen atom forming the imine group in benzimidazole compounds.
OH is the polar hydrogen in the 2′-ribose OH of NADH.
The occupancy ratios from MD simulations and MM-GBSA calculations also suggest that compounds with the S-configuration, corresponding to (-)-rotation in the experimental study, are the more efficient binding inhibitors.
Conclusions
Determining the AC for these optical compounds and elucidating their binding modes is important for further designing active benzimidazole compounds as FtFabI inhibitors. The AC for optical benzimidazole compounds 1 and 2 was assigned by ECD comparison between theoretical and experimental spectra. The MM-GBSA results for these compounds with FtFabI also support the conclusion from spectral analysis that the S-configuration corresponds to (-)-rotation and the R-configuration corresponds to the (+)-rotation. Furthermore, the distinct binding mode for enantiomers with FtFabI was elaborated during MD simulations, helping to explain the differing efficacies. Thus, this approach has allowed us to define the AC for chiral products without the effort required for chiral purification and crystallization. Moreover, this study can be expected to provide significant input to the molecular basis for the design of more potent FtFabI inhibitors.
Supplementary Material
Absolute configurations of benzimidazole enantiomers were determined
Theoretical electronic circular dichroism is consistent with experimental data
MM-GBSA calculations provide strong discrimination between enantiomers
MM-GBSA supports configuration determination from electronic circular dichroism
Acknowledgments
This work was supported by the National Institutes of Health Grants AI110090 & AI077949. The authors thank the Academic Computing and Communications Center of the University of Illinois at Chicago for access to the high-performance computing cluster, Extreme, to perform the computational work. We also thank ChemAxon for access to their programs, including JChem, Chemicalize and Marvin, used in the preceding analysis. Molecular graphics and analyses for Fig. 5 were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
Footnotes
A. Supplementary data
Supplementary data associated with this article can be found in the online version
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Spellberg B, Powers JH, Brass EP, Miller LG, Edwards JE. Trends in antimicrobial drug development: Implications for the future. Clin Infect Dis. 2004;38(9):1279–1286. doi: 10.1086/420937. [DOI] [PubMed] [Google Scholar]
- 2.Deleo FR, Chambers HF. Reemergence of antibiotic-resistant Staphylococcus aureus in the genomics era. J Clin Invest. 2009;119(9):2464–2474. doi: 10.1172/JCI38226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silver S. Laboratory-acquired lethal infections by potential bioweapons pathogens including Ebola in 2014. FEMS Microbiology Letters. 2015;362(1):1–6. doi: 10.1093/femsle/fnu008. [DOI] [PubMed] [Google Scholar]
- 4.Payne DJ, Warren PV, Holmes DJ, Ji YD, Lonsdale JT. Bacterial fatty-acid biosynthesis: a genomics-driven target for antibacterial drug discovery. Drug Discov Today. 2001;6(10):537–544. doi: 10.1016/s1359-6446(01)01774-3. [DOI] [PubMed] [Google Scholar]
- 5.Bergler H, Fuchsbichler S, Hogenauer G, Turnowsky F. The enoyl-[acyl-carrier-protein] reductase (FabI) of Escherichia coli, which catalyzes a key regulatory step in fatty acid biosynthesis, accepts NADH and NADPH as cofactors and is inhibited by palmitoyl-CoA. Eur J Biochem. 1996;242(3):689–694. doi: 10.1111/j.1432-1033.1996.0689r.x. [DOI] [PubMed] [Google Scholar]
- 6.Zhang YM, White SW, Rock CO. Inhibiting bacterial fatty acid synthesis. J Biol Chem. 2006;281(26):17541–17544. doi: 10.1074/jbc.R600004200. [DOI] [PubMed] [Google Scholar]
- 7.Wright HT, Reynolds KA. Antibacterial targets in fatty acid biosynthesis. Curr Opin Microbiol. 2007;10(5):447–453. doi: 10.1016/j.mib.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heath RJ, White SW, Rock CO. Lipid biosynthesis as a target for antibacterial agents. Prog Lipid Res. 2001;40(6):467–497. doi: 10.1016/s0163-7827(01)00012-1. [DOI] [PubMed] [Google Scholar]
- 9.Heath RJ, Rock CO. Microbiology - A triclosan-resistant bacterial enzyme. Nature. 2000;406(6792):145–146. doi: 10.1038/35018162. [DOI] [PubMed] [Google Scholar]
- 10.Heath RJ, Su N, Murphy CK, Rock CO. The enoyl-[acyl-carrier-protein] reductases FabI and FabL from Bacillus subtilis. J Biol Chem. 2000;275(51):40128–40133. doi: 10.1074/jbc.M005611200. [DOI] [PubMed] [Google Scholar]
- 11.Massengo-Tiasse RP, Cronan JE. Vibrio cholerae FabV defines a new class of enoyl-acyl carrier protein reductase. J Biol Chem. 2008;283(3):1308–1316. doi: 10.1074/jbc.M708171200. [DOI] [PubMed] [Google Scholar]
- 12.Kingry LC, Cummings JE, Brookman KW, Bommineni GR, Tonge PJ, Slayden RA. The Francisella tularensis FabI Enoyl-Acyl Carrier Protein Reductase Gene Is Essential to Bacterial Viability and Is Expressed during Infection. J Bacteriol. 2013;195(2):351–358. doi: 10.1128/JB.01957-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerusz V, Denis A, Faivre F, et al. From Triclosan toward the Clinic: Discovery of Nonbiocidal, Potent FabI Inhibitors for the Treatment of Resistant Bacteria. J Med Chem. 2012;55(22):9914–9928. doi: 10.1021/jm301113w. [DOI] [PubMed] [Google Scholar]
- 14.Oyston PCF, Sjostedt A, Titball RW. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat Rev Micro. 2004;2(12):967–978. doi: 10.1038/nrmicro1045. [DOI] [PubMed] [Google Scholar]
- 15.Hepburn MJ, Simpson AJH. Tularemia: current diagnosis and treatment options. Expert Rev Anti-Infe. 2008;6(2):231–240. doi: 10.1586/14787210.6.2.231. [DOI] [PubMed] [Google Scholar]
- 16.Loveless BM, Yermakova A, Christensen DR, et al. Identification of ciprofloxacin resistance by SimpleProbe (TM), High Resolution Melt and Pyrosequencing (TM) nucleic acid analysis in biothreat agents: Bacillus anthracis, Yersinia pestis and Francisella tularensis. Mol Cell Probe. 2010;24(3):154–160. doi: 10.1016/j.mcp.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 17.Hevener KE, Mehboob S, Su PC, et al. Discovery of a Novel and Potent Class of F. tularensis Enoyl-Reductase (FabI) Inhibitors by Molecular Shape and Electrostatic Matching. J Med Chem. 2012;55(1):268–279. doi: 10.1021/jm201168g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mehboob S, Hevener KE, Truong K, Boci T, Santarsiero BD, Johnson ME. Structural and Enzymatic Analyses Reveal the Binding Mode of a Novel Series of Francisella tularensis Enoyl Reductase (FabI) Inhibitors. J Med Chem. 2012;55(12):5933–5941. doi: 10.1021/jm300489v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mehboob S, Song JH, Hevener KE, et al. Structural and biological evaluation of a novel series of benzimidazole inhibitors of Francisella tularensis enoyl-ACP reductase (FabI) Bioorg Med Chem Lett. 2015;25(6):1292–1296. doi: 10.1016/j.bmcl.2015.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mistry TL, Truong L, Ghosh AK, Johnson ME, Mehboob S. Benzimidazole-Based FabI Inhibitors: A Promising Novel Scaffold for Anti-staphylococcal Drug Development. ACS Infectious Diseases. 2017;3(1):54–61. doi: 10.1021/acsinfecdis.6b00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang YY, Liu Y, Mehboob S, et al. Metabolism-directed structure optimization of benzimidazole-based Francisella tularensis enoyl-reductase (FabI) inhibitors. Xenobiotica. 2014;44(5):404–416. doi: 10.3109/00498254.2013.850553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Polavarapu PL, Covington CL. Comparison of Experimental and Calculated Chiroptical Spectra for Chiral Molecular Structure Determination. Chirality. 2014;26(9):539–552. doi: 10.1002/chir.22316. [DOI] [PubMed] [Google Scholar]
- 23.Santoro E, Mazzeo G, Petrovic AG, et al. Absolute configurations of phytotoxins seiricardine A and inuloxin A obtained by chiroptical studies. Phytochemistry. 2015;116:359–366. doi: 10.1016/j.phytochem.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 24.Sardella R, Ianni F, Di Michele A, et al. Enantioresolution and stereochemical characterization of two chiral sulfoxides endowed with COX-2 inhibitory activity. Chirality. 2017;29(9):536–540. doi: 10.1002/chir.22724. [DOI] [PubMed] [Google Scholar]
- 25.Su PC, Tsai CC, Mehboob S, Hevener KE, Johnson ME. Comparison of radii sets, entropy, QM methods, and sampling on MM-PBSA, MM-GBSA, and QM/MM-GBSA ligand binding energies of F-tularensis enoyl-ACP reductase (FabI) J Comput Chem. 2015;36(25):1859–1873. doi: 10.1002/jcc.24011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Polavarapu PL. Optical rotation: recent advances in determining the absolute configuration. Chirality. 2002;14(10):768–781. doi: 10.1002/chir.10145. [DOI] [PubMed] [Google Scholar]
- 27.Evidente M, Santoro E, Petrovic AG, et al. Absolute configurations of phytotoxic inuloxins B and C based on experimental and computational analysis of chiroptical properties. Phytochemistry. 2016;130:328–334. doi: 10.1016/j.phytochem.2016.07.012. [DOI] [PubMed] [Google Scholar]
- 28.Schrödinger LLC, New York NY. 2016 [Google Scholar]
- 29.Jorgensen WL, Maxwell DS, TiradoRives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118(45):11225–11236. [Google Scholar]
- 30.Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09, Revision B.01. Wallingford CT: 2009. [Google Scholar]
- 31.Bruhn T, Schaumloffel A, Hemberger Y, Bringmann G. SpecDis: Quantifying the Comparison of Calculated and Experimental Electronic Circular Dichroism Spectra. Chirality. 2013;25(4):243–249. doi: 10.1002/chir.22138. [DOI] [PubMed] [Google Scholar]
- 32.Tzoupis H, Leonis G, Mavromoustakos T, Papadopoulos MG. A Comparative Molecular Dynamics, MM-PBSA and Thermodynamic Integration Study of Saquinavir Complexes with Wild-Type HIV-1 PR and L10I, G48V, L63P, A71V, G73S, V82A and I84V Single Mutants. Journal of Chemical Theory and Computation. 2013;9(3):1754–1764. doi: 10.1021/ct301063k. [DOI] [PubMed] [Google Scholar]
- 33.Leonis G, Steinbrecher T, Papadopoulos MG. A Contribution to the Drug Resistance Mechanism of Darunavir, Amprenavir, Indinavir, and Saquinavir Complexes with HIV-1 Protease Due to Flap Mutation I50V: A Systematic MM-PBSA and Thermodynamic Integration Study. Journal of Chemical Information and Modeling. 2013;53(8):2141–2153. doi: 10.1021/ci4002102. [DOI] [PubMed] [Google Scholar]
- 34.Genheden S, Nilsson I, Ryde U. Binding Affinities of Factor Xa Inhibitors Estimated by Thermodynamic Integration and MM/GBSA. Journal of Chemical Information and Modeling. 2011;51(4):947–958. doi: 10.1021/ci100458f. [DOI] [PubMed] [Google Scholar]
- 35.Chen J, Wang X, Zhu T, Zhang Q, Zhang JZH. A Comparative Insight into Amprenavir Resistance of Mutations V32I, G48V, I50V, I54V, and I84V in HIV-1 Protease Based on Thermodynamic Integration and MM-PBSA Methods. Journal of Chemical Information and Modeling. 2015;55(9):1903–1913. doi: 10.1021/acs.jcim.5b00173. [DOI] [PubMed] [Google Scholar]
- 36.Duan LL, Liu X, Zhang JZH. Interaction Entropy: A New Paradigm for Highly Efficient and Reliable Computation of Protein-Ligand Binding Free Energy. J Am Chem Soc. 2016;138(17):5722–5728. doi: 10.1021/jacs.6b02682. [DOI] [PubMed] [Google Scholar]
- 37.Aldeghi M, Bodkin MJ, Knapp S, Biggin PC. Statistical Analysis on the Performance of Molecular Mechanics Poisson-Boltzmann Surface Area versus Absolute Binding Free Energy Calculations: Bromodomains as a Case Study. Journal of Chemical Information and Modeling. 2017 doi: 10.1021/acs.jcim.7b00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duan LL, Feng GQ, Wang XW, Wang LZ, Zhang QG. Effect of electrostatic polarization and bridging water on CDK2-ligand binding affinities calculated using a highly efficient interaction entropy method. Phys Chem Chem Phys. 2017;19(15):10140–10152. doi: 10.1039/c7cp00841d. [DOI] [PubMed] [Google Scholar]
- 39.Duan LL, Feng GQ, Zhang QG. Large-scale molecular dynamics simulation: Effect of polarization on thrombin-ligand binding energy. 2016;6:31488. doi: 10.1038/srep31488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaplan N, Albert M, Awrey D, et al. Mode of Action, In Vitro Activity, and In Vivo Efficacy of AFN-1252, a Selective Antistaphylococcal FabI Inhibitor. Antimicrobial Agents and Chemotherapy. 2012;56(11):5865–5874. doi: 10.1128/AAC.01411-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schiebel J, Chang A, Shah S, et al. Rational Design of Broad Spectrum Antibacterial Activity Based on a Clinically Relevant Enoyl-Acyl Carrier Protein (ACP) Reductase Inhibitor. The Journal of Biological Chemistry. 2014;289(23):15987–16005. doi: 10.1074/jbc.M113.532804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.