

Abstract
We have previously identified and reported several potent piperidine-derived amide inhibitors of the human soluble epoxide hydrolase (sEH) enzyme. The inhibition of this enzyme leads to elevated levels of epoxyeicosatrienoic acids (EETs), which are known to possess anti-inflammatory, vasodilatory, and anti-fibrotic effects. Herein, we report the synthesis of 9 analogs of the lead sEH inhibitor and the follow-up structure-activity relationship and liver microsome stability studies. Our findings show that isosteric modifications that lead to significant alterations in the steric and electronic properties at a specific position in the molecule can reduce the efficacy by up to 75-fold. On the other hand, substituting hydrogen with deuterium produces a notable increase (~30%) in the molecules’ half-lives in both rat and human microsomes, while maintaining sEH inhibition potency. These data highlight the utility of isosteric replacement for improving bioavailability, and the newly-synthesized inhibitor structures may thus, serve as a starting point for preclinical development. Our docking study reveals that in the catalytic pocket of sEH, these analogs are in proximity of the key amino acids involved in hydrolysis of EETs.
Keywords: Soluble epoxide hydrolase (sEH), Non-urea sEH inhibitors, Liver microsomal stability assay, Structure activity relationship (SAR) study, Isosteres
Introduction
There are three main metabolic pathways involved in the metabolism of arachidonic acid (AA): those catalyzed by the cyclooxygenase (COX), the lipoxygenase (LOX) and the cytochrome P450 (CYP) enzymes (1). Of these, the COX- and LOX-mediated reactions have been studied in detail, providing us with inhibitor molecules such as aspirin(2) and zileuton(3), respectively, for the treatment of various inflammatory diseases. On the other hand, the bioactivity of epoxyeicosatrienoic acids (EETs, Figure 1), a metabolite produced in the CYP-dependent reaction, is comparatively less understood than the prostaglandins and leukotrienes.(4)Studies into EET’s biological role have led to the discovery of the importance of sEH (soluble epoxide hydrolase) in regulating EET; inhibition of the enzyme causes an increase in EET concentration, which could have beneficial effects.(5)It is important to note, however, that inhibition of sEH alone may not necessarily yield the desired results, as this could shunt AA to the COX and LOX pathways.(6) Careful in vivo studies will be necessary to address this possibility, in order to achieve the best therapeutic results.
Figure 1.
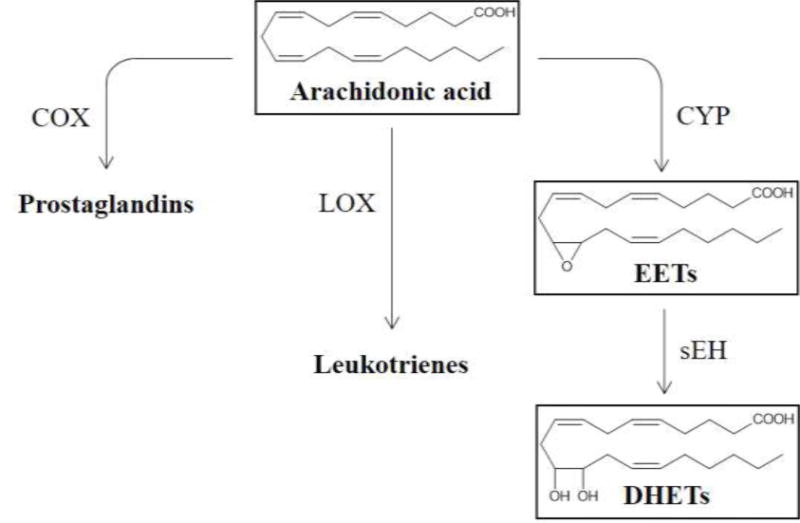
Main metabolic pathways of Arachidonic acid and the role of soluble epoxide hydrolase.
In the CYP- and sEH-mediated biotransformation (Figure 1), AA is first converted by CYP to EETs, which arean important class of lipid mediatorsexhibitingvasodilatory effects(7)as well as anti-inflammatory (8), anti-fibrotic (9), and potent pro-fibrinolytic properties(10). The ensuing sEH-catalyzed addition of a water molecule to EETs leads to the accumulation of the corresponding diols (DHETs), which have diminished biological activity and possibly increased toxicity.(7) sEH is found in many mammalian tissues and has the highest activity in the lungs, kidneys and cardiovascular system (8) and its inhibition has been shown to cause an elevated level of EETs in airway, which helps in alleviating airway hyperresponsiveness.(6)
Thus far, a large body of work has focused on a class of urea-based inhibitors for sEH, e.g. AUDA (Figure 2).(4)Many of these compounds suffer from high melting points, poor solubility, and they require careful and complex formulation.(4)We decided to explore other non-urea scaffolds and our initial screening from the collection provided by the NIH Roadmap Project (Pubchem, AID: 1026) at University of California Davis and Columbia University Medical Center led to the identification of a variety of compounds. (11) Among those that do not contain the urea moiety, the most potent inhibitor was found to be the derivative of isonipectoic acid, 1 (Figure 2).
Figure 2.
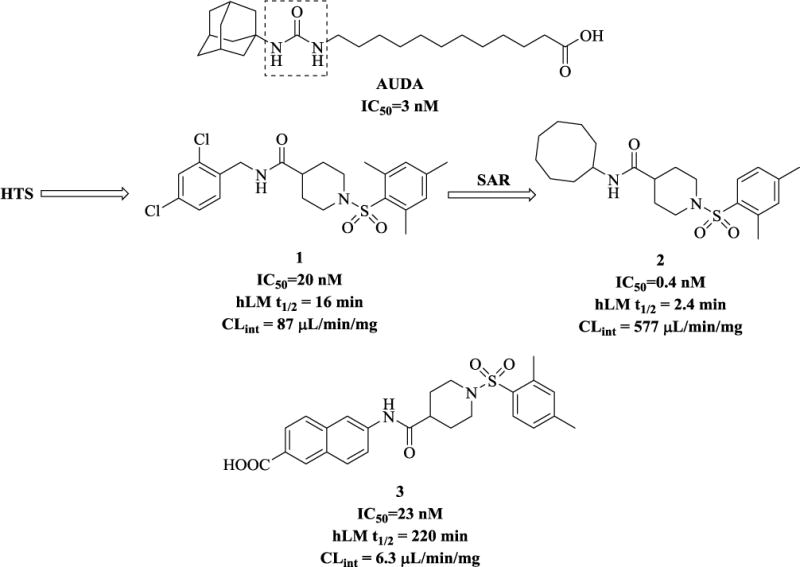
Representative urea and non-urea sEH inhibitors. Dotted box shows urea moiety.
Besides the structure-activity relationship (SAR) studies, (11, 12) we have also successfully co-crystallized one of these non-urea based inhibitors with human sEH (the X-ray crystallographic structure can be found under PDB code: 4HAI).(13) Following careful examination of the human sEH binding pocket, we hypothesized that the left-hand part of the inhibitor molecules could be optimized to further improve their potency. Our previous synthetic efforts yielded several inhibitors with efficacy in the low nanomolar and even picomolar range (e.g. compound 2 in Fig. 2) (12, 13). Our liver microsomal stability assays (which we used as a predictor of in vivo metabolism) revealed that compounds with hydrophobic cycloalkyl substituents on the left of the non-urea piperidine scaffold have poor metabolic profile (e.g. inhibitor 2 that has a half-life (t1/2) of only2.4 min). On the other hand, placing an aromatic moiety in this position improves the molecules’ microsomal stability significantly, while allowing them to maintain a good inhibition profile (Figure 2, inhibitor3, a carboxylic acid derivative, t1/2= 220 min). (12, 13) We therefore decided that this compound (inhibitor 3) would serve as a suitable starting point for further SAR study.
Scheme 2.
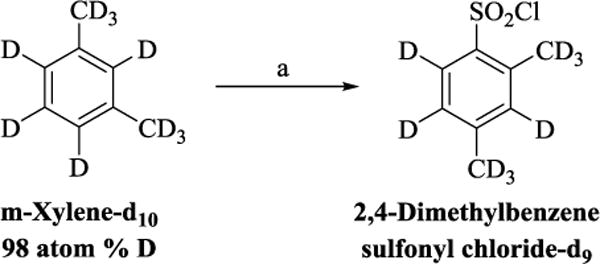
Reagents and condotions (a) CISO3H, CHCL3, 0 °C-rt,2 h, 65%.
Improving metabolic stability of the drug candidates may be achieved via several strategies. These include reducing the molecular weight and lipophilicity of the molecules, replacing H- on the α-carbon atoms with −CH3 to avoid racemization of a stereocenter, modifying labile functional groups, deactivating aromatic rings toward oxidation, and introducing isosteric groups. (14–17) It is important to note, however, that these are just examples of some common approaches and may not be universally applicable to all molecules. This study focuses on the effect of isosteric replacement. Substituting H with F can considerably alter several characteristics of the molecules.(15) Likewise, deuterium, although highly similar to hydrogen, may produce notable changes in the pharmacokinetic parameters. (15)
Herein, we report the design and synthesis of 9 analogs of the selected inhibitor 3, along with the SAR and biological evaluation of these compounds. In addition, we performed human and rat liver microsomal stability assay to investigate the effect of these structural modifications on the metabolic stability of the derivatives of our lead compound 3.
Material and methods
Chemistry
All solvents and reagents were obtained from Sigma–Aldrich, TCI America and Matrix Scientific and used without further purification. Analytical thin-layer chromatography (TLC) was performed on aluminum plates precoated with silica gel, also obtained from Sigma–Aldrich. Column chromatography was carried out on Merck 938S silica gel. Proton and carbon NMR spectra were recorded with a Varian 400 MHz NMR spectrometer. Spectra were referenced to the residual solvent peak, and chemical shifts are expressed in ppm from the internal reference peak. All compounds described were of >95% purity. Purity was confirmed by analytical LC/MS recorded with a Shimadzu system. Elution started with water (95%, +0.1% formic acid) and acetonitrile (5%, +0.1% formic acid) and ended with acetonitrile (95%, 0.1% formic acid) and water (5%, 0.1% formic acid) and used a linear gradient at a flow rate of 0.2 mL/min. The molecular ions [M]+, with intensities in parentheses, are given, followed by peaks corresponding to major fragment losses. Melting points were measured with a MEL-TEMP II melting point apparatus and are reported uncorrected.
sEHand FAAH IC50 Assay Conditions
Cyano(2-methoxynaphthalen-6-yl)methyl trans-(3-phenyloxyran-2-yl) methyl carbonate (CMNPC) was used as the fluorescent substrate. Human sEH (1 nM) was incubated with the inhibitor for 5 min in pH 7.0 Bis-Tris/HCl buffer (25 mM) containing 0.1 mg/mL of BSA at 30 °C prior to substrate introduction ([S] = 5 μM). Activity was determined by monitoring the appearance of 6-methoxy-2-naphthaldehyde over 10 min by fluorescence detection with an excitation wavelength of 330 nm and an emission wavelength of 465 nm. Reported IC50values are the average of the three replicates with at least two data points above and at least two below the IC50.
Measurement of FAAH potency was performed using the substrate N-(6-methoxypyridin-3-yl) octanamide (OMP) ([S]final = 50 μM) in sodium phosphate buffer (0.1 M, pH = 8, 0.1 mg/mL BSA) and progress of the reaction was measured at λexcitation= 303 nm, λemission= 394 nm, 37°C. All experiments were run in triplicate and values reported as average ± SD.(18)
In vitro human liver microsomal metabolic stability
Microsomalstability was assessed in mixed-gender human and rat liver microsomes purchased from XenoTech. The microsomes were incubated with the test compound and internal standard for 240 minutes for human and 60 minutes for rat microsomal assay, respectively at 37 °C. The reaction was initiated by the addition of NADPH generating system containing glucose 6-phosphate, glucose 6-phosphate dehydrogenase, NADP+ and MgCl2(Sigma, St. Louis, MO) in PBS buffer. Positive control incubations proceeded with 7-ethoxycoumarin as the substrate. Aliquots (100 μL) were withdrawn at 0, 10, 30, 60, 120 and 240 minutes for human microsomal assay and at 0, 10, 20, 30 and 60 minutes for the rat microsomal assay. Reactions were terminated by adding methanol. The mixtures were centrifuged and the supernatants were evaporated. The residues were reconstituted in mobile phase (85% ACN; 15% H2O) and subjected to LC/MS analysis. The peak arearesponse ratio (PARR) to internal standard was compared to the PARR at time 0 to determine the percent remaining at each time point. Half-lives were calculated using GraphPad software, fitting to a single-phase exponential decay equation.(19)
Molecular modeling
Lead compound 3andanalog 3i were drawn as 2D structures with ChemDraw Professional version 15.1.0.144, and then energy minimized through Chem3D version 15.1/MM2, Job Type: Minimum RMS Gradient of 0.010 kcal/mol and RMS distance of 0.1 Å, and saved as MDL MolFiles (*.mol) for purpose of docking with ICM Pro(20). To perform the ICM Pro small molecule docking the following steps were executed according to the program guidelines:crystal structure of human soluble epoxide hydrolase complexed with N-cycloheptyl-1-(mesitylsulfonyl)piperidine-4-carboxamide (PDB file: 4HAI)(13)was converted to ICM file. The inhibitor N-cycloheptyl-1-(mesitylsulfonyl)piperidine-4-carboxamidewas removed and docking experiment was performed:(21, 22) interactive docking was used to dock 3 and 3i. The thoroughness level was set to the maximum value of 10. ICM scores were obtained after this procedure.
Results and Discussion
The lead compound 3 has been previously identified as a potent inhibitor of sEH with a moderate half-life in human liver microsomes.(13) In this study, our goal is to improve the in vitro metabolic stability of 3 via isosteric modifications. Optimizing metabolic stability in the early phase of drug discovery is key to prevent the waste of resources associated with developing labile and potentially toxic molecules to an advanced stage.(23) Several additional advantages of improved stability include first, increased bioavailability and longer half-life of the drug candidate that will reduce the need for frequent dosing and promote better patient compliance.(23, 24)Second, the lower doses and reduced metabolism will diminish the amount of active metabolites that may potentially interact with other enzymes and proteins.(23) Finally, the less frequent administration will lead to significant reduction in cost.
To evaluate the effect of bioisosteric modification on the inhibitory capacity of the molecules, we first resynthesized the previously identified lead compound 3in four steps, as shown in Scheme 1. In short, starting from commercially available methyl isonipecotate and 2,4-dimethylbenzenesulfonyl chloride, we obtained compound 4 in 80% yield via a coupling reaction with triethylamine as the base and tetrahydrofuran as the solvent. Saponification of this methyl ester with 2M aqueous solution of lithium hydroxide furnished the carboxylic acid 5in moderate yield. The acid 5was subsequently coupled withmethyl-6-amino-2-naphthoate under standard EDC peptide coupling conditions. The product of this reaction, methyl ester 6 was again subjected to saponification yielding the lead compound 3.
Scheme 1.
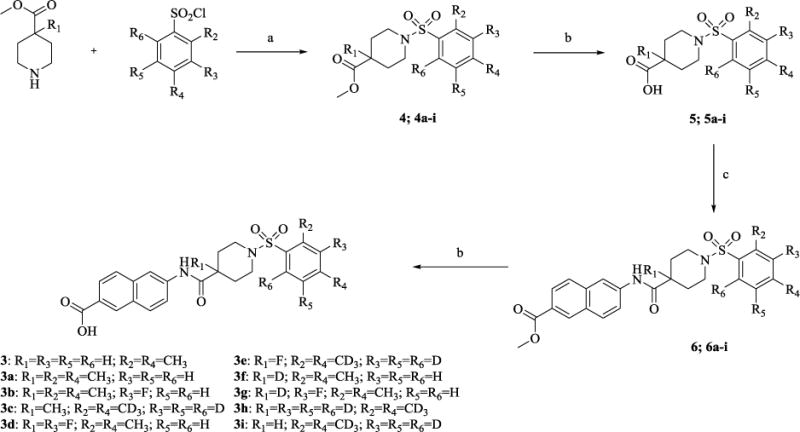
Reagents and condotions: (a)Et3N, THF, rt, 24 h, 70-84%; (b)LiOH, THF/H2O, rt, 24 h, 86-91%: (c) Methyl 6-amino 2-naphthoate, EDC, THF, rt, 24 h, 88-81%.
Subsequently, we synthesized the newly designed analogs 3a-i(Table 1) and assessed their potencyin humansEH inhibition assays. Several selected inhibitors (compounds that showed sEH inhibitory profiles relatively similar to the lead compound 3) were then further tested for their metabolic stability. Following the determination of half-lives in both human and rat liver microsomal enzymatic assays, we calculated the intrinsic clearance of the molecules as predictors of their first-pass metabolism.
Table 1.
The biological results for analogs 3a-i.
| Compound | Structure | sEH IC50 (nM) | FAAH IC50 (nM) | Compound | Structure | sEH IC50 (nM) | FAAH IC50 (nM) |
|---|---|---|---|---|---|---|---|
| 3 |
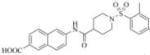
|
85(23) | 23000 | 3e |
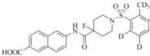
|
1461 | 19000 |
| 3a |
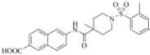
|
3882 | 52000 | 3f |
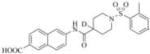
|
53 | 20000 |
| 3b |
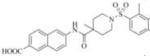
|
6351 | 56000 | 3g |
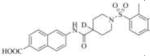
|
129 | 24000 |
| 3c |
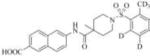
|
3501 | 56000 | 3h |
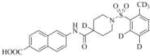
|
58 | 6800 |
| 3d |
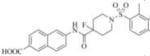
|
1683 | 24000 | 3i |
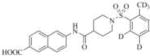
|
106 | 8800 |
Previously published values in parenthesis
Our design of 3a-i was guided by two rationales. First, our previously reportedSAR studies(12, 13)did not examine the central part of the structure comprised of amide bondconnecting the piperidine ring and the naphthoic acid. Second, solid-state structure of the human sEH revealed the involvement of three keyamino acids located in the enzyme’s catalytic pocket (tyrosine, Tyr383 and Tyr466 and aspartic acid, Asp335), in the degradation of EET. The tyrosine residues act as hydrogen bond donors to promote the epoxide ring opening by Asp335.(1)Further analysis of the structure showed theparticipation of the amide moiety (of our non-urea sEH inhibitor) in hydrogen bonding with the tyrosine and aspartic acid residues in this catalytic pocket.(13)
Therefore, in order to explore the importance of the amide functionality and the adjunct piperidine ring on the activity, we prepared 4 different classes of analogs. The first group consists of compounds that modulate the steric bulk and piperidine ring conformationin the central region with a methyl substituent instead of H atom in position 4 of the ring. 3a(the methyl analog of 3) represents the first structure within this category. Compounds 3b and 3c are subsequent derivatives of 3a, with fluorination and perdeuteration, respectively, on the phenyl group, in order to investigate their effects on the overall metabolic stability. Compounds 3d and 3e represent the second set where the electronic environment is explored by placinga fluorine substituent in lieu of the hydrogen atomin position 4 of the piperidine ring, and the same modification on the phenyl moiety as that in the first group. The thirdisosteric replacement is deuteration at position 4 (3f)and substitution of hydrogen with fluorine and deuterium on the benzene ring (3gand 3h). Last, but not least, to investigate the role played by the phenyl ring in determining the overall potency and stability, we synthesized 3i.
We proceeded to prepare these nine compounds following the general procedures outlined in Scheme 1, usinga different commercially available benzenesulfonyl chloride as the starting material for each analog. One exception is 2,4-bis(methyl-d3)benzenesulfonyl chloride-3,5,6-d3, which is the precursor for 3c, 3e, 3h, and 3iandmethyl piperidine-4-carboxylate-4-d which is the precursor for 3f, 3g and 3h. Theperdeuterated molecule was synthesized following the route shown in Scheme 2 and deuterated isonipecotate following the synthetic route shown in Scheme 3, Supplemental data.
Scheme 3.
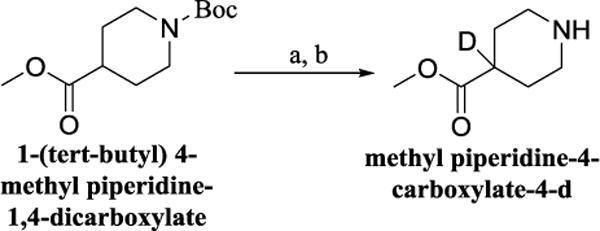
Reagents and condotions (a) LHMDS, THF, then CD3OD, −78°C, 2 h: (b) TFA, CH2CL2, rt, 16 h, 71% overall.
Through the inhibition assay using human sEH, we found that placing a methyl group at position 4 of the piperidine ring leads to >40-fold reduction or even complete loss of activity (IC50>3000 nM), as demonstrated by the compounds within the first class of analogs. The corresponding fluorination (3dand 3e), also significantly reduces the potency. This suggests that the steric effect coupled with conformational change and electronic environment of methyl and fluorine play significant roles and contribute negatively to the molecular recognition of the enzyme. By comparison, the replacement of hydrogen in this region with deuterium yields analogs with potency profiles similar to that of the lead compound3(Table 1, compounds 3f, 3g, and 3h). Deuterium is highly similar to hydrogen electronically and sterically; interchanging one with the otherdoes not result in marked differences in the IC50 values. For the phenyl ring, fluorination on the para position of the phenyl ringlowers the potency slightly (3a vs. 3b and 3f vs. 3g), whileperdeuterationdoes not seem to have any effect (3i vs. 3, 3c vs. 3a, and 3h vs. 3f). As a last study, we tested compound 3 using rat sEH, as studies on various sEH inhibitors have shown some variations among species.(25) The IC50 for 3 in rat sEH enzyme is 141 ±20 nM(experiment run in triplicate), indicating the compound can serve an inhibitor for sEH from both species.
The 9 analogs were subsequently screened against the enzyme fatty acid amide hydrolase (FAAH) to determine their selectivity for sEH. Our choice of FAAH was spurred by an earlier study(26) which reported molecules with a central core of aromatic ring-amide-piperidine-sulfonyl moiety (similar to our compounds) to be excellent inhibitors of this enzyme. FAAH, although has a different endogenous substrate from sEH, is also involved in the hydrolysis of fatty acids and both sEH and FAAH can act synergistically.(27) Our results show that none of the compounds 3a-iare inhibitors of FAAH (Table 1).
The four most potent analogs (3f, 3g, 3h and 3i) were then selected for comparison against the lead compound 3 in in vitro evaluation of both human and rat liver microsomes assays (Table 2). The stability of 3 in rat liver microsomes was assessed for the first time herein (we previously reported the stability in human liver microsome only), and found to be relatively short(26 min). The inclusion of deuterium resulted in significant improvements in metabolic stabilities, illustrated by 3hand 3i, which displayed a half-life >240 min in human liver and above 60 minutes in rat liver microsomes. The metabolic stability of 3f and 3g was close to 3(around 200 min and 30 min in human and rat liver miscrosomes, respectively), and both are shorter than those of 3h and 3i. The increased microsomal stability with deuterium atom is more significant when substitution occurs on the phenyl ring (as exemplified by 3h and 3i), rather than on the piperidine moiety (structure 3f). This data suggests that this particular part of the piperidine ring (near position 4) does not seem to be involved in the initial degradative reaction. The observed improvement with perdeuterationmay have arisen from kinetic isotope effect (KIE), a strategy often used in drug design, especially when hydrogen abstraction is the rate-limiting step in the metabolism.(28)The introduction of fluorine in position 3 of the phenyl ring (analog 3g) yielded only modest changes in inhibition potency and stability (3g compared to 3 and 3f).
Table 2.
Stability in Human and Rat Liver Microsomes (t1/2) for Selected Compounds
| Compound | hLM t1/2 (min)a |
CLint, app (μL/min/mg)b |
rLM t1/2 (min)a |
CLint, app (μL/min/mg)b |
|---|---|---|---|---|
| 3 | 186 (220)c |
7.5 (6.3) |
26 | 53.3 |
| 3f | 193 | 7.2 | 32 | 43.3 |
| 3g | 232 | 6.0 | 29 | 47.8 |
| 3h | >240 | <5.8 | >60 | <23.1 |
| 3i | >240 | <5.8 | >60 | <23.1 |
Data represents averages of duplicate determination. hLMt1/2 is the half-life in human liver microsomes. When the calculated half-life is longer than the duration of the experiment, the half-life is expressed as > the longest incubation time.
Apparent intrinsic clearance (CLint, app) was calculated based on CLint = k/P, where k is the elimination rate constant and P is the protein concentration in the incubation.
Previously published values in parenthesis.
The stability of these analogs may be further assessed using their apparent intrinsic clearance, which may be calculated from the elimination rate (Table 2). Comparing our results and the accepted classification bands shown in Table 3 compounds that have intrinsic clearance in rat microsomal assay below 13.2 μL/min/mg are considered as low clearance, while those above 71.9 are classified as high clearance compounds.(29) Based on this, all the tested analogs can be classified as low clearance in human microsome liver enzyme (<8.6 μL/min/mg), and two analogs (3h and 3i), at ~20 μL/min/mg has a low to medium clearance rate in rat liver.
Table 3.
Classification bands typically used for categorizing compounds into low, medium or high clearance. (29)
| Clearance Category | Intrinsic Clearance (μL/min/mg protein) | ||||
|---|---|---|---|---|---|
| Human | Monkey | Dog | Rat | Mouse | |
| Low | < 8.6 | < 12.5 | < 5.3 | < 13.2 | < 8.8 |
| High | > 47.0 | > 67.8 | > 28.9 | > 71.9 | > 48.0 |
Finally, we conducted a molecular modeling study in order to better understand the binding modes of this series of inhibitors within the catalytic site of human sEH (Figure 3). Our docking experiment revealed that inhibitor 3 binds in the proximity of the keyamino acids within the catalytic pocket that are involved in the degradation of EETs. As shown in Figure 3, two tyrosines (Tyr383 andTyr466) and one aspartic acid (Asp335), based on their distances, have the potential to form hydrogen bonds with the amide moiety of the inhibitor, which may prevent the substrate EET from binding.
Figure 3.
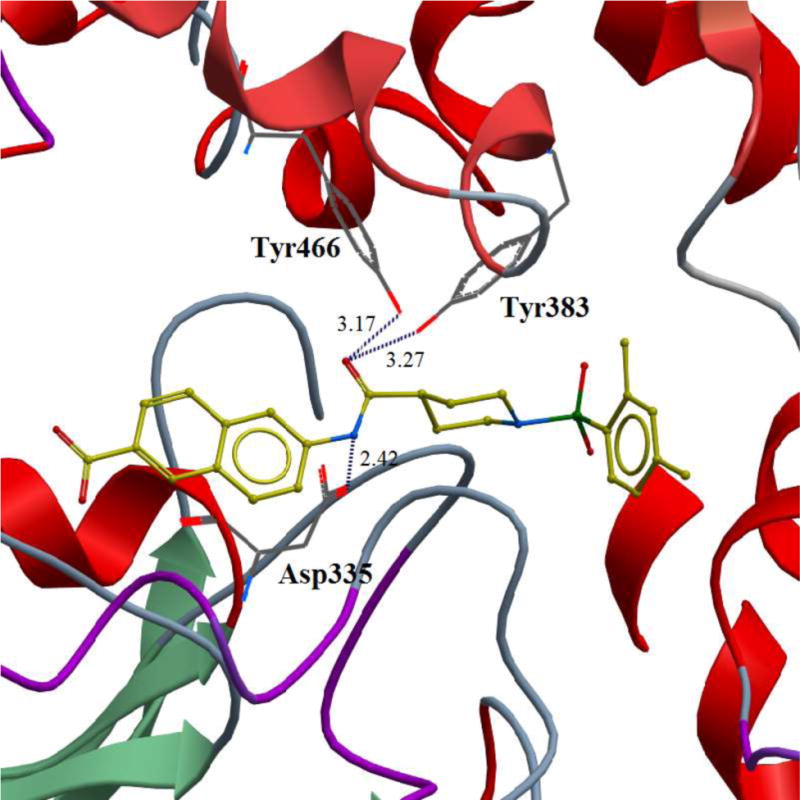
Binding pose of the inhibitor 3 in the catalytic site of the human sEH enzyme obtained in docking experiment. Potential hydrogen bonds are indicated by dashes, distances are in Å.
Conclusion
Our rational design to discover new sEH inhibitors was guided by a dual approach: the information obtained from earlier performed SAR studies and the solid-state structure of cocrystallization of non-urea analogs and human sEH. Analysis of these precedent sallowed us to design compounds with increased metabolic stability while maintaining potency profiles similarto the previously identified lead compound 3. Evaluation of the in vitro stability for selected analogs 3f, 3g, 3hand 3i (Table 2) indicated slower metabolism in both human and rat liver microsomes compared to compound 3. We hypothesize that the observed slower in vitro metabolism would translate to prolonged clearance in vivo, and thus improving theplasma retention of these compounds. Based on our results, we propose the amide-piperidinemoiety to be essential for molecular recognition and exposure of the phenylring to microsomal degradation. Therefore, introduction of isosteres in these regions could potentially improve the overall metabolic stability of this series of non-urea amide inhibitors. This hypothesis can be proved by further examination of the in vivo metabolites generated from these analogs in rats. The data from this SAR study also reveals relatively strict requirements for the central amide-piperidine moiety, suggesting a tight fit of this pharmacophore in the binding pocket and emphasizing the importance of this motif as a molecular recognition element for sEH. Significant changes in the steric and electronic environments (3a, 3b, 3c, 3d and 3e) lead to reduced potency. Overall, deuterium serves as the best bioisostere of hydrogen at position 4 of the piperidine ring.
In conclusion, sinces EH plays an important role in the lipid metabolism (specifically arachidonic acid), continued research on this pathway may be of great interest from the clinical standpoint. The inhibition of this enzyme has the potential to impact many different chronic inflammatory conditions.(4)Improved drug stability in humans and minimum toxicity and adverse effects are essential criteria to fulfill before embarking upon randomized clinical trials. Taking into account the inhibition profile and intrinsic clearance, analogs 3h and 3i would serve as appropriate candidates for future in vivo pharmacological profiling.
Highlights.
Novel soluble epoxide hydrolase (sEH) inhibitors are discovered; these inhibitors are playing an important role in the epoxyeicosatrienoic acids (EETs) metabolism.
Isosteric replacements are improving bioavailability of the newly-synthesized soluble epoxide hydrolase inhibitors.
Docking experiments reveal that novel inhibitors bind in the catalytic pocket of the human sEH enzyme, in the proximity of the key amino acids involved in hydrolysis of EETs.
The inhibition of this enzyme has the potential to impact many different chronic inflammatory conditions.
Acknowledgments
This work is supported by funding from National Institute of Environmental Health Science (NIEHS) grant R01 ES002710, and NIEHS Superfund Research Program grant P42 ES004699. SKD was supported by NIGMS-funded Pharmacology Training Program (T32GM099608).
Appendix. Supplementary data
Modified isonipecotates: 1-(tert-butyl) 4-methyl 4-methylpiperidine-1,4-dicarboxylate and 1-(tert-butyl) 4-methyl 4-fluoropiperidine-1,4-dicarboxylate were purchased from commercial sources and used without further purification.
Procedure for the preparation of 2,4-bis(methyl-d3)benzenesulfonyl chloride-3,5,6-d3 (Scheme 2):(30
Commercially available m-xylene-d10(500 mg, 4.3 mmol) was dissolved in chloroform (5 mL), in a50 mL round-bottom flask and stirred in an ice-bath. The solution was treated dropwise with chlorosulfonic acid (1.43 mL, 21.5 mmol). After 15 minutes, the reaction mixture was warmed to room temperature and stirred for additional 30 minutes. The content of the round-bottom flask was poured into a 50 mL beaker filled with crushed ice, the organic layer separated, washed with cold water, dried over anhydrous sodium sulfate and the solvent evaporated. The crude product was purified in 1:1 EtOAc/Hexane solvent system and 600 mg (65% yield) of pure product was obtained as a colorless oil.
Procedure for the preparation of methyl piperidine-4-carboxylate-4-d (Scheme 3): (31)
A solution of 1-(tert-butyl) 4-methyl piperidine-1,4-dicarboxylate (75 mg, 0.30 mmol) in THF (5 mL) was added over 45 min to a 1 M solution of LHMDS in THF (0.36 mL, 0.36 mmol) at −78 °C. The reactionmixture was stirred for 30 min, and quenched with CD3ODat −78 °C. The reaction was warmed to room temperature and diluted with water (10 mL). The aqueous layer was extracted with EtOAc (3×35 mL). The organic layers were combined and dried over MgSO4, filtered, and concentrated in vacuo. The crude was purified by flash chromatography (1:1 ethyl acetate/hexane solvent system), and colorless oil was obtained. This product was dissolved in dichloromethane (25 mL) and treated with trifluoroacetic acid and stirred over night at room temperature. The reaction was concentrated and water added. The product (32 mg; 71% over two steps)was extracted with ethyl acetate, dried over sodium sulfate and used for next step without further purification.
General procedure for the preparation of the lead compound 3 and target analogs 3a-i (examples given for the lead compound 3 and the analog 3i, which showed the best profile in liver microsomes assay)
2,4-dimethylbenzenesulfonyl chloride (200 mg, 9.8 mmol) was added to a stirred solution of methyl isonipecotate (200 μL, 1.46 mmol) and triethylamine (350 μL, 2.91 mmol) in anhydrous tetrahydrofuran (20 mL). The reaction mixture was stirred overnight under argon atmosphere. After removal of solvent under reduced pressure, the residue was dissolved in ethyl acetate, the organic layer was washed with 1N HCl (25 mL), followed by aqueous solution of saturated sodium bicarbonate (25 mL), and was then dried over anhydrous sodium sulfate, filtered and concentrated. The crude product, a yellowish oil, was purified by flash chromatography (1:4 ethyl acetate/hexane solvent system) and the final product 4 was obtained as a colorless oil. Saponification of the methyl ester 4 was achieved via following procedure: a stirred solution of 4 (150 mg, 0.48 mmol) in tetrahydrofuran (25 mL) was treated with 2M aqueous solution of lithium hydroxide (2 mL), and the reaction was stirred overnight at room temperature. Following concentration in vacuo, water (15 mL) and ethyl acetate were added (35 mL). The mixture was then cooled to 0 °C and 1N HCl was added dropwise, while stirring, until the reaction became acidic. The organic layer was separated, dried over anhydrous sodium sulfate, filtered, and concentrated. The crude product was recrystallized in diethyl ether, and 5 was obtained as a white solid. The intermediate 6 was prepared using standard EDC peptide coupling reaction conditions (32); acid 5 (20 mg, 0.07 mmol) and EDC (26 mg, 0.13 mmol) were dissolved in anhydrous dichloromethane (10 mL). Methyl 6-amino-2-naphthoate (26 mg, 0.13 mmol) was added and the reaction mixture stirred overnight at room temperature under argon atmosphere. Solvent was removed under reduced pressure and the residue dissolved in ethyl acetate (20 mL), washed with 1N HCl (25 mL), followed by aqueous solution of saturated sodium bicarbonate (25 mL). The organic layer was separated, dried over anhydrous sodium sulfate, filtered, and concentrated. The crude product was purified by flash chromatography using 1:1 ethyl acetate/hexane solvent system and 6 was obtained as an off-white solid. The final product 3 was obtained under the same saponification conditions as described above.
Although specification of commercially available m-xylene-d10 (Aldrich: 175919) stated that this compound has 98% of D atoms, our NMR analysis of products 3c, 4c, 5c and 6c, shows that certain percentage (~30%) of H-protons are present in the molecule in position 3, 5 and 6 of aromatic ring. Based on NMR analysis, hydrogens are not present in methyl groups on aromatic ring.
Methyl 1-((2,4-dimethylphenyl)sulfonyl)piperidine-4-carboxylate (4)
Yield 80% (0.98 mmol, 243 mg), colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.77 (d, J = 8.4Hz, 1H), 7.09 (d, J = 7.2 Hz, 2H), 3.66 (s, 3H), 3.63-3.59 (m, 2H), 2.75 (t, J = 11 Hz, 2H), 2.56 (s, 3H), 2.40-2.37 (m, 1H), 2.36 (s, 3H), 1.95 (dd, J = 13.6, 3.2 Hz, 2H), 1.78-1.68 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 174.5, 143.6, 137.9, 133.6, 132.9, 130.5, 126.7, 51.9, 44.5, 40.3, 27.7, 21.3, 20.6. ESI-MS (m/z+1)312.
Methyl 1-((2,4-bis(methyl-d3)phenyl-3,5,6-d3)sulfonyl)piperidine-4-carboxylate (4i)
Yield 66% (1.43 mmol, 461 mg), colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.75 (m, 0.3 H), 7.09 (m, 0.6 H), 3.66 (s, 3H), 3.63-3.59 (m, 2H), 2.75 (t, J = 11 Hz, 2H), 2.38-2.31 (m, 1H), 1.95-1.91 (m, 2H), 1.75-1.66 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 174.4, 143.4, 137.6, 133.5, 132.8, 130.4, 126.7, 51.9, 44.5, 40.3, 27.7.
1-((2,4-dimethylphenyl)sulfonyl)piperidine-4-carboxylic acid (5)
Yield 93% (0.48 mmol, 132 mg), white solid, mp: 122-124 °C. 1H NMR (400 MHz, CDCl3): δ 7.75 (d, J = 8.8Hz, 1H), 7.10 (d, J = 7.2 Hz, 2H), 3.65-3.60 (m, 2H), 2.76 (t, J = 10.4 Hz, 2H), 2.56 (s, 3H), 2.45-2.38 (m, 1H), 2.36 (s, 3H), 1.98 (dd, J = 10.8, 2.8 Hz, 2H), 1.81-1.71 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 179.6, 143.7, 137.9, 133.6, 132.9, 130.6, 126.8, 44.4, 40.1, 27.5, 21.3, 20.6. ESI-MS (m/z-1)296.
1-((2,4-bis(methyl-d3)phenyl-3,5,6-d3)sulfonyl)piperidine-4-carboxylic acid (5i)
Yield 77% (0.76 mmol, 235 mg), white solid, mp: 135-138 °C. 1H NMR (400 MHz, CDCl3): δ 7.77 (d, J = 8 Hz, 0.3H), 7.10 (s, 0.6H), 3.62 (d, J = 12.4 Hz, 2H), 2.74 (t, J = 10.8 Hz, 2H), 2.43-2.38 (m, 1H), 1.97 (dd, J = 11.2, 2.4 Hz, 2H), 1.79-1.70 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 180.2, 143.5, 137.8, 133.6, 132.8, 130.5, 126.8, 44.4, 40.1, 27.4, 21. ESI-MS (m/z-2)304.
methyl 6-(1-((2,4-dimethylphenyl)sulfonyl)piperidine-4-carboxamido)-2-naphthoate (6)
Yield 53% (0.04 mmol, 18 mg), gray solid, mp 178-180 °C. 1H NMR (400 MHz, CDCb): δ 8.50 (s, 1H), 8.28 (s, 1H), 8.00 (d, J = 8.4Hz, 1H), 7.86-7.75 (m, 4H), 7.48 (d, J = 8.8 Hz, 1H), 7.11 (d, J = 10 Hz, 1H), 3.95 (s, 3H), 3.75 (d, J = 12 Hz, 1H), 2.76 (t, J = 12 Hz, 2H), 2.58 (s, 3H), 2.42-2.35 (m, 1H), 2.37 (s, 3H) 2.01-1.98 (m, 2H), 1.93-1.86 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 172.7, 167.3, 144.0, 138.1, 137.4, 136.3, 133.7, 132.6, 130.7, 130.6, 130.4, 129.6, 127.9, 126.8, 126.6, 126.1, 120.4, 116.3, 52.3, 44.4, 43.6, 28.4, 21.4, 20.5. ESI-MS (m/z+1)481.
methyl 6-(1-((2,4-bis(methyl-d3)phenyl-3,5,6-d3)sulfonyl)piperidine-4-carboxamido)-2-naphthoate (6i)
Yield 70% (0.46 mmol, 224 mg), white solid, mp 189-191 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.24 (s, 1H), 8.50 (s, 1H), 8.38 (s, 1H), 8.03 (d, J = 8.8Hz, 1H), 7.88 (dd, J = 9.2, 5.2 Hz, 2H), 7.69 (s, 0.3H), 7.63 (d, J = 8.8 Hz, 1H), 7.22 (d, J = 14.4 Hz, 0.6H), 3.87 (s, 3H), 3.62 (d, J= 12.4 Hz, 2H), 2.65 (t, J = 12 Hz, 2H), 2.48 (s, 1H), 1.89 (d, J = 12 Hz, 2H), 1.68-1.59 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 173.2, 166.3, 143.1, 139.1, 136.9, 135.8, 133.4, 132.6, 130.1, 130.0, 129.6, 128.5, 127.7, 126.7, 125.3, 125.2, 120.7, 114.6, 52.0, 44.3, 41.7, 27.8. ESI-MS (m/z+1)491.
6-(1-((2,4-dimethylphenyl)sulfonyl)piperidine-4-carboxamido)-2-naphthoic acid (3)
Yield 86% (0.03 mmol, 15 mg), white solid, mp 220-224°C. 1H NMR (400 MHz, DMSO-d6): δ 10.30 (s, 1H); 8.45 (s, 1H), 8.35 (s, 1H), 7.98 (d, J = 9.2Hz, 1H), 7.87 (d, J = 8.4Hz, 1H), 7.81 (d, J = 8.4Hz, 1H), 7.67 (d, J = 8Hz, 1H), 7.61 (d, J = 9.2Hz, 1H), 7.2 (d, J = 8 Hz, 2H), 3.60 (d, J = 12 Hz, 2H), 2.62 (t, J = 12 Hz, 2H), 2.46 (s, 2H), 2.31 (s, 3H), 1.87 (d, J = 12 Hz, 2H), 1.64-1.55 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 173.1, 167.3, 143.2, 138.7, 137.0, 135.5, 133.3, 132.6, 130.0, 129.8, 129.7, 128.5, 127.4, 126.8, 126.5, 125.5, 120.4, 114.5, 44.2, 41.6, 27.7, 20.6, 19.9. ESI-MS (m/z-1)465.
6-(1-((2,4-bis(methyl-d3)phenyl-3,5,6-d3)sulfonyl)piperidine-4-carboxamido)-2-naphthoic acid (3i)
Yield 43% (0.15 mmol, 75 mg), white solid, mp 279-281 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.53 (s, 1H); 8.40 (s, 1H), 8.32 (s, 1H), 7.92 (d, J = 9.2 Hz, 1H), 7.82 (d, J = 8.4 Hz, 1H), 7.76 (d, J = 8.8 Hz, 1H), 7.63 (d, J = 10 Hz, 1H), 7.61 (s, 0.3H), 7.16 (d, J = 14.8 Hz, 0.6H), 3.55 (d, J = 12.4 Hz, 2H), 2.53 (t, J = 11.6 Hz, 2H), 2.41 (s, 1H), 1.83 (d, J = 11.2 Hz, 2H), 1.58-1.48 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 173.4, 167.5, 143.1, 139.0, 136.9, 135.7, 133.5, 132.6, 130.2, 129.7, 128.6, 127.5, 126.8, 126.6, 125.7, 120.6, 114.6, 44.4, 41.7, 27.9. ESI-MS (m/z-1)474.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest
The authors declare the existence of a patent filed by Columbia University on the sEH inhibitors used in this study.
References
- 1.Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov. 2009;8(10):794–805. doi: 10.1038/nrd2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fuster V, Sweeny JM. Aspirin: a historical and contemporary therapeutic overview. Circulation. 2011;123(7):768–78. doi: 10.1161/CIRCULATIONAHA.110.963843. [DOI] [PubMed] [Google Scholar]
- 3.McGill KA, Busse WW. Zileuton. Lancet. 1996;348(9026):519–24. doi: 10.1016/S0140-6736(95)12297-4. [DOI] [PubMed] [Google Scholar]
- 4.Wagner KM, McReynolds CB, Schmidt WK, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharmacol Ther. 2017;180:62–76. doi: 10.1016/j.pharmthera.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen HC, Hammock BD. Discovery of inhibitors of soluble epoxide hydrolase: a target with multiple potential therapeutic indications. J Med Chem. 2012;55(5):1789–808. doi: 10.1021/jm201468j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang J, Bratt J, Franzi L, Liu JY, Zhang G, Zeki AA, et al. Soluble epoxide hydrolase inhibitor attenuates inflammation and airway hyperresponsiveness in mice. Am J Respir Cell Mol Biol. 2015;52(1):46–55. doi: 10.1165/rcmb.2013-0440OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harris TR, Li N, Chiamvimonvat N, Hammock BD. The potential of soluble epoxide hydrolase inhibition in the treatment of cardiac hypertrophy. Congest Heart Fail. 2008;14(4):219–24. doi: 10.1111/j.1751-7133.2008.08430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004;43(1):55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- 9.Kim J, Yoon SP, Toews ML, Imig JD, Hwang SH, Hammock BD, et al. Pharmacological inhibition of soluble epoxide hydrolase prevents renal interstitial fibrogenesis in obstructive nephropathy. Am J Physiol Renal Physiol. 2015;308(2):F131–9. doi: 10.1152/ajprenal.00531.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou Y, Liu T, Duan JX, Li P, Sun GY, Liu YP, et al. Soluble Epoxide Hydrolase Inhibitor Attenuates Lipopolysaccharide-Induced Acute Lung Injury and Improves Survival in Mice. Shock. 2017;47(5):638–45. doi: 10.1097/SHK.0000000000000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie Y, Liu Y, Gong G, Smith DH, Yan F, Rinderspacher A, et al. Discovery of potent non-urea inhibitors of soluble epoxide hydrolase. Bioorg Med Chem Lett. 2009;19(8):2354–9. doi: 10.1016/j.bmcl.2008.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pecic S, Deng SX, Morisseau C, Hammock BD, Landry DW. Design, synthesis and evaluation of non-urea inhibitors of soluble epoxide hydrolase. Bioorg Med Chem Lett. 2012;22(1):601–5. doi: 10.1016/j.bmcl.2011.10.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pecic S, Pakhomova S, Newcomer ME, Morisseau C, Hammock BD, Zhu Z, et al. Synthesis and structure-activity relationship of piperidine-derived non-urea soluble epoxide hydrolase inhibitors. Bioorg Med Chem Lett. 2013;23(2):417–21. doi: 10.1016/j.bmcl.2012.11.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blanchard SG, Andrews RC, Brown PJ, Gan LS, Lee FW, Sinhababu AK, et al. Discovery of bioavailable inhibitors of secretory phospholipase A2. Pharm Biotechnol. 1998;11:445–63. doi: 10.1007/0-306-47384-4_19. [DOI] [PubMed] [Google Scholar]
- 15.Dragovich PS, Prins TJ, Zhou R, Johnson TO, Hua Y, Luu HT, et al. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 8. Pharmacological optimization of orally bioavailable 2-pyridone-containing peptidomimetics. Journal of medicinal chemistry. 2003;46(21):4572–85. doi: 10.1021/jm030166l. [DOI] [PubMed] [Google Scholar]
- 16.Victor F, Brown TJ, Campanale K, Heinz BA, Shipley LA, Su KS, et al. Synthesis, antiviral activity, and biological properties of vinylacetylene analogs of enviroxime. Journal of medicinal chemistry. 1997;40(10):1511–8. doi: 10.1021/jm960718i. [DOI] [PubMed] [Google Scholar]
- 17.Tagat JR, Steensma RW, McCombie SW, Nazareno DV, Lin SI, Neustadt BR, et al. Piperazine-based CCR5 antagonists as HIV-1 inhibitors. II. Discovery of 1-[(2,4-dimethyl-3-pyridinyl)carbonyl]-4- methyl-4-[3(S)-methyl-4-[1(S)-[4-(trifluoromethyl)phenyl]ethyl]-1-piperazinyl]- piperidine N1-oxide (Sch-350634), an orally bioavailable, potent CCR5 antagonist. Journal of medicinal chemistry. 2001;44(21):3343–6. doi: 10.1021/jm0155401. [DOI] [PubMed] [Google Scholar]
- 18.Kodani SD, Overby HB, Morisseau C, Chen J, Zhao L, Hammock BD. Parabens inhibit fatty acid amide hydrolase: A potential role in paraben-enhanced 3T3-L1 adipocyte differentiation. Toxicol Lett. 2016;262:92–9. doi: 10.1016/j.toxlet.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terzic N, Konstantinovic J, Tot M, Burojevic J, Djurkovic-Djakovic O, Srbljanovic J, et al. Reinvestigating Old Pharmacophores: Are 4-Aminoquinolines and Tetraoxanes Potential Two-Stage Antimalarials? J Med Chem. 2016;59(1):264–81. doi: 10.1021/acs.jmedchem.5b01374. [DOI] [PubMed] [Google Scholar]
- 20.Cardozo T, Totrov M, Abagyan R. Homology modeling by the ICM method. Proteins. 1995;23(3):403–14. doi: 10.1002/prot.340230314. [DOI] [PubMed] [Google Scholar]
- 21.Pecic S, McAnuff MA, Harding WW. Nantenine as an acetylcholinesterase inhibitor: SAR, enzyme kinetics and molecular modeling investigations. J Enzyme Inhib Med Chem. 2011;26(1):46–55. doi: 10.3109/14756361003671078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pecic S, Makkar P, Chaudhary S, Reddy BV, Navarro HA, Harding WW. Affinity of aporphines for the human 5-HT2A receptor: insights from homology modeling and molecular docking studies. Bioorg Med Chem. 2010;18(15):5562–75. doi: 10.1016/j.bmc.2010.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thompson TN. Optimization of metabolic stability as a goal of modern drug design. Med Res Rev. 2001;21(5):412–49. doi: 10.1002/med.1017. [DOI] [PubMed] [Google Scholar]
- 24.Nassar AE, Kamel AM, Clarimont C. Improving the decision-making process in the structural modification of drug candidates: enhancing metabolic stability. Drug Discov Today. 2004;9(23):1020–8. doi: 10.1016/S1359-6446(04)03280-5. [DOI] [PubMed] [Google Scholar]
- 25.Eldrup AB, Soleymanzadeh F, Taylor SJ, Muegge I, Farrow NA, Joseph D, et al. Structure-based optimization of arylamides as inhibitors of soluble epoxide hydrolase. J Med Chem. 2009;52(19):5880–95. doi: 10.1021/jm9005302. [DOI] [PubMed] [Google Scholar]
- 26.Wang X, Sarris K, Kage K, Zhang D, Brown SP, Kolasa T, et al. Synthesis and evaluation of benzothiazole-based analogues as novel, potent, and selective fatty acid amide hydrolase inhibitors. Journal of medicinal chemistry. 2009;52(1):170–80. doi: 10.1021/jm801042a. [DOI] [PubMed] [Google Scholar]
- 27.Sasso O, Wagner K, Morisseau C, Inceoglu B, Hammock BD, Piomelli D. Peripheral FAAH and soluble epoxide hydrolase inhibitors are synergistically antinociceptive. Pharmacol Res. 2015;97:7–15. doi: 10.1016/j.phrs.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meanwell NA. Synopsis of some recent tactical application of bioisosteres in drug design. J Med Chem. 2011;54(8):2529–91. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 29.Houston JB. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem Pharmacol. 1994;47(9):1469–79. doi: 10.1016/0006-2952(94)90520-7. [DOI] [PubMed] [Google Scholar]
- 30.Jha T, Basu S, Halder AK, Adhikari N, Samanta S. Possible anticancer agents: synthesis, pharmacological activity, and molecular modeling studies on some 5-N-Substituted-2-N-(substituted benzenesulphonyl)-L(+)Glutamines. Med Chem Res. 2017;26:1437–58. [Google Scholar]
- 31.Huard K, Bagley SW, Menhaji-Klotz E, Preville C, Southers JA, Jr, Smith AC, et al. Synthesis of spiropiperidine lactam acetyl-CoA carboxylase inhibitors. J Org Chem. 2012;77(22):10050–7. doi: 10.1021/jo3014808. [DOI] [PubMed] [Google Scholar]
- 32.Grabarek Z, Gergely J. Zero-length crosslinking procedure with the use of active esters. Anal Biochem. 1990;185(1):131–5. doi: 10.1016/0003-2697(90)90267-d. [DOI] [PubMed] [Google Scholar]