

Abstract
In an attempt to refine a CAN-mediated synthesis of 1,3,4,6-tetra-O-acetyl-α-D-glucopyranose (2-OH glucose) we unexpectedly discovered that this reaction proceeds via the intermediacy of glycosyl nitrates. Improved mechanistic understanding of this reaction led to the development of a more versatile synthesis of 2-OH glucose from a variety of precursors. Also demonstrated is the conversion of 2-OH glucose into a variety of building blocks differentially protected at C-2, a position that is generally hard to protect regioselectively in the glucopyranose series.
Graphical abstract

Introduction
Poor accessibility to monosaccharide building blocks hampers development of all methods for oligosaccharide synthesis. In particular, the development of automation platforms for oligosaccharide assembly is affected. As Seeberger, the developer of the first oligosaccharide synthesizer, notes “differentially protected monosaccharide building blocks is currently the bottleneck for chemical synthesis”.1 Indeed, most bench-time is dedicated to making building blocks. Since a large glycosyl donor excess is still needed for the automated solid phase synthesis, researchers experience significant setbacks having to make and remake building blocks. Things are further complicated by the large variety of carbohydrate structures and sequences. “Unlike the synthesis of peptides and oligonucleotides, there are no universal building blocks or methods for the synthesis of all glycans.”2
D-Glucose, the predominant monosaccharide in plant polysaccharides and bacterial glycans, is also one of the major components of the mammalian glycome.3 Building blocks of the D-gluco series are often first compounds tested in practically all new glycosylation reactions and applications. Every synthetic glycoscience lab makes glucose building blocks. Paradoxically, glucose 1 remains the hardest sugar for selective protection due to its trans–trans–trans all-equatorial 2,3,4-triol arrangement (Scheme 1). Methods for selective protection of different positions of glucose exist, but many suffer from relaxed regioselectivity and/or require multiple steps with tedious separations of isomers.4–6 Even excellent concepts including tin-mediated alkylation,7–10 phase-transfer reactions,11–13 TMS-mediated regioselective protection in one-pot,14,15 etc.16 all suffer from a limited scope and/or fair selectivity.
Scheme 1.

Previous methods for the synthesis of 2-OH glucose 2.
The C-2 is the most important position in synthesis because protecting groups at C-2 have a profound effect on stereoselectivity of glycosylation. The C-2 can be either alkylated or acylated for the synthesis of either 1,2-cis or 1,2-trans-linked glycosides, respectively. For instance, a vast majority of all syntheses of α-(1,2-cis)glucosides use 2-O-benzylated donors.17,18 Paradoxically, C-2 is the most difficult position to benzylate selectively. A few approaches are known, but all provide fair selectivity. Regioselectivity suffers during the phase-transfer differentiation of 2- vs. 3-OH,13 whereas stereo-selectivity suffers during glycal-based syntheses that often lead to mixtures of Glc/Man isomers.19–21 Hung’s excellent method for the one-pot regioselective protection provides no access to 2-benzyl derivatives at all.22
In addition to selective protections, acyl group migration or other selective deprotection techniques have proven to be useful reaction pathways to access partially substituted building blocks for oligosaccharide synthesis.23–25 A few approaches to employ such rearrangements to obtain 2-hydroxyl sugars have been developed, but many have limitations and their scope remains narrow. The classical Helferich’s three-step in situ conversion of glucose 1 to 1,3,4,6-tetra-O-acetyl-α-D-glucopyranose 2 depicted in Scheme 1 is in practice very elaborate. It requires strict control of the reaction temperature and reactant addition order, but still provides a low yield of 27%.26 Somewhat more straightforward procedures have been developed for the synthesis of 2-OH galactose27 and mannose28 derivatives, but these approaches do not work for the synthesis of 2-OH glucose 2. Our reinvestigation of the synthesis of 2-OH glucose led to the discovery of a scalable one-step protocol with cerium(IV) ammonium nitrate (CAN). This procedure afforded compound 2 in 51% isolated yield in one step from acetobromoglucose 3 and was cross-validated via the Proven Methods book series.29
Since both Helferich’s and our CAN-mediated reaction provide a mixture of regioisomers, 2-OH 2 and its 1-OH counterpart 4, the reaction was thought to proceed via the intermediacy of an acyloxonium ion. Nevertheless, both the reaction mechanism and the modes to enhance the 2-OH selectivity remain largely unknown. The lack of regiocontrol hampers the yields of these reactions. In addition, 2-OH glucose 2 can be purified by crystallization only because the chromatographic purification is impractical due to the propensity of product 2 to rapidly isomerize into its more stable hemiacetal counterpart 4. Reported herein is our first attempt to understand the driving forces of this reaction and its expansion to other substrates and targets.
Results and discussion
The benchmark experiment for the synthesis of 2-OH glucose 2 was performed under the previously established reaction conditions. CAN (1.6 equiv.) was added to a stirring solution of 330 in nitromethane and the reaction mixture was stirred for 18 h at room temperature. After that, the mixture was quenched with water, extracted with CH2Cl2, the extract was neutralized, dried, and concentrated. The product was purified by crystallization from diethyl ether to afford white crystalline compound 2 in 51% yield. It is noteworthy that stoichiometric amount of water (as a source of a proton) is needed to ensure that this reaction proceeds to completion. This requirement is supported by the fact that no reaction occurred in the presence of molecular sieves in anhydrous nitromethane. The addition of a large excess of water helps to accelerate the reaction, but it also significantly diminishes the regioselectivity towards the formation of 2-OH isomer 2.
Our attempts to improve the CAN-mediated reaction rates and the yield of product 2 began with the investigation of various precursors shown in Fig. 1. The reaction with glucose penta-acetate 5 was very sluggish, and only a trace amount of the desired product 2 was observed. The addition of BF3–OEt2 helped to accelerate this reaction, but also diminished the regioselectivity of product 2 vs. hemiacetal 4. The outcome of the reaction with S-ethyl glycoside 631 was similar to that of the reaction with bromide 3 and 2-OH glucose 2 was isolated in 52% yield. Accordingly, benzoylated compounds 7–932,33 were investigated as starting materials. However, no reactions occurred under the standard CAN-mediated activation conditions. Attempts to investigate building blocks bearing benzylidene protecting group failed because acetals are readily removed in the presence of CAN.
Fig. 1.
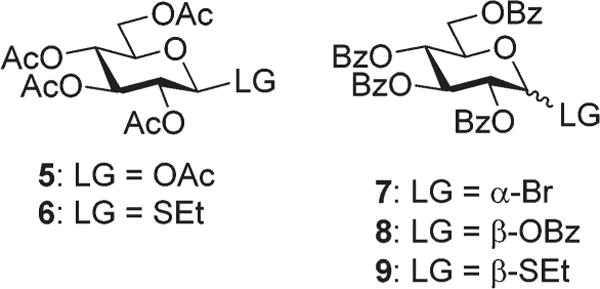
Preliminary investigation of other precursors 5, 6 for the synthesis of 2 and 7–9 for the attempted synthesis of 2-OH glucose tetrabenzoate.
Results of this preliminary study showed that only reactions with acetylated bromide 3 and thioglycoside 6 as precursors provide acceptable yields for the formation of 2 (around 50%). Still, these reactions are rather sluggish and require at least 16–18 h to complete. This is possibly in part due to a fairly low solubility of CAN in nitromethane, the reaction solvent used in all preliminary experiments. Hence, as the next step forward we screened other solvents that may provide a more suitable environment for reactions with CAN. Indeed, acetonitrile, DMF and DMSO, all accelerated the reaction with glycosyl bromide precursor 3 (14 h, 5 h, and 1 h, respectively). However, only reactions in acetonitrile maintained the regioselectivity of product 2 vs. 4, similar to that achieved in reactions in nitro-methane. With this incremental success, we added acetonitrile in future experiments. Interestingly, nitromethane was the only solvent in this series that worked well with S-ethyl glycoside 6 precursor.
The most commonly proposed mechanism of cerium(IV)-mediated organic reactions involves generation of the radicalcation species along with the reduction of Ce(IV) to Ce(III) with subsequent regeneration of Ce(IV) in the presence of an oxidant specifically added for this purpose.34 To investigate whether the CAN-mediated formation of 2-OH glucose 2 follows this radical-regenerative pathway, we investigated whether catalytic amounts of CAN would be sufficient to convert precursors 3 and 6 to the desired product. As a result of this experiment, we determined that compound 3 indeed can be converted to 2-OH glucose 2 in the presence of sub-stoichiometric amounts of CAN (range investigated 10–90 mol%) in acetonitrile. The optimal balance between the reaction time, yield of 2, and regioselectivity of 2-OH/1-OH was achieved in the presence of 0.5 equiv. of CAN. This reaction allowed us to maintain the same selectivity of product 2 (2/4 = 4/1) as that achieved with stoichiometric amount of CAN. We also noted that typical reactions of bromide 3 were accompanied by the formation of foul-smelling brown-red fumes, which were determined to be bromine by a standard test reaction with AgNO3 solution. Bromine that was produced by the oxidation of bromide 3, along with the reduction of Ce(IV) to Ce(III), can act as the co-oxidant to regenerate Ce(IV) from Ce(III) and thus sustain the catalytic cycle of the reaction.
In strong contrast, the conversion of thioglycoside 6 to the 2-OH derivative 2 was incomplete in the presence of catalytic amounts of CAN, but could be driven to completion by adding at least stoichiometric CAN. This reaction was accompanied by the formation of a white precipitate that was determined to be (NH4)2[Ce(ffl)(NO3)5]·2H2O 10 by X-ray crystallography (Fig. 2). Based on the preliminary in situ NMR studies (see the ESI†), we believe that the thioglycoside activation proceeds via the oxidation with CAN, which requires quantitative CAN. The reaction originated from bromide 3 performed in the presence of added ethanethiol also produced salt 10, and required stoichiometric CAN to go to completion. A reaction of ethane thiol alone with CAN in acetonitrile also produced 10 and the NMR showed a strong shift of the methylene signal in comparison with ethanethiol (see the ESI†). Alongside, phenyl 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranoside 1135 was investigated providing comparable results (see the ESI†). Based on the chemical shifts recorded, we believe that thiols are oxidized to the corresponding sulfonic acids, which requires stoichiometric CAN. For this reason, catalytic amounts of CAN are insufficient to drive the conversion of S-ethyl glycoside 6 into 2-OH glucose 2 to completion.
Fig. 2.

Crystal Structure of (NH4)2[Ce(III)(NO3)5]-2H2O 10.
Having determined the involvement of CAN and the beneficial role of acetonitrile in accelerating the reaction of bromide 3, we turned our attention to identifying more effective modes for accelerating the rates of the formation of 2-OH glucose 2. For this reason, we chose a more reactive glycosyl iodide 1236 as the starting material. Indeed, the reaction of iodide 12 in the presence of 0.5 equiv. of CAN in acetonitrile was much faster (3 h), but it also led to a reduced regioselectivity for the formation of 2 vs. 4 (2/4 = 2/1). Intriguingly, an additional spot, right underneath the spot corresponding to the starting material 12, was observed by TLC during the reaction. This additional compound was not present in the final reaction mixture, which was indicative of the presence of a quasi-stable reaction intermediate en route to the products. All initial attempts to isolate this additional compound/inter-mediate from the prematurely quenched reaction mixtures by column chromatography have failed. Persistent in situ tracking of reaction mixtures with NMR, gave us a hypothesis that this additional compound might have been glycosyl nitrate 13 (Scheme 2). This elusive intermediate was reported pre-viously,37 but we needed the additional proof to support this hypothesis. On the basis of this discovery, reactions initiated from bromide 3 were reinvestigated and the same intermediate 13 was detected by NMR (Scheme 2a). The formation of nitrate 13 was previously overlooked because this compound has the same retention factor (Rf) as that of bromide 3. Based on this evidence, we concluded that the synthesis of 2 with CAN undergoes the pathway via the intermediacy of reactive glycosyl nitrate 13 that gets hydrolyzed into 2-OH 2 (and its 1-OH counterpart 4).
Scheme 2.
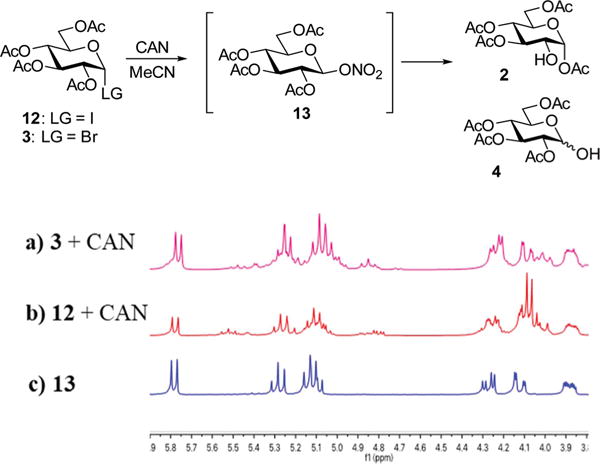
The discovery of glycosyl nitrate intermediate 13.
Since the synthesis of 2-OH glucose 2 was found to proceed via glycosyl nitrate intermediate 13, we investigated other nitrate salt reagents with the general idea of investigating their applicability to the synthesis of glycosyl nitrates and, by extension, compound 2. Among screened common ammonium, potassium, silver(I), copper(II), magnesium(II), lead(II), barium(II), and ferric nitrates, only AgNO3 provided the enhancement of the regioselectivity towards 2-OH product over the original procedure with CAN. Thus, the highest ratio of 2 to 4 (5/1) was obtained from bromide 3 in the presence of 50 mol% of AgNO3. This approach allowed us to obtain nitrate 13 by the reaction of bromide 3 with stoichiometric AgNO3 in the presence of molecular sieves in dry acetonitrile (Scheme 3). As a result, nitrate 13 was obtained in a pure crystalline form in 77% yield and its structure was confirmed by spectroscopic and crystallographic techniques. It should be noted that AgNO3 (in combination with γ-collidine in benzene) was previously used for the synthesis of nitrate 13.37
Fig. 3.

X-Ray structures of acetylated nitrate 13 and its benzoylated counterpart 14.
The proton NMR spectrum of 13, partially depicted in Scheme 2c, is consistent with the previously recorded reaction mixtures resulting from halides 3 and 12 and CAN (Scheme 2a and b). X-ray crystal structure of nitrate 13 is depicted in Fig. 3. In a similar fashion, benzoylated glycosyl nitrate 14 was synthesized by reaction of bromide 7 with stoichiometric AgNO3 in the presence of molecular sieves in dry acetonitrile (Scheme 3). As a result, nitrate 14 was obtained in a pure crystalline form in 91% yield and its structure was confirmed by spectroscopic and crystallographic techniques. X-ray crystal structure of nitrate 14 is depicted in Fig. 3.
Scheme 3.

Synthesis of acetylated and benzoylated glucosyl nitrates 13 and 14.
When the crystalline glycosyl nitrate 13 was re-dissolved in acetonitrile, 2-OH derivative 2 was readily formed in 6 h in the absence of any additional reagents. Even better regioselectivity (2/4 = 6/1) was recorded in this reaction. A preparative-scale (3–5 g) synthesis followed by the purification by crystallization from ether gave 2-OH glucose 2 in 75% yield. Based on the experimental evidence obtained by the in situ reaction monitoring by NMR and eventually the isolation and characterization of the reaction intermediate, we propose the following mechanism of this reaction. Silver(I)-assisted bromide leaving group departure leads to the formation of the glycosyl nitrate. This may proceed via the intermediacy of oxacarbenium and acyloxonium intermediates. However, this could also proceed via a concerted displacement via a six-membered intermediate A shown in Scheme 4. While we do not yet have sufficient experimental evidence to unambiguously prove this Walden-like inversion pathway,38 this hypothesis is supported by previous observations. As shown by many since the pioneering work by Isbell,39 the displacement of α-bromides under Koenigs–Knorr conditions40 can proceed via the SN2-like mechanism. We believe that the complete β-selectivity obtained in the synthesis of glycosyl nitrates is also due to the occurrence of Walden inversion, not the participatory effect of the 2-O-acetyl substituent. The glycosyl nitrate intermediate can readily dissociate to form the acyloxonium ion intermediate B. The latter is then attacked by the molecule of water to form the tetrahedral intermediate C upon the loss of the proton. This hydrate intermediate C is too unstable to be isolated, but if essentially the same transformation in performed in the presence of methanol and molecular sieves a stable methyl 1,2-orthoacetate is formed instead. Since the O-2 oxygen in C is more basic than its anomeric counterpart the protonation is preferentially occurring at C-2 leading to the 2-OH glucose as the kinetic product. The alternative pathway via the protonation of O-1 is slower, but it can be favored in the presence of a large excess of water. In these cases, decreased regioselectivity towards the formation of the 2-OH product is observed.
Scheme 4.

A plausible reaction mechanism for the synthesis of 2-OH with silver nitrate.
It has become common knowledge that aqueous acidic conditions promote isomerization of 2-OH glucose 2 into hemiacetal 4. However, when compounds 2 and 4 were treated individually with AgNO3 or with HNO3 in acetonitrile no product interchange was detected. This implies that these reaction conditions do not promote the isomerization of 2-OH glucose 2 into its thermodynamically more stable 1-OH counterpart. Besides, bromide 3 is very stable in acetonitrile and no hydrolysis would occur without adding nitrate reagents. Bromide 3 would slowly hydrolyze in the presence of silver(I) oxide producing hemiacetal 4, but not 2-OH glucose 2. Similarly, the methyl orthoester would remain stable under the reaction conditions, but would slowly decompose into a mixture of a hemiacetal and methyl glycoside, but not 2-OH glucose, in the acidic medium. Based on these experiments, we conclude that the formation of nitrate 13 is fundamental to the high regioselectivity obtained for the formation of 2-OH glucose 2.
Having achieved certain success in the synthesis of 2-OH glucose 2, we turned our attention towards further modification of this regioselectively protected intermediate. Benzylation of 2-OH glucose 2 with benzyl triflate41 or 2,2,2-trichloroacetimidate42–45 was previously established. Other reagents including the Dudley reagent46,47 and benzyl N-aryltrifluoroacetimidate48 may also be suitable for this purpose. Therefore, herein we predominantly investigated ester-type protecting groups. This study included the comparative investigation of conventional stepwise approaches and more streamlined one-pot conversion-protection reactions. Levulinoyl group was introduced by using levulinic acid in the presence of EDC coupling reagent and DMAP. A sequential two-step conversion of bromide 3 in the presence of CAN with the subsequent treatment with LevOH/EDC/DMAP afforded 2-O-levulinoyl derivative 15 in 63% yield (entry 1, Table 1). When essentially the same sequence was performed in the presence of AgNO3 instead of CAN, product 15 was obtained in 71% yield (entry 2). The direct conversion of nitrate 13 into 2-O-levulinoyl tetraacetate 15 in the presence of LevOH/EDC/DMAP produced the product in 85% yield (entry 3). Picoloyl group was introduced in a similar fashion using picolinic acid, EDC and DMAP. Reactions of bromide 3 with CAN, bromide 3 with AgNO3, and nitrate 13 followed by esterification with PicoOH/EDC/DMAP afforded 2-O-picoloyl tetraacetate 16 in 49%, 51% and 67% yield, respectively (entries 4–6). Along similar lines, we have also investigated the introduction of trifluoromethane-sulfonyl ester, a possible intermediate for the synthesis of mannosamine and its derivatives. Mannosamine is a common component of bacterial glycans49–51 and a biosynthetic precursor of sialic acids.52–54 Reactions of bromide 3 with CAN, bromide 3 with AgNO3, and nitrate 13 followed by sulfonylation with triflic anhydride in the presence of pyridine afforded 2-O-trifluoromethanesulfonyl tetraacetate 17 in 54%, 52% and 67% over-all yield, respectively (entries 7–9). Benzoylated glucosyl nitrate 14 could also be converted into 2-O-triflyl tetrabenzoate 18 in 54% yield (entry 10).
Table 1.
The synthesis of selectively 2-O-esterified derivatives 15–18

| |||||
|---|---|---|---|---|---|
| Entry | Starting material | Additive | Conditions | Product | Yield |
| 1 | 3 | CAN (0.5 equiv.) | A | 15 | 63% |
| 2 | 3 | AgNO3 (0.5 equiv.) | A | 15 | 71% |
| 3 | 13 | None | A | 15 | 85% |
| 4 | 3 | CAN (0.5 equiv.) | B | 16 | 49% |
| 5 | 3 | AgNO3 (0.5 equiv.) | B | 16 | 51% |
| 6 | 13 | None | B | 16 | 67% |
| 7 | 3 | CAN (0.5 equiv.) | C | 17 | 54% |
| 8 | 3 | AgNO3 (0.5 equiv.) | C | 17 | 52% |
| 9 | 13 | None | C | 17 | 67% |
| 10 | 14 | None | C | 18 | 54% |
Conclusions
Reported herein is an improved method for the synthesis of 2-OH glucose, which is an important synthon for production of differentially protected building blocks of D-glucose. It was discovered that the synthesis of 2-OH glucose proceeds via the intermediacy of glycosyl nitrates. Glycosyl nitrates of the 2-aminosugars have been investigated quite extensively as intermediates of the azidonitration reaction of glycals.55 Glycosyl nitrates of neutral, per-oxygenated sugar series, are much more rare, and their utility in synthesis remains practically unexplored beyond a few isolated examples.56–58 Esterification of C-2 hydroxyl group could be performed without the isolation of the 2-OH glucose intermediate. This, along with the previously established methods for 2-O-benzylations under acidic or neutral conditions, offers a convenient route for the synthesis of important intermediates and building blocks for oligosaccharide synthesis.
Experimental
General
The reactions were performed using commercial reagents. The ACS grade solvents used for reactions were purified and dried in accordance with standard procedures. Cerium(IV) ammonium nitrate (CAN) was dried for 6 h under reduced pressure. CH2Cl2 was distilled from CaH2 directly prior to application. Molecular sieves (3 Å) used for reactions, were crushed and activated in vacuo at 390 °C for 8 h in the first instance and then for 2–3 h at 390 °C directly prior to application. Reactions were monitored by TLC on Kieselgel 60 F254 (EM Science). The compounds were detected by examination under UV light and by charring with 10% sulfuric acid in methanol. Column chromatography was performed on silica gel 60 (70–230 mesh). Solvents were removed under reduced pressure at <40 °C. Optical rotations were measured with a Jasco P-2000 polarimeter. 1H NMR spectra were recorded at 300 MHz and 13C NMR spectra were recorded at 75 MHz. The 1H chemical shifts are referenced to the signal of the residual CHCl3 (δH = 7.26 ppm) for solutions in CDCl3. The 13C chemical shifts are referenced to the central signal of CDCl3 (δC = 77.23 ppm) for solutions in CDCl3. HRMS were recorded with a JEOL MStation (JMS-700) Mass Spectrometer.
General procedure for the synthesis of 1,3,4,6-tetra-O-acetyl-α-D-glucopyranose (2) with CAN (Method 1)
CAN (33.3 mg, 0.061 mmol) was added to a solution of bromide 3 (50 mg, 0.121 mmol) in acetonitrile (1.0 mL) and water (2.2 μL), and the resulting reaction mixture was stirred for 16–20 h at rt. After that, the volatiles were removed under reduced pressure. The residue was dissolved in CH2Cl2 (~10 mL) and washed with water (5 mL), 10% aq. Na2S2O3 (5 mL), and water (3 × 5 mL). The organic layer was separated, dried over MgSO4, and concentrated in vacuo. The residue was crystallized from dry diethyl ether to afford the title compound as colorless crystals. Analytical data for 2: Rf = 0.40 (ethyl acetate/hexanes, 1/1, v/v); m.p. 98–99 °C (diethyl ether); (c = 1, CHCl3); Lit. data:26 m.p. 98–100 °C, [α]D +141 (c = 3.2, CHCl3); 1H n.m.r.: δ, 2.03, 2.06, 2.08, 2.18 (4 s, 12H, 4 × COCH3), 2.66 (d, 1H, J2,OH = 8.2 Hz, OH), 3.88 (ddd, 1H, J2,3 = 9.8 Hz, H-2), 3.98–4.04 (m, 1H, J5,6a = 2.5 Hz, J5,6b = 4.3 Hz, H-5), 4.05 (dd, 1H, J6a,6b = 12.6 Hz, H-6a), 4.25 (dd, 1H, H-6b), 5.07 (dd, 1H, J4,5 = 9.8 Hz, H-4), 5.25 (dd, 1H, J3,4 = 9.8 Hz, H-3), 6.22 (d, 1H, J1,2 = 3.8 Hz, H-1) ppm; 13C n.m.r.: δ, 20.7, 20.8, 20.9, 21.1, 61.7, 67.6, 69.7, 69.8, 73.2, 91.4, 169.4, 169.7, 170.9, 171.5 ppm; HR-FAB MS [M + Na]+ calcd for C14H20O10Na 371.0954, found 371.0970.
General procedure for the synthesis of 1,3,4,6-tetra-O-acetyl-α-D-glucopyranose (2) with AgNO3 (Method 2)
AgNO3 (10.3 mg, 0.061 mmol) was added to a solution of bromide 3 (50 mg, 0.121 mmol) in acetonitrile (1.0 mL) and water (2.2 μL), and the resulting reaction mixture was stirred for 16–20 h at rt. After that, the volatiles were removed under reduced pressure. The residue was dissolved in CH2Cl2 (~10 mL) and washed with water (5 mL), 10% aq. Na2S2O3 (5 mL), and water (3 × 5 mL). The organic layer was separated, dried over MgSO4, and concentrated in vacuo. The residue was crystallized from dry diethyl ether to afford the title compound as colorless crystals.
General procedure for the synthesis of 1,3,4,6-tetra-O-acetyl-α-D-glucopyranose (2) from nitrate 13 (Method 3)
Compound 13 (50 mg, 0.127 mmol) was dissolved in acetonitrile (1.0 mL) and water (2.2 μL) and the reaction mixture was stirred for 6 h at rt. After that, the volatiles were removed under reduced pressure. The residue was dissolved in CH2Cl2 (~10 mL) and washed with water (5 mL), sat. aq. NaHCO3 (5 mL), and water (3 × 5 mL). The organic layer was separated, dried over MgSO4, and concentrated in vacuo. The residue was crystallized from dry diethyl ether to afford the title compound as colorless crystals.
2.3.4.6-Tetra-O-acetyl-β-D-glucopyranosyl nitrate (13)
AgNO3 (2.2 g, 12.9 mmol) was added to a solution of bromide 3 (5.0 g, 12.2 mmol) in dry acetonitrile (30 mL) and the resulting mixture was stirred for 5 min at rt. After that, the solids were filtered off through a pad of Celite and the filtrate was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (~250 mL) and washed with water (50 mL), 1% aq. NaOH (50 mL), and water (3 × 50 mL). The organic layer was separated, dried over MgSO4, and concentrated in vacuo. The residue was crystallized from dry diethyl ether (~40 mL) to afford the title compound (3.68 g, 77% yield) as colorless crystals. Analytical data for 13: Rf = 0.75 (ethyl acetate/hexanes, 1/1, v/v); (c = 1, CHCl3); Lit. data:37 m.p. 96–97 °C, [α]D −4 (c 0.4, acetonitrile), −8 (c 1.0, carbon tetrachloride); 1H n. m.r.: δ, 2.02, 2.04, 2.07, 2.09 (4 s, 12H, 4 × COCH3), 3.87–3.93 (m, 1H, J5,6a = 2.3 Hz, J5,6b = 4.6 Hz, H-5), 4.14 (dd, 1H, J6a,6b = 12.5 Hz, H-6a), 4.29 (dd, 1H, H-6b), 5.13 (m, 2H, J2,3 = 8.4 Hz, J4,5 = 9.2 Hz, H-2, 4), 5.30 (dd, 1H, J3,4 = 9.2 Hz, H-3), 5.80 (d, 1H, J1,2 = 8.4 Hz, H-1) ppm; 13C n.m.r.: δ, 20.5, 20.6, 20.7, 21.0, 61.2, 67.3, 67.8, 72.6, 72.8, 96.9, 168.9, 169.3, 170.1, 170.6 ppm; HR-FAB MS [M + Na]+ calcd for C14H19NO12Na 416.0805, found 416.0785.
Preparative-scale synthesis of 1,3,4,6-tetra-O-acetyl-α-D-glucopyranose (2) from nitrate 13
Nitrate 13 (3.68 g, 9.36 mmol) was dissolved in acetonitrile (35 mL) and water (0.15 mL), and the resulting reaction mixture was stirred for 6 h at rt. After that, the volatiles were removed under reduced pressure. The residue was dissolved in CH2Cl2 (~100 mL) and washed with water (30 mL), 10% aq. Na2S2O3 (30 mL), and water (3 × 10 mL). The organic layer was separated, dried over MgSO4, and concentrated in vacuo. The residue was crystallized from dry diethyl ether (~30 mL) to afford the title compound (2.44 g, 75% yield) as colorless crystals.
2.3.4.6-Tetra-O-benzoyl-β-D-glucopyranosyl nitrate (14)
AgNO3 (0.27 g, 1.60 mmol) was added to a solution of bromide 7 (1.00 g, 1.52 mmol) in dry acetonitrile (10 mL) and the reaction mixture was stirred for 5 min at rt. After that, the solids were filtered off through a pad of Celite and the filtrate was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (~75 mL) and was washed with water (30 mL), 1% aq. NaOH (30 mL), and water (3 × 30 mL). The organic layer was separated, dried over MgSO4, and concentrated in vacuo. The residue was crystallized from dry diethyl ether (~10 mL) to afford the title compound (0.88 g, 91% yield) as colorless crystals. Analytical data for 14: Rf = 0.60 (ethyl acetate/hexanes, 3/7, v/v); (c = 1, CHCl3); m.p. 94.6–96.0 °C; 1H n.m.r.: δ, 4.38 (m, 1H, J5,6a = 5.3 Hz, J5,6b = 3.0 Hz, H-5), 4.50 (dd, 1H, J6a,6b = 12.3 Hz, H-6a), 4.66 (dd, 1H, H-6b), 5.66 (dd, 1H, J2,3 = 9.3 Hz, H-2), 5.73 (dd, 1H, J4,5 = 9.3 Hz, H-4), 6.01 (dd, 1H, J3,4 = 9.3 Hz, H-3), 6.15 (d, 1H, J1,2 = 8.2 Hz, H-1), 7.27–7.61 (m, 12H, aromatic), 7.80–8.07 (m, 8H, aromatic) ppm; 13C n.m.r.: δ, 62.6, 68.5, 68.7, 72.7, 73.5, 97.4, 128.4 (×2), 128.5 (×2), 128.6 (×2), 128.7 (×6), 129.9 (×4), 130.1 (×2), 130.2 (×2), 133.4, 133.7, 133.8, 133.9, 164.8, 165.1, 165.7, 166.2 ppm; HR-FAB MS [M + Na]+ caled for C34H27NO12Na caled: 664.1431, found 664.1425.
1.3.4.6-Tetra-O-acetyl-2-O-levulinoyl-α-D-glucopyranose (15). From bromide 3 (Methods 1 and 2)
CAN (33.3 mg, 0.061 mmol, Method 1) or AgNO3 (10.3 mg, 0.061 mmol, Method 2) was added to a solution of bromide 3 (50 mg, 0.121 mmol) in acetonitrile (1.0 mL) and water (2.2 μL), and the resulting reaction mixture was stirred for 16–20 h at rt. After that, the volatiles were removed under reduced pressure. The residue was dissolved in CH2Cl2 (~10 mL) and washed with water (5 mL), 10% aq. Na2S2O3 (5 mL), and water (3 × 5 mL). The organic layer was separated, dried over MgSO4, and concentrated in vacuo. Crude residues obtained by Method 1 or 2 were dissolved in dry CH2Cl2 (1.0 mL) and levulinic acid (24.9 mg, 0.214 mmol), EDC (55.0 mg, 0.287 mmol) and DMAP (3.5 mg, 0.029 mmol) were added. The resulting reaction mixture was stirred under argon for 1 h at rt. After that, the reaction mixture was diluted with CH2Cl2 (~50 mL) and washed with water (10 mL), sat. aq. NaHCO3 (10 mL), and water (3 × 10 mL). The organic layer was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–toluene gradient elution) to afford the title compound in 63% (from 3 by Method 1) or 71% (from 3 by Method 2), respectively, as a clear syrup. From nitrate 13 (Method 3). Nitrate 13 (50 mg, 0.127 mmol) was dissolved in acetonitrile (1.0 mL) and water (2.2 pL), and the resulting reaction mixture was stirred for 6 h at rt. After that, the volatiles were removed under reduced pressure. The residue was dissolved in CH2Cl2 (~10 mL) and washed with water (5 mL), sat. aq. NaHCO3 (5 mL), and water (2 × 5 mL). The organic layer was separated, dried over MgSO4, and concentrated in vacuo. Crude residue was dissolved in dry CH2Cl2 (1.0 mL) and levulinic acid (24.9 mg, 0.214 mmol), EDC (55.0 mg, 0.287 mmol) and DMAP (3.5 mg, 0.029 mmol) were added. The resulting reaction mixture was stirred under argon for 1 h at rt. After that, the reaction mixture was diluted with CH2Cl2 (~50 mL) and washed with water (10 mL), sat. aq. NaHCO3 (10 mL), and water (3 × 10 mL). The organic layer was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate-toluene gradient elution) to afford the title compound in 85% yield as a clear syrup. Analytical data for 15: Rf = 0.25 (ethyl acetate/hexanes, 1/1, v/v); (c = 1, CHCl3); 1H n.m.r.: δ, 2.00, 2.03, 2.04, 2.11, 2.14 (5 s, 15H, 5 × COCH3), 2.29–2.80 (m, 4H, CH2CH2), 3.99–4.12 (m, 2H, J5,6b = 4.4 Hz, J6a,6b = 12.7 Hz, H-5, H-6a), 4.22 (dd, 1H, H-6b), 5.01–5.14 (m, 2H, J2,3 = 9.8 Hz, H-2, 4), 5.44 (dd, 1H, J3,4 = 9.8 Hz, H-3), 6.26 (d, 1H, J1,2 = 3.7 Hz, H-1) ppm; 13C n.m.r.: δ, 20.6, 20.7 (×2), 20.9, 27.6, 29.7, 37.6, 61.5, 67.8, 69.3, 69.6, 69.8, 89.0, 168.8, 169.4, 170.4, 170.6, 171.5, 206.0 ppm; HR-FAB MS [M + Na]+ calcd for C19H26O12Na 469.1322, found 469.1298.
1.3.4.6-Tetra-O-acetyl-2-O-picoloyl-α-D-glucopyranose (16). From bromide 3 (Method 1 or 2) or nitrate 13 (Method 3)
Crude residues obtained from bromide 3 or nitrate 13, as described for the synthesis of 15, were dissolved in dry CH2Cl2 (1.0 mL) and picolinic acid (26.5 mg, 0.214 mmol), EDC (55.0 mg, 0.287 mmol), and DMAP (3.5 mg, 0.029 mmol) were added. The resulting reaction mixture was stirred under argon for 1 h at rt. After that, the reaction mixture was diluted with CH2Cl2 (~50 mL) and washed with water (10 mL), sat. aq. NaHCO3 (10 mL), and water (3 × 10 mL). The organic layer was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate-toluene gradient elution) to afford the title compound in 49% (from 3 by Method 1), 51% (from 3 by Method 2), or 67% (from 13 by Method 3) yield, respectively, as a white foam. Analytical data for 16: Rf = 0.20 (ethyl acetate/hexanes, 1/1, v/v); (c = 1, CHCl3); 1H n.m.r.: δ, 1.94, 2.03, 2.08, 2.14 (4 s, 12H, 4 × COCH3), 4.03–4.21 (m, 2H, J5,6b = 3.8 Hz, J6a,6b = 12.3 Hz, H-5, 6a), 4.28 (dd, 1H, H-6b), 5.21 (dd, 1H, J4,5 = 9.8 Hz, H-4), 5.36 (dd, 1H, J2,3 = 10.3 Hz, H-2), 5.67 (dd, 1H, J3,4 = 9.8 Hz, H-3), 6.46 (d, 1H, J1,2 = 3.7 Hz, H-1), 7.46–8.73 (m, 4H, aromatic) ppm; 13C n.m.r.: δ, 20.6, 20.7, 20.8, 20.9, 61.4, 67.7, 69.8, 69.9, 70.4, 89.0, 125.3, 127.4, 137.2, 146.6, 150.4, 163.5, 168.7, 169.4, 170.2, 170.7 ppm; HR-FAB MS [M + Na]+ calcd for C20H23NO11Na 476.1169, found 476.1125.
1.3.4.6-Tetra-O-acetyl-2-O-trifluoromethanesulfonyl-α-D-glucopyranose (17). From bromide 3 (Method 1 or 2) or nitrate 13 (Method 3)
Crude residues obtained from bromide 3 or nitrate 13, as described for the synthesis of 15, were dissolved in pyridine (1.0 mL) and CH2Cl2 (0.1 mL) and triflic anhydride (36 μL, 0.214 mmol) was added dropwise under argon at 0 °C. The resulting reaction mixture was stirred for 1 h at 0 °C rt. After that, the volatiles were removed under reduced pressure, and the residue was co-evaporated with toluene. The residue was diluted with CH2Cl2 (~50 mL) and washed with water (10 mL), sat. aq. NaHCO3 (10 mL), and water (3 × 10 mL). The organic layer was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate-toluene gradient elution) to afford the title compound in 54% (from 3 by Method 1), 52% (from 3 by Method 2), or 67% (from 13) yield, respectively, as a white foam. Analytical data for 17 was in accordance with that reported previously.59 HR-FAB MS [M + Na]+ calcd for C15H19F3O10SNa 503.0447, found 503.0460.
1.3.4.6-Tetra-O-benzoyl-2-O-trifluoromethanesulfonyl-α-D-glucopyranose (18)
Nitrate 14 (811 mg, 1.26 mmol) was dissolved in acetonitrile (8.0 mL) and water (23 μL) and the resulting mixture was stirred for 24 h at rt. After that, the volatiles were removed under reduced pressure. The residue was dissolved in CH2Cl2 (~100 mL) and washed with water (30 mL), sat. aq. NaHCO3 (30 mL), and water (3 × 30 mL). The organic layer was separated, dried over MgSO4, and concentrated in vacuo. Crude residue was dissolved in pyridine (10 mL) and CH2Cl2 (1.0 mL) and triflic anhydride (0.32 mL, 1.90 mmol) was added drop-wise under argon at 0 °C. The resulting reaction mixture was stirred for 1 h at 0 °C. After that, the volatiles were removed under reduced pressure, and the residue was co-evaporated with toluene. The residue was diluted with CH2Cl2 (~100 mL) and washed with water (30 mL), sat. aq. NaHCO3 (30 mL), and water (3 × 30 mL). The organic layer was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–hexanes gradient elution) to afford the title compound in 54% yield as a white amorphous solid. Analytical data for 18: Rf = 0.50 (ethyl acetate/hexanes, 3/7, v/v); (c = 1, CHCl3); 1H n. m.r.: δ, 4.40–4.50 (m, 1H, H-6a), 4.65–4.52 (m, 2H, H-5, 6b), 5.29 (dd, 1H, J2,3 = 10.0 Hz, H-2), 5.76 (dd, 1H, J4,5 = 9.9 Hz, H-4), 6.25 (dd, 1H, J3,4 = 9.9 Hz, H-3), 6.82 (d, 1H, J1,2 = 3.8 Hz, H-1), 7.31–7.75 (m, 12H, aromatic), 7.88–8.04 (m, 6H, aromatic), 8.14–8.22 (m, 2H, aromatic) ppm; 13C n.m.r.: δ, 62.0, 68.7, 69.5, 70.4, 80.2, 89.1, 118.3 (q), 128.1, 128.2, 128.3, 128.5 (×2), 128.6 (×2), 129.1 (×2), 129.4, 129.9 (×2), 130.0 (×4), 130.3 (×2), 130.4 (×2), 133.9 (×2), 134.5 (×2), 164.0, 165.1, 165.5, 166.1 ppm; HR-FAB MS [M + Na]+ calcd for C35H27F3O12SNa 751.1073, found 751.1064.
X-ray structure determination
Crystals of appropriate dimension were obtained by slow evaporation. Single crystals of appropriate dimensions were mounted on MiTeGen cryoloops in random orientations. Preliminary examination and data collection were performed using a Bruker X8 Kappa Apex II Charge Coupled Device (CCD) Detector system single crystal X-ray diffractometer equipped with an Oxford Cryostream LT device. All data were collected using graphite monochromated Mo K radiation (λ = 0.71073 Å) from a fine focus sealed tube X-ray source. Preliminary unit cell constants were determined with a set of 36 narrow frame scans. Typical data sets consist of combinations of and ϕ scan frames with typical scan width of 0.5 and counting time of 15 seconds/frame at a crystal to detector distance of 4.0 cm. The collected frames were integrated using an orientation matrix determined from the narrow frame scans. Apex II and SAINT software packages (Bruker Analytical X-Ray, Madison, WI, 2010) were used for data collection and data integration. Analysis of the integrated data did not show any decay. Final cell constants were determined by global refinement of reflections harvested from the complete data set. Collected data were corrected for systematic errors using SADABS (Bruker Analytical X-Ray, Madison, WI, 2010) based on the Laue symmetry using equivalent reflections.
Crystal data and intensity data collection parameters are listed in Tables 1S–6S for compound 10, 7S–13S for compound 13, and 14S–20S for compound 14.† Structure solution and refinement were carried out using the SHELXTL-PLUS software package.60 The structures were solved by direct methods and refined successfully with full matrix least-squares refinements by minimizing Σw(Fo2 − Fc2)2. The non-hydrogen atoms were refined anisotropically to convergence. All hydrogen atoms were treated using appropriate riding model (AFIX m3). The final residual values and structure refinement parameters are listed in Table 1S for compound 10, 7S for compound 13, 14S for compound 14.† Absolute structure determinations were carried out using Parson’s method61 for compounds 13 and 14. Twin data reduction and twin refinement (HKLF 5) refinement was carried out for compound 10. Disordered water molecules could not be modeled for compound 14 and squeezed data (Platon-Squeeze) was used for the final refinement. As the crystals for compound 14 were weakly diffracting we shipped the crystals for synchrotron data collection at ALS. However, no data set could be collected with the crystals as the crystals had decomposed in shipping, even under dry ice.
Complete listings of positional and isotropic displacement coefficients for hydrogen atoms, anisotropic displacement coefficients for the non-hydrogen atoms are listed as ESI (Tables 2S, 4S and 5S for compound 10; 8S, 10S and 11S for compound 13; 15S, 17S and 18S for compound 14†). Bond lengths [Å] and angles [°] as well as torsion angles [°] are listed as ESI (Tables 3S and 6S for compound 10; 9S and 12S for compound 13; 16S and 19S for compound 14†).
Supplementary Material
Acknowledgments
Funding sources
This work was supported by grants from the National Institute of General Medical Sciences (GM111835 and GM120673).
Footnotes
Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for all new compounds have been supplied as the ESI. Table of calculated and observed X-ray structure factors for compounds 10, 13, and 14. CCDC 1814198–1814200. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ob00477c
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.Borman S. Chem Eng News. 2007;85:9. [Google Scholar]
- 2.Hsieh-Wilson LC, Griffin ME. Science. 2013;342:1332. doi: 10.1126/science.1247615. [DOI] [PubMed] [Google Scholar]
- 3.Herget S, Toukach PV, Ranzinger R, Hull WE, Knirel YA, von der Lieth CW. BMC Struct Biol. 2008;8:35. doi: 10.1186/1472-6807-8-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oscarson S. In: The organic chemistry of sugars. Levi DE, Fugedi P, editors. CRC, Taylor & Francis; Boca Raton, London, New York: 2006. p. 53. [Google Scholar]
- 5.Jager M, Minnaard AJ. Chem Commun. 2016;52:656. doi: 10.1039/c5cc08199h. [DOI] [PubMed] [Google Scholar]
- 6.Lawandi J, Rocheleau S, Moitessier N. Tetrahedron. 2016;72:6283. [Google Scholar]
- 7.David S, Thieffry A, Veyrieres A. J Chem Soc, Perkin Trans. 1981;1:1796. [Google Scholar]
- 8.Halila S, Banazza M, Demailly G. J Carbohydr Chem. 2001;20:467. [Google Scholar]
- 9.Roelens S. J Org Chem. 1996;61:5257. [Google Scholar]
- 10.Bredenkamp MW, Spies HSC. Tetrahedron Lett. 2000;41:543. [Google Scholar]
- 11.Pozsgay V. Carbohydr Res. 1979;69:284. [Google Scholar]
- 12.Szeja W. Carbohydr Res. 1983;115:240. [Google Scholar]
- 13.Garegg PJ, Kvarnstrom I, Niklasson A, Niklasson G, Svensson SCT. J Carbohydr Chem. 1993;12:933. [Google Scholar]
- 14.Wang CC, Lee JC, Luo SY, Fan HF, Pai CL, Yang WC, Lu LD, Hung SC. Angew Chem, Int Ed. 2002;41:2360. doi: 10.1002/1521-3773(20020703)41:13<2360::AID-ANIE2360>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 15.Wang CC, Lee JC, Luo SY, Kulkarni SS, Huang YW, Lee CC, Chang KL, Hung SC. Nature. 2007;446:896. doi: 10.1038/nature05730. [DOI] [PubMed] [Google Scholar]
- 16.Gangadharmath UB, Demchenko AV. Synlett. 2004;2191 [Google Scholar]
- 17.Demchenko AV. Curr Org Chem. 2003;7:35. [Google Scholar]
- 18.Nigudkar SS, Demchenko AV. Chem Sci. 2015;6:2687. doi: 10.1039/c5sc00280j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murray RW, Jeyaraman R. J Org Chem. 1985;50:2847. [Google Scholar]
- 20.Halcomb RL, Danishefsky SJ. J Am Chem Soc. 1989;111:6661. [Google Scholar]
- 21.Danishefsky SJ, Bilodeau MT. Angew Chem, Int Ed Engl. 1996;35:1380. [Google Scholar]
- 22.Huang TY, Zulueta MM, Hung SC. Org Biomol Chem. 2014;12:376. doi: 10.1039/c3ob42097c. [DOI] [PubMed] [Google Scholar]
- 23.Tanner DD, Law FCP. J Am Chem Soc. 1969;91:7535. [Google Scholar]
- 24.Barclay LRC, Griller D, Ingold KU. J Am Chem Soc. 1982;104:4399. [Google Scholar]
- 25.Barclay LRC, Lusztyk J, Ingold KU. J Am Chem Soc. 1984;106:1793. [Google Scholar]
- 26.Helferich B, Zirner J. Chem Ber. 1962;95:2604. [Google Scholar]
- 27.Chittenden GJF. Carbohydr Res. 1988;183:140. [Google Scholar]
- 28.Becker D, Galili N. Carbohydr Res. 1993;248:129. doi: 10.1016/0008-6215(93)84121-l. [DOI] [PubMed] [Google Scholar]
- 29.Nigudkar SS, Hasty SJ, Varese M, Pornsuriyasak P, Demchenko AV. In: Carbohydrate Chemistry: Proven Synthetic Methods. van der Marel G, Codee J, editors. Vol. 2. CRC Press; 2014. p. 183. [Google Scholar]
- 30.Lemieux RU. In: Methods in Carbohydrate Chemistry. Whistler RL, Wolform ML, editors. Vol. 2. Academic Press Inc; New York and London: 1963. p. 221. [Google Scholar]
- 31.Contour MO, Defaye J, Little M, Wong E. Carbohydr Res. 1989;193:283. [Google Scholar]
- 32.Lemieux RU. In: Methods in Carbohydrate Chemistry. Whistler RL, Wolform ML, editors. Vol. 2. Academic Press Inc; New York and London: 1963. p. 226. [Google Scholar]
- 33.Lonn H. J Carbohydr Chem. 1987;6:301. [Google Scholar]
- 34.Sridharan V, Menendez JC. Chem Rev. 2010;110:3805. doi: 10.1021/cr100004p. [DOI] [PubMed] [Google Scholar]
- 35.Ferrier RJ, Furneaux RH. In: Methods in Carbohydrate Chemistry. Whistler RL, BeMiller JN, editors. Vol. 8. Academic Press; New York - London: 1980. p. 251. [Google Scholar]
- 36.Gervay J, Nguyen TN, Hadd MJ. Carbohydr Res. 1997;300:119. [Google Scholar]
- 37.Zurabyan SE, Tikhomirov MM, Nesmeyanov VA, Khorlin AY. Carbohydr Res. 1973;26:117. [Google Scholar]
- 38.Walden P. Ber Dtsch Chem Ges. 1896;29:133. [Google Scholar]
- 39.Isbell HS. Annu Rev Biochem. 1940;9:65. doi: 10.1146/annurev.bi.22.070153.000543. [DOI] [PubMed] [Google Scholar]
- 40.Koenigs W, Knorr E. Ber Dtsch Chem Ges. 1901;34:957. [Google Scholar]
- 41.Lemieux RU, Kondo T. Carbohydr Res. 1974;35:C4. [Google Scholar]
- 42.Iversen T, Bundle DR. J Chem Soc, Chem Commun. 1981;1240 [Google Scholar]
- 43.Dahmen J, Frejd T, Magnusson G, Noori G, Carlstrom AS. Carbohydr Res. 1984;125:237. doi: 10.1016/0008-6215(84)85159-9. [DOI] [PubMed] [Google Scholar]
- 44.Kochetkov NK, Klimov EM, Malysheva NN, Demchenko AV. Bioorg Khim. 1990;16:701. [Google Scholar]
- 45.Eckenberg P, Groth U, Huhh T, Richter N, Schmeck C. Tetrahedron. 1993;49:1619. [Google Scholar]
- 46.Poon KWC, Dudley GB. J Org Chem. 2006;71:3923. doi: 10.1021/jo0602773. [DOI] [PubMed] [Google Scholar]
- 47.Lopez SS, Dudley GB. Beilstein J Org Chem. 2008;4:44. doi: 10.3762/bjoc.4.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsabedze SB, Kabotso DEK, Pohl NLB. Tetrahedron Lett. 2013;54:6983. [Google Scholar]
- 49.Drew MGB, Ennis SC, Gridley JJ, Osborn HMI, Spackman DG. Tetrahedron. 2001;57:7919. [Google Scholar]
- 50.Walvoort MTC, Lodder G, Overkleeft HS, Codee JDC, van der Marel GA. J Org Chem. 2010;75:7990. doi: 10.1021/jo101779v. [DOI] [PubMed] [Google Scholar]
- 51.Visansirikul S, Yasomanee JP, Pornsuriyasak P, Kamat MN, Podvalnyy NM, Gobble CP, Thompson M, Kolodziej SA, Demchenko AV. Org Lett. 2015;17:2382. doi: 10.1021/acs.orglett.5b00899. [DOI] [PubMed] [Google Scholar]
- 52.Yarema KJ, Mahal LK, Bruehl RE, Rodriguez EC, Bertozzi CR. J Biol Chem. 1998;273:31168. doi: 10.1074/jbc.273.47.31168. [DOI] [PubMed] [Google Scholar]
- 53.Saxon E, Luchansky SJ, Hang HC, Yu CS, Lee SC, Bertozzi CR. J Am Chem Soc. 2002;124:14893. doi: 10.1021/ja027748x. [DOI] [PubMed] [Google Scholar]
- 54.Varki A. Trends Mol Med. 2008;14:351. doi: 10.1016/j.molmed.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lemieux RU, Ratcliffe RM. Can J Chem. 1979;57:1244. [Google Scholar]
- 56.Illarionov PA, Torgov VI, Shibaev VN. Russ Chem Bull Int Ed. 2000;49:1895. [Google Scholar]
- 57.Francisco CG, Leon EI, Martin A, Moreno P, Rodriguez MS, Suarez E. J Org Chem. 2001;66:6967. doi: 10.1021/jo0156565. [DOI] [PubMed] [Google Scholar]
- 58.Rice FAH, Inatome M. J Am Chem Soc. 1958;80:4709. [Google Scholar]
- 59.Popelova A, Kefurt K, Hlavackova M, Moravcova J. Carbohydr Res. 2005;340:161. doi: 10.1016/j.carres.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 60.Sheldrick GM. Acta Crystallogr, Sect A: Fundam Crystallogr. 2008;64:112. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 61.Parsons SFH. Acta Crystallogr, Sect A: Found Crystallogr. 2004;s61 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.