

Abstract
Previously, we communicated 3,3-difluoroxindole (HOFox)-mediated glycosylations wherein 3,3-difluoro-3H-indol-2-yl (OFox) imidates were found to be key intermediates. Both the in situ synthesis from the corresponding glycosyl bromides and activation of the OFox imidates could be conducted in a regenerative fashion. Herein, we extend this study to the synthesis of various glycosidic linkages using different sugar series. The main outcome of this study relates to enhanced yields and/or reduced reaction times of glycosylations. The effect of HOFox-mediated reactions is particularly pronounced in case of unreactive glycosyl donors and/or glycosyl acceptors. A multistep regenerative synthesis of oligosaccharides is also reported.
Graphical abstract
INTRODUCTION
Complex carbohydrates are paving the way to a variety of novel applications, most prominently in the areas of therapeutic-agent, diagnostic-platform, and functional-food development. Practically all glycans are connected via O-glycosidic linkages, but the chemical synthesis of these linkages remains challenging. Many methods for glycoside synthesis have been developed,1 and a vast majority of all glycosylations reported in the literature are performed with halides,2 thioglycosides,3 or O-imidates.4–6 Nevertheless, even these common methods have drawbacks. For instance, although all thioglycosides and many halides are stable, their activation requires stoichiometric activators. Conversely, O-imidates can be activated with catalytic amounts of a Lewis acid, but these reactive donors cannot be stored. If not properly controlled, glycosidation of O-imidates can give poor yields due to competing rapid hydrolysis. With enhanced understanding of the reaction mechanism,7–10 new methods and concepts would be a very timely addition to the arsenal of methods used for chemical glycosylation. Versatile new methods making use of stable precursors that could be activated under catalytic conditions11–13 would be of high significance to supplement other recently developed concepts that fall into this general category.14–17
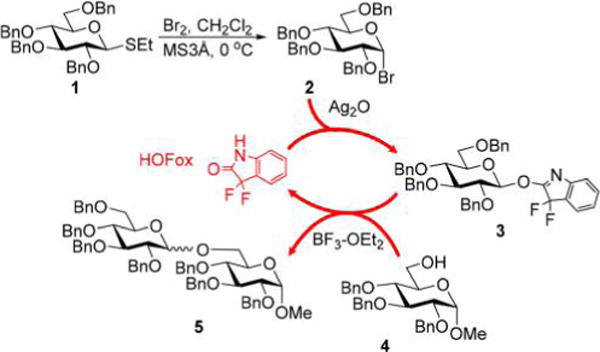
Recently, our laboratory reported O-benzoxazolyl (OBox)18 and 3,3-difluoro-3H-indol-2-yl (OFox) imidates.19 While investigating approaches to the synthesis of OFox imidates, we discovered that these compounds can be generated and glycosidated in a regenerative fashion in situ. This created the basis for discovering 3,3-difluoroxindole (HOFox)-mediated regenerative glycosylations wherein the OFox imidates were found to be key intermediates.19 In our preliminary study we first reacted thioglycoside 120 with stoichiometric bromine to form glycosyl bromide 2. The latter can be glycosidated slowly, but more readily gets converted into highly reactive OFox imidate 3 if HOFox is added. The amount of the reactive glycosyl donor present in the reaction medium can be controlled by the amount of HOFox added. OFox imidates have reasonable shelf life, but are readily activated with various Lewis acids (5–10 mol %). Previously, BF3·Et2O was used for this purpose, and we managed to achieve reasonable glycosylation rates between 2 and 4 (2–3 h) with as little as 10–25 mol % of HOFox aglycone to form disaccharide 5 in commendable yields (Scheme 1). Described herein is a continuation of this study with the main focus on broadening the scope of this reaction and its application to a variety of glycosidic linkages and sugar series.
Scheme 1.
Concept of Regenerative Glycosylation
RESULTS AND DISCUSSION
All glycosyl bromide donors employed in this study were obtained from the corresponding ethylthio glycosides with stoichiometric bromine. HOFox was obtained in one step from the commercially available Isatin by reaction with diethylaminosulfur trifluoride (DAST) in anhydrous CH2Cl2 at room temperature.19,21 A number of other approaches for the synthesis of HOFox have been developed.22,23 O-Imidates can be activated for glycosylation in the presence of catalytic amounts of TMSOTf,24,25 BF3·Et2O,26 p-TsOH,27 Bi(OTf)3,28 Yb(OTf)3,29 Sm(OTf)3,29 AgOTf,30 or MeOTf.18 For the further expansion of the regenerative glycosylation reaction we chose TMSOTf, which is arguably the most commonly used promoter.
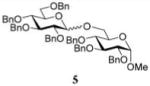
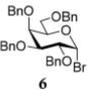
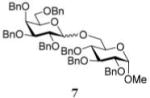
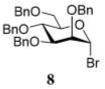
In the first round of experiments, we investigated perbenzylated bromides 2, 6, and 8 that were conveniently generated from the corresponding ethylthio glycosides20,31,32 by reaction with bromine in the presence of molecular sieves.33 A series of reactions between perbenzylated bromides with glycosyl acceptor 434 was performed as depicted in Table 1 (entries 1–3). First, a reaction of bromide 2 with acceptor 4 was performed in the presence of TMSOTf (0.08 equiv) as the activator and Ag2O (3.0 equiv) as the HBr scavenger. This sluggish reaction was stopped after 4 h, and disaccharide 518 was isolated in 18% yield (α/β = 1/5.4, entry 1). When essentially the same reaction was performed in the presence of HOFox catalyst (0.25 equiv) disaccharide 5 was isolated in 81% yield (α/β = 1/2.5, entry 1). The latter reaction was significantly faster and completed within 2 h. Galactosyl bromide 6 swiftly reacted with acceptor 4, but the effect of HOFox was negligible with this highly reactive donor. Irrespective of whether the reaction was performed without or with HOFox, disaccharide 716 was rapidly produced in 45 min in a similar yield (79% or 84%, respectively) with decent β-stereoselectivity in both cases (α/β = 1/18 or 1/10, respectively, entry 2). The HOFox catalyst was more efficient in the case of the less reactive mannosyl bromide 8. Thus, the reaction without HOFox was fairly slow (4 h) but still gave disaccharide 918 in a respectable yield of 74% (α/β > 2.9/1, entry 3). The same glycosylation in the presence of HOFox (0.15 equiv) was significantly faster (2 h), and disaccharide 9 was obtained in an excellent yield of 94%, albeit with no stereoselectivity (entry 3).
Table 1.
Establishing the Benchmark and Defining the Basic Scope of the HOFox-Catalyzed Glycosylations
| |||||
|---|---|---|---|---|---|
| Entry | Glycosyl Donor | HOFox (equiv.) | Time | Product | Yield (α/β ratio) |
| 1a |
|
0 0.25 |
4h 2h |
|
18% (1/5.4) 81% (1/2.5) |
| 2b |
|
0 0.25 |
0.75 h 0.75 h |
|
79% (1/18) 84% (1/10) |
| 3c |
|
0 0.15 0.25 |
4h 2h 3h |
|
74% (2.9/1) 94% (1.1/1) 88% (1.2/1) |
| 4c |
|
0 0.25 |
h h |
|
35% (β-only) 85% (β-only)d |
| 5b |
|
0 0.25 |
3h 3h |
|
19% (β-only) 90% (β-only) |
| 6a |
|
0 0.25 |
5 h 2 h |
|
13% (α-only) 98% (α-only) |
| 7b |
|
0 0.25 |
0.5 h 0.5 h |
|
21% (9.8/1) 95% (4.3/1) |
TMSOTf (0.08 equiv).
TMSOTf (0.05 equiv).
TMSOTf (0.10 equiv).
The corresponding 1,2-ortho ester byproduct was also formed.
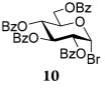
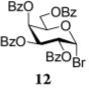
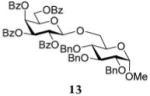
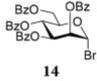
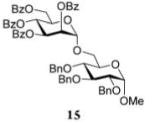
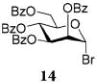
After that, we investigated the effect of HOFox on reactions of deactivated, perbenzoylated bromides. To endure fair comparison of the results, perbenzoylated bromides were also generated from the corresponding S-ethyl glycoside precursors35 by the reaction with bromine.33 However, perbenzoylated bromides can also be obtained from the corresponding perbenzoates.36 We were pleased to observe a very prominent effect of HOFox in all reactions of deactivated bromides. Thus, glycosidation of perbenzoylated glucosyl bromide 10 with glycosyl acceptor 4 without HOFox produced disaccharide 1118 in a poor yield of 35%. When the same reaction was performed in the presence of HOFox (0.25 equiv), disaccharide 11 was obtained in a significantly improved yield of 85% (Table 1, entry 4). A 1,2-orthoester was also produced as the side product. Glycosidation of perbenzoylated galactosyl bromide donor 12 with glycosyl acceptor 4 without HOFox produced disaccharide 1318 in 3 h in a poor yield of 19%. When the same reaction was performed in the presence of HOFox (0.25 equiv), disaccharide 13 was obtained in an excellent yield of 90% (entry 5). No 1,2-orthoester formation has been detected in this case. The HOFox effect was even more pronounced with mannosyl bromide donor 14. When the latter was glycosidated with acceptor 4 without HOFox, disaccharide 1518 was obtained in a poor yield of 13% after 5 h. When the same reaction was performed in the presence of HOFox (0.25 equiv) disaccharide 15 was obtained in an excellent yield of 98% in only 2 h (entry 6).
Peracetylated glycosyl bromide donors showed a similar trend. However, the efficiency of these reactions was hampered by a high propensity of the acetyl protecting groups to migrate to the hydroxyl of the glycosyl acceptors leading to poor yields. The latter could be improved in the presence of SnCl4 as the promoter (results are not shown). All reactions with peracylated donors were entirely 1,2-trans stereoselective due to the participatory effect of the ester substituent at C-2.
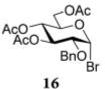
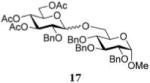
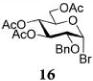
The results presented in entries 1–6 of Table 1 indicate that the role of catalytic HOFox is far more significant in the electronically deactivated, benzoylated, disarmed donors. To explore this further, we also investigated unreactive glucosyl bromide 1633,37 equipped with the 2-benzyl-3,4,6-triacetyl super-disarming protecting group pattern.38,39 Glycosidation of bromide donor 16 with glycosyl acceptor 4 without HOFox produced disaccharide 1737 in a poor yield of 21%. When the same reaction was performed in the presence of HOFox (0.25 equiv), disaccharide 17 was obtained in an excellent yield of 95% (entry 7). It should be noted that both reactions were quenched after 30 min. In another set of experiments, the reactions were continued for 2.5 h with/without HOFox, but no observable change in yields of disaccharide 17 was recorded in either case.
As evident from previous results, HOFox catalyzes reactions of both perbenzylated armed and perbenzoylated disarmed glycosyl donors. Also noticed was a significantly more profound effect of the HOFox catalysis on glycosidations of less reactive disarmed donors. The fact that nearly all reactions of 2-O-benzylated donors were more stereoselective without HOFox than those performed in the presence of HOFox has not remained unnoticed either. To understand the basis for these effects, we turned our focus to screening the effect of the amount of HOFox on the reactivity of armed and disarmed donors.40 For this study, we chose per-benzylated mannosyl donor 8 and its perbenzoylated counterpart 14 both of which provided high yield in the preliminary experiments in the presence of 25 mol % of HOFox. The study of the effect of different amounts of HOFox on the reactivity of donors 8 and 14 is summarized in Table 2.
Table 2.
Effect of the Catalyst on the Rate and the Yield of Reactions
| |||
|---|---|---|---|
| entry | HOFox (equiv) | 8→9, time (h), yield of 9 (%) (α/β ratio) | 14→15, time (h), yield of 15a (%) |
| 1 | 0 | 4, 74 (2.9/1) | 5, 13 |
| 2 | 0.05 | 2, 92 (1.1/1) | 5, 52 |
| 3 | 0.10 | 2, 91 (1.4/1) | 3, 90 |
| 4 | 0.25 | 3, 88 (1.2/1) | 2, 98 |
| 5 | 0.50 | 3, 89 (1.2/1) | 0.5, 98 |
| 6 | 0.75 | 3, 87 (1.1/1) | 0.5, 96 |
| 7 | 1.00 | 3, 88 (1.0/1) | 0.5, 74 |
Disaccharide 15 is obtained as a pure α-diastereomer.
Glycosidation of benzylated mannosyl bromide 8 without HOFox progressed slowly (4 h) to afford disaccharide 9 in 74% yield (entry 1). When glycosidation of 8 was performed in the presence of 5 or 10 mol % of HOFox practically identical results have been obtained. Disaccharide 9 was produced in excellent yields (92 or 91%, respectively) within 2 h (entries 2 and 3). These results with perbenzylated donor 8 indicate that the impact of HOFox is immediately evident even with very small amounts of the catalyst.
Increasing the amount of HOFox (25, 50, 75, or 100 mol %) did not show any enhancement in the rate of the reaction with donor 8 or yields that seemed to reach the plateau (entries 4–7). A gradual decrease of stereoselectivity was observed along with the increase of HOFox.
The effect of different amounts of HOFox on the reactivity of donor 14 was more predictable, with a steady decrease of the reaction time and increase of yields. The glycosylation reaction without HOFox proceeded slowly and afforded disaccharide 15 in poor yield (13%) in 5 h (Table 2, entry 1). A similar reaction performed with 5 mol % of HOFox produced disaccharide 15 in a moderate yield (52%) in 5 h (entry 2). Increasing the HOFox amount to 10 mol % further decreased the reaction time. The reaction was completed in 3 h, and disaccharide 15 was produced in a commendable yield of 90% (entry 3). Further overall enhancement was achieved in the presence of 25 mol % of HOFox. Disaccharide 15 was cleanly produced in 2 h in 98% yield (entry 4). Further increments in the amount of HOFox to 50 and 75 mol % also afforded disaccharide in excellent yields, and in these cases the reactions were even faster (30 min, entries 5 and 6). When the amount of HOFox was increased to 100 mol % the reaction was still swift, but the yield of disaccharide 15 dropped to 74% due to competing hydrolysis of the OFox imidate intermediate that was formed in large amounts from the beginning.
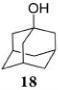
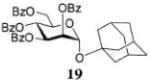
After investigating a series of glycosyl donors and optimization of the reaction conditions for glycosylation of primary acceptor 4, we decided to investigate the efficacy of the HOFox catalysis in application to other glycosyl acceptors. The reactivity of glycosyl acceptors depends on the nature of the hydroxyl group, i.e., primary or secondary position on sugar units. The reactivity can also be affected by the electronic nature of protecting groups and steric hindrance around the hydroxyl group. For this comparison study we chose unreactive glycosyl donors 14 for the synthesis of 1,2-trans linkages and 16 for the synthesis of 1,2-cis linkages along with the set of standard glycosyl acceptors. This study is summarized in Table 3. A reaction of the disarmed mannosyl bromide donor 14 with 1-adamantanol 18 in the presence of TMSOTf (5 mol %) without/with HOFox afforded glycoside 19 in excellent yields of 93/99% (entry 1). Although the numeric outcome of the HOFox catalysis may seem negligible in this swift (1 h) reaction, the reaction in the presence of HOFox was notably cleaner.
Table 3.
Glycosidation of Donors 14 and 16 with Different Acceptors in the Presence of TMSOTfa

| ||||||
|---|---|---|---|---|---|---|
| Entry | Donor | Acceptor (R”) | HOFox (equiv.) | Time | Product | Yield (α/β ratio) |
| 1 |
|
|
0 0.25 |
1 h 1 h |
|
93% (α-only) 99% (α-only) |
| 2 | 14 |
|
0 0,25 |
3 h 3 h |
|
27% (α-only) 85% (α-only) |
| 3 | 14 |
|
0 0,25 |
6 h 2 h |
|
13% (α-only) 98% (α-only) |
| 4 | 14 |
|
0 0.25 |
3 h 3 h |
|
14% (α-only) 86% (α-only) |
| 5 | 14 |
|
0 025 |
3 h 3 h |
|
25% (α-only) 92% (α-only) |
| 6a | 14 |

|
0 0.25 |
6 h 2 h |
|
20% (α-only) 68% (α-only) |
| 7 |
|
18 | 0 025 |
0.5 h 0.5 h |
|
61% (10/1) 90% (10/1) |
| 8 | 16 | 20 | 0 0.25 |
0.75 h 0.75 h |
|
26% (α-only) 92% (α-only) |
| 9 | 16 | 22 | 0 0.25 |
0.75 h 0.75 h |
|
27% (36/1) 91% (20/1) |
| 10 | 16 | 24 | 0 0.25 |
0.75 h 0.75 h |
|
14% (12/1) 85% (12/1) |
TMSOTf (0.15 equiv).
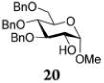
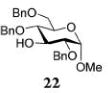
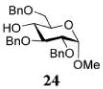
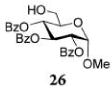
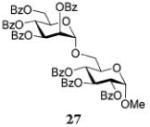
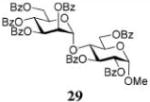
Glycosylations of sugar acceptors, which are typically much slower than those of aliphatic alcohols, were affected by HOFox much more strongly. Thus, glycosidation of donor 14 with 2-OH acceptor 2034 to form disaccharide 21 was enhanced from 27% (no HOFox) to 85% (25 mol % HOFox, entry 2). Both reactions were stopped after 3 h. Glycosidation of donor 14 with 3-OH acceptor 2234 to form disaccharide 2341 was enhanced from 13% (6 h, no HOFox) to 98% (2 h, 25 mol % HOFox, entry 3). Glycosidation of donor 14 with 4-OH acceptor 2434 to form disaccharide 2518 was enhanced from 14% (3 h, no HOFox) to 86% (3 h, 25 mol % HOFox, entry 4). After this initial success, we investigated benzoylated acceptors that are generally less reactive than their benzylated counterparts, which often translates into reduced yields. HOFox catalysis was found to be very effective in these cases as well. Thus, glycosidation of donor 14 with benzoylated 6-OH acceptor 2642 to form disaccharide 2743 was enhanced from 25% (3 h, no HOFox) to 92% (3 h, 25 mol % HOFox, entry 5). Glycosidation of donor 14 with sterically hindered and deactivated C4-OH glycosyl acceptor 2844 to form disaccharide 29 was enhanced from <2% (6 h, no HOFox) to 44% (2 h, 25 mol % HOFox). To improve this result, we conducted these reactions in the presence of larger quantities of the activator, 0.15 equiv TMSOTf. In this case, the formation of disaccharide 29 was enhanced from 20% (6 h, no HOFox) to 68% (2 h, 25 mol % HOFox, entry 6). All reactions with donor 14 were entirely 1,2-trans stereoselective due to the participatory effect of the ester substituent at C-2.
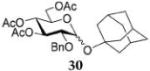
After that, we turned our attention to glucosyl bromide 16 equipped with the 2-O-benzyl-3,4,6-tri-O-acyl protecting group pattern. The glycosylation reactions of glucosyl donor 16 with a variety of glycosyl acceptors were conducted in a fashion similar to that mentioned earlier. Glycosidation of donor 16 with 1-adamantanol acceptor 18 to form glycoside 3045 was enhanced from 61% (30 min, no HOFox) to 90% (30 min, 25 mol % HOFox, entry 7, Table 3). The stereoselectivity of both reactions was the same, α/β = 10/1. Glycosidation of donor 16 with 2-OH acceptor 20 to form disaccharide 3137 was enhanced from 26% (45 min, no HOFox) to 92% (45 min, 25 mol % HOFox, entry 8). The stereoselectivity of both reactions was complete, α-only. Glycosidation of donor 16 with 3-OH acceptor 22 to form disaccharide 3237 was enhanced from 27% (no HOFox) to 91% (25 mol % HOFox, entry 9). The stereoselectivity of the reaction without HOFox was higher (α/β = 36/1) than that of the HOFox-catalyzed reaction (α/β = 20/1). Finally, glycosidation of donor 16 with 4-OH acceptor 24 to form disaccharide 3337 was enhanced from 14% (no HOFox) to 85% (25 mol % HOFox, entry 10). The stereoselectivity of both reactions was the same, α/β = 12/1.
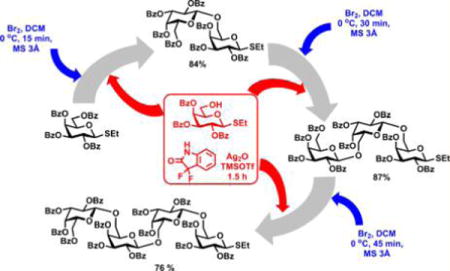
Encouraged by these results, we decided to investigate the applicability of this regenerative method to multistep oligosaccharide synthesis. For this endeavor, we chose perbenzoylated galactosyl thioglycosides: compound 3435 as the precursor for bromide 12 and galactosyl acceptor 35.46 Bromination of 34 with bromine produced galactosyl bromide donor 12 quantitatively. The latter was reacted with acceptor 35 (0.8 equiv) in the presence of TMSOTf (8 mol %) and HOFox (25 mol %) to afford 1,2-trans-linked disaccharide 3646 in 84% yield. For comparison, the same reaction in the absence of HOFox produced disaccharide 36 in only 26% yield. For the trisaccharide synthesis, bromination of disaccharide 36 with bromine (1.5 equiv) followed by glycosidation with acceptor 35 (0.8 equiv) in the presence of TMSOTf (8 mol %) and HOFox (25 mol %) afforded 1,2-trans-linked trisaccharide 37 in 87% yield. Further, bromination of trisaccharide 37 with bromine (1.5 equiv) followed by glycosidation with acceptor 35 (0.9 equiv) in the presence of TMSOTf (8 mol %) and HOFox (25 mol %) afforded 1,2-trans-linked tetrasaccharide 38 in 76% yield.
The HOFox-assisted regenerative glycosylation for oligosaccharide synthesis involves the donor and acceptor with the same leaving group. The concept may look similar to the previously developed two-step47–49 or preactivation approaches.50–52 The similarity is undeniable, but there is a key conceptual difference. In two-step or preactivation approaches, glycosyl donor is entirely converted to a reactive intermediate which is then reacted with a glycosyl acceptor. This may lead to high rates of competitive hydrolysis that become more pronounced at the advanced stages of the synthesis. As a result, a significant drop in yields can be observed with the increase in the size of the oligosaccharides. In the regenerative concept, the glycosyl bromide donor reacts with a catalytic amount of HOFox to furnish OFox imidates in a catalytic amount. The latter is then coupled with the S-ethyl glycosyl acceptor to afford disaccharide. During oligosaccharide synthesis, the actual reactive species which reacted with S-ethyl glycosyl acceptor are imidates which are obtained in small quantities and get regenerated only after the first batch has been consumed. We believe, that the regenerative approach helps to endure that the yields remain high throughout the entire duration of the oligosaccharide synthesis.
CONCLUSIONS
Presented herein is our first attempt to extend the HOFox-catalyzed regenerative glycosylation to other sugar series and targets. The versatility of this approach has been demonstrated in application to the regenerative oligosaccharide synthesis. The reason why nearly all reactions of 2-O-benzylated donors were less stereoselective in the presence of HOFox remains unknown. Further in-depth mechanistic study may help to explain this phenomenon and provide tools for the enhancement of the stereoselectivity.
Further implementation of the OFox-based regenerative glycosylation into the HPLC-assisted automated oligosaccharide synthesis on solid phases is currently underway in our laboratories. The regenerative glycosylation has a conservatively estimated 3-fold benefit for these reactions. First, the reactive OFox imidate donor is generated in small amounts, which helps to minimize side reactions. Second, the OFox donor is constantly regenerated ensuring continuous feeding of the system with the “fresh” donor. Third, a stable precursor can be used, and careful monitoring of glycosylation will ensure that only the necessary amounts of reagents is used.
EXPERIMENTAL SECTION
General Remarks
The reactions were performed using commercial reagents, and the ACS grade solvents used for reactions were purified and dried in accordance with standard procedures. Column chromatography was performed on silica gel 60 (70–230 mesh); reactions were monitored by TLC on Kieselgel 60 F254. The compounds were detected by examination under UV light and by charring with 10% sulfuric acid in methanol. Solvents were removed under reduced pressure at <40 °C. CH2Cl2 was distilled from CaH2 directly prior to application. Molecular sieves (3 Å), used for reactions, were crushed and activated in vacuo at 390 °C for 8 h in the first instance and then for 2–3 h at 390 °C directly prior to application. Optical rotations were measured at “JASCO P-2000” polarimeter. 1H NMR spectra were recorded at 300 MHz, 13C NMR spectra were recorded at 75 MHz. The 1H NMR chemical shifts are referenced to tetramethylsilane (TMS) for 1H NMR spectra for solutions in CDCl3. The 13C NMR chemical shifts are referenced to the central signal of CDCl3 (δC = 77.00 ppm) for solutions in CDCl3. Mass analysis was performed in a Waters Synapt G2 HDMS mass spectrometer with 10000 resolution, calibrated with CsI clusters in the mass range of 100–3000 m/z. The sample was infused using a syringe pump at a flow rate of 500 nL/min directly to the nano ESI source operated in positive mode. Conditions: Capillary voltage 2.0 kV, cone 20 V, extraction cone 2 V, source temperature 30 °C.
Typical Glycosylation Procedure and the Synthesis of Glycosides
A thioglycoside precursor (0.022–0.047 mmol) and freshly activated molecular sieves (3 Å, 100–150 mg) in CH2Cl2 (1–1.5 mL) was stirred under argon for 1 h at rt. Br2 (1.3 equiv or as indicated in tables and Scheme 2) was added at 0 °C, and the resulting mixture was stirred for 15 min or as mentioned in Scheme 2. Solvent was removed under reduced pressure, and the residue was dried in vacuo for 1 h. Silver oxide (3 equiv), glycosyl acceptor (0.018–0.037 mmol), and HOFox (0–1.0 equiv) were added, and the resulting solid was additionally dried in vacuo for 1 h. CH2Cl2 (1.5 mL) was added, and the resulting mixture was stirred for 30 min at rt. The mixture was cooled to 0 °C, TMSOTf (0.05–0.15 equiv) was added, and the resulting mixture was stirred for the time specified in the tables and Scheme 2. The solids were filtered off through a pad of Celite and rinsed successively with CH2Cl2. The combined filtrate (∼40 mL) was washed with 1% aq NaOH (10 mL) and water (2 × 10 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate – hexane gradient elution) to afford a glycoside derivative in yields listed in tables and Scheme 2. Anomeric ratios (if applicable) were determined by comparison of the integral intensities of relevant signals in 1H NMR spectra.
Scheme 2.
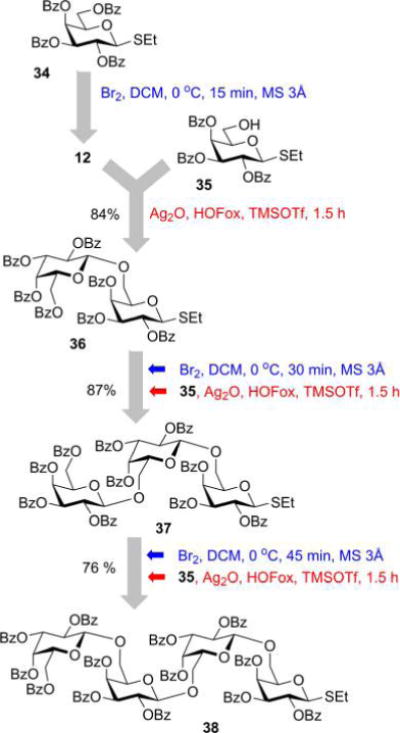
Oligosaccharide Synthesis through Regenerative Glycosylation
1-Adamantyl 2,3,4,6-Tetra-O-benzoyl-α-D-mannopyranoside (19)
The title compound was obtained from mannosyl donor 14 and acceptor 18 by the glycosylation method described earlier in 99% yield as a thick transparent syrup. Analytical data for α-19: Rf = 0.62 (ethyl acetate/hexane, 25/75, v/v); [α]D21 −65.9 (c = 1, CHCl3); 1H NMR (300 MHz) δ, 1.54–2.16 (m, 15H, 1-adamantyl), 4.48 (dd, 1H, J5,6a = 6.0 Hz, J6a,6b = 12.4 Hz, H-6a), 4.60–4.69 (m, 2H, H-5,6b), 5.50–5.55 (m, 2H, J2,3 = 2.5 Hz, H-1,2), 5.98 (dd, 1H, J3,4 = 10.0 Hz, H-3), 6.05 (dd, 1H, J4,5 = 10.1 Hz, H-4), 7.12–8.14 (m, 20H, aromatic) ppm; 13C NMR (75 MHz) δ 30.6 (×3), 36.1 (×3), 42.3 (×3), 63.4, 67.3, 68.4, 70.2, 72.2, 76.0, 90.9, 128.2 (×2), 128.3 (×2), 128.4 (×2), 128.5 (×2), 129.0, 129.1, 129.5, 129.7 (×4), 129.8 (×5), 133.0, 133.1, 133.4 (×2), 165.5, 162.6, 162.7, 166.2 ppm; HR-ES MS [M + NH4]+ calcd for [C44H42O10 + NH4] 748.3116, found 748.3146.
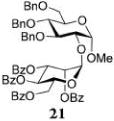
Methyl 2-O-(2,3,4,6-Tetra-O-benzoyl-α-D-mannopyranosyl)-3,4,6-tri-O-benzyl-α-D-glucopyranoside (21)
The title compound was obtained from mannosyl donor 14 and acceptor 20 by the glycosylation method described earlier in 85% yield as a white amorphous solid. Analytical data for α-21: Rf = 0.83 (ethyl acetate/hexanes, 35/65, v/v); [α]D22 +36.9 (c = 2.0, CHCl3); 1H NMR (300 MHz): δ 3.45 (s, 3H), 3.68–3.84 (m, 4H, H-4,5,6a,6b), 4.02 (dd, 1H, J2,3 = 9.9 Hz, H-2), 4.10 (dd, 1H, J3,4 = 8.4 Hz, H-3), 4.21 (dd, 1H, J5′,6′a = 4.3 Hz, J6′a,6′b = 12.4 Hz, H-6′a), 4.48–4.69 (m, 5H, H-5′, 6′b, 1.5 x CH2Ph), 4.79 (d, 1H, 2J = 10.7 Hz, 1/2 CH2Ph), 4.98 (d, 1H, J1,2 = 3.2 Hz, H-1), 5.08 (dd, 2H, 2J = 11.4 Hz, CH2Ph), 5.22 (d, 1H, J1′,2′ = 1.5 Hz, H-1′), 5.78 (dd, 1H, J2′,3′ = 3.2 Hz, H-2′), 5.94 (dd, 1H, J3′,4′ = 10.2 Hz, H-3′), 6.09 (dd, 1H, J4′,5′ = 10.2 Hz, H-4′), 7.10–8.13 (m, 35H, aromatic) ppm; 13C NMR (75 MHz): δ 55.3, 62.3, 66.4, 68.3, 68.9, 69.9, 70.2, 70.5, 73.5, 74.9, 75.1, 75.7, 78.1, 80.4, 94.5, 96.2, 127.2 (×2), 127.4, 127.7 (×2), 127.8 (×2), 127.9 (×2), 128.3 (×4), 128.4 (×9), 128.5 (×2), 128.8, 129.0, 129.2, 129.7 (×5), 129.8 (×2), 129.9, 132.9, 133.1, 133.3, 133.5, 137.8, 137.9, 138.4, 165.3 (×3), 166.0 ppm; HR-ES MS [M + NH4]+ calcd for [C62H58O15 + NH4]+ 1060.4114, found 1060.4126.
Methyl 4-O-(2,3,4,6-Tetra-O-benzoyl-α-D-mannopyranosyl)-2,4,6-tri-O-benzoyl-α-D-glucopyranoside (29)
The title compound was obtained from mannosyl donor 14 and acceptor 28 by the glycosylation method described earlier in 68% yield as a white amorphous solid. Analytical data for α-29: Rf = 0.60 (ethyl acetate/toluene, 15/85, v/v); [α]D22 +27.1 (c = 1.2, CHCl3); 1H NMR (300 MHz) δ 3.48 (s, 3H), 4.28–4.41 (m, 3H, H-4, 5, 6′a), 4.46–4.54 (m, 1H, J5′,6′b = 2.3 Hz, H-5′), 4.58 (dd, 1H, J6′a,6′b = 12.3 Hz, H-6′b), 4.72 (dd, 1H, J6a,6b = 11.8 Hz, H-6a), 4.87 (br d, 1H, H-6b), 5.15 (dd, 1H, J2,3 = 10.1 Hz, H-2), 5.20 (d, 1H, J1,2 = 3.6 Hz, H-1), 5.42 (d, 1H, J1′,2′ = 1.7 Hz, H-1′), 5.47 (dd, 1H, J2′,3′ = 3.1 Hz, H-2′), 5.86 (dd, 1H, J3′,4′ = 10.2 Hz, H-3′), 6.05 (dd, 1H, J4′,5′ = 10.1 Hz, H-4′), 6.22 (br dd, 1H, J3,4 = 9.9 Hz, H-3), 7.08–8.15 (m, 35H, aromatic) ppm; 13C NMR (75 MHz) δ 55.5, 62.3, 63.1, 66.3, 68.2, 69.2, 70.2 (×2), 72.1, 72.4, 75.8, 96.8, 99.1, 128.0 (×3), 128.2 (×4), 128.4 (×5), 128.6 (×3), 128.8, 128.9 (×3), 129.0, 129.4, 129.6 (×2), 129.7 (×4), 129.8 (×5), 129.9 (×3), 132.9 (×2), 133.0, 133.1, 133.4 (×3), 164.2, 165.1, 165.3 (×2), 165.9, 166.0, 166.2 ppm; HR-ES MS [M + NH4]+ calcd for [C62H52O18 + NH4]+ 1102.3492, found 1102.3503.
Ethyl O-(2,3,4-Tri-O-benzoyl-β-D-galactopyranosyl)-(1→6)-O-(2,3,4-tri-O-benzoyl-β-D-galactopyranosyl)-(1 → 6)-2,3,4-tri-O-ben-zoyl-1-thio-β-D-galactopyranoside (37)
The title compound was obtained from mannosyl donor 36 and acceptor 35 by the glycosylation method described earlier in 87% yield as a colorless amorphous solid. Analytical data for 37: Rf = 0.57 (ethyl acetate/toluene, 12/88, v/v); [α]D22 +150.2 (c = 1.5, CHCl3); 1H NMR (300 MHz) δ 1.14 (t, 3H, 3J = 7.4 Hz, –CH3), 2.50–2.72 (m, 2H, –CH2–), 3.58–4.30 (m, 9H, H-5, 5′, 5″, 6a, 6a′, 6a″, 6b, 6b′, 6b″), 4.63–4.81 (3d, 3H, H-1, 1′, 1″), 5.45–5.60 (m, 3H, H-3, 3′, 3″), 5.63–5.79 (m, 3H, H-2, 2′, 2″), 5.84–5.97 (m, 3H, H-4, 4′, 4″) 7.17–8.12 (m, 50H, aromatic) ppm; 13CNMR (75 MHz) δ 14.7, 24.1, 61.4, 66.2, 67.5, 67.7, 67.8, 68.2, 68.7, 69.7 (×2), 71.0, 71.5, 71.6, 72.4, 72.6, 76.6, 83.8, 100.6, 100.9, 128.1 (×2), 128.2 (×4), 128.3 (×4), 128.4 (×5), 128.5 (×5), 128.6 (×2), 128.7, 128.8, 128.9 (×2), 129.1, 129.2 (×3), 129.3, 129.6 (×4), 129.7 (×4), 129.8 (×4), 129.9 (×2), 130.0 (×3), 130.1 (×2), 133.0, 133.2 (×6), 133.3, 133.4, 133.5, 164.9, 165.1 (×2), 165.2, 165.3 (×2), 165.4, 165.5 (×2), 165.7 ppm; HR-ES MS [M + NH4]+ calcd for [C90H76O25S + NH4]+ 1606.4735, found 1606.4744.
Ethyl O-(2,3,4-Tri-O-benzoyl-β-D-galactopyranosyl)-(1→6)-O-(2,3,4,6-tetra-O-benzoyl-β-D-galactopyranosyl)-(1→6)-O-(2,3,4-tri-O-benzoyl-β-D-galactopyranosyl)-(1→6)-2,3,4-tri-O-benzoyl-1-thio-β-D-galactopyranoside (38)
The title compound was obtained from mannosyl donor 37 and acceptor 35 by the glycosylation method described earlier in 76% yield as a colorless amorphous solid. Analytical data for 38: Rf = 0.48 (ethyl acetate/toluene, 12/88, v/v); [α] 22D +111.1 (c = 1, CHCl3); 1H NMR (300 MHz): δ 1.13 (t, 3H, 3J = 7.5 Hz, –CH3), 2.46–2.71 (m, 2H, –CH2–), 3.22–4.26 (m, 12H, H-5, 5′, 5″, 5‴, 6a, 6a′, 6a″, 6a‴, 6b, 6b′, 6b″, 6b‴), 4.42–4.83 (4 d, 4H, H-1, 1′, 1″, 1‴), 5.38–5.77 (m, 8H, H-2, 2′, 2″, 2‴, 3, 3′, 3″, 3‴), 5.84–5.97 (m, 4H, H-4, 4′, 4″, 4‴), 7.12–8.13 (m, 65H, aromatic) ppm; 13C NMR (75 MHz): δ 14.7, 24.1, 61.2, 66.1 (×2), 67.5, 67.6, 67.7 (×2), 68.2, 68.7, 69.7, 69.8 (×2), 70.9, 71.5, 71.6, 71.7, 72.1 (×2), 72.7, 76.6, 83.8, 100.7 (×2), 100.9, 128.1 (×5), 128.2 (×6), 128.3 (×5), 128.4 (×6), 128.5 (×3), 128.6 (×2), 128.7, 128.8, 128.9, 129.0 (×4), 129.1, 129.2 (×4), 129.3 (×2), 129.5, 129.7 (×8), 129.8 (×7), 129.9 (×2), 130.0 (×2), 130.1 (×4), 133.0 (×2), 133.1 (×4), 133.2 (×5), 133.3, 133.4, 164.9, 165.0 (×2), 165.1 (×2), 165.2, 165.3 (×2), 165.4 (×2), 165.5 (×), 165.7 ppm; HR-ES MS [M + NH4]+ calcd for [C117H98O33S + NH4]+ 2080.6049, found 2080.6047.
Methyl 6-O-(2,3,4,6-Tetra-O-benzyl-β-D-glucopyranosyl)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (5)
The title compound was obtained from donor 2 and acceptor 4 by typical glycosylation method in 18% yield (α/β = 1/5.4) with no HOFox and 81% yield (α/β = 1/2.5) with 25 mol % of HOFox as a white amorphous solid. The analytical data for the title compound was in accordance to that reported previously.18
Methyl 6-O-(2,3,4,6-Tetra-O-benzyl-β-D-galacopyranosyl)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (7)
The title compound was obtained from donor 6 and acceptor 4 by typical glycosylation method in 79% yield (α/β = 1/18) with no HOFox and 84% yield (α/β = 1/10) with 25 mol % of HOFox as a white amorphous solid. The analytical data for the title compound was in accordance to that reported previously.16
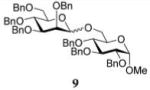
Methyl 2,3,4-Tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α-D-man-nopyranosyl)-α-D-glucopyranoside (9)
The title compound was obtained from donor 8 and acceptor 4 by typical glycosylation method in 74% yield (α/β = 2.9/1) with no HOFox and 94% yield (α/β = 1.1/1) with 15 mol % of HOFox as a white amorphous solid. The analytical data for the title compound was in accordance to that reported previously.18
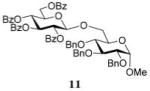
Methyl 6-O-(2,3,4,6-Tetra-O-benzoyl-β-D-glucopyranosyl)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (11)
The title compound was obtained from donor 10 and acceptor 4 by typical glycosylation method in 35% yield (β only) with no HOFox and 85% yield (β only) with 25 mol % of HOFox as a white amorphous solid. The analytical data for the title compound was in accordance to that reported previously.18
Methyl 6-O-(2,3,4,6-Tetra-O-benzoyl-β-D-galactopyranosyl)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (13)
The title compound was obtained from donor 12 and acceptor 4 by typical glycosylation method in 19% yield (β only) with no HOFox and 90% yield (β only) with 25 mol % of HOFox as a white amorphous solid. The analytical data for the title compound was in accordance to that reported previously.18
Methyl 2,3,4-Tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzoyl-α-D-mannopyranosyl)-α-D-glucopyranoside (15)
The title compound was obtained from donor 14 and acceptor 4 by typical glycosylation method in 13% yield (α-only) with no HOFox and 98% yield (α-only) with 25 mol % of HOFox as a white amorphous solid. The analytical data for the title compound was in accordance to that reported previously.18
Methyl 6-O-(3,4,6-Tri-O-acetyl-2-O-benzyl-α-D-glucopyranosyl)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (17)
The title compound was obtained from donor 16 and acceptor 4 by typical glycosylation method in 21% yield (α/β = 9.8/1) with no HOFox and 95% yield (α/β = 4.3/1) with 25 mol % of HOFox as a clear film. The analytical data for the title compound was in accordance to that reported previously.37
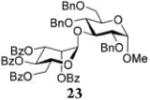
Methyl 2,4,6-Tri-O-benzyl-3-O-(2,3,4,6-tetra-O-benzoyl-α-D-mannopyranosyl)-α-D-glucopyranoside (23)
The title compound was obtained from donor 14 and acceptor 22 by typical glycosylation method in 13% yield (α-only) with no HOFox and 98% yield (α-only) with 25 mol % of HOFox as white amorphous solid. The analytical data for the title compound was in accordance to that reported previously.41
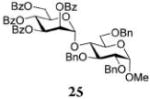
Methyl 2,3,6-Tri-O-benzyl-4-O-(2,3,4,6-tetra-O-benzoyl-α-D-mannopyranosyl)-α-D-glucopyranoside (25)
The title compound was obtained from donor 14 and acceptor 24 by typical glycosylation method in 14% yield (α-only) with no HOFox and 86% yield (α-only) with 25 mol % of HOFox as a white amorphous solid. The analytical data for the title compound was in accordance to that reported previously.18
Methyl 2,3,4-Tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzoyl-α-D-mannopyranosyl)-α-D-glucopyranoside (27)
The title compound was obtained from donor 14 and acceptor 26 by typical glycosylation method in 25% yield (α-only) with no HOFox and 92% yield (α-only) with 25 mol % of HOFox as a white amorphous solid. The analytical data for the title compound was in accordance to that reported previously.43
1-Adamantyl 3,4,6-Tri-O-acetyl-2-O-benzyl-α-D-glucopyranoside (30)
The title compound was obtained from donor 16 and acceptor 18 by typical glycosylation method in 61% yield (α/β = 10/1) with no HOFox and 90% yield (α/β = 10/1) with 25 mol % of HOFox as a clear film. The analytical data for the title compound was in accordance to that reported previously.45
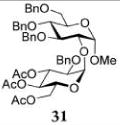
Methyl 2-O-(3,4,6-Tri-O-acetyl-2-O-benzyl-α-D-glucopyranosyl)-3,4,6-tri-O-benzyl-α-D-glucopyranoside (31)
The title compound was obtained from donor 16 and acceptor 20 by typical glycosylation method in 26% yield (α only) with no HOFox and 92% yield (α-only) with 25 mol % of HOFox as a clear film. The analytical data for the title compound was in accordance to that reported previously.37
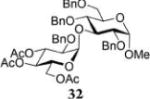
Methyl 3-O-(3,4,6-Tri-O-acetyl-2-O-benzyl-α-D-glucopyranosyl)-2,4,6-tri-O-benzyl-α-D-glucopyranoside (32)
The title compound was obtained from donor 16 and acceptor 22 by typical glycosylation method in 27% yield (α/β = 36/1) with no HOFox and 91% yield (α/β = 20/1) with 25 mol % of HOFox as a clear film. The analytical data for the title compound was in accordance to that reported previously.37
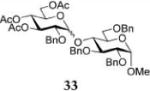
Methyl 4-O-(3,4,6-Tri-O-acetyl-2-O-benzyl-α-D-glucopyranosyl)-2,3,6-tri-O-benzyl-α-D-glucopyranoside (33)
The title compound was obtained from donor 16 and acceptor 24 by typical glycosylation method in 14% yield (α/β = 12/1) with no HOFox and 85% yield (α/β = 12/1) with 25 mol % of HOFox as a clear film. The analytical data for the title compound was in accordance to that reported previously.37
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institute of General Medical Sciences (GM111835 and GM120673).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.7b02768.
1H and 13C NMR spectra for all new compounds (PDF)
ORCID
Alexei V. Demchenko: 0000-0003-3515-212X
Notes
The authors declare no competing financial interest.
References
- 1.Zhu X, Schmidt RR. Angew Chem, Int Ed. 2009;48:1900. doi: 10.1002/anie.200802036. [DOI] [PubMed] [Google Scholar]
- 2.Kulkarni SS, Gervay-Hague J. In: Handbook of Chemical Glycosylation. Demchenko AV, editor. Wiley-VCH; Weinheim: 2008. p. 59. [Google Scholar]
- 3.Zhong W, Boons GJ. In: Handbook of Chemical Glycosylation. Demchenko AV, editor. Wiley-VCH; Weinheim: 2008. p. 261. [Google Scholar]
- 4.Zhu X, Schmidt RR. In: Handbook of Chemical Glycosylation. Demchenko AV, editor. Wiley-VCH; Weinheim: 2008. p. 143. [Google Scholar]
- 5.Yu B, Tao H. Tetrahedron Lett. 2001;42:2405. [Google Scholar]
- 6.Yu B, Sun J. Chem Commun. 2010;46:4668. doi: 10.1039/c0cc00563k. [DOI] [PubMed] [Google Scholar]
- 7.Crich D. Acc Chem Res. 2010;43:1144. doi: 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]
- 8.Crich D. J Org Chem. 2011;76:9193. doi: 10.1021/jo2017026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ranade SC, Demchenko AV. J Carbohydr Chem. 2013;32:1. [Google Scholar]
- 10.Mydock LK, Demchenko AV. Org Biomol Chem. 2010;8:497. doi: 10.1039/b916088d. [DOI] [PubMed] [Google Scholar]
- 11.Li X, Zhu J. Eur J Org Chem. 2016;2016:4724. [Google Scholar]
- 12.McKay MJ, Nguyen HM. ACS Catal. 2012;2:1563. doi: 10.1021/cs3002513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams R, Galan MC. Eur J Org Chem. 2017;2017:6247. [Google Scholar]
- 14.Goswami M, Ellern A, Pohl NL. Angew Chem, Int Ed. 2013;52:8441. doi: 10.1002/anie.201304099. [DOI] [PubMed] [Google Scholar]
- 15.Park Y, Harper KC, Kuhl N, Kwan EE, Liu RY, Jacobsen EN. Science. 2017;355:162. doi: 10.1126/science.aal1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vibhute AM, Dhaka A, Athiyarath V, Sureshan KM. Chem Sci. 2016;7:4259. doi: 10.1039/c6sc00633g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun L, Wu X, Xiong DC, Ye XS. Angew Chem, Int Ed. 2016;55:8041. doi: 10.1002/anie.201600142. [DOI] [PubMed] [Google Scholar]
- 18.Nigudkar SS, Parameswar AR, Pornsuriyasak P, Stine KJ, Demchenko AV. Org Biomol Chem. 2013;11:4068. doi: 10.1039/c3ob40667a. [DOI] [PubMed] [Google Scholar]
- 19.Nigudkar SS, Stine KJ, Demchenko AV. J Am Chem Soc. 2014;136:921. doi: 10.1021/ja411746a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andersson F, Fugedi P, Garegg PJ, Nashed M. Tetrahedron Lett. 1986;27:3919. [Google Scholar]
- 21.Torres JC, Garden SJ, Pinto AC. Tetrahedron. 1999;55:1881. [Google Scholar]
- 22.Zhu J, Zhang W, Hu J. J Org Chem. 2010;75:5505. doi: 10.1021/jo1005262. [DOI] [PubMed] [Google Scholar]
- 23.Yu L-C, Gu J-W, Zhang S, Zhang X. J Org Chem. 2017;82:3943. doi: 10.1021/acs.joc.7b00111. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt RR. Angew Chem, Int Ed Engl. 1986;25:212. [Google Scholar]
- 25.Morelli L, Bernardi A, Sattin S. Carbohydr Res. 2014;390:33. doi: 10.1016/j.carres.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt RR, Jung KH. In: Preparative Carbohydrate Chemistry. Hanessian S, editor. Marcel Dekker; New York: 1997. p. 283. [Google Scholar]
- 27.Schmidt RR, Michel J. Angew Chem, Int Ed Engl. 1980;19:731. [Google Scholar]
- 28.Adinolfi M, Iadonisi A, Ravida A, Valerio S. Tetrahedron Lett. 2006;47:2595. [Google Scholar]
- 29.Adinolfi M, Barone G, Guariniello L, Iadonisi A. Tetrahedron Lett. 2000;41:9005. [Google Scholar]
- 30.Du Y, Wei G, Linhardt RJ. Tetrahedron Lett. 2003;44:6887. [Google Scholar]
- 31.Weïwer M, Sherwood T, Linhardt RJ. J Carbohydr Chem. 2008;27:420. [Google Scholar]
- 32.Wang C, Sanders B, Baker DC. Can J Chem. 2011;89:959. [Google Scholar]
- 33.Kaeothip S, Yasomanee JP, Demchenko AV. J Org Chem. 2012;77:291. doi: 10.1021/jo2019174. [DOI] [PubMed] [Google Scholar]
- 34.Ranade SC, Kaeothip S, Demchenko AV. Org Lett. 2010;12:5628. doi: 10.1021/ol1023079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sail D, Kovac P. Carbohydr Res. 2012;357:47. doi: 10.1016/j.carres.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lemieux RU. In: Methods in Carbohydrate Chemistry. Whistler RL, Wolform ML, editors. Vol. 2. Academic Press; New York: 1963. p. 226. [Google Scholar]
- 37.Nigudkar SS, Wang T, Pistorio SG, Yasomanee JP, Stine KJ, Demchenko AV. Org Biomol Chem. 2017;15:348. doi: 10.1039/c6ob02230h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamat MN, Demchenko AV. Org Lett. 2005;7:3215. doi: 10.1021/ol050969y. [DOI] [PubMed] [Google Scholar]
- 39.Mydock LK, Kamat MN, Demchenko AV. Org Lett. 2011;13:2928. doi: 10.1021/ol2009818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bandara MD, Yasomanee JP, Demchenko AV. In: Selective Glycosylations: Synthetic Methods and Catalysts. Bennett CS, editor. Wiley; 2017. p. 29. [Google Scholar]
- 41.Baek JY, Lee B-Y, Pal R, Lee W-Y, Kim KS. Tetrahedron Lett. 2010;51:6250. [Google Scholar]
- 42.Zhang F, Zhang W, Zhang Y, Curran DP, Liu G. J Org Chem. 2009;74:2594. doi: 10.1021/jo9000993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mishra B, Neralkar M, Hotha S. Angew Chem, Int Ed. 2016;55:7786. doi: 10.1002/anie.201511695. [DOI] [PubMed] [Google Scholar]
- 44.Bandara MD, Yasomanee JP, Rath NP, Pedersen CM, Bols M, Demchenko AV. Org Biomol Chem. 2017;15:559. doi: 10.1039/c6ob02498j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kowalska K, Pedersen CM. Chem Commun. 2017;53:2040. doi: 10.1039/c6cc10076g. [DOI] [PubMed] [Google Scholar]
- 46.Pornsuriyasak P, Demchenko AV. Tetrahedron: Asymmetry. 2005;16:433. [Google Scholar]
- 47.Nicolaou KC, Ueno H. In: Preparative Carbohydrate Chemistry. Hanessian S, editor. Marcel Dekker; New York: 1997. p. 313. [Google Scholar]
- 48.Halcomb RL, Danishefsky SJ. J Am Chem Soc. 1989;111:6661. [Google Scholar]
- 49.Smoot JT, Demchenko AV. Adv Carbohydr Chem Biochem. 2009;62:161. doi: 10.1016/S0065-2318(09)00005-5. [DOI] [PubMed] [Google Scholar]
- 50.Wang Z, Zhou L, El-Boubbou K, Ye XS, Huang X. J Org Chem. 2007;72:6409. doi: 10.1021/jo070585g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang L, Qin Q, Ye X-S. Asian J Org Chem. 2013;2:30. [Google Scholar]
- 52.Bouhall SK, Sucheck SJ. J Carbohydr Chem. 2014;33:347. doi: 10.1080/07328303.2014.931964. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.