

Abstract
A dearomative 1,4-carboamination of arenes has been achieved using arenophile cycloaddition and subsequent palladium-catalyzed substitution with non-stabilized lithium enolates. This protocol delivers products with exclusive syn-1,4-selectivity and can be also conducted in an asymmetric fashion. The method allows rapid dearomative difunctionalization of simple aromatic compounds into functional small molecules amenable to further diversification.
The development of functionalization methods that convert simple building blocks to more complex, value-added compounds is at the heart of contemporary organic synthesis. Particularly, functional elaboration of arenes has received significant attention,1 given their ample availability and application within all areas of molecular sciences. While most of the recent focus has been in the area of C–H activation,2 functionalization with concurrent loss of aromatization3 (i.e., dearomative functionalization) is significantly less developed, despite the unique synthetic and functional versatility of the products.4
Alternatively, formal dearomative difunctionalization of polynuclear arenes has been established using transition-metal-catalyzed ring-opening reactions of heterobicyclic alkenes.5 Accordingly, azabenzonorbornadienes can be opened with a variety of nucleophiles delivering mainly 1,2-amino functionalized dihydronaphthalenes (Scheme 1A).6 While exceptionally powerful, and with notable applications in natural product and drug synthesis,7 these transformations do require more elaborate starting precursors that are prepared through benzyne–pyrrole cycloaddition chemistry. Here, we document dearomative carboamination involving visible-light-mediated arene–arenophile cycloaddition and in situ Pd-catalyzed substitution of the resulting cycloadducts (Scheme 1B). The reaction delivers syn-1,4-difunctionalized products directly from feedstock or readily available arenes with the lithium enolate employed as nucleophiles. In addition, this process can be used for asymmetric desymmetrization, delivering products with high enantioselectivity. Finally, the synthetic utility of the method is demonstrated by a rapid preparation of functionalized small molecules.
Scheme 1.
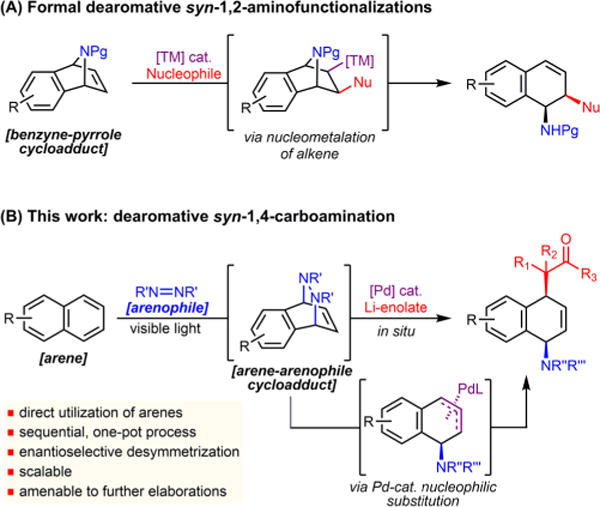
Dearomative Amino Functionalizations
Following the seminal achievements of Lautens, heterobicyclic alkenes are now widely employed in catalytic ring-opening reactions. In the case of formal dearomative 1,2-amino functionalizations (Scheme 1A), the observed syn-1,2-selectivity is a result of syn-nucleometalation.8 We have recently explored an arene–arenophile cycloaddition process9 that delivers similar bicyclic structures directly from aromatic precursors (Scheme 1B).10 In subsequent studies, we have sought to examine whether the intermediate cycloadducts would directly engage in a Pd-catalyzed substitution process11 with enolates.12 Since arene–arenophile adducts have different intrinsic strain as well as electrophilic character compared to benzyne-derived heterobicycles, they could operate under a mechanistically distinct regime, proceeding through an oxidative addition and allylic substitution process. Overall, this would result in a dearomative amino functionalization, a highly step- and atom-economical transformation that would deliver syn-1,4-carbo-aminated products directly from simple arenes.
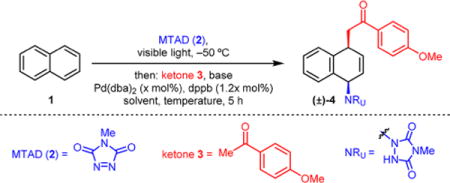
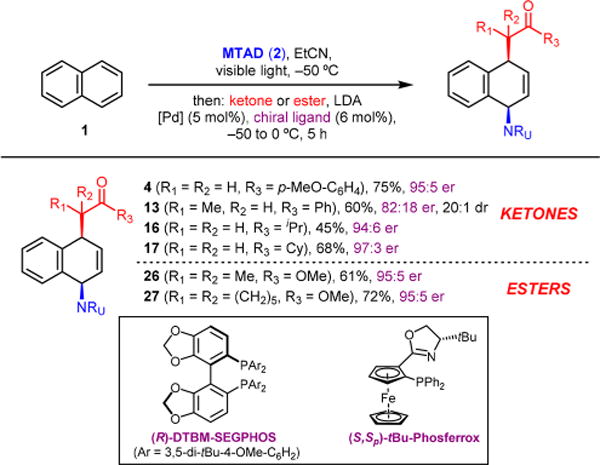
At the outset of our work, we focused on the identification of suitable reaction conditions that would readily elaborate naphthalene (1) to the corresponding carboaminated product 4 using N-methyl-1,2,4-triazoline-3,5-dione (MTAD, 2) as an arenophile and ketone 3 as an enolate source (Table 1). Initial investigations (see Supporting Information (SI) for full details) revealed that bidentate ligands in combination with Pd(dba)2 as Pd source were critical for good conversions. Thus, conducting the cycloaddition reaction at −50 °C in dichloromethane followed by subsequent addition of the lithium enolate of 3, generated using LDA, and catalyst [Pd(dba)2/dppb] provided the product 4 in 43% yield (Table 1, entry 1). Temperature played a significant role, as conducting the substitution step at −30 °C resulted in noticeably higher yield (72%, entry 2). However, cycloreversion of the cycloadduct was observed at higher temperatures (−10 °C), providing a moderate yield of the desired product (entry 3). While the use of other bases was not beneficial (entries 4 and 5), switching the solvent to propionitrile increased the yield to 85% (entry 6). Moreover, we found that the reaction progressed much more efficiently when it was allowed to slowly warm up, and even under reduced catalyst loading led to the formation of 4 in 92% isolated yield (entry 7). Importantly, decreasing the amount of enolate or arene had little effect on the reaction outcome (entries 8 and 9).
Table 1.
Optimization of Reaction Conditionsa
| |||||
|---|---|---|---|---|---|
| entry | [Pd] (mol%) | base | solvent | temp (°C) | yield (%)b |
| 1 | 5.0 | LDA | CH2CI2 | −50 | 43 |
| 2 | 5.0 | LDA | CH2CI2 | −30 | 72 |
| 3 | 5.0 | LDA | CH2CI2 | −10 | 47 |
| 4 | 5.0 | LiTMP | CH2CI2 | −30 | 44 |
| 5 | 5.0 | LiHMDS | CH2CI2 | −30 | 58 |
| 6 | 5.0 | LDA | EtCN | −30 | 85 |
|
| |||||
| 7 | 2.5 | LDA | EtCN | −50 to 0 | 91 (92)c |
|
| |||||
| 8 | 2.5 | LDA | EtCN | −50 to 0 | 82d |
| 9 | 2.5 | LDA | EtCN | −50 to 0 | 85e |
Standard reaction conditions: MTAD (2, 1.0 mmol, 1.0 equiv), naphthalene (2.0 mmol, 2.0 equiv), solvent (0.13 M), visible light, −50 °C, 12 h; then solution of enolate [p-methoxyacetophenone (2.0 mmol, 2.0 equiv), base (1.9 mmol, 1.9 equiv)], solution of catalyst [Pd(dba)2 (x mol%), dppb (1.2x mol%), THF], 5 h.
Determined by 1H NMR integration relative to the internal standard.
Isolated yield after purification by flash chromatography.
1.4 equiv of enolate used.
1.5 equiv of naphthalene used.
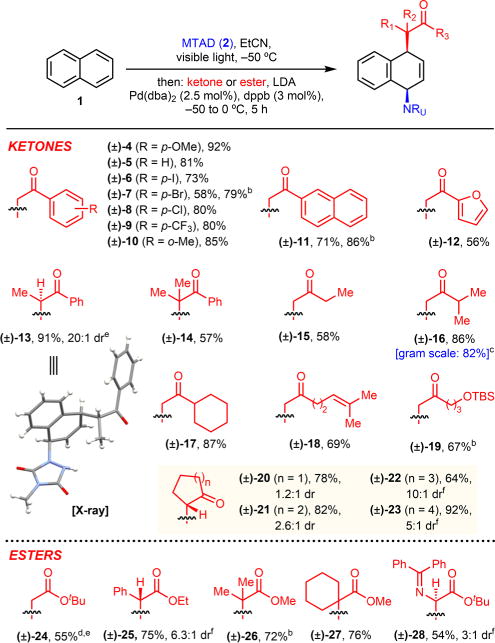
Having found reaction conditions which resulted in efficient dearomative difunctionalization of naphthalene, we set out to investigate the scope of this process using different ketones and esters (Table 2). Thus, acetophenone (5) and a range of diverse analogues bearing halogens (6–8), trifluoromethyl (9), and methyl groups (10) gave the corresponding products in high yields. Moreover, the phenyl motif on the enolate could be substituted with polynuclear or heteronuclear arenes, as exemplified with naphthalene (11) and furan (12). Sterically demanding substrates, represented by different levels of enolate substitution, were efficiently tolerated (13 and 14), providing product with high diastereoselectivity (>20:1 for 13, see X-ray data in SI including CIF file). In addition to aryl ketones, acyclic aliphatic ketones could be incorporated (15–19), including more elaborated substrates encompassing an alkene or protected hydroxyl group (18 and 19). Cyclic ketones proved to be feasible substrates as well (20–23), though the diastereoselectivity was highly dependent on the ring size. At the same time, the carboamination method could be extended to esters as substrates. In addition to acetate (24) and derivatives with different substitution patterns (25–27), protected glycine was also tolerated, delivering α-amino acid analogue 28. Finally, the robustness and scalability of this dearomative difunctionalization was tested by conducting this protocol on a larger scale, and we observed only a slight decrease in the yield of product 16. Importantly, in all cases, the products were obtained as a single constitutional isomer.
Table 2.
Ketone and Ester Scope of the Dearomative Carboaminationa
|
All reactions were run on 1.0 mmol scale under the standard conditions. Reported yields are of isolated products, and dr was determined by 1H NMR of the crude reaction mixtures.
Run at −30 °C for 12 h.
Run on 8.8 mmol scale.
Reaction was conducted in CH2Cl2 with LiCl as additive.
Pd(dba)2/rac-BINAP (5/6 mol%) used.
Relative stereochemistry determined by X-ray.
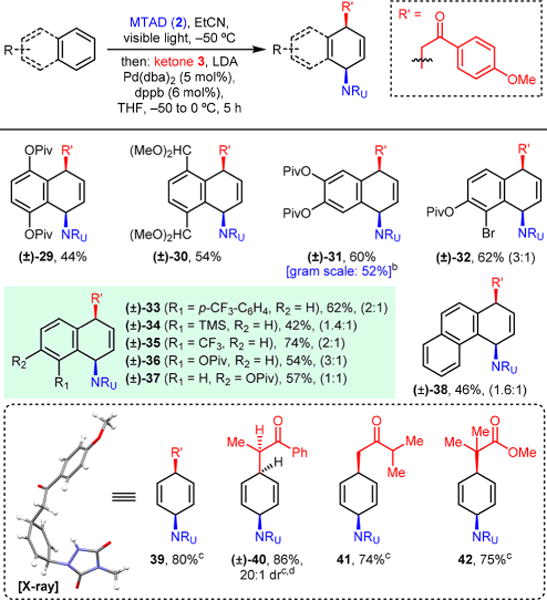
We next investigated the scope of arenes by using acetophenone 3 as an enolate source, as depicted in Table 3. In addition to symmetrically 1,4- and 2,3-disubstituted naphthalene derivatives (29–31), unsymmetrical substrates also function well, though constitutional isomers were observed resulting from ring opening at either site of arene–arenophile cycloadducts. Thus, 1,2-disubstituted naphthalene-based product 32 was obtained in a 3:1 ratio, and 1-substituted products 33–36 were obtained in 1.4:1 to 3:1 ratios. Only naphthalene with a more remote substituent at the 2-position resulted in nonselective difunctionalization (37), and diminished selectivity was also observed for higher order polyaromatic phenanthrene (38, 46%, 1.6:1). In general, this process displayed broad functional group compatibility and showed a general insensitivity to the electronic nature of substituents.
Table 3.
Arene Scope of the Dearomative Carboaminationa
|
All reactions were run on 1.0 mmol scale under the standard conditions. Reported yields are of isolated products, with ratio of constitutional isomers (in parentheses), and dr was determined by 1H NMR of the crude reaction mixtures.
Run on 8.8 mmol scale.
Run with benzene (10 mmol, 10 equiv) under the standard conditions and at −30 °C for 12 h.
Relative stereochemistry assigned by analogy.
We also examined the feasibility of using benzene as a substrate for this dearomative process (Table 3, inset). Gratifyingly, by performing the substitution step at −30 °C, the corresponding syn-1,4-carboaminated cyclohexadiene product 39 was obtained in 80% yield. Moreover, using this strategy, benzene also reacted with other enolates, providing products derived from propiophenone (40), an aliphatic ketone (41), and an ester (42) with comparable efficiency. Unfortunately, the analogous substituted benzene derivatives react poorly under the current reaction conditions.
This catalytic dearomative difunctionalization is also amenable to enantioselective desymmetrization13 as demonstrated with naphthalene (Table 4). Thus, two reaction conditions were identified depending upon the nature of the enolate.14 For example, a range of ketones under the standard protocol, using (S,Sp)-tBu-Phosferrox as a ligand,15 delivered carboaminated products 4, 13, 16, and 17 in high optical purity (82:18 to 97:3 er). On the other hand, enantioselective carboamination with esters required [Pd(allyl)Cl]2 and (R)-DTBM-SEGPHOS as a precatalyst to achieve good conversion and high selectivities, as exemplified with 26 and 27 (95:5 er).
Table 4.
Pd-Catalyzed Enantioselective Dearomative Carboamination of Naphthalenea
|
All reactions were run on 0.5 mmol scale under the standard conditions. For ketones, (S,Sp)-tBu-Phosferrox was used as a ligand. For esters, [Pd(allyl)Cl]2 and (R)-DTBM-SEGPHOS were used. Reported yields are of isolated products, and dr was determined by 1H NMR of the crude reaction mixtures.
The dearomatized products contain several regions amenable to further functionalization. As demonstrated with product 16, the alkene, urazole, and ketone moieties can undergo a series of selective and orthogonal transformations, enabling rapid diversification (Figure 1). For example, catalytic hydrogenation with Pd/C resulted in complete reduction of olefin and urazole, furnishing ketone 43. Chemoselective hydrogenation of the double bond using Rh/alumina or Upjohn dihydroxylation,16 followed by urazole oxidation,17 delivered ketones 44 and 45. Finally, chemoselective alkene reduction and ketone protection (16 → 46) enabled preparation of amine 47,18 ketone 48, and α-chloroketone 49.
Figure 1.
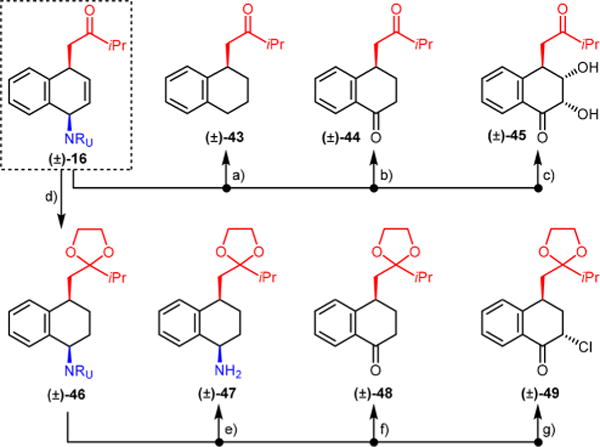
Derivatization of product 16. Reagents and conditions: (a) Pd/C (cat.), H2, 60%; (b) (i) Rh/Al2O3 (cat.), H2, 93%; (ii) tBuOCl, AcOH, 79%; (c) (i) OsO4 (cat.), NMO, 87%; (ii) NaOCl, 40%; (d) (i) Rh/Al2O3 (cat.), H2, 93%; (ii) glycol, TsOH, 85%; (e) (i) α-bromoacetophenone, NaH, 78%; (ii) KOH, 97%; (f) NaOCl, 53%; (g) tBuOCl, 77%, 20:1 dr.
In conclusion, we have developed a dearomative strategy for 1,4-carboamination of simple arenes. Our approach features an arenophile-based cycloaddition followed by Pd-catalyzed nucleophilic opening of the resulting cycloadducts using ketone- or ester-derived lithium enolates. The method delivers products with exclusive syn-1,4-selectivity and high enantioselectivity in the case of naphthalene. Finally, the products are amenable to further elaboration, enabling controlled access to a wide range of small, polyfunctionalized building blocks. Further studies regarding the expansion of the substrate scope and related transformations, as well as further applications of the method, are ongoing and will be reported in due course.
Supplementary Material
Acknowledgments
Financial support for this work was provided by the University of Illinois, the National Science Foundation (CAREER Award No. CHE-1654110), and the NIH/National Institute of General Medical Sciences (R01 GM122891). D.S. is an Alfred P. Sloan Fellow. M.O. acknowledges the Honjo International Scholarship Foundation. We also thank Dr. D. Olson and Dr. L. Zhu for NMR spectroscopic assistance, Dr. D. L. Gray and Dr. T. J. Woods for X-ray crystallographic analysis assistance, and F. Sun for mass spectrometric assistance.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b11663.
Experimental procedures and spectral data for all new compounds (PDF)
X-ray crystallographic data for (±)-4 (CIF)
X-ray crystallographic data for (±)-13 (CIF)
X-ray crystallographic data for 17 (CIF)
X-ray crystallographic data for (±)-22 (CIF)
X-ray crystallographic data for (±)-23 (CIF)
X-ray crystallographic data for (±)-25 (CIF)
X-ray crystallographic data for (±)-28 (CIF)
X-ray crystallographic data for (±)-39 (CIF)
ORCID
Alexander S. Shved: 0000-0001-5979-179X
David Sarlah: 0000-0002-8736-8953
Notes
The authors declare no competing financial interest.
References
- 1.Mortier J, editor. Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds. Wiley-VCH; Weinheim: 2016. [Google Scholar]
- 2.For selected reviews, see:; (a) Colby DA, Bergman RG, Ellman JA. Chem Rev. 2010;110:624. doi: 10.1021/cr900005n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]; (c) Lyons TW, Sanford MS. Chem Rev. 2010;110:1147. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yamaguchi J, Yamaguchi AD, Itami K. Angew Chem, Int Ed. 2012;51:8960. doi: 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]; (e) Arockiam PB, Bruneau C, Dixneuf PH. Chem Rev. 2012;112:5879. doi: 10.1021/cr300153j. [DOI] [PubMed] [Google Scholar]
- 3.For recent reviews, see:; (a) Roche SP, Porco JA. Angew Chem, Int Ed. 2011;50:4068. doi: 10.1002/anie.201006017. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pouységu L, Deffieux D, Quideau S. Tetrahedron. 2010;66:2235. [Google Scholar]; (c) Lewis SE. Chem Commun. 2014;50:2821. doi: 10.1039/c3cc49694e. [DOI] [PubMed] [Google Scholar]; (d) Pape AR, Kaliappan KP, Kündig EP. Chem Rev. 2000;100:2917. doi: 10.1021/cr9902852. [DOI] [PubMed] [Google Scholar]; (e) See also ref 13.
- 4.For selected recent examples, see:; (a) Good SN, Sharpe RJ, Johnson JS. J Am Chem Soc. 2017;139:12422. doi: 10.1021/jacs.7b07745. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wilson KB, Myers JT, Nedzbala HS, Combee LA, Sabat M, Harman WD. J Am Chem Soc. 2017;139:11401. doi: 10.1021/jacs.7b05118. [DOI] [PubMed] [Google Scholar]; (c) Nakayama H, Harada S, Kono M, Nemoto T. J Am Chem Soc. 2017;139:10188. doi: 10.1021/jacs.7b04813. [DOI] [PubMed] [Google Scholar]; (d) Zheng J, Wang SB, Zheng C, You SL. J Am Chem Soc. 2015;137:4880. doi: 10.1021/jacs.5b01707. [DOI] [PubMed] [Google Scholar]; (e) García-Fortanet J, Kessler F, Buchwald SL. J Am Chem Soc. 2009;131:6676. doi: 10.1021/ja9025193. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zhuo CX, You SL. Angew Chem, Int Ed. 2013;52:10056. doi: 10.1002/anie.201304591. [DOI] [PubMed] [Google Scholar]; (g) Phipps RJ, Toste FD. J Am Chem Soc. 2013;135:1268. doi: 10.1021/ja311798q. [DOI] [PubMed] [Google Scholar]; (h) Oguma T, Katsuki T. J Am Chem Soc. 2012;134:20017. doi: 10.1021/ja310203c. [DOI] [PubMed] [Google Scholar]
- 5.For selected reviews, see:; (a) Lautens M, Fagnou K, Hiebert S. Acc Chem Res. 2003;36:48. doi: 10.1021/ar010112a. [DOI] [PubMed] [Google Scholar]; (b) Rayabarapu DK, Cheng CH. Acc Chem Res. 2007;40:971. doi: 10.1021/ar600021z. [DOI] [PubMed] [Google Scholar]; (c) Woo S, Keay BA. Synthesis. 1996;6:669. [Google Scholar]; (d) Chiu P, Lautens M. Top Curr Chem. 1997;190:190. [Google Scholar]
- 6.For selected examples, see:; (a) Cho YH, Zunic V, Senboku H, Olsen M, Lautens M. J Am Chem Soc. 2006;128:6837. doi: 10.1021/ja0577701. [DOI] [PubMed] [Google Scholar]; (b) Nishimura T, Tsurumaki E, Kawamoto T, Guo XX, Hayashi T. Org Lett. 2008;10:4057. doi: 10.1021/ol801549t. [DOI] [PubMed] [Google Scholar]; (c) Ogura T, Yoshida K, Yanagisawa A, Imamoto T. Org Lett. 2009;11:2245. doi: 10.1021/ol900533h. [DOI] [PubMed] [Google Scholar]; (d) Nakao Y, Kashihara N, Kanyiva KS, Hiyama T. Angew Chem, Int Ed. 2010;49:4451. doi: 10.1002/anie.201001470. [DOI] [PubMed] [Google Scholar]; (e) Lautens M, Dockendorff C. Org Lett. 2003;5:3695. doi: 10.1021/ol035369i. [DOI] [PubMed] [Google Scholar]; (f) Chen CL, Martin SF. J Org Chem. 2006;71:4810. doi: 10.1021/jo060299p. [DOI] [PubMed] [Google Scholar]
- 7.For recent applications, see:; (a) McManus HA, Fleming MJ, Lautens M. Angew Chem, Int Ed. 2007;46:433. doi: 10.1002/anie.200603945. [DOI] [PubMed] [Google Scholar]; (b) Fleming MJ, McManus HA, Rudolph A, Chan WH, Ruiz J, Dockendorff C, Lautens M. Chem – Eur J. 2008;14:2112. doi: 10.1002/chem.200701775. [DOI] [PubMed] [Google Scholar]; (c) Lautens M, Fagnou K, Zunic V. Org Lett. 2002;4:3465. doi: 10.1021/ol026579i. [DOI] [PubMed] [Google Scholar]
- 8.Lautens M, Hiebert S, Renaud JL. J Am Chem Soc. 2001;123:6834. [Google Scholar]
- 9.(a) Hamrock SJ, Sheridan RS. J Am Chem Soc. 1989;111:9247. [Google Scholar]; (b) Kjell DP, Sheridan RS. J Am Chem Soc. 1984;106:5368. [Google Scholar]
- 10.(a) Southgate EH, Pospech J, Fu J, Holycross DR, Sarlah D. Nat Chem. 2016;8:922. doi: 10.1038/nchem.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Okumura M, Nakamata-Huynh SM, Pospech J, Sarlah D. Angew Chem, Int Ed. 2016;55:15910. doi: 10.1002/anie.201609686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For reviews on Pd-catalyzed allylic substitution, see:; (a) Trost BM, Van Vranken DL. Chem Rev. 1996;96:395. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Crawley ML. Chem Rev. 2003;103:2921. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]; (c) Lu Z, Ma S. Angew Chem, Int Ed. 2008;47:258. doi: 10.1002/anie.200605113. [DOI] [PubMed] [Google Scholar]; (d) Weaver JD, Recio A, III, Grenning AJ, Tunge JA. Chem Rev. 2011;111:1846. doi: 10.1021/cr1002744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For recent examples using lithium enolates as nucleophiles, see:; (a) Braun M, Laicher F, Meier T. Angew Chem, Int Ed. 2000;39:3494. doi: 10.1002/1521-3773(20001002)39:19<3494::aid-anie3494>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]; (b) Zheng WH, Zheng BH, Zhang Y, Hou XL. J Am Chem Soc. 2007;129:7718. doi: 10.1021/ja071098l. [DOI] [PubMed] [Google Scholar]; (c) Lei BL, Ding CH, Yang XF, Wan XL, Hou XL. J Am Chem Soc. 2009;131:18250. doi: 10.1021/ja9082717. [DOI] [PubMed] [Google Scholar]
- 13.For recent reviews on catalytic asymmetric dearomatizations, see:; (a) Zhuo CX, Zhang W, You SL. Angew Chem, Int Ed. 2012;51:12662. doi: 10.1002/anie.201204822. [DOI] [PubMed] [Google Scholar]; (b) Zheng C, You SL. Chem. 2016;1:830. [Google Scholar]
- 14.See SI for details.
- 15.Dai LX, Tu T, You SL, Deng WP, Hou XL. Acc Chem Res. 2003;36:659. doi: 10.1021/ar020153m. [DOI] [PubMed] [Google Scholar]
- 16.(a) Schneider WP, McIntosh AV. Upjohn. 2,769,824. US Patent. 1956; (b) VanRheenen V, Kelly RC, Cha DY. Tetrahedron Lett. 1976;17:1973. [Google Scholar]
- 17.For direct oxidation of urazole to ketone, see:0; (a) Wilson RM, Hengge AC. J Org Chem. 1990;55:197. [Google Scholar]; (b) Wilson RM, Hengge AC, Ataei A, Ho DM. J Am Chem Soc. 1991;113:7240. [Google Scholar]
- 18.Adam W, Pastor A, Wirth T. Org Lett. 2000;2:1295. doi: 10.1021/ol000044c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.