

Abstract
Background
Recent studies have shown that epigenetic differences can increase the risk of spontaneous preterm birth (PTB). However, little is known about heterogeneity underlying such epigenetic differences, which could lead to hypotheses for biological pathways in specific patient subgroups, and corresponding targeted interventions critical for precision medicine. Using bipartite network analysis of fetal DNA methylation data we demonstrate a novel method for classification of PTB.
Method
The data consisted of DNA methylation across the genome (HumanMethylation450 BeadChip) in cord blood from 50 African-American subjects consisting of 22 cases of early spontaneous PTB (24-34 weeks of gestation) and 28 controls (>39 weeks of gestation). These data were analyzed using a combination of (1) a supervised method to select the top 10 significant methylation sites, (2) unsupervised “subject-variable” bipartite networks to visualize and quantitatively analyze how those 10 methylation sites co-occurred across all the subjects, and across only the cases with the goal of analyzing subgroups and their underlying pathways, and (3) a simple linear regression to test whether there was an association between the total methylation in the cases, and gestational age.
Results
The bipartite network analysis of all subjects and significant methylation sites revealed statistically significant clustering consisting of an inverse symmetrical relationship in the methylation profiles between a case-enriched subgroup and a control-enriched subgroup: the former was predominantly hypermethylated across seven methylation sites, and hypomethylated across three methylation sites, whereas the latter was predominantly hypomethylated across the above seven methylation sites and hypermethylated across the three methylation sites. Furthermore, the analysis of only cases revealed one subgroup that was predominantly hypomethylated across seven methylation sites, and another subgroup that was hypomethylated across all methylation sites suggesting the presence of heterogeneity in PTB pathophysiology. Finally, the analysis found a strong inverse linear relationship between total methylation and gestational age suggesting that methylation differences could be used as predictive markers for gestational length.
Conclusions
The results demonstrate that unsupervised bipartite networks helped to identify a complex but comprehensible data-driven hypotheses related to patient subgroups and inferences about their underlying pathways, and therefore were an effective complement to supervised approaches currently used.
Keywords: Epigenetics, preterm, bipartite networks, network analysis, visual analytics, network analysis, visualization
Introduction
An estimated 13 million children are born annually through preterm deliveries, accounting for 9.6% of all births worldwide [1]. Preterm births (PTB; less than 37 weeks of gestation) account for approximately 70% of infant mortality and morbidity resulting in high personal and financial costs [1]. For example, compared to children born at term, those born preterm have a higher incidence of conditions such as cerebral palsy, sensory deficits, learning disabilities, and respiratory illnesses [2]. Furthermore, as preterm children tend to have reduced fetal growth and numerous adverse intrauterine conditions, they are highly prone to the late onset of chronic diseases such as diabetes, hypertension, coronary heart disease, and stroke [3, 4]. Being born preterm therefore not only imparts a difficult start to life, but also confers considerable risk for a disease-burdened life [2–6].
What causes PTBs, and how can they be prevented? Reviews on this topic (e.g., [7]) cite numerous studies which have identified risk factors for PTB including socioeconomic status [8, 9], pre-existing comorbidities [10, 11], and smoking [12] that predispose a mother to a PTB. For example, African-American women have approximately twice the risk of PTB compared to other races [13]. However, given the narrow window of the gestation period, few of these risk factors can be easily modified, and therefore do not provide practical targets for effective and timely interventions.
Recent studies [14–16] have begun to focus on the genetic and epigenetic changes that could be implicated in triggering preterm deliveries, and which could potentially provide more practical targets for preventing them. As these studies have suggested the existence of epigenetic components in the biological pathways that trigger PTB, we recently analyzed methylated sites in fetal leukocyte DNA using whole genome analysis of cord blood from mothers who had early spontaneous PTB (gestational age 24-34 weeks) with intact membranes [16]. Methylated sites are locations on the DNA where methyl groups are added to the DNA, resulting in modification in the function of genes. In humans, DNA methylation typically occurs where a cytosine nucleotide occurs next to a guanine nucleotide (often referred to as a CpG site). Our analysis identified more than 9,000 differentially methylated sites representing several potential pathophysiologic pathways including inflammation, oxidative stress, matrix metabolisms, and myometrial activation. While such studies have proposed several biological pathways, little is known about how they trigger early spontaneous PTB with intact membranes.
One possible limitation of many such studies is that they have used primarily supervised methods to conduct a univariable analysis of the genes or methylation sites. Such methods typically generate a list of significant methylated sites (after correcting for multiple testing) based on their differential methylation levels between cases and controls. While such univariable analyses are powerful for narrowing genome-wide data to a small set of significant methylation sites, the methods potentially conceal patient subgroups that share similar methylation profiles caused by underlying molecular heterogeneity. Identifying and comprehending such patient subgroups based on their methylation profiles could enable inference for pathways triggering PTBs in each subgroup. Such results are a critical step in the design of targeted interventions, a corner stone of precision medicine.
One promising approach for identifying and comprehending such complex patterns of co-occurrence is through unsupervised bipartite network analysis [17]. For example, we have demonstrated that subject-variable bipartite networks [18] (which represent both subjects and variables in the same representation) can enable (1) the rapid identification of significant patient subgroups, and the variables (e.g., genes) that are strongly associated with them, and (2) the comprehension of those relationships resulting in hypotheses for processes (e.g., biological mechanisms) underlying those subgroups. Here we demonstrate the use of bipartite network analysis and visualization for re-analyzing data from our previous study [16] with the goal of enabling new insights into molecular heterogeneity and potential mechanisms that underlie PTB.
We begin by briefly describing current methods that have been used to identify patient subgroups in biomedical data, and our motivation for using bipartite networks to analyze subgroups based on PTB methylation. Next, we describe our network-based analytical method and how it enabled a domain expert in PTB to rapidly arrive at a complex but comprehensible understanding of heterogeneity in cases and in controls, in addition to heterogeneity within the cases which could be critical to the design of future targeted interventions. We conclude with a discussion on why subject-variable bipartite networks enabled a deeper comprehension of the data, resulting in data-driven hypotheses about the mechanisms underlying PTB.
The Role of Bipartite Networks in Identifying and Comprehending Patient Subgroups and Underlying Mechanisms
Current Approaches for Identifying Patient Subgroups
A patient subgroup is defined as a subset of patients drawn from a population (e.g., PTB patients) that share one or more characteristics (e.g., a combination of methylation sites). Patients have been divided into subgroups by using (a) investigator-selected variables such as using race for developing stratified regression models,[19] or assigning patients to different arms of a clinical trial, (b) existing classification systems such as by using the Medicare Severity-Diagnosis Related Group (MS-DRG) [20] to assign patients into a disease category for purposes of billing, or (c) computational methods such as classification [21–23] and clustering [24, 25] to discover patient subgroups from data.
One of the simplest unsupervised methods for computationally identifying patient subgroups is by enumerating conjunctions of variables, such as by analyzing all dyads and triads of co-occurring comorbidities in the Medicare database [26], and then examining the most prevalent subgroups. Other methods attempt to partition a dataset of patients and characteristics into sets that are relatively homogenous. These sets can either be one-sided clusters (clusters of patients, or clusters of characteristics) or co-clusters [25, 27, 28] (clusters of patients and characteristics). K-means and hierarchical clustering [23, 25] are among the most commonly used one-sided clustering methods and require inputs such as a similarity measure (e.g., Jaccard similarity) and the expected number of subgroups, but with no agreed-upon approaches to automatically determine them. More recently, co-clustering [25, 27, 28] methods (also called biclustering methods) have been developed to automatically identify non-overlapping or overlapping submatrices consisting of both patients and characteristics. Compared to the above partitioning methods that use similarity measures to identify clusters, dimensionality reduction methods attempt to find a reduced dimensional space where differences among patients is maximized. For example, principle component analysis [23] (PCA) attempts to identify principal components which are weighted combinations of characteristics along which patients have the maximum variance. The patients are projected onto a plane typically defined by the two most important principal components. Methods such as k-means are then used to identify clusters of patients in this reduced dimensional space.
In contrast to the above unsupervised methods, supervised methods focus on identifying patient subgroups by taking into consideration outcome variables (e.g., responders and non-responders in a treatment arm). For example, classification and regression trees (CART) [23] (and enhancements such as random forests [29] and bump hunting [22]) progressively divides patients into subgroups based on the outcome variable by using conjunctions of patient characteristics at each step. The method outputs a tree, and each path from the root node to a leaf node defines a patient subgroup.
Strengths and Limitations of Existing Methods
Although the above methods have improved our understanding of heterogeneity in different populations, they have important limitations with respect to enabling the identification and comprehension of patient subgroups. While all share the goal of identifying patient subgroups based on characteristics, they either (a) consider only some characteristics at a time when defining subgroups (e.g., methods using variable conjunctions), (b) output one-sided clusters such as patient subgroups without their characteristics (e.g., k-means, hierarchical clustering, PCA), or (c) cannot reveal the relationship among patient subgroups (e.g., co-clustering, CART).
As stated in the introduction, a central goal of precision medicine is not only to identify patient subgroups, but also to enable stakeholders to comprehend the processes underlying those subgroups. This comprehension of disease processes underlying patient subgroups enables stakeholders to design interventions that are targeted for each subgroup.
Bipartite Network Analysis and Visualization
One approach that achieves the goals of analysis and comprehension of multivariable is unsupervised bipartite networks [17]. Network visualization and analysis [17] is an advanced form of visual analytics defined as “the science of analytical reasoning facilitated by interactive visual interfaces” [30]. Visual analytical methods such as unsupervised network analysis are designed to augment cognitive reasoning by transforming symbolic and numeric data into visualizations, which can be manipulated through interaction [30]. Networks have been used to analyze a wide range of complex clinical, molecular, and social phenomena such as the co-occurrence of multimorbidities across patients [31], protein-protein interactions [32], and the spread of infections across a social group [33].
A network (also called a graph) [17] consists of a set of nodes, connected in pairs by edges; nodes represent one or more types of entities (e.g., subjects or methylation sites). Edges between nodes represent a specific relationship between the entities (e.g., a subject has a specific methylation difference at a methylation site). Figure 1A shows a unipartite network where nodes are of the same type (commonly used to analyze co-occurrence of genes across patients, or to analyze protein-protein interaction networks [32]). In contrast, Figure 1B shows a bipartite network where nodes are of two types, and edges exist only between different types of nodes such as between subject nodes (circles) and methylation site nodes (triangles).
Figure 1.
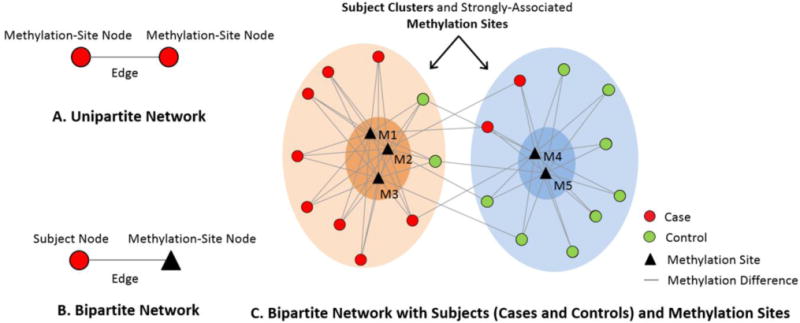
The distinction between a unipartite network (A), a bipartite network (B), and how the latter can be used to identify clusters of subjects and strongly associated methylation sites (C).
Networks are typically laid out using force-directed algorithms that pull together nodes that are strongly connected, and push apart nodes that are not. The result is that nodes with a similar pattern of connections are placed close to each other, and those that are dissimilar are pushed apart. As shown in Figure 1C, the application of the Kamada Kawai force-directed algorithm [34] to a bipartite network has revealed two clusters of subjects (one on the left and one on the right), each strongly associated with different methylation sites.
In prior work [35–40] we have shown that such “subject-variable” [18] bipartite networks are especially effective in helping to comprehend subject subgroups because they not only help to identify how subjects cluster with each other, but also how they are related to variables such as methylation sites. This feature enables an understanding of intra-cluster associations (e.g., the left cluster in enriched with cases associated predominantly with three methylation sites) and inter-cluster associations (e.g. the degree to which the subjects in the case-enriched cluster shares methylation sites with the control-enriched cluster as reevaled by the inter-cluster edges). This feature distinguishes bipartite networks from other unsupervised methods [41] such as unipartite clustering (e.g., k-means and hierarchical clustering), and dimensionality reduction methods (e.g., principal component analysis) that cluster either subjects or variables, but not both simultaneously.
Method
Data Selection
For the current study, we reanalyzed samples drawn from the Nashville Birth Cohort (NBC) described in a previous study [16]. Briefly, the NBC consists of samples of spontaneous preterm birth (cases), and of normal term birth (controls). In this cohort, maternal demographic and clinical data were recorded from medical records or through interviews during the consenting process; demographic and clinical data specific to the fetus were extracted from clinical records; gestational age of the neonate was determined by maternal reporting of the last menstrual period and corroborated through ultrasound dating; race was identified by self-reporting tracing back to three generations from the maternal and paternal sides of the fetus; and maternal self-reports were used to determine socioeconomic (education, household income, marital status, and insurance status), and behavioral (cigarette smoking) factors. A detailed description of these and other variables such as infections are described in past publications [42–44].
As described in our primary study [16], the samples used for the current analysis consisted of 50 African-American subjects of non-Hispanic ethnicity consisting of 22 cases of early spontaneous PTB (gestational age 24-34 weeks) and 28 controls (gestational age > 39 weeks). These cases and controls did not differ significantly in demographic or clinical factors [16, 45–50]. A detailed description of assay methods, analytical approaches, and data quality control measures can be accessed from the primary study [16]. To limit the influence of technical artifacts, beta values for each methylation site were residualized to account for chip and row. Similarly, to limit sex-specific effects, the effects of sex were also residualized using a multiple regression. This retrospective study was approved by the Institutional Review Board (IRB) at the University of Texas Medical Branch. The data files used for the study were in the research identifiable format (RIF), and the records were anonymized and de-identified prior to analysis. As analysis of such data does not require informed consent, it was therefore not done.
Bipartite Network Analysis
Our analysis consisted of three steps [18]: (1) exploratory visual analysis to identify emergent bipartite relationships such as patterns of how methylation sites co-occur across subjects; (2) quantitative analysis to quantitatively verify and statistically evaluate the emergent patterns such as clusters; (3) inference of the biological mechanisms underlying different emergent clusters of subjects. This three-step method used in our earlier studies has revealed complex but comprehensible visual patterns, leading to inferences about the biomarkers and underlying mechanisms involved.
1. Exploratory Visual Analysis
We constructed two bipartite networks to analyze patient subgroups based on methylation profiles with the goal of comprehending the molecular pathways involved in PTB: (1) a case-control bipartite network of 22 cases, 28 controls, and significant methylation sites, and (2) a case-only bipartite network of only the 22 PTB cases and significant methylation sites.
Nodes in the above bipartite networks represented subjects or methylation sites. To analyze the association of subjects to methylation sites that had strong signal for PTB, we (a) ranked all methylation sites identified in our earlier study based on their univariable significance, (b) removed all methylation sites that were not on a gene, and that had a SNP under the probe, and (c) selected the top-10 ranked (FDR≤7.41× 10−8) methylation sites. The resulting case-control network consisted of 50 subjects (22 cases and 28 controls) and 10 methylation sites, and the case-only network consisted of 22 cases and the same 10 methylation sites. Furthermore, we used node color to distinguish cases (red) from controls (green) in the case-control network, and to distinguish emergent subgroups (case-subgroup-1 = pink, case-subgroup-2 = blue) in the case-only network.
Edge weights in the networks were used to represent the degree of methylation differences for each subject-methylation site pair. Because DNA methylation was measured on different chips, and methylation is already known to be strongly associated with gender of the fetus, the beta values for each methylation site were residualized to account for chip, row, and sex-specific effects using a multiple regression. As regression residuals can range from negative to positive, and network layout algorithms require positive distances to position nodes, we shifted all residual values into the positive range. This was done by adding the least residual value for each methylation site to all its values, an approach which preserved the relative distances between subjects, and therefore enabled laying out the network using a standard force-directed algorithm.
Global patterns related to subjects and variables in the network were visualized and analyzed using the Kamada-Kawai [34] layout algorithm in Pajek (version 3.02) [51]. As shown in Figure 1C, the algorithm pulls together nodes that are strongly connected, and pushes apart nodes that are not. This algorithm is fast but approximate and is well-suited for medium sized networks consisting of between 100-1000 nodes [51]. The result is that nodes with a similar pattern of connections (e.g., M1 and M2 strongly associated with the left cluster in Figure 1C) are placed close to each other.
A key advantage of a bipartite network representation is the simultaneous visualization of subjects and variables, relationships between them (methylation differences), node type (cases and controls), and emergent global patterns (clusters) in a uniform visual representation. Such a representation enables domain experts such as clinicians and biologists to comprehend explicit associations such as how subject nodes are connected to methylation site nodes, in addition to emergent associations such as intra and inter cluster associations, leading to the rapid generation of hypotheses based on complex multivariable relationships.
2. Quantitative Analysis
We used three measures to quantitatively verify and statistically evaluate patterns derived from the exploratory visual analysis. These methods were selected based on their appropriateness to the emergent patterns in the network.
(a) Agglomerative Hierarchical Clustering
Because the network layout suggested a clustered topology for subjects and for methylation sites, we used the agglomerative hierarchical clustering method [41], which is best suited for networks that have small clusters [18, 35]. The clustering was done using the Manhattan dissimilarity measure with the Ward linkage function, and the number of clusters and their boundaries were determined based on natural breaks in the subject and methylation site dendrograms. The dendrograms were also combined with the heatmaps to aid in the visual analysis of the results.
(b) Clusteredness
To test whether the clusters in the network could have occurred by chance, we compared the variance, skewness, and kurtosis of the dissimilarities in the data, to 1000 random permutations of the dataset. For each network permutation, we preserved the size of the network, in addition to the edge weight distribution across patients when analyzing the patient dendrogram, and the edge weight distribution across methylation sites when analyzing the methylation dendrogram. Significant breaks in the subject or methylation site dendrograms would result in a significantly larger variance, skewness, and kurtosis of the dissimilarity measures, compared to the same measures generated from random permutations of the networks. Furthermore, we tested whether the proportion of cases and controls in the emergent subject clusters were significant using chi-square.
(c) Association between Methylation and Gestational Age
As the two clusters of nodes in the case-only network appeared to have a wide range in methylation differences, we used simple linear regression to test whether there was an association between total methylation of each subject, and gestational age. This was done by calculating the weighted degree centrality [17] for each patient node (sum of all its methylation differences across all the methylation sites), and testing its association (binned in increments of 0.25) to gestational age.
3. Inference of Biological Mechanisms
The verified clusters of subjects and methylation sites were used to identify hypotheses for biological pathways. This was done by (a) identifying the methylation sites that were strongly associated with each subject cluster, (b) mapping the methylation sites to their respective genes, and (c) identifying the biological pathways that are represented by the differentially methylated genes through the use of Ingenuity Pathway Analysis (IPA). IPA (Ingenuity® Systems www.ingenuity.com) is a widely used database and retrieval system designed to help researchers map a given set of molecules to biological pathways published in the literature.
Results
The bipartite network analyses revealed distinct patterns of methylation differences between cases and controls, in addition to distinct patterns of methylation differences between subsets of cases.
DNA Methylation Differences between Cases and Controls
The bipartite network visualization of 50 subjects (22 cases and 28 controls), and 10 methylation sites revealed a complex but understandable clustered pattern. As shown in Figure 2, there were two major clusters of subjects and methylation sites, one to the left, and another to the right.
Figure 2.
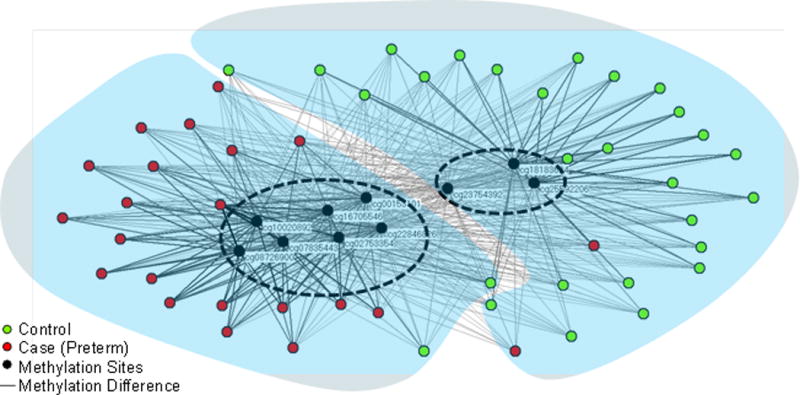
Bipartite network visualization of 50 subjects (22 cases, 28 controls) and methylation sites. The network revealed a significant separation between cases and controls, and the methylation sites that were strongly associated with each cluster. The blue shapes and dashed ovals denote cluster boundaries of subjects and methylation sites respectively identified through agglomerative hierarchical clustering.
To quantitatively verify the number of clusters and their members, we used agglomerative hierarchical clustering for the subjects, and for the methylation sites. As shown in Figure 3, the dendrogram shows a substantial break at 2 clusters both for the subjects, and for the methylation sites. The clusteredness of the subjects in the case-control network was statistically significant when compared to 1000 random permutations of the networks based on variance of the dissimilarities (case-control network = 9.25, Random Mean = 1.09, p<.001 two-tailed test), skewness of the distribution of dissimilarities (case-control network = 6.06, Random Mean = 2.77, p<.001 two-tailed test), and kurtosis of the distribution of dissimilarities (case-control network = 40.08, Random Mean = 12.40, p<.001 two-tailed test). Similarly, the clusteredness of the methylation sites in the case-control network was also statistically significant when compared to 1000 random permutations of the networks based on variance of the dissimilarities (case-control network = 38.01, Random Mean = 3.61, p<.001 two-tailed test), skewness of the distribution of dissimilarities (case-control network = 2.32, Random Mean = −0.24, p<.001 two-tailed test), and kurtosis of the distribution of dissimilarities (case-control network = 6.68, Random Mean = 1.89, p<.001 two-tailed test).
Figure 3.
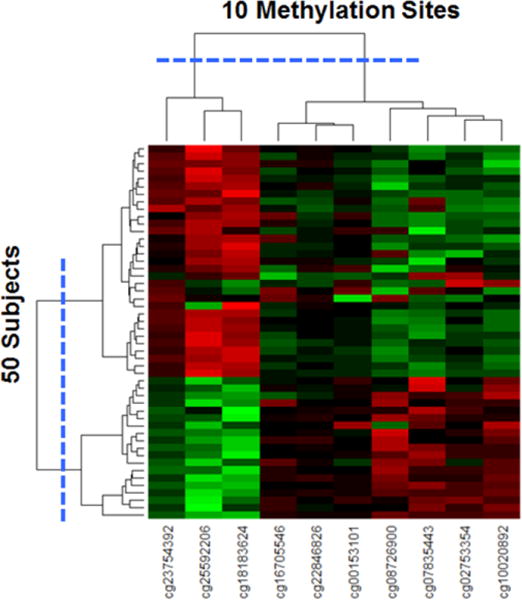
Heatmap with dendrograms of 50 subjects and 10 methylation sites generated through agglomerative hierarchical clustering. The largest break in the dendrogram is shown with the blue dotted lines, resulting in two clusters of methylation sites, and two clusters of subjects.
The cluster boundaries of subjects were superimposed onto the network using translucent blue shapes, and the cluster boundaries of methylation sites were superimposed on the network using dashed ovals. This superimposition of cluster boundaries on the network revealed that the subject cluster on the left contained mainly cases, but also included two controls; the subject cluster on the right had mainly controls, but also included two cases. Despite this cross-over of phenotypes, the proportion of cases and controls in each subject cluster was significantly different (χ2 Yates (1, N=50) = 35.0757, p<.001), suggesting an overall strong separation in cases and controls based on their methylation profiles.
The subjects in the left case-dominated cluster (red nodes) were hypermethylated at 7 methylation sites and their respective genes: cg10020892 (BCL9), cg22846826 (FOXK1), cg08726900 (ANKRD11), cg02753354 (HMHA1), cg07835443 (C16orf55), cg16705546 (IRF8), cg00153101 (PLCH2). They were also hypomethylated at the following 3 methylation sites and their respective genes: cg23754392 (BMI1) and cg25592206 (CDKN2C), and cg18183624 (IGF2BP1). In contrast, the subjects in the right control-dominated cluster (green nodes) had the opposite pattern: the subjects were hypomethylated at the above 7 methylation sites, and hypermethylated at the above 3 methylation sites.
DNA Methylation Differences among Cases
Because there was an overall strong and significant separation of cases from controls, this separation could have concealed sub-patterns within the cases. We therefore removed all the controls from the network to inspect possible patterns among only the cases. Figure 4 shows the resulting network of 22 cases and the 10 methylation sites which was laid out and analyzed using the same approach as was used for the previous case-control network.
Figure 4.
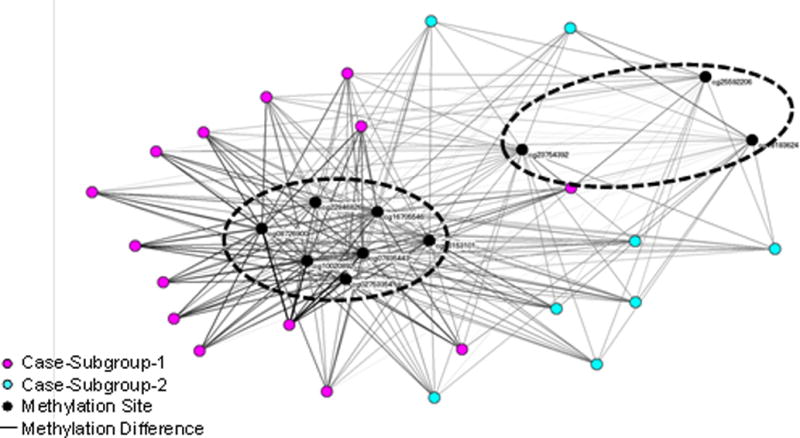
Bipartite network visualization of 22 cases and 10 methylation sites. The network revealed two clusters of cases, and the methylation sites that were strongly associated with each. The dashed ovals denote boundaries of case clusters identified through agglomerative hierarchical clustering.
As shown in Figure 4, there was a cluster of cases on the left that was strongly hypermethylated at the same 7 methylated sites as in the case-control network. However, as shown on the right, there was a dispersed group of cases with mainly thin edges connecting them to all the methylation sites, suggesting that this subgroup was hypomethelated at all the 10 methylation sites.
Similar to the previous analysis, we quantitatively determined the boundaries of the clusters in the case-only network through agglomerative hierarchical clustering for the subjects and for the methylation sites. As shown in Figure 5, the dendrogram showed a substantial break at 2 clusters both for the subjects, as well as for the methylation sites. The cluster boundaries of the subjects were superimposed on the network using node color, and the cluster boundaries of the methylation sites were superimposed on the network using dashed ovals.
Figure 5.
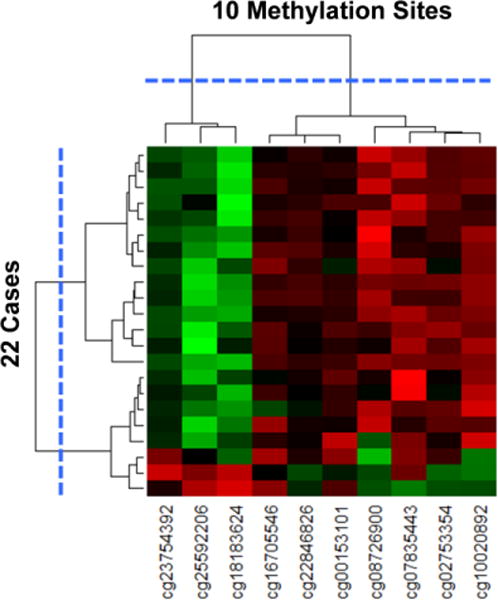
Heatmap with dendrograms of 22 cases and 10 methylation sites generated through agglomerative hierarchical clustering. The largest break in the dendrogram is shown with the blue dotted lines, resulting in two clusters of methylation sites, and two clusters of cases.
The clusteredness of the subjects in the case-only network was statistically significant when compared to 1000 random permutations of the network based on variance of the dissimilarities (case-only network = 1.31, Random Mean = 0.89, p<.001 two-tailed test), skewness of the distribution of dissimilarities (case-only network = 2.80, Random Mean = 1.60, p<.001 two-tailed test), and kurtosis of the distribution of dissimilarities (case-control network = 10.47, Random Mean = 5.20, p<.001 two-tailed test). Similarly, the clusteredness of the methylation sites in the case-only network was also statistically significant when compared to 1000 random permutations of the network based on variance of the dissimilarities (case-only network = 10.05, Random Mean = 3.76, p<.001 two-tailed test), skewness of the distribution of dissimilarities (case-only network = 2.30, Random Mean = 1.96, p<.001 two-tailed test), and kurtosis of the distribution of dissimilarities (case-only network = 6.62, Random Mean = 5.66, p<.001 two-tailed test). These results suggest the existence of two PTB subgroups. The first subgroup on the left was hypermethylated at 7 sites, and hypomethylated at 3 sites. In contrast, the second subgroup on the right was hypomethylated on all 10 sites.
In summary, the bipartite network visualizations and analyses led to two key findings. (1) There existed an inverse symmetrical relationship in the methylation profiles between the cases and controls: cases were predominantly hypermethylated at 7 methylation sites, and hypomethylated at 3 methylation sites, whereas controls were predominantly hypomethylated at the above 7 methylation sites and hypermethylated at the above 3 methylation sites. (2) There was strong evidence for heterogeneity in the profiles of the cases, where one subgroup was predominantly hypermethylated across 7 methylation sites, and another subgroup was hyomethylated across all 10 methylation sites.
Relationship between Methylation and Gestational Age
Because the two subgroups in the case-only network had a wide range in overall methylation differences, we tested if there was an association between total methylation difference in each case, and gestational age. The results showed an inverse linear relationship between the total methylation for each subject (binned in increments of 0.25), and gestational age (best fitted by y = −1.6222x + 36.052, R2 = 0.8488).
Inferences for Biological Mechanisms in Preterm Birth
An important goal of network visualization and analysis is to enable the comprehension of complex patterns in the data leading to hypotheses for the underlying processes such as biological mechanisms. Accordingly, given the significant separation of cases and controls in the case-control network, and the significant heterogeneity in the case-only network, our goal was to infer the possible biological pathways underlying those patterns. We therefore used IPA to identify known pathways related to genes represented by the 7 methylation sites, and related to the genes represented by the 3 methylation sites (shown on the left and the right of both networks respectively). The two bipartite networks along with the pathways identified from IPA were provided to a domain expert in PTB, who was asked to infer the potential mechanisms leading to PTB.
For the case-control network, he first attempted to analyze the IPA-identified pathways related to the 7 methylation sites that were hypermethylated in the left case-dominated cluster. Unfortunately, none of the pathways appeared to be meaningful for PTB. Next, he analyzed the IPA-identified pathways related to the 3 sites that were hypermethylated in the right control-dominated cluster. Here he inferred that two of the hypermethylated sites (cg23754392, cg25592206) were likely downregulating their respective genes (BMI1 and CDKN2C) leading to the upregulation of TP53 (a known tumor suppressor), resulting in normal cell senescence required for the normal rupture of the placenta during labor. Because these very methylation sites were hypomethylated (represented explicitly by the thin edges that connected most of the subjects in the left case-dominated cluster, to these two methylation sites on the right), he hypothesized that the opposite might hold for the cases: hypomethylation of the same two sites would lead to upregulation of BMI1 and CDKN2C, leading to the suppression of TP53 potentially resulting in decreased or absent cellular senescence.
Having determined a plausible role of cellular senescence in PTB, he reexamined the genes related to the hypermethylated sites in the case-dominated cluster on the left. This led to a focus on BCL9 and IRF8, which he noted were both cell cycle promoters. He therefore unified the two insights by hypothesizing that the decrease or absence of the pathway related to normal cellular senescence (inferred from the methylation sites strongly associated with the control-dominated cluster on the right), in combination with the presence of the pathway that promoted cell cycle (inferred from the methylation sites strongly associated with the case-dominated cluster on the left) might potentially be responsible for triggering PTB in the cases.
Next, he attempted to infer the plausible mechanisms underlying the heterogeneity in the case-only network (Figure 4). As the left subgroup had mostly uniform hypermethylation of 7 sites and hypomethylation of 3 sites, he inferred that the mechanisms underlying this subgroup also related to senescence. In contrast, as the right subgroup had mostly uniform hypomethylation of all 10 sites, he inferred that while they did not have a strong signature for senescence like the left subgroup, they also did not have as strong a signature as that of the control-dominated subgroup identified in Figure 3. This implied the existence of a continuum in methylation profiles, which could be the result of interaction with other risk factors triggering a PTB. Furthermore, the inverse relationship between total methylation and gestational age suggested that the fetal methylation associated epigenetic signature may be a useful predictor of an adverse pregnancy outcome such as PTB.
Recent studies provide corroborative evidence for the above mechanistic inferences. For example, existence of senescence as a mechanism was recently reported in fetal membranes and in fetal DNA [52–55]. However, because the precise mechanisms or functional pathways cannot be identified from differential methylation profiling, additional functional studies need to be conducted on these identified genes. Furthermore, methylation differences (hyper or hypo) are not always unidirectional [14], and many of the functional changes are linked to the type of cell or tissue, and the environment to which they are associated. Therefore, while the above inferences of pathways and heterogeneity derived from the visual analytics provide promising hypotheses related to senescence of fetal cells, these results need to be closely examined through future hypothesis-testing studies.
Discussion
From a biological perspective, even though the data had been previously rigorously analyzed, both networks revealed complex but comprehensible patterns leading to novel data-driven hypotheses. The case-control network revealed an inverse symmetrical relationship between cases and controls leading to biological inferences related to senescence. Furthermore, as discussed in the methods section, we used stringent criteria for the inclusion and exclusion of subjects, resulting in a relatively homogeneous group of cases and of controls with no significant demographic and clinical differences between them. However, despite these stringent criteria, the case-only network revealed patient subgroups based on methylation differences alone, demonstrating the important role that methylation changes in fetal DNA can play in revealing meaningful heterogeneities among cases.
From a methodological perspective, there were four features of the network representation that together contributed to the rapid inferences related to the pathophysiology in preterm births:
Representation of Node Similarity in a Euclidean Plane. Because a force-directed algorithm positions nodes in a Euclidean plane, it can use two degrees of freedom and continuous distances to more accurately represent inter-node similarity. For example, in a Euclidean plane, a node can have an identical relationship to many other nodes simultaneously. This feature enabled the rapid detection of node associations such as clusters in both networks, in addition to revealing the degree of similarity of nodes within each cluster in the case-only network. In contrast, heatmaps (Figures 3 and 5) position all nodes along a line either on the x- or y-axes at discrete distances determined by the widths of the rows and columns, which constrains how distance can be used to represent similarity between nodes. For example, a node can have an identical distance to a maximum of two nodes (one on either side), making it more difficult to accurately represent and comprehend complex inter- and intra-cluster relationships. Therefore, while heatmaps are useful for verifying patterns once identified, bipartite networks laid out in a Euclidean plane are more effective to support the process of discovery and inference of inter-node associations between and within clusters [35].
Representation of Subjects and Variables Using Two Sets of Nodes. Because we represented subjects and variables simultaneously in the network, they helped to comprehend inter and intra cluster relationships. This feature facilitated the inspection of which subject clusters were or were not strongly associated with which methylation clusters enabling inference of pathways.
Representation of Subject Type Using Node Color. Because we chose to distinguish cases and controls in the case-control network by coloring them red and green respectively, they enabled rapid detection of the composition of clusters. This feature resulted in the identification of a case-dominated cluster, and a control-dominated cluster in the case-control network.
Representation of Variable Values Using Continuous Edge Thickness. Because we chose to represent variable values as edge thicknesses, they enabled comprehension of the strength of associations within and across clusters. This feature enabled detection of hypo- and hypermethylation associations within and between clusters in both networks.
While each of the above representational features made specific contributions to the comprehension of the data, it is their simultaneous visualization which enabled the complex inference of the underlying biology. Therefore, while the network topology with two subject and two methylation site clusters looked deceptively simple, the combination of the above four representational features precipitated a plausible hypothesis of mechanisms and heterogeneity in PTB. Such a result would be difficult to derive if we had used only supervised methods such as univariable significance of the methylation sites, or by just analyzing a textual description of node membership in subject and methylation clusters.
The above process of comprehending visual patterns and inferring their meaning is based on well-known cognitive processes related to information visualization. Cognitively, visualizations such as subject-variable networks map multiple data elements to externalized visual representations. When this mapping to visual elements is aligned with cognitive principles [30, 56–59], the resulting visual representation enables comprehension of complex patterns because of two key cognitive processes: (1) The visual representation leverages the massively parallel architecture of the human visual system consisting of the eye and the visual cortex of the brain [56]. This parallel cognitive architecture enables the rapid comprehension of multiple graphical elements simultaneously, which often leads to insights about relationships in complex data such as similarities, trends, and anomalies [30]. (2) The externalized representation reduces working memory load needed to process the data [60], enabling the freed-up working memory to be used for higher-level processing such as the interpretation of patterns, requiring access of domain-knowledge in long-term memory.
Furthermore, while visualizations enable rapid comprehension of associations that are made explicit by the nodes, edges, and their properties, they also enable detection of implicit associations [61] resulting from the layout in an Euclidean plane including emergent multivariable patterns such as clusters. As demonstrated in the current analyses, these cognitive advantages conferred by appropriately designed subject-variable networks are critical for projects at early stages of discovery (such as the epigenetic analysis of PTB) as they enable complex reasoning about subjects and the variables. Often this process results in the discovery of novel multivariable patterns in the data [35–40], such as heterogeneity based on methylation differences, and hypotheses for their underlying mechanisms, an early but crucial step in the design of targeted interventions.
Conclusion
Although several studies have analyzed epigenetic changes in preterm, little is known about the mechanisms that trigger PTB. Here we demonstrated how bipartite networks revealed an inverse symmetrical relationship in the methylation profiles between PTB cases and controls, resulting in a complex but comprehensible hypothesis of the mechanisms precipitating PTB. Furthermore, the analysis revealed statistically significant heterogeneity within the methylation profiles of PTB cases, which is an early step towards the design of targeted interventions, a critical goal of precision medicine.
Although we don’t yet know how methylation affects the function of genes involved in PTB, our analysis suggests distinct mechanisms of PTB that involve the presence or absence of senescence. These pathways are most likely mediated by exposure to different risks which can impact methylation patterns leading to PTB. Bipartite network analyses therefore enabled us to derive data-driven hypotheses of pathways in PTB, which should be tested in future functional methylation studies. Our current research focuses on extending the subject-variable network analysis approach to process big datasets consisting of thousands of subjects and variables.
The limitation of this study is that our samples were derived from cord blood, and therefore the data cannot be used to establish causation based on fetal DNA methylation patterns at the time of birth. Accordingly, in our future work we will use maternal samples and prospective samples through which we will test the validity of our approach to further delineate cause and effect in patient subgroups, with the ultimate goal of developing targeted interventions to reduce the risk of preterm deliveries.
Acknowledgments
We thank Rohit Divekar for feedback and support on the analysis and results.
Funding: This study was funded in part by a Clinical and Translational Science Award (UL1TR000071) from the National Center for Advancing Translational Sciences, National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Footnotes
Data Availability: The data used in this study were extracted from epigenetic studies conducted by Dr. Menon (ram.menon@utmb.edu), and Dr. Smith (alicia.smith@emory.edu). These data can be obtained through email request.
Competing interests: The authors have declared that no competing interests exist.
Author Contributions
Conceived and designed the analysis: SKB BD RM. Extracted the data: RM VK. Analyzed the data: SKB BD VK AKS. Wrote the paper: SKB BD VK MC SV GS SEP AKS RM.
References
- 1.Beck S, Wojdyla D, Say L, Betran AP, Merialdi M, Requejo JH, et al. The worldwide incidence of preterm birth: a systematic review of maternal mortality and morbidity. Bulletin of the World Health Organization. 2010;88(1):31–8. doi: 10.2471/blt.08.062554. Epub 2010/04/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chmurzynska A. Fetal programming: link between early nutrition, DNA methylation, and complex diseases. Nutrition reviews. 2010;68(2):87–98. doi: 10.1111/j.1753-4887.2009.00265.x. Epub 2010/02/09. [DOI] [PubMed] [Google Scholar]
- 3.Barker DJ. The fetal and infant origins of adult disease. BMJ (Clinical research ed) 1990;301(6761):1111. doi: 10.1136/bmj.301.6761.1111. Epub 1990/11/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barker DJ, Gelow J, Thornburg K, Osmond C, Kajantie E, Eriksson JG. The early origins of chronic heart failure: impaired placental growth and initiation of insulin resistance in childhood. European journal of heart failure. 2010;12(8):819–25. doi: 10.1093/eurjhf/hfq069. Epub 2010/05/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Champagne FA, Curley JP. Epigenetic mechanisms mediating the long-term effects of maternal care on development. Neuroscience and biobehavioral reviews. 2009;33(4):593–600. doi: 10.1016/j.neubiorev.2007.10.009. Epub 2008/04/24. [DOI] [PubMed] [Google Scholar]
- 6.Burdge GC, Hanson MA, Slater-Jefferies JL, Lillycrop KA. Epigenetic regulation of transcription: a mechanism for inducing variations in phenotype (fetal programming) by differences in nutrition during early life? The British journal of nutrition. 2007;97(6):1036–46. doi: 10.1017/s0007114507682920. Epub 2007/03/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menon R. Spontaneous preterm birth, a clinical dilemma: etiologic, pathophysiologic and genetic heterogeneities and racial disparity. Acta obstetricia et gynecologica Scandinavica. 2008;87(6):590–600. doi: 10.1080/00016340802005126. Epub 2008/06/24. [DOI] [PubMed] [Google Scholar]
- 8.Koullali B, Oudijk MA, Nijman TA, Mol BW, Pajkrt E. Risk assessment and management to prevent preterm birth. Seminars in fetal & neonatal medicine. 2016;21(2):80–8. doi: 10.1016/j.siny.2016.01.005. Epub 2016/02/26. [DOI] [PubMed] [Google Scholar]
- 9.Mortensen LH, Helweg-Larsen K, Andersen AM. Socioeconomic differences in perinatal health and disease. Scandinavian journal of public health. 2011;39(7 Suppl):110–4. doi: 10.1177/1403494811405096. Epub 2011/08/04. [DOI] [PubMed] [Google Scholar]
- 10.Yee LM, Truong YN, Caughey AB, Cheng YW. The association between interdelivery interval and adverse perinatal outcomes in a diverse US population. Journal of perinatology : official journal of the California Perinatal Association. 2016 doi: 10.1038/jp.2016.54. Epub 2016/04/01. [DOI] [PubMed] [Google Scholar]
- 11.Romero R, Dey SK, Fisher SJ. Preterm labor: one syndrome, many causes. Science (New York, NY) 2014;345(6198):760–5. doi: 10.1126/science.1251816. Epub 2014/08/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coleman T, Chamberlain C, Davey MA, Cooper SE, Leonardi-Bee J. Pharmacological interventions for promoting smoking cessation during pregnancy. The Cochrane database of systematic reviews. 2012;9:Cd010078. doi: 10.1002/14651858.cd010078. Epub 2012/09/14. [DOI] [PubMed] [Google Scholar]
- 13.Messer LC, Kaufman JS, Mendola P, Laraia BA. Black-white preterm birth disparity: a marker of inequality. Annals of epidemiology. 2008;18(11):851–8. doi: 10.1016/j.annepidem.2008.06.007. Epub 2008/10/23. [DOI] [PubMed] [Google Scholar]
- 14.Behnia F, Parets SE, Kechichian T, Yin H, Dutta EH, Saade GR, et al. Fetal DNA methylation of autism spectrum disorders candidate genes: association with spontaneous preterm birth. American journal of obstetrics and gynecology. 2015;212(4):533e1-9. doi: 10.1016/j.ajog.2015.02.011. Epub 2015/02/18. [DOI] [PubMed] [Google Scholar]
- 15.Monangi NK, Brockway HM, House M, Zhang G, Muglia LJ. The genetics of preterm birth: Progress and promise. Seminars in perinatology. 2015;39(8):574–83. doi: 10.1053/j.semperi.2015.09.005. Epub 2015/10/16. [DOI] [PubMed] [Google Scholar]
- 16.Parets SE, Conneely KN, Kilaru V, Fortunato SJ, Syed TA, Saade G, et al. Fetal DNA Methylation Associates with Early Spontaneous Preterm Birth and Gestational Age. PloS one. 2013;8(6):e67489. doi: 10.1371/journal.pone.0067489. Epub 2013/07/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newman MEJ. Networks: An Introduction. Oxford United Kingdom: Oxford University Press; 2010. [Google Scholar]
- 18.Bhavnani SK, Dang B, Bellala G, Divekar R, Visweswaran S, Brasier A, et al. Unlocking proteomic heterogeneity in complex diseases through visual analytics. Proteomics. 2015 doi: 10.1002/pmic.201400451. Epub 2015/02/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lacy ME, Wellenius GA, Carnethon MR, Loucks EB, Carson AP, Luo X, et al. Racial Differences in the Performance of Existing Risk Prediction Models for Incident Type 2 Diabetes: The CARDIA Study. Diabetes care. 2015 doi: 10.2337/dc15-0509. Epub 2015/12/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baker JJ. Medicare payment system for hospital inpatients: diagnosis-related groups. Journal of health care finance. 2002;28(3):1–13. Epub 2002/06/25. [PubMed] [Google Scholar]
- 21.Lipkovich I, Dmitrienko A, Denne J, Enas G. Subgroup identification based on differential effect search–a recursive partitioning method for establishing response to treatment in patient subpopulations. Statistics in medicine. 2011;30(21):2601–21. doi: 10.1002/sim.4289. Epub 2011/07/26. [DOI] [PubMed] [Google Scholar]
- 22.Kehl V, Ulm K. Responder identification in clinical trials with censored data. Comput Stat Data Anal. 2006;50(5):1338–55. doi: 10.1016/j.csda.2004.11.015. [DOI] [Google Scholar]
- 23.Hastie T, Tibshirani R, Friedman J. The Elements of Statistical Learning. New York, NY, USA: Springer New York Inc.; 2001. [Google Scholar]
- 24.Fitzpatrick AM, Teague WG, Meyers DA, Peters SP, Li X, Li H, et al. Heterogeneity of severe asthma in childhood: confirmation by cluster analysis of children in the National Institutes of Health/National Heart, Lung, and Blood Institute Severe Asthma Research Program. The Journal of allergy and clinical immunology. 2011;127(2):3829e1–13. doi: 10.1016/j.jaci.2010.11.015. Epub 2011/01/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abu-jamous B, Fa R, Nandi AK. Integrative Cluster Analysis in Bioinformatics. 2015 [Google Scholar]
- 26.Lochner KA, Cox CS. Prevalence of multiple chronic conditions among Medicare beneficiaries, United States, 2010. Preventing chronic disease. 2013;10:E61. doi: 10.5888/pcd10.120137. Epub 2013/04/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shabalin AA, Weigman VJ, Perou CM, Nobel AB. Finding large average submatrices in high dimensional data. 2009:985–1012. doi: 10.1214/09-AOAS239. [DOI] [Google Scholar]
- 28.Odibat O, Reddy CK. Efficient Mining of Discriminative Co-clusters from Gene Expression Data. Knowledge and information systems. 2014;41(3):667–96. doi: 10.1007/s10115-013-0684-0. Epub 2015/02/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casanova R, Saldana S, Chew EY, Danis RP, Greven CM, Ambrosius WT. Application of Random Forests Methods to Diabetic Retinopathy Classification Analyses. PloS one. 2014;9(6):e98587. doi: 10.1371/journal.pone.0098587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomas JJ, Cook KA, editors. Illuminating the path: the R&D agenda for visual analytics. IEEE Press; 2005. [Google Scholar]
- 31.Cramer AO, Waldorp LJ, van der Maas HL, Borsboom D. Comorbidity: a network perspective. The Behavioral and brain sciences. 2010;33(2–3):137–50. doi: 10.1017/s0140525x09991567. discussion 50-93. Epub 2010/06/30. [DOI] [PubMed] [Google Scholar]
- 32.Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, et al. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic acids research. 2009;37(Database issue):D412–6. doi: 10.1093/nar/gkn760. Epub 2008/10/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Christakis NA, Fowler JH. Social Network Sensors for Early Detection of Contagious Outbreaks. PloS one. 2010;5(9):e12948. doi: 10.1371/journal.pone.0012948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamada T, Kawai S. An algorithm for drawing general undirected graphs. Information Processing Letters. 1989;31:7–15. [Google Scholar]
- 35.Bhavnani SK, Bellala G, Victor S, Bassler KE, Visweswaran S. The role of complementary bipartite visual analytical representations in the analysis of SNPs: a case study in ancestral informative markers. Journal of the American Medical Informatics Association : JAMIA. 2012;19(e1):e5–e12. doi: 10.1136/amiajnl-2011-000745. Epub 2012/06/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhavnani SK, Dang B, Caro M, Bellala G, Visweswaran S, Mejias A, et al. Heterogeneity within and across Pediatric Pulmonary Infections: From Bipartite Networks to At-Risk Subphenotypes. AMIA Joint Summits on Translational Science proceedings AMIA Summit on Translational Science. 2014;2014:29–34. Epub 2015/02/27. [PMC free article] [PubMed] [Google Scholar]
- 37.Bhavnani SK, Dang B, Visweswaran S, Divekar R, Tan A, Karmarkar A, et al. How Comorbidities Co-Occur in Readmitted Hip Fracture Patients: From Bipartite Networks to Insights for Post-Discharge Planning. AMIA Joint Summits on Translational Science proceedings AMIA Summit on Translational Science. 2015;2015:36–40. Epub 2015/08/26. [PMC free article] [PubMed] [Google Scholar]
- 38.Bhavnani SK, Drake J, Bellala G, Dang B, Peng BH, Oteo JA, et al. How Cytokines Co-occur across Rickettsioses Patients: From Bipartite Visual Analytics to Mechanistic Inferences of a Cytokine Storm. AMIA Joint Summits on Translational Science proceedings AMIA Summit on Translational Science. 2013;2013:15–9. Epub 2013/12/05. [PMC free article] [PubMed] [Google Scholar]
- 39.Bhavnani SK, Drake J, Divekar R. The role of visual analytics in asthma phenotyping and biomarker discovery. Advances in experimental medicine and biology. 2014;795:289–305. doi: 10.1007/978-1-4614-8603-9_18. Epub 2013/10/29. [DOI] [PubMed] [Google Scholar]
- 40.Bhavnani SK, Victor S, Calhoun WJ, Busse WW, Bleecker E, Castro M, et al. How cytokines co-occur across asthma patients: from bipartite network analysis to a molecular-based classification. Journal of biomedical informatics. 2011;44(Suppl 1):S24–30. doi: 10.1016/j.jbi.2011.09.006. Epub 2011/10/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson RA, Wichern DW, editors. Applied multivariate statistical analysis. Prentice-Hall, Inc; 1988. [Google Scholar]
- 42.Parets SE, Conneely KN, Kilaru V, Menon R, Smith AK. DNA methylation provides insight into intergenerational risk for preterm birth in African Americans. Epigenetics. 2015;10(9):784–92. doi: 10.1080/15592294.2015.1062964. Epub 2015/06/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Menon R, Velez DR, Simhan H, Ryckman K, Jiang L, Thorsen P, et al. Multilocus interactions at maternal tumor necrosis factor-alpha, tumor necrosis factor receptors, interleukin-6 and interleukin-6 receptor genes predict spontaneous preterm labor in European-American women. American journal of obstetrics and gynecology. 2006;194(6):1616–24. doi: 10.1016/j.ajog.2006.03.059. Epub 2006/05/30. [DOI] [PubMed] [Google Scholar]
- 44.Menon R, Williams SM, Fortunato SJ. Amniotic fluid interleukin-1beta and interleukin-8 concentrations: racial disparity in preterm birth. Reproductive sciences (Thousand Oaks, Calif) 2007;14(3):253–9. doi: 10.1177/1933719107301336. Epub 2007/07/20. [DOI] [PubMed] [Google Scholar]
- 45.Menon R, Fortunato SJ, Edwards DR, Williams SM. Association of genetic variants, ethnicity and preterm birth with amniotic fluid cytokine concentrations. Annals of human genetics. 2010;74(2):165–83. doi: 10.1111/j.1469-1809.2010.00562.x. Epub 2010/04/08. [DOI] [PubMed] [Google Scholar]
- 46.Menon R, Pearce B, Velez DR, Merialdi M, Williams SM, Fortunato SJ, et al. Racial disparity in pathophysiologic pathways of preterm birth based on genetic variants. Reproductive biology and endocrinology : RB&E. 2009;7:62. doi: 10.1186/1477-7827-7-62. Epub 2009/06/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Menon R, Velez DR, Morgan N, Lombardi SJ, Fortunato SJ, Williams SM. Genetic regulation of amniotic fluid TNF-alpha and soluble TNF receptor concentrations affected by race and preterm birth. Human genetics. 2008;124(3):243–53. doi: 10.1007/s00439-008-0547-z. Epub 2008/09/23. [DOI] [PubMed] [Google Scholar]
- 48.Velez DR, Fortunato SJ, Thorsen P, Lombardi SJ, Williams SM, Menon R. Preterm birth in Caucasians is associated with coagulation and inflammation pathway gene variants. PloS one. 2008;3(9):e3283. doi: 10.1371/journal.pone.0003283. Epub 2008/09/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Velez DR, Fortunato SJ, Morgan N, Edwards TL, Lombardi SJ, Williams SM, et al. Patterns of cytokine profiles differ with pregnancy outcome and ethnicity. Human reproduction (Oxford, England) 2008;23(8):1902–9. doi: 10.1093/humrep/den170. Epub 2008/05/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fortunato SJ, Menon R, Velez DR, Thorsen P, Williams SM. Racial disparity in maternal-fetal genetic epistasis in spontaneous preterm birth. American journal of obstetrics and gynecology. 2008;198(6):666e1–9. doi: 10.1016/j.ajog.2008.02.003. discussion .e9-10. Epub 2008/06/10. [DOI] [PubMed] [Google Scholar]
- 51.Nooy W, Mrvar A, Batagelj V. Exploratory Social Network Analysis with Pajek 2nd ed. Cambridge University Press; 2011. [Google Scholar]
- 52.Menon R, Behnia F, Polettini J, Saade GR, Campisi J, Velarde M. Placental membrane aging and HMGB1 signaling associated with human parturition. Aging. 2016;8(2):216–30. doi: 10.18632/aging.100891. Epub 2016/02/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Behnia F, Taylor BD, Woodson M, Kacerovsky M, Hawkins H, Fortunato SJ, et al. Chorioamniotic membrane senescence: a signal for parturition? American journal of obstetrics and gynecology. 2015;213(3):359.e1–16. doi: 10.1016/j.ajog.2015.05.041. Epub 2015/05/31. [DOI] [PubMed] [Google Scholar]
- 54.Dutta EH, Behnia F, Boldogh I, Saade GR, Taylor BD, Kacerovsky M, et al. Oxidative stress damage-associated molecular signaling pathways differentiate spontaneous preterm birth and preterm premature rupture of the membranes. Molecular human reproduction. 2016;22(2):143–57. doi: 10.1093/molehr/gav074. Epub 2015/12/23. [DOI] [PubMed] [Google Scholar]
- 55.Menon R, Yu J, Basanta-Henry P, Brou L, Berga SL, Fortunato SJ, et al. Short fetal leukocyte telomere length and preterm prelabor rupture of the membranes. PloS one. 2012;7(2):e31136. doi: 10.1371/journal.pone.0031136. Epub 2012/02/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Readings in information visualization: using vision to think. Morgan Kaufmann Publishers Inc; 1999. [Google Scholar]
- 57.Halford GS, Baker R, McCredden JE, Bain JD. How many variables can humans process? Psychological science. 2005;16(1):70–6. doi: 10.1111/j.0956-7976.2005.00782.x. Epub 2005/01/22. [DOI] [PubMed] [Google Scholar]
- 58.Wagemans J, Elder JH, Kubovy M, Palmer SE, Peterson MA, Singh M, et al. A Century of Gestalt Psychology in Visual Perception I. Perceptual Grouping and Figure-Ground Organization. Psychological bulletin. 2012;138(6):1172–217. doi: 10.1037/a0029333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tufte ER. The visual display of quantitative information. Graphics Press; 1986. p. 197. [Google Scholar]
- 60.Zhang JA. ND. Representations in distributed cognitive tasks. Cognitive Science. 1994;18:87–122. [Google Scholar]
- 61.JH L, HA S. Why a Diagram is (Sometimes) Worth Ten Thousand Words. Cognitive Science. 1987;11:65–9. [Google Scholar]