

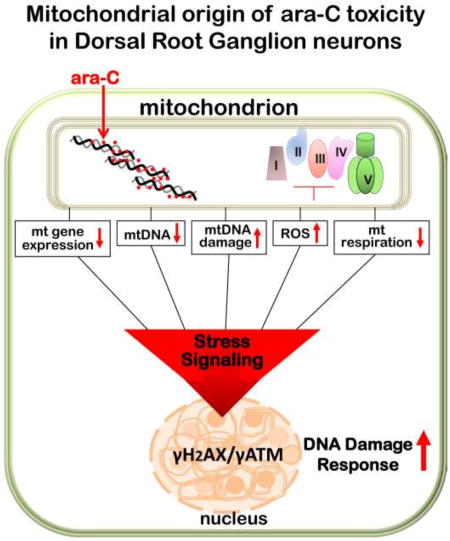
Abstract
Peripheral Nervous System (PNS) neurotoxicity caused by cancer drugs hinders attainment of chemotherapy goals. Due to leakiness of the blood nerve barrier, circulating chemotherapeutic drugs reach PNS neurons and adversely affect their function. Chemotherapeutic drugs are designed to target dividing cancer cells and mechanisms underlying their toxicity in postmitotic neurons remain to be fully clarified. The objective of this work was to elucidate progression of events triggered by antimitotic drugs in postmitotic neurons. For proof of mechanism study, we chose cytarabine (ara-C), an antimetabolite used in treatment of hematological cancers. Ara-C is a cytosine analog that terminates DNA synthesis. To investigate how ara-C affects postmitotic neurons, which replicate mitochondrial but not genomic DNA, we adapted a model of Dorsal Root Ganglion (DRG) neurons. We showed that DNA polymerase γ, which is responsible for mtDNA synthesis, is inhibited by ara-C and that sublethal ara-C exposure of DRG neurons leads to reduction in mtDNA content, ROS generation, oxidative mtDNA damage formation, compromised mitochondrial respiration and diminution of NADPH and GSH stores, as well as, activation of the DNA damage response. Hence, it is plausible that in ara-C exposed DRG neurons, ROS amplified by the high mitochondrial content shifts from physiologic to pathologic levels signaling stress to the nucleus. Combined, the findings suggest that ara-C neurotoxicity in DRG neurons originates in mitochondria and that continuous mtDNA synthesis and reliance on oxidative phosphorylation for energy needs sensitize the highly metabolic neurons to injury by mtDNA synthesis terminating cancer drugs.
Keywords: mitochondria, mtDNA, DNA polymerase γ, DNA damage response, dorsal root ganglion neurons, neurotoxicity, cytarabine (ara-C)
Graphical abstract
1. INTRODUCTION
Peripheral nervous system neurotoxicities incidental to cancer therapies hinder effective drug dosing and compromise efficacy of treatments [1–3]. To address this issue, the mechanisms by which antimitotic drugs injure postmitotic neurons need to be clarified. To this end, we developed an in vitro model of anti cancer drug toxicity to the peripheral nervous system, using cultured dorsal root ganglion (DRG) neurons. Unlike the central nervous system neurons, DRG neurons are not protected by tight blood barrier and readily accessible to circulating compounds. For a proof of mechanism study, we chose the chemotherapeutic drug cytarabine (ara-C), a cytosine analog, which works as ‘chain terminator’ of DNA synthesis and leads to blockade of cancer cell proliferation [4–7]. Ara-C serves as first line chemotherapy in treatment of acute myeloid leukemia as well as other hematological cancers [6, 8]. While ara-C mode of action in proliferating cells is well understood [9, 10] and germane to other classes of antimitotic drugs which also hinder DNA synthesis [11, 12], limited work was done to address mechanisms underlying adverse effects of ara-C in postmitotic cells, which do not replicate DNA, and particularly in the context of the peripheral nervous system [13–15]. To act as effective ‘chain terminator’ ara-C must first be incorporated into DNA. While postmitotic neurons do not replicate their genomic DNA, they continuously replicate mtDNA in support of vigorous mitochondrial biogenesis necessitated by high-energy demands in highly metabolic neurons.
Here we investigated whether features inherent to DRG neurons, namely their high mitochondrial content, reliance on mitochondria for energy needs and limited shielding from circulating compounds by the blood nerve barrier [16], make DRGs particularly sensitive targets for chemotherapeutic drugs. We show in vitro that the mitochondrial DNA polymerase γ, which is responsible for mtDNA synthesis [17], is strongly inhibited by incorporated ara-C suggesting that in vivo induced ara-C DRG toxicity might be initiated by impediment of mtDNA synthesis [18] and resultant impairments of mitochondrial function. We found that in DRG neurons, sublethal ara-C exposure leads to increased production of reactive oxygen species (ROS), formation of oxidative adducts in mtDNA, reduction in mtDNA copy number and mitochondrial content, as well as, compromised mitochondrial respiration. Recent evidence supports the notion of retrograde cross talk from mitochondria to nucleus that involves stress signaling and activation of the DNA damage response [19, 20]. Elevated mitochondrial ROS has been previously implicated in induction of oxidative DNA damage and genomic instability [21, 22]. Here, consistently with this scenario, we observe chromatin modifications detectable by immunoreactivity of phosphorylated forms of the histone variant H2AX (γH2AX) and of ataxia-telangiectasia mutated (ATM), a PI3-family kinase [23, 24]. Phosphorylated γH2AX and γATM proteins are involved in oxidative stress and damage signaling and nuclear foci containing the phosphorylated forms of these proteins are considered biomarkers of modifications that facilitate chromatin decondensation and activation of the DNA damage response (DDR). Considered together, our findings support causal link between impediment of mtDNA synthesis, resultant respiratory compromise and subsequent activation of the DNA damage response in DRG neurons exposed to sublethal ara-C treatment.
2. METHODS
2.1. Culture and treatment of mouse Dorsal Root Ganglion (DRG) neurons
The University of Texas Medical Branch Institutional Animal Care and Use Committee approved all mouse-handling procedures. Dorsal root ganglion neurons were isolated from 3–4 months old C57BL/6 male mice (Envigo Laboratories, USA) according to established protocols [25–27] and as we described [28–30]. Briefly, ganglia were collected from all spinal levels, placed in cold dissecting solution (130 mM NaCl, 5 mM KCl, 2 mM KH2PO4, 1.5 mM CaCl2, 6 mM MgCl2, 10 mM glucose and 10 mM Hepes, pH 7.2), incubated with collagenase A (Roche) and trypsin (1 h/37°C) followed by DNase I (Roche), dissociated by 20 gentle triturations and spun (175 g/3 min). Pellets were passed through 70-µm strainer and re-suspended in DMEM/F12 (Sigma) with 10% FBS, 10 ng/ml nerve growth factor (Sigma) and penicillin/streptomycin. Cells were seeded on pre-coated (10 µg/ml laminin and 100 µg/ml poly-L-ornithine, Sigma) glass coverslips or wells at (4–5)×103 and 3×104/cm2, respectively. Treatments were initiated 24 h post seeding when neurite network has been established. Cytarabine (ara-C) purchased from MP Biomedicals (Solon, OH) was dissolved in DMSO and stored as 10 mM stock solution at −20°C. Stock was diluted 1:10 with complete culture medium prior to supplementation at 50 µM for indicated incubation times.
2.2. Immunofluorescent staining
DRG neurons seeded on coverslips were treated as indicated and fixed in 4% paraformaldehyde as we described [28–30]. Cells were permeabilized with 0.1% Triton X-100/0.1% sodium citrate in PBS and incubated with 3% BSA (w/v)/1% Donkey serum (v/v) in PBS. Primary antibodies were: rabbit anti-Neurofilament 200 (1:20,000, Sigma-N4142); mouse anti-COX1 (1:1500, Millipore-459600); mouse anti-8-oxoguanine (1:200, Millipore #MAB3560); rabbit anti-TFAM (1:2000, Genetex #GTX103231); mouse anti γH2AX (1:4,000, Millipore #05-636) or rabbit anti γH2AX (1:1000, Santa Cruz SC-101696) and mouse anti-ATM phosphorylated at serine 1981 (1:1500, Cell Signaling #4526L). For detection of 8-oxoG immunoreactivity, a published protocol [31] was used as follows: Coverslips were fixed in 100% methanol (−20°C), pretreated for one hour with RNase A (100 µg/ml), denatured with 50 mM NaOH (10 min) followed by 3× 3 min washes in PBS and incubation with primary antibody. Subsequently, coverslips were washed 3× with 1% BSA and incubated 45 min with goat-anti mouse 488 and goat anti-rabbit 594 AlexaFluor secondary antibodies, mounted with Prolong® Gold Anti-fade with DAPI and viewed with 40× objective on Olympus IX71 with QIC-F-M-12-C cooled camera (QImaging, Surrey, BC) and QCapture Pro (QImaging) software.
2.3. In situ imaging of superoxide mediated dihydroethidium oxidation
Changes in superoxide levels were assessed in situ, in live DRG neurons by imaging superoxide-mediated oxidation of dihydroethidium (#D23107, Invitrogen) to 2-hydroxyethidium [32, 33] and as we previously described [28, 29, 34]. Briefly, upon termination of ara-C exposure, dihydroethidium was added at final concentration of 100 nM for 20 min in the dark. Incubation was terminated by quick washes with PBS and coverslips were mounted with DAPI containing mounting medium. Coverslips were observed with Olympus IX71 fluorescence microscope and images were captured sequentially with QIC-F-M-12-C cooled camera fitted with the QCapture Pro software. Fluorescence intensity of individual DRG neurons demarcated by circular boundaries was scored with ImageJ software (NIH) and exported to Excel for determination of average intensity and further analysis. At least 30 DRG neurons obtained in 3 independent experimental sets were scored for each condition.
2.4. mtDNA copy number determination by Real-Time quantitative PCR
Total DNA was isolated from ~3×104 DRG neurons using easy DNA isolation kit (Life Technologies) and RNA was digested with RNase A (40 µg/ml/30 min/37°C). Real-time qPCR reactions were assembled in duplicates with 10 ng total DNA and SSO FAST Evagreen supermix (Biorad, Hercules, CA). The single-copy nuclear gene beta-2-microglobulin (B2M) was used as reference for determination of mtDNA copy number. A 180-nucleotide fragment of mitochondrial gene encoding cytochrome oxidase subunit 3 (COX3) was amplified after validation of qPCR data by targeting two additional mitochondria encoded genes, COX1 and ND1 that yielded similar results. mtDNA copy number was calculated using the formula: mtDNA copy number =2×2(CTCoxIII − CTB2M) [28, 35, 36]. Primers for COX3 (ID 17710) were F-caattacatgagctcatcatagc and R-ccatggaatccagtagcca and for B2M (NM_009735) F-atccaaatgctgaagaacgg and R-atcagtctcagtgggggtga. Three sets of independent experiments were carried out and mean±SEM values for mtDNA copy number were calculated.
2.5. Measurement of mitochondrial gene expression by Real-Time quantitative PCR
Total cellular RNA was isolated from cultured DRG neurons (4×104/3.5 cm dish) using RNeasy plus mini kit (Qiagen) and reverse transcribed with iScript RT supermix (Biorad), which contains random as well as oligo dT primers. Real-time PCR was done with CFX96 Real-Time System (Biorad) as we described [28–30]. B2M and 18S gene transcripts were used as internal controls. PCR reactions were assembled in duplicates with SSO FAST Evagreen supermix (Biorad). PCR program was: 95°C 2 min, 40 cycles of 95°C 5 sec, 55°C 15 sec. Data represent averages of at least 3 sets of independent biological experiments. The relative amount of target gene RNA was calculated as described [37] using the formula: −ΔΔCt=[(CT gene of interest − CT internal control) sample − (CT gene of interest − CT internal control) control]. Primer sequences are listed in Table 1.
Table 1.
IDs and sequences of mouse primers.
| Gene Symbol |
Sequence | Accession Number | Gene Name | |
|---|---|---|---|---|
|
| ||||
| Forward | Reverse | |||
| COX1 | cagaccgcaacctaaacaca | ttctgggtgcccaaagaat | JF286601 | cytochrome c oxidase I |
| COX3 | caattacatgagctcatcatagc | ccatggaatccagtagcca | ID 17710 | cytochrome c oxidase III |
| ND1 | Catgatctaggaggctgctgac | cgtttaccttctataaggctatga | ID 17716 | NADH dehydrogenase 1 |
| B2M | atccaaatgctgaagaacgg | atcagtctcagtgggggtga | NM_009735 | Beta 2 macroglobulin |
| 18S | gtaacccgttgaaccccatt | ccatccaatcggtagtagcg | NR_003278.3 | 18S ribosomal RNA |
2.6. Measurement of oxygen consumption rates (OCR)
Oxygen consumption rates (OCR) were measured using XF24 extracellular flux analyzer (Seahorse, Agilent) according to established protocols [38–40] and as we previously described [28, 29, 41]. DRG neurons were seeded in XF24 plates ([0.8–1]×104/well) and cultured as above. Culture medium was replaced with un-buffered Dulbecco’s Modified Eagle’s medium (Sigma, D5030) supplemented with 2 mM pyruvate/15 mM glucose/2 mM GlutaMAX (#35050, Invitrogen) adjusted to pH 7.4 and equilibrated in CO2 free incubator at 37°C. Sequential additions of mitochondrial effectors were through ports of XF24 cartridges; effectors concentrations were optimized previously for DRG neurons to 2 µM oligomycin (O4876, Sigma), 2 µM carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), (C2920, Sigma) and 1.8 µM antimycin A (A8674, Sigma). Changes in OCR invoked by injections of mitochondrial effectors served to compare respiratory parameters between control and test groups [38, 39]. The addition of 25 mM 2-deoxyglucose (2DG) serves to demonstrate that in control cultures inhibition of the glycolytic pathway leads to a small increase in OCR consistent with compensatory utilization of pyruvate or glutamine to fuel mitochondrial oxygen consumption. Baseline OCR was calculated by subtraction of non-mitochondrial OCR, i.e., oxygen consumption retained after the addition of mitochondrial respiratory complex lll inhibitor, antimycin A. Each parameter calculated for control cultures was assigned the value of 100%; effects of treatment were calculated as percent change relative to each respective control. Data are presented as mean±SEM of four independent biological experiments.
2.7. DNA polymerase γ activity assays
Extension and bypass reactions catalyzed by mitochondrial DNA polymerase γ were assembled with 5’-end labeled annealed duplex oligonucleotide substrates containing a single cytarabine (ara-C) residue either at the 3’-end of the extended strand or in the template strand opposite incoming nucleotide, respectively. Extension substrate was prepared by annealing the 5’-end labeled ara-C containing 21-oligonucleotide (5’-ATT ACG AAT GCC CAC ACC GC[ara-C]-3’) with complementary 26-mer oligonucleotide (3’-TAA TGC TTA CGG GTG TGG CGG CGT GA-5’) and the bypass substrate by annealing the 5’-end labeled 21-mer (5’-ATT ACG AAT GCC CAC ACC GCC-3’) with template 26-mer containing single ara-C residue at position 22 (3’-TAA TGC TTA CGG GTG TGG CGG [ara-C]GT GA-5’). Control substrates were assembled with identical oligonucleotides carrying non-modified cytosine residues. Reactions were assembled with 20 nM recombinant human DNA polymerase γ (#85, EnzyMax, LLC), end-labeled duplex substrate and 5 µM dGTP in 20 µl reaction buffer (50 mM Tris–HCl pH 8.0, 10 mM KCl, 10 mM MgCl2, 1 mM DTT, 10% glycerol and 5 µg BSA) and incubated at 37°C for 30–120 min. At indicated time points reactions were terminated with loading buffer (96% formamide, 20 mM EDTA, pH 8, 0.025% bromophenol blue and 0.025% xylene cyanol) and heated at 95°C for 5 min prior to loading. Reaction aliquots were resolved in 14% polyacrylamide, 7 M urea gels in Tris-Borate buffer, pH 8.3 at 14 mA; products were visualized by autoradiography and quantified by a Phosphorimager as we described [41–43]. Reaction yields generated in at least three independent experiments were quantified and values were used to calculate mean±SEM yields of the [+1] product.
2.8. Measurement of NADP+/NADPH and GSH/GSSG ratios
Intracellular NADP+/NADPH ratios were measured using NADP+/NADPH-Glo™ kit (#G9081, Promega) according to the manufacturer. DRGs were seeded in 96-well plates. Following treatment, DRGs were lysed in NADP+ or NADPH lysis buffer (60°C/15 min) and spun at 13,000 g. Supernatants were loaded into white wall 96-well plate, mixed with detection reagent and incubated in the dark (22°C/30 min). NADP+ and NADPH concentrations were derived from standard curves (Magellan™ software, TECAN, San Jose, CA). Measurements of total and reduced glutathione levels were carried out using the GSH/GSSG-Glo™ luminescence based kit (V6611, Promega™) following manufacturer’s guide using 96-well white wall-plate format. Three sets of independent experiments were carried out and luminescence was read on TECAN FL200 plate reader.
2.9 Statistical analyses
Data are given as mean±SEM obtained from 3–4 independent biological experiments, as indicated. Student’s t-test was employed to compare the means among groups followed by post-test Tukey’s analysis to determine differences in means of multiple groups or as indicated. P<0.05 was considered statistically significant. MegaStat® package for Excel was used.
3. RESULTS
3.1. Ara-C inhibits DNA polymerase γ (pol γ) catalyzed DNA synthesis
To examine the effects of incorporated ara-C residues on mtDNA transactions, DNA pol γ-catalyzed extension and bypass synthesis, as well as exonuclease activity were examined using accordingly designed ara-C residue-containing DNA substrates [Fig. 1]. The 5’-end labeled oligonucleotide duplex substrates containing a single ara-C residue either at the 3’-end of the extended strand or in the template strand opposite the incoming nucleotide were assembled with recombinant pol γ and the templated dGTP, as we previously described [41–43]. One-nucleotide extension reactions on non-modified substrates yielded [+1] products in a time dependent manner [Fig. 1, lanes 2–4 and 8–10]. Formation of [+1] product was abolished on substrate containing an ara-C residue at the 3’-end [Fig. 1, lanes 5–7], likewise, the 3’ to 5’ exonuclease proofreading activity of pol γ (yielding the [−1] product) was eliminated on the 3’-ara-C containing substrate [lanes 5–7]. Similarly, the bypass reaction on template strand containing an ara-C residue opposite the incoming nucleotide was blocked [Fig. 1, lanes 11–13]. In combination, the data demonstrate inability of pol γ to eliminate incorporated ara-C and overcome the ara-C-imposed impediment of mtDNA synthesis.
Figure 1. mtDNA polymerase γ activity is inhibited by the cytosine analog ara-C.

Reactions were assembled with end-labeled oligonucleotide substrates, dGTP and recombinant pol γ. (A) Substrate design is shown above the relevant lanes; * indicates 5’-end label; R - indicates ara-C residue. The one nucleotide extension [+1] product generated on non-modified substrate [lanes 2–4] is abolished on 3’-ara-C containing substrate [lanes 5–7]. Polymerase γ 3’-exonuclease activity [−1 product, lanes 3–4] is also abolished on 3’-ara-C containing substrate [lanes 5–7]. Likewise bypass extension reaction opposite ara-C residue incorporated in the template strand is inhibited [lanes 11–13]. 21-mer substrate (S) [lane #1] and the 20-mer and 22-mer products [P] are indicated. (B) Yields of [+1] extension products generated by pol γ on modified and non-modified substrates are presented as percent mean±SEM of 3 reactions sets. ***P<0.001 indicates different from either extension or bypass reaction assembled with non-modified substrates.
3.2. Evidence for ROS formation and induction of oxidative mtDNA damage in ara-C treated DRG neurons
We used in situ detection of superoxide-mediated oxidation as surrogate for monitoring changes in ROS levels in DRG neurons incubated with ara-C [Fig. 2]. Superoxide levels were assessed by imaging superoxide-mediated oxidation of dihydroethidium to 2-hydroxyethidium, observed as red fluorescence as we previously described [28, 29, 34]. Increases in dihydroethidium oxidation following incubation in the presence of 50 µM ara-C, were observed as intensification of red fluorescence in DRGs somas; fluorescence intensity normalized to cell surface area was quantified by ImageJ [Fig. 2].
Figure 2. Ara-c exposure increases ROS in DRG neurons.

In situ imaging of superoxide-mediated oxidation of dihydroethidium to 2-hydroxyethidium (2-HE) in ara-C exposed DRG neurons. Representative images: 2-HE is observed as red fluorescence and nuclei stain blue with DAPI; scale bar = 10 µm. Fluorescence intensity normalized to soma surface area and quantified by ImageJ, is presented as mean±SEM of three biological experiments; in each set #8 neurons were sequentially scored per each condition; *P<0.05.
Detection of elevated ROS levels in ara-C treated DRG neurons, prompted us to ask whether mtDNA might be oxidatively damaged under these conditions. Because guanine is the most readily oxidizable DNA base, formation of 8-oxoguanine, an oxidized form of guanine was assessed as surrogate for oxidative damage to mtDNA [Fig. 3]. For immunofluorescent detection of 8-oxoguanine (8-oxoG) in mtDNA coverslips were pre-treated with RNase A and pre-incubated with NaOH. Cytoplasmic 8-oxoG IF was observed in green and large DRG neurons were identified by the DRG specific cytoskeleton neurofilament 200 (NF200) protein [red] in somas and neurites [Fig. 3A]. Green 8-oxoG IF was observed also in small DRG neurons, which do not react with NF200. Significant 8-oxoG immunoreactivity in majority of DRG neurons was observed by 16 h and persisted during the 48 h ara-C incubation period [Fig 3B]. To ascertain that the observed 8-oxoG immunoreactivity co-localizes with mtDNA, coverslips were double stained for 8-oxoG and mitochondrial transcription factor A (TFAM), which binds to mtDNA. TFAM immunoreactivity [Fig 3C, red] largely coincided with 8-oxoG IF (green), localizing 8-oxoG immunoreactivity to mtDNA [Fig 3C, yellow, merged].
Figure 3. Detection of oxidative DNA damage in mtDNA of ara-C exposed DRG neurons.

Representative images show immunofluorescence of 8-oxoguanine (8-oxoG) in DRG neurons: (A) In DRG cultures exposed to ara-C for 24 h cytoplasmic punctate 8-oxoG immunoreactivity is observed (green); DRG neurons are identified by immunoreactivity of NF200 (red). 8-oxoG IF is observed also in small DRG neurons, which do not react with anti NF200 antibody (center panel, green; scale bar = 10 µm). (B) Bar graph shows percent of 8-oxoG positive DRG neurons as a function of exposure time. Values represent mean+SEM for four biological experiments. (C) Representative images of DRG neurons double stained with antibodies reacting with 8-oxoG (green) and mtDNA-binding mitochondrial transcription factor A (TFAM) (red). In ara-C exposed DRG neurons, TFAM largely co-localizes with the punctate green 8-oxoG immunofluorescence (merged image).
3.3. Evidence for mitochondrial content reduction, depletion of mtDNA and decreased mitochondrial gene expression in ara-C exposed DRG neurons
Next, mitochondrial distribution pattern in DRG neurons was evaluated by immunofluorescence (IF) of the mitochondria encoded mitochondrial protein, cytochrome oxidase subunit 1 (COX1) [Fig. 4A, green]. DRG neurons are identified by the DRG specific NF200 cytoskeleton protein [red]. Following 48-h ara-C exposure, COX1 intensity is diminished showing an aggregated pattern suggestive of reduced mitochondrial content [Fig. 4A, green, arrowhead]. This observation was corroborated by temporal depletion of mtDNA measured using Real-Time (RT)-qPCR analyses, which revealed ~20% and 50% reduction in mtDNA copy number after 48 h and 72 h exposure to 50 µM ara-C, respectively [Fig. 4B]. In parallel experiments, RT-qPCR assays revealed ~20% and ~50% reduction in the expression of mitochondria encoded genes, ND1, COX1 and COX3, after 48 h and 72 h exposures to ara-C [Fig. 4C].
Figure 4. Depletion of mitochondrial content, reduction of mtDNA copy number and reduced expression of mtDNA encoded genes in ara-C exposed DRG neurons.

(A) Following ara-C exposure IF of mitochondrial cytochrome oxidase 1 (COX1) showed reduced signal and aggregated distribution in soma of DRG neurons (green, arrowhead); DRGs are identified by NF200 (red), scale bar = 10 µm. (B) Reduced mtDNA copy number in DRG neurons following ara-C exposure. (C) Expression of mitochondria encoded genes was reduced in the course of ara-C exposure. Bars show changes relative to respective controls. Values represent mean±SEM for four biological experiments; *P<0.05 versus control.
3.4. Ara-C exposure compromises mitochondrial respiration in DRG neurons
To examine whether ara-C mediated compromise of mtDNA in DRG neurons affects mitochondrial function, mitochondrial respiration was assessed using the Seahorse XF24 extracellular flux analyzer. Analyses revealed adverse changes in mitochondrial respiratory parameters following a 48 h exposure to 50 µM ara-C [Fig. 5]. Mitochondrial respiratory profiles in control DRG cultures (black) show baseline OCR of ~180 pmoles O2/min/8×103 DRG neurons. Sequential, in port additions of mitochondrial effectors (vertical arrows) reveal that in DRGs under normal conditions nearly 90% of O2 consumption feeds mitochondrial respiration: of this baseline OCR, ~70% support ATP synthesis, with ~30% lost to proton leak. Spare respiratory capacity (SRC), which is defined as FCCP-induced increase over baseline OCR and considered an indicator of mitochondrial respiratory capacity, was calculated as about 250% for control cultures. As expected this steep increase in OCR following FCCP addition to control cultures, was reflected in a drastic reduction of oxygen tension in culture wells [Fig. 5A, bottom graph]. Interestingly, the addition of 2-deoxyglucose an inhibitor of the glycolytic pathway, results in a slight increase in OCR, suggestive of compensatory augmentation of ATP production via mitochondrial oxidative phosphorylation fueled by pyruvate and/or glutamine. This response to 2DG is diminished in the ara-C exposed DRGs, plausibly due to mitochondrial compromise. The respiratory profile of DRG neurons was markedly changed following exposure to ara-C. In exposed cultures baseline OCR [Fig. 5a, red] dropped by ~30% versus control. A nearly 50% reduction in maximal respiration, as well as ~30% decrease in spare respiratory capacity (SRC) were measured [Fig. 5B]. These changes were concomitant with a ~20% increase in proton leak when compared to control cultures. In sum, the XF24 implemented ‘mitochondrial stress test’ revealed that the inherently high spare respiratory capacity of DRG neurons, which under normal conditions helps meet changing metabolic demands, was significantly compromised by ara-C exposures.
Figure 5. Ara-C exposure compromises mitochondrial respiration and decreases NADPH and GSH levels in DRG neurons.

(A) Respiratory profile for non-treated control DRG neurons (black) shows baseline OCR of ~180 pmoles O2/min. Sequential, in port additions of mitochondrial effectors (vertical arrows), reveal that ~90% of total oxygen consumption feed mitochondrial respiration: of this OCR ~70% support ATP synthesis and ~30% is lost to proton leak. Spare respiratory capacity (SRC), i.e., FCCP-induced increase in OCR over the baseline is ~250% for control DRGs. Following ara-C exposure baseline OCR (red) drops by ~30% and maximal respiration by nearly 50% (FCCP-induced), with ~30% reduction in SRC and ~ 20% increase in OCR feeding the proton leak. (Bottom) Oxygen tension in control wells is reduced by the addition of FCCP and even further by 2-deoxyglucose (2DG). Oxygen tension is reduced to a lesser extent in ara-C exposed DRGs, reflecting diminished mitochondrial OCR. (B) Each parameter calculated for control cultures was assigned the value of 100%; effects of treatment were calculated as percent change relative to each respective control. Data are presented as mean±SEM of four independent biological experiments. (C) NADPH levels are decreased and NADP+/NADPH ratios increased by ara-C. Concomitant addition of 6AN increased these effects. Data are mean±SEM for 3 independent experiments; * different from control; **different from ara-C; P< 0.05. (D) GSH levels were reduced by ara-C and further depleted by 6AN. Data are given as mean±SEM calculated from 3 independent experiments; * different from control; **different from ara-C; P< 0.05
3.5 NADPH and GSH stores are reduced by ara-C exposure in DRG neurons
To determine to what extent ara-C induced oxidative stress and mitochondrial compromise are associated with unfavorable shift in NADP+/NADPH ratio, NADP+ and NADPH levels were measured in cultures following 48-h exposure to ara-C [Fig. 5C]. Exposure decreased NADPH levels by ~35%, indicative of diminished antioxidant capacity in the ara-C challenged DRG neurons [Fig. 5C]. As expected, supplementation of 6-aminonicotinamide (6AN), a competitive inhibitor of glucose-6-phosphate dehydrogenase and 6-phosphogluconate, the first two enzymes of the oxidative branch of pentose phosphate pathway [44, 45], further diminished NADPH levels and increased NADP+/NADPH ratio. The glutathione system that uses NADPH to maintain reduced glutathione stores was similarly affected [Fig. 5D]. GSH levels were depleted and GSH/GSSG ratio was reduced with further reduction in the presence of 6AN, consistently with involvement of the oxidative branch of pentose phosphate pathway in generation of reducing equivalents in DRG neurons.
3.6 Nuclear γH2AX/γATM foci are temporally induced in DRG neurons by ara-C exposure and partially cleared in the course of post ara-C recovery
To determine to what extent ara-C induced mitochondrial compromise might be accompanied by nuclear manifestations, we monitored immunoreactivity of phosphorylated histone variant γH2AX, which in its phosphorylated form enables chromatin rearrangements that facilitate the DNA damage response. Following 24 h ara-C exposure, nuclear γH2AX foci were detected in ~35% of DRG neurons. Foci density and the number of γH2AX positive DRGs increased to nearly 75% by 48 h and to ~95% by 72 h of ara-C exposure [Fig. 6A, green]. DRG neurons are identified by immunoreactivity of the cytoskeleton protein NF200 (red). Ara-c induced nuclear γH2AX foci (green) show relatively low density at 24 h with increasing foci density in time dependent manner [Fig. 6A & B]. Formation of γATM positive foci, which largely coincided with γH2AX [Fig. 7, green], was also observed in the course of ara-C exposure. To assess colocalization of γH2AX and γATM positive foci and the extent of their clearance in the course of recovery, after 48 h incubation in the presence of ara-C, culture media were replaced and DRG neurons were let recover for 24, 48, 72 or 96 h. In the course of recovery, nuclear foci density gradually decreased with just few large foci per nucleus detected after 48 h recovery with continued reduction in nuclear foci number throughout the 96 h recovery period [Fig. 7A, merge]. Interestingly, nuclear γH2AX and γATM foci co-localized not only during exposure but also in the course of post ara-C recovery [Fig. 7A & C], plausibly reflecting ongoing nuclear DNA repair processes en route to containment of the remaining DNA damage within a few larger chromatin domains, with γH2AX/γATM co-localization retained.
Figure 6. Temporal induction of nuclear γH2AX foci in ara-C exposed DRG neurons.

Representative images of time dependent formation of nuclear γH2AX foci during ara-C exposure of DRG neurons are shown (A). DRG neurons are identified by NF200 (red) and γH2AX foci are observed in green; sparse foci emerge by 12 h with density increasing in the course of 72 h ara-C exposure (scale bar = 20 µm). B) Percent of γH2AX positive nuclei increased over time with positive DRGs reaching ~35%, 75% and 95% by 24 h, 48 h and 72 h, respectively. Data are obtained from three independent biological experiments and presented as mean±SEM percent of γH2AX positive nuclei versus time.
Figure 7. Nuclear γH2AX/γATM foci induced by ara-C exposure of DRG neurons are gradually cleared in the course of post ara-C recovery.

Representative images of γH2AX/γATM foci formed following 48 h ara-C exposure and foci clearance in the course of 96 h recovery. (A) Merged images show largely colocalized γH2AX [red]/γATM [green] foci with gradual reduction in foci density in the course of post exposure recovery [scale bar = 10 µm]. (B) Bar graphs show the number of co-localized γH2AX/γATM foci per DRG nucleus as a function of recovery time. Data are mean±SEM values obtained from four independent biological experiments.
4. DISCUSSION
We report formation of mtDNA damage, ROS generation, reduced mitochondrial content and compromised mitochondrial respiration in DRG neurons following sublethal exposure to the antimetabolite cancer drug, ara-C. A delayed formation of nuclear γH2AX/γATM foci indicative of oxidative stress signaling and activation of the DNA damage response was also observed. When considered together, these in vitro findings reveal a potential mechanism of unintended consequences of antimetabolite drugs, which while designed to abolish nuclear DNA synthesis in dividing cancer cells, inadvertently block mtDNA synthesis in post mitotic neurons. In the highly metabolic and mitochondria rich DRG neurons, impediment of mtDNA synthesis and other mtDNA transactions leads to ROS generation, mitochondrial content reduction and impairments of mitochondrial function, with subsequent initiation of the DNA damage response.
Using recombinant mitochondrial DNA polymerase γ and oligonucleotide substrates engineered to contain ara-C residues, we demonstrated that catalytic activities of DNA polymerase γ are blocked by ara-C residues incorporated into either the extending or templating DNA strand, suggesting that ara-C works as ‘chain terminator’ also in the mitochondrial compartment. DNA polymerase γ is responsible for mtDNA synthesis [17, 18] and a critical determinant of mitochondrial biogenesis and function [46, 47]. Because in postmitotic neurons, mtDNA is continuously replicated [48], it follows that circulating ara-C that enters DRG neurons can be incorporated into mtDNA [9, 49, 50] leading to inhibition of polymerase γ-catalyzed mtDNA synthesis and subsequent reduction in mitochondrial content and compromised function. Notably, in patients carrying loss of function mutations of the catalytic subunit of mitochondrial DNA polymerase γ, sensory neuropathies [51, 52], as well as other neurodegenerative disorders [53, 54] have been reported, highlighting the link between mitochondrial dysfunction and neurotoxicity.
Interestingly, however, the magnitude of PNS neurotoxicity induced by the different chemotherapeutic drugs varies and may depend, at least in part, on the type of DNA damage the drugs cause and availability of specific DNA repair capacities in corresponding subcellular compartments. For example, here we show that ara-C leads to formation of oxidative mtDNA damage. Oxidative DNA damage is repaired primarily by the base excision repair (BER) pathway [55]. Ample evidence confirms that the BER pathway is active in the mitochondrial compartment [56–58]. Hence, it is plausible that in DRG neurons, mtDNA damage caused by ara-C is at least partially repairable, accounting for the rather limited evidence for long-term ara-C induced PNS neurotoxicity in the clinic [59, 60]. In contrast, a different-cisplatin based class of antimitotic drugs, which form crosslinks with DNA, has been implicated in severe lasting PNS neurotoxicity [61]. Formation of DNA:cisplatin crosslinks does not require DNA synthesis and therefore, cisplatin crosslinks rapidly form in nuclear as well as mtDNA. Crosslinks removal is catalyzed mainly by the nucleotide excision repair (NER) pathway [62], which while active in nuclei, is absent from the mitochondrial compartment [63], suggesting that cisplatin adducts may persist in mtDNA hampering post treatment neuronal recovery. Hence, the lack of mitochondrial NER pathway might contribute to the lasting cisplatin toxicity observed in experimental models of DRG neurons [28, 30, 64] as well as in the clinic [65].
We propose that features inherent to DRG neurons, namely the high mitochondrial content, reliance on mitochondria for energy needs and limited shielding from circulating substances by the blood nerve barrier [16], sensitize DRGs to drugs whose injurious effects are mediated by impediment of mtDNA transactions and impairment of mitochondrial function. Mitochondrial compromise in DRG neurons exposed to ara-C is manifested in formation of oxidative mtDNA damage, gradual reduction in mtDNA copy number and in mitochondria-encoded gene expression and reduced mitochondrial content. These changes impair mitochondrial function as reflected in compromised respiration. Analyses of mitochondrial respiratory parameters also reveal that ara-C exposure leads to increased proton leak and diminution of spare respiratory capacity. This is significant since unlike cells with substantial glycolytic capacity, DRG neurons rely mainly on oxidative phosphorylation for meeting routine and sudden energy needs, such that mitochondria attempting to maintain ATP levels under compromised conditions, may enter futile cycle where ROS generation escalates, overwhelms defense mechanisms and underlies subsequent formation of nuclear DNA damage. The observed depletion of NADPH and GSH stores and resultant unfavorable shifts in NADP+/NADPH and GSH/GSSG ratios indicative of redox imbalance supports this possibility. Interestingly, concomitant supplementation of 6-aminonicotinamide, an inhibitor of oxidative branch of the pentose phosphate pathway [44, 45] further reduced NADPH and increased NADP+/NADPH ratios. This additive effect might suggest that depletion of NADPH following ara-C exposure, results from increased consumption of NADPH, rather than dysregulation of the pentose phosphate pathway.
Consistently with the scenario of elevated mitochondrial ROS, we detected 8-oxoguanine in mtDNA. Accumulation of 8-oxoguanine in mouse cortical neurons mtDNA under oxidative stress conditions has been reported previously [66]. Interestingly, unlike in the case of mtDNA, our analyses failed to detect 8-oxoguanine immunoreactivity in nuclear DNA of ara-C exposed DRG neurons. Because in neurons, base excision repair capacity is substantial [67, 68] and likely to support rapid DNA glycosylase OGG1-catalyzed removal of oxidized guanine from nuclear DNA [69], it is plausible that extant levels of 8-oxoguanine are below immunodetection limits, while BER repair intermediates, such as single strand breaks persist longer. We detect gradual induction of nuclear γH2AX foci, which are indicative of chromatin rearrangements and activation of the DNA damage response [70]. Importantly, sluggish induction of γH2AX foci in the terminally differentiated DRG neurons sharply differs from rapid induction of γH2AX foci in dividing primary astrocytes, where foci form within one hour of ara-C exposure (not shown), further supporting the scenario that in post-mitotic neurons ara-C targets mtDNA, whereas nuclear manifestations of exposure are secondary to mtDNA damage. Our findings also reveal involvement of ATM in the DNA damage response in DRG neurons, since substantial nuclear γATM immunoreactivity is also observed. In addition to its role in the DNA damage response via phosphorylation of H2AX, ATM has been implicated in redox sensing, ROS control and as target for activation by oxidative stress [71–73]. Interestingly, in DRG neurons γH2AX and γATM foci co-localize not only in the course of ara-C treatment, but also in the course of the post exposure recovery. Moreover, in the course of post ara-C recovery, foci density gradually decreases with just few large colocalized γH2AX/γATM foci remaining in DRG nuclei in the course of 48–96-h period of post exposure recovery, plausibly reflecting containment of unrepaired DNA in specialized modified chromatin domains. Importantly, clearance of γH2AX/γATM foci indicates that after drug removal, nuclear DNA damage in DRG neurons is being repaired, suggesting that DRG neurons are able to sustain their basic cellular functions in the aftermath of sublethal ara-C challenge and hence, are destined for recovery.
HIGHLIGHTS.
Antimetabolite cancer drug ara-C blocks DNA polymerase γ-catalyzed DNA synthesis
In DRG neurons, sublethal ara-C exposure leads to formation of mtDNA damage
Exposure impairs mitochondrial respiration, elevates ROS and reduces NADPH and GSH
Nuclear γH2AX/γATM foci indicative of activated DNA damage response also form
In the course of post ara-C DRG recovery density of γH2AX/γATM foci declines
Acknowledgments
This work was supported by grants from the National Institutes of Health (ES014613) and Shriners Hospitals for Children (86700) to EWE. We thank Steve Schuenke and Eileen Figueroa for assistance with manuscript preparation.
Abbreviations
- DRG
dorsal root ganglion
- mtDNA
mitochondrial DNA
- BER
base excision repair
- NER
nucleotide excision repair
- DDR
DNA damage response
- OCR
oxygen consumption rate
- PNS
peripheral nervous system
- pol γ
DNA polymerase γ
- ROS
reactive oxygen species
- 8-oxoG
8-oxoguanine
- GSH
reduced glutathione
- NADP+
nicotinamide adenine dinucleotide phosphate
- NADPH
nicotinamide adenine dinucleotide phosphate reduced form
- 6AN
6-aminonicotinamide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest/Disclosure: Authors declare no conflicts of interest.
References
- 1.Argyriou AA, Bruna J, Marmiroli P, Cavaletti G. Chemotherapy-induced peripheral neurotoxicity (CIPN): an update. Crit Rev Oncol Hematol. 2012;82:51–77. doi: 10.1016/j.critrevonc.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 2.Beijers AJ, Jongen JL, Vreugdenhil G. Chemotherapy-induced neurotoxicity: the value of neuroprotective strategies. Neth J Med. 2012;70:18–25. [PubMed] [Google Scholar]
- 3.Cavaletti G, Marmiroli P. Chemotherapy-induced peripheral neurotoxicity. Nat Rev Neurol. 2010;6:657–666. doi: 10.1038/nrneurol.2010.160. [DOI] [PubMed] [Google Scholar]
- 4.Grant S. Ara-C: cellular and molecular pharmacology. Adv Cancer Res. 1998;72:197–233. doi: 10.1016/s0065-230x(08)60703-4. [DOI] [PubMed] [Google Scholar]
- 5.Iliakis G, Bryant PE. Effects of the nucleoside analogues alpha-ara A, beta-ara A and beta-ara C on cell growth and repair of both potentially lethal damage and DNA double strand breaks in mammalian cells in culture. Anticancer Res. 1983;3:143–149. [PubMed] [Google Scholar]
- 6.Lamba JK. Genetic factors influencing cytarabine therapy. Pharmacogenomics. 2009;10:1657–1674. doi: 10.2217/pgs.09.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Major PP, Egan EM, Beardsley GP, Minden MD, Kufe DW. Lethality of human myeloblasts correlates with the incorporation of arabinofuranosylcytosine into DNA. Proc Natl Acad Sci U S A. 1981;78:3235–3239. doi: 10.1073/pnas.78.5.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yee KW, Cortes J, Ferrajoli A, Garcia-Manero G, Verstovsek S, Wierda W, Thomas D, Faderl S, King I, O'Brien S M, Jeha S, Andreeff M, Cahill A, Sznol M, Giles FJ. Triapine and cytarabine is an active combination in patients with acute leukemia or myelodysplastic syndrome. Leuk Res. 2006;30:813–822. doi: 10.1016/j.leukres.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 9.Heintz NH, Hamlin JL. In vivo effects of cytosine arabinoside on deoxyribonucleic acid replication in Chinese hamster ovary cells. 1. Resolution of differential effects on mitochondrial and nuclear deoxyribonucleic acid synthesis. Biochemistry. 1983;22:3552–3557. doi: 10.1021/bi00284a003. [DOI] [PubMed] [Google Scholar]
- 10.Snyder RD, van Houten B, Regan JD. The inhibition of ultraviolet radiation-induced DNA repair in human diploid fibroblasts by arabinofuranosyl nucleosides. Chem Biol Interact. 1984;50:1–14. doi: 10.1016/0009-2797(84)90127-3. [DOI] [PubMed] [Google Scholar]
- 11.Atsumi Y, Inase A, Osawa T, Sugihara E, Sakasai R, Fujimori H, Teraoka H, Saya H, Kanno M, Tashiro F, Nakagama H, Masutani M, Yoshioka K. The Arf/p53 protein module, which induces apoptosis, down-regulates histone H2AX to allow normal cells to survive in the presence of anticancer drugs. J Biol Chem. 2013;288:13269–13277. doi: 10.1074/jbc.M112.402560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang N, Wang X, Yang Y, Dai W. Advances in mitotic inhibitors for cancer treatment. Mini Rev Med Chem. 2006;6:885–895. doi: 10.2174/138955706777934955. [DOI] [PubMed] [Google Scholar]
- 13.Anand U, Otto WR, Bountra C, Chessell I, Sinisi M, Birch R, Anand P. Cytosine arabinoside affects the heat and capsaicin receptor TRPV1 localisation and sensitivity in human sensory neurons. J Neurooncol. 2008;89:1–7. doi: 10.1007/s11060-008-9585-6. [DOI] [PubMed] [Google Scholar]
- 14.Geller HM, Cheng KY, Goldsmith NK, Romero AA, Zhang AL, Morris EJ, Grandison L. Oxidative stress mediates neuronal DNA damage and apoptosis in response to cytosine arabinoside. J Neurochem. 2001;78:265–275. doi: 10.1046/j.1471-4159.2001.00395.x. [DOI] [PubMed] [Google Scholar]
- 15.Wallace TL, Johnson EM., Jr Cytosine arabinoside kills postmitotic neurons: evidence that deoxycytidine may have a role in neuronal survival that is independent of DNA synthesis. J Neurosci. 1989;9:115–124. doi: 10.1523/JNEUROSCI.09-01-00115.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weerasuriya A, Mizisin AP. The blood-nerve barrier: structure and functional significance. Methods Mol Biol. 2011;686:149–173. doi: 10.1007/978-1-60761-938-3_6. [DOI] [PubMed] [Google Scholar]
- 17.Lestienne P. Evidence for a direct role of the DNA polymerase gamma in the replication of the human mitochondrial DNA in vitro. Biochem Biophys Res Commun. 1987;146:1146–1153. doi: 10.1016/0006-291x(87)90767-4. [DOI] [PubMed] [Google Scholar]
- 18.Young MJ, Copeland WC. Human mitochondrial DNA replication machinery and disease. Curr Opin Genet Dev. 2016;38:52–62. doi: 10.1016/j.gde.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaughnessy DT, McAllister K, Worth L, Haugen AC, Meyer JN, Domann FE, Van Houten B, Mostoslavsky R, Bultman SJ, Baccarelli AA, Begley TJ, Sobol RW, Hirschey MD, Ideker T, Santos JH, Copeland WC, Tice RR, Balshaw DM, Tyson FL. Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ Health Perspect. 2014;122:1271–1278. doi: 10.1289/ehp.1408418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimura T, Kunugita N. Mitochondrial reactive oxygen species-mediated genomic instability in low-dose irradiated human cells through nuclear retention of cyclin D1. Cell Cycle. 2016;15:1410–1414. doi: 10.1080/15384101.2016.1170271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crowe SL, Movsesyan VA, Jorgensen TJ, Kondratyev A. Rapid phosphorylation of histone H2A.X following ionotropic glutamate receptor activation. Eur J Neurosci. 2006;23:2351–2361. doi: 10.1111/j.1460-9568.2006.04768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saki M, Prakash A. DNA damage related crosstalk between the nucleus and mitochondria. Free Radic Biol Med. 2017;107:216–227. doi: 10.1016/j.freeradbiomed.2016.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14:197–210. [PubMed] [Google Scholar]
- 24.Carlessi L, De Filippis L, Lecis D, Vescovi A, Delia D. DNA-damage response, survival and differentiation in vitro of a human neural stem cell line in relation to ATM expression. Cell Death Differ. 2009;16:795–806. doi: 10.1038/cdd.2009.10. [DOI] [PubMed] [Google Scholar]
- 25.Lindsay RM. Nerve growth factors (NGF, BDNF) enhance axonal regeneration but are not required for survival of adult sensory neurons. J Neurosci. 1988;8:2394–2405. doi: 10.1523/JNEUROSCI.08-07-02394.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malin SA, Davis BM, Molliver DC. Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and plasticity. Nat Protoc. 2007;2:152–160. doi: 10.1038/nprot.2006.461. [DOI] [PubMed] [Google Scholar]
- 27.Owen DE, Egerton J. Culture of dissociated sensory neurons from dorsal root ganglia of postnatal and adult rats. Methods Mol Biol. 2012;846:179–187. doi: 10.1007/978-1-61779-536-7_16. [DOI] [PubMed] [Google Scholar]
- 28.Gorgun MF, Zhuo M, Englander EW. Cisplatin toxicity in dorsal root ganglion neurons is relieved by meclizine via diminution of mitochondrial compromise and improved clearance of DNA damage. Mol Neurobiol. 2017;54:7883–7895. doi: 10.1007/s12035-016-0273-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhuo M, Gorgun MF, Englander EW. Augmentation of glycolytic metabolism by meclizine is indispensable for protection of dorsal root ganglion neurons from hypoxia-induced mitochondrial compromise. Free Radic Biol Med. 2016;99:20–31. doi: 10.1016/j.freeradbiomed.2016.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhuo M, Gorgun MF, Englander EW. Translesion synthesis DNA polymerase kappa is indispensable for DNA repair synthesis in cisplatin exposed dorsal root ganglion neurons. Mol Neurobiol. 2017 Apr 8; doi: 10.1007/s12035-017-0507-5. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohno M, Oka S, Nakabeppu Y. Quantitative analysis of oxidized guanine, 8-oxoguanine, in mitochondrial DNA by immunofluorescence method. Methods Mol Biol. 2009;554:199–212. doi: 10.1007/978-1-59745-521-3_13. [DOI] [PubMed] [Google Scholar]
- 32.Bindokas VP, Jordan J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forkink M, Willems PH, Koopman WJ, Grefte S. Live-cell assessment of mitochondrial reactive oxygen species using dihydroethidine. Methods Mol Biol. 2015;1264:161–169. doi: 10.1007/978-1-4939-2257-4_15. [DOI] [PubMed] [Google Scholar]
- 34.Englander EW, Hu Z, Sharma A, Lee HM, Wu ZH, Greeley GH. Rat MYH, a glycosylase for repair of oxidatively damaged DNA, has brain-specific isoforms that localize to neuronal mitochondria. J Neurochem. 2002;83:1471–1480. doi: 10.1046/j.1471-4159.2002.01259.x. [DOI] [PubMed] [Google Scholar]
- 35.Dimmock D, Tang LY, Schmitt ES, Wong LJ. Quantitative evaluation of the mitochondrial DNA depletion syndrome. Clin Chem. 2010;56:1119–1127. doi: 10.1373/clinchem.2009.141549. [DOI] [PubMed] [Google Scholar]
- 36.Venegas V, Wang J, Dimmock D, Wong LJ. Real-time quantitative PCR analysis of mitochondrial DNA content. Curr Protoc Hum Genet Chapter. 2011;19(Unit 19):17. doi: 10.1002/0471142905.hg1907s68. [DOI] [PubMed] [Google Scholar]
- 37.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 38.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dranka BP, Benavides GA, Diers AR, Giordano S, Zelickson BR, Reily C, Zou L, Chatham JC, Hill BG, Zhang J, Landar A, Darley-Usmar VM. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic Biol Med. 2011;51:1621–1635. doi: 10.1016/j.freeradbiomed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gerencser AA, Neilson A, Choi SW, Edman U, Yadava N, Oh RJ, Ferrick DA, Nicholls DG, Brand MD. Quantitative microplate-based respirometry with correction for oxygen diffusion. Anal Chem. 2009;81:6868–6878. doi: 10.1021/ac900881z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh S, Englander EW. Nuclear depletion of apurinic/apyrimidinic endonuclease 1 (Ape1/Ref-1) is an indicator of energy disruption in neurons. Free Radic Biol Med. 2012;53:1782–1790. doi: 10.1016/j.freeradbiomed.2012.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H, Swiercz R, Englander EW. Elevated metals compromise repair of oxidative DNA damage via the base excision repair pathway: implications of pathologic iron overload in the brain on integrity of neuronal DNA. J Neurochem. 2009;110:1774–1783. doi: 10.1111/j.1471-4159.2009.06271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei W, Englander EW. DNA polymerase beta-catalyzed-PCNA independent long patch base excision repair synthesis: a mechanism for repair of oxidatively damaged DNA ends in postmitotic brain. J Neurochem. 2008;107:734–744. doi: 10.1111/j.1471-4159.2008.05644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hothersall JS, Gordge M, Noronha-Dutra AA. Inhibition of NADPH supply by 6-aminonicotinamide: effect on glutathione, nitric oxide and superoxide in J774 cells. FEBS Lett. 1998;434:97–100. doi: 10.1016/s0014-5793(98)00959-4. [DOI] [PubMed] [Google Scholar]
- 45.Tyson RL, Perron J, Sutherland GR. 6-Aminonicotinamide inhibition of the pentose phosphate pathway in rat neocortex. Neuroreport. 2000;11:1845–1848. doi: 10.1097/00001756-200006260-00009. [DOI] [PubMed] [Google Scholar]
- 46.Clayton DA. Replication of animal mitochondrial DNA. Cell. 1982;28:693–705. doi: 10.1016/0092-8674(82)90049-6. [DOI] [PubMed] [Google Scholar]
- 47.Copeland WC. Defects in mitochondrial DNA replication and human disease. Crit Rev Biochem Mol Biol. 2012;47:64–74. doi: 10.3109/10409238.2011.632763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Korr H, Kurz C, Seidler TO, Sommer D, Schmitz C. Mitochondrial DNA synthesis studied autoradiographically in various cell types in vivo. Braz J Med Biol Res. 1998;31:289–298. doi: 10.1590/s0100-879x1998000200012. [DOI] [PubMed] [Google Scholar]
- 49.Lewis W, Levine ES, Griniuviene B, Tankersley KO, Colacino JM, Sommadossi JP, Watanabe KA, Perrino FW. Fialuridine and its metabolites inhibit DNA polymerase gamma at sites of multiple adjacent analog incorporation, decrease mtDNA abundance, and cause mitochondrial structural defects in cultured hepatoblasts. Proc Natl Acad Sci U S A. 1996;93:3592–3597. doi: 10.1073/pnas.93.8.3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu C, Johansson M, Karlsson A. Incorporation of nucleoside analogs into nuclear or mitochondrial DNA is determined by the intracellular phosphorylation site. J Biol Chem. 2000;275:26727–26731. doi: 10.1074/jbc.M002304200. [DOI] [PubMed] [Google Scholar]
- 51.Lax NZ, Whittaker RG, Hepplewhite PD, Reeve AK, Blakely EL, Jaros E, Ince PG, Taylor RW, Fawcett PR, Turnbull DM. Sensory neuronopathy in patients harbouring recessive polymerase gamma mutations. Brain. 2012;135:62–71. doi: 10.1093/brain/awr326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cassereau J, Codron P, Funalot B. Inherited peripheral neuropathies due to mitochondrial disorders. Rev Neurol (Paris) 2014;170:366–374. doi: 10.1016/j.neurol.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 53.Sohl CD, Kasiviswanathan R, Copeland WC, Anderson KS. Mutations in human DNA polymerase gamma confer unique mechanisms of catalytic deficiency that mirror the disease severity in mitochondrial disorder patients. Hum Mol Genet. 2013;22:1074–1085. doi: 10.1093/hmg/dds509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tzoulis C, Tran GT, Coxhead J, Bertelsen B, Lilleng PK, Balafkan N, Payne B, Miletic H, Chinnery PF, Bindoff LA. Molecular pathogenesis of polymerase gamma-related neurodegeneration. Ann Neurol. 2014;76:66–81. doi: 10.1002/ana.24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ide H. DNA substrates containing defined oxidative base lesions and their application to study substrate specificities of base excision repair enzymes. Prog Nucleic Acid Res Mol Biol. 2001;68:207–221. doi: 10.1016/s0079-6603(01)68101-7. [DOI] [PubMed] [Google Scholar]
- 56.Bohr VA, Dianov GL. Oxidative DNA damage processing in nuclear and mitochondrial DNA. Biochimie. 1999;81:155–160. doi: 10.1016/s0300-9084(99)80048-0. [DOI] [PubMed] [Google Scholar]
- 57.Prasad R, Caglayan M, Dai DP, Nadalutti CA, Zhao ML, Gassman NR, Janoshazi AK, Stefanick DF, Horton JK, Krasich R, Longley MJ, Copeland WC, Griffith JD, Wilson SH. DNA polymerase beta: A missing link of the base excision repair machinery in mammalian mitochondria. DNA Repair (Amst) 2017;60:77–88. doi: 10.1016/j.dnarep.2017.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu P, Demple B. DNA repair in mammalian mitochondria: Much more than we thought? Environ Mol Mutagen. 2010;51:417–426. doi: 10.1002/em.20576. [DOI] [PubMed] [Google Scholar]
- 59.Cavaliere R, Schiff D. Neurologic toxicities of cancer therapies. Curr Neurol Neurosci Rep. 2006;6:218–226. doi: 10.1007/s11910-006-0009-8. [DOI] [PubMed] [Google Scholar]
- 60.Sioka C, Kyritsis AP. Central and peripheral nervous system toxicity of common chemotherapeutic agents. Cancer Chemother Pharmacol. 2009;63:761–767. doi: 10.1007/s00280-008-0876-6. [DOI] [PubMed] [Google Scholar]
- 61.Dzagnidze A, Katsarava Z, Makhalova J, Liedert B, Yoon MS, Kaube H, Limmroth V, Thomale J. Repair capacity for platinum-DNA adducts determines the severity of cisplatininduced peripheral neuropathy. J Neurosci. 2007;27:9451–9457. doi: 10.1523/JNEUROSCI.0523-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sancar A. Excision repair in mammalian cells. J Biol Chem. 1995;270:15915–15918. doi: 10.1074/jbc.270.27.15915. [DOI] [PubMed] [Google Scholar]
- 63.Clayton DA, Doda JN, Friedberg EC. The absence of a pyrimidine dimer repair mechanism in mammalian mitochondria. Proc Natl Acad Sci U S A. 1974;71:2777–2781. doi: 10.1073/pnas.71.7.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yan F, Liu JJ, Ip V, Jamieson SM, McKeage MJ. Role of platinum DNA damage-induced transcriptional inhibition in chemotherapy-induced neuronal atrophy and peripheral neurotoxicity. J Neurochem. 2015;135:1099–1112. doi: 10.1111/jnc.13355. [DOI] [PubMed] [Google Scholar]
- 65.Ewertz M, Qvortrup C, Eckhoff L. Chemotherapy-induced peripheral neuropathy in patients treated with taxanes and platinum derivatives. Acta Oncol. 2015;54:587–591. doi: 10.3109/0284186X.2014.995775. [DOI] [PubMed] [Google Scholar]
- 66.Leon J, Sakumi K, Castillo E, Sheng Z, Oka S, Nakabeppu Y. 8-Oxoguanine accumulation in mitochondrial DNA causes mitochondrial dysfunction and impairs neuritogenesis in cultured adult mouse cortical neurons under oxidative conditions. Sci Rep. 2016;6:22086. doi: 10.1038/srep22086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Englander EW. DNA damage response in peripheral nervous system: coping with cancer therapy-induced DNA lesions. DNA Repair (Amst) 2013;12:685–690. doi: 10.1016/j.dnarep.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Englander EW. Brain capacity for repair of oxidatively damaged DNA and preservation of neuronal function. Mech Ageing Dev. 2008;129:475–482. doi: 10.1016/j.mad.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aleksandrov R, Dotchev A, Poser I, Krastev D, Georgiev G, Panova G, Babukov Y, Danovski G, Dyankova T, Hubatsch L, Ivanova A, Atemin A, Nedelcheva-Veleva MN, Hasse S, Sarov M, Buchholz F, Hyman AA, Grill SW, Stoynov SS. Protein Dynamics in Complex DNA Lesions. Mol Cell. 2018;69:1046–1061. e1045. doi: 10.1016/j.molcel.2018.02.016. [DOI] [PubMed] [Google Scholar]
- 70.Lukas J, Lukas C, Bartek J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol. 2011;13:1161–1169. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- 71.Ditch S, Paull TT. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem Sci. 2012;37:15–22. doi: 10.1016/j.tibs.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010;330:517–521. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 73.Lee JH, Mand MR, Kao CH, Zhou Y, Ryu SW, Richards AL, Coon JJ, Paull TT. ATM directs DNA damage responses and proteostasis via genetically separable pathways. Sci Signal. 2018;11 doi: 10.1126/scisignal.aan5598. [DOI] [PMC free article] [PubMed] [Google Scholar]