

Abstract
Human papillomaviruses (HPVs) cause benign lesions that can lead to malignancy. How cellular changes induced by viral oncogenes contribute to the progeny virion production is not always clear. Stromally-derived growth factors and their receptors are critical for development of malignancy, but their impact on the pre-malignant HPV life cycle is unknown. We show that HPV16 increases levels of Met, a growth factor receptor critical for tumor cell invasion, motility, and cancer metastasis. The viral oncogene E5 is primarily responsible for Met upregulation, with E6 playing a minor role. Met induction by E5 requires the epidermal growth factor receptor, which is also increased by E5 at the mRNA level. E5-induced Met contributes motility of HPV-containing cells. Finally, Met signaling is necessary for viral gene expression, particularly in the differentiation-dependent phase of the viral life cycle. These studies show a new role for E5 in epithelial-stromal interactions, with implications for cancer development.
Keywords: Human papillomavirus type 16, Met, E5, E6, HGF, EGFR
Introduction
Stromal cells in the tumor microenvironment have been increasingly recognized as major sources of paracrine mediators that contribute to the growth and invasiveness of a cancer (Kankuri et al., 2005; Lopes-Bastos et al., 2016; Matsumoto and Nakamura, 2006; Polanska and Orimo, 2013; Steffan et al., 2011). Work by many investigators has uncovered a complex array of interactions between epithelial cells and fibroblasts, endothelial cells, and immune cells in the tissue stroma during cancer development (Cirri and Chiarugi, 2012; Lopes-Bastos et al., 2016; Matsumoto and Nakamura, 2006; Steffan et al., 2011; Woodby et al., 2016). One stromally-derived growth factor, hepatocyte growth factor (HGF), activates signaling pathways to promote cellular proliferation, sustained cell motility, and invasiveness by binding to Met, a receptor tyrosine kinase (Boccaccio and Comoglio, 2006; Gherardi et al., 2012; Steffan et al., 2011; Trusolino et al., 2010). Met is important for survival and migration of epithelial cells during development and is required for keratinocyte mobilization during wound healing (Chmielowiec et al., 2007; Gherardi et al., 2012; Thiery and Sleeman, 2006). Cell motility induced by HGF/Met in cell culture resembles an epithelial-to-mesenchymal transition (EMT), which is often a precursor to metastasis (Boccaccio and Comoglio, 2006; Craene and Berx, 2012; Drake and Macleod, 2014; Hanahan and Weinberg, 2011; Steffan et al., 2011; Thiery and Sleeman, 2006). Met is frequently overexpressed in cancers, resulting in many cellular changes that underlie tumor invasion (Boccaccio and Comoglio, 2006; Steffan et al., 2011). Met levels can be increased by activation of oncogenes or inactivation of tumor suppressors; consequently, early oncogenic changes can facilitate later progression to malignancy (Boccaccio and Comoglio, 2006; Gherardi et al., 2012; Hwang et al., 2011). Due to the importance of metastasis for cancer mortality, Met is also a promising therapeutic target (Gherardi et al., 2012).
Human papillomaviruses (HPVs) infect the keratinocytes of stratified squamous epithelia, causing benign hyperproliferations which sometimes progress to malignancy. Certain “high risk” HPV types, including HPV16, are associated with cervical, oropharyngeal, and other cancers. By contrast, “low risk” HPV types, including HPV11, are rarely associated with malignancy, despite having broadly similar life cycles (Egawa and Doorbar, 2017; Klingelhutz and Roman, 2012; Moody and Laimins, 2010). HPV is maintained in cells of the basal epithelial layer, where viral copy number and gene expression levels are kept low. As host cells detach from the basement membrane and differentiate, viral gene expression and replication are activated, culminating in the formation of progeny virus (Doorbar et al., 2015; Doorbar et al., 2012; Moody and Laimins, 2010). To make cellular DNA synthesis machinery available in differentiating cells, HPV encodes the oncogenes E6 and E7 which induce degradation of p53 and pRb family members, respectively, resulting in activation of cell cycling and inhibition of cell cycle arrest and apoptosis (Howie et al., 2009; Nees et al., 2000; Patel et al., 1999; Roman and Munger, 2013). E6 and E7 are the major viral factors that promote and sustain HPV-associated cancers (Doorbar et al., 2015; Munger et al., 2004). In addition, HPV16 encodes E5, a small, hydrophobic, weakly immortalizing membrane protein primarily found in the Golgi complex and endoplasmic reticulum (Dimaio and Petti, 2013). This protein is not needed for HPV-induced immortalization or maintenance of episomal genome replication, but it does contribute to the efficiency of genome amplification and late gene expression upon differentiation (Fehrmann et al., 2003; Genther et al., 2003). In mice, E5 expressed alone can synergize with estrogen to cause cervical cancers and also exacerbate tumor formation when co-expressed with E6 and/or E7 (Genther Williams et al., 2005; Maufort et al., 2010). E5 expression in human cervical tumors correlates with worse prognosis and more aggressive tumor behavior (Hsieh et al., 2000; Venuti et al., 2011), perhaps through the ability of E5 to promote motility and invasion (Barbaresi et al., 2010; Liao et al., 2013). The best understood molecular activity of HPV16 E5 is to augment signaling from the epidermal growth factor receptor (EGFR) by increasing levels of EGFR on the cell surface, thereby enhancing the ability of the receptor to bind to EGF (Dimaio and Petti, 2013). However, the molecular basis for the effect of E5 on EGFR and the significance in the HPV life cycle or to cancer development remain to be fully understood.
HPVs infect epithelial cells exclusively; however epithelial cells are influenced by their stromal microenvironment. HPV modifies a wide array of epithelial-stromal communication systems (den Boon et al., 2015; Gius et al., 2007; Spurgeon and Lambert, 2017; Woodby et al., 2016), but how stromally-derived factors such as HGF or EGF affect the normal, benign life cycle of HPVs in infected epithelia, and how these factors influence progression from benign infection to cancer, is unclear. HGF experimentally increases the invasiveness of cervical cancer cells (Shimabukuro et al., 2001). Met levels are upregulated in early, low grade lesions caused by high risk HPV types, and increase progressively as lesions develop into carcinomas (Walker et al., 2003; Zhang et al., 2014). Met expression in cervical cancers is associated with poor prognosis (Baykal et al., 2003; Shimabukuro et al., 2001). Several studies have shown that HPV oncogenes can promote some aspects of EMT (Caberg et al., 2008; Cheng et al., 2012; D’Costa et al., 2012; Duffy et al., 2003; Faghihloo et al., 2016; Geiger et al., 2008; Hellner et al., 2009; Jung et al., 2013; Latorre et al., 2005; Lee et al., 2008; Myong, 2012; Pim et al., 2002) but the involvement of Met in these changes has not been investigated. Here we show that Met protein and mRNA levels are increased by HPV16. E5 is primarily responsible for this upregulation, although E6 may play a minor role. Induction of Met by E5 requires EGFR, which is also upregulated at the mRNA and protein levels in an E5-dependent manner. E5-induced Met upregulation contributes to increased motility of HPV-containing cells. Finally, Met activity and expression support the expression of HPV transcripts, especially upon differentiation, showing that Met contributes to the HPV life cycle. These studies uncover a mechanism by which HPV16 can alter the response of infected cells to factors in the stromal microenvironment and suggest a novel mechanism by which E5 could contribute to the viral life cycle and to the development of malignancy.
Results
HPV16 upregulates Met mRNA and protein
To determine whether the presence of HPV16 can influence Met levels, uninfected human foreskin keratinocyte (HFK) cells or HFKs containing episomal HPV16 genomes under normal growth conditions were examined for Met mRNA and protein levels by RT-qPCR and western blotting. Since these HPV16-containing keratinocytes represent conditions in a benign, low grade lesion and Met is usually a feature of higher grade cancers, we did not expect that Met levels would be significantly different as compared to uninfected HFK cells. However, we found that total MET transcript levels were markedly higher in the HPV-containing cells than in HFKs, whether the cells were grown under undifferentiating (monolayer) or differentiating (methylcellulose, MC) conditions (Figure 1a). Increased MET transcript levels were reflected in higher Met protein levels in monolayer culture (Figure 1b). Met protein levels declined significantly upon differentiation in both cell types, suggesting post-translational regulation during differentiation (not shown). Phosphorylated/activated Met (Y1234/1235) levels were increased to a degree similar to total Met (Figure 1b and c). Immunofluorescence staining of HFKs or HPV16-containing keratinocytes showed higher levels of Met on the cell surface in cells containing HPV16 (Figure 2). After treatment with HGF, Met staining was present in early endosome antigen 1 (EEA1)-positive vesicles, suggesting internalization to early endosomes. The pattern of localization was similar between HFKs and HPV-containing keratinocytes regardless of HGF treatment. These data indicate that cells stably maintaining HPV16 have increased levels of MET transcripts, which is reflected in higher levels of both total and phosphorylated Met protein, but the subcellular distribution of Met under basal and stimulated conditions is not grossly altered.
Figure 1. HPV increases Met transcript and protein.

a. HFKs or keratinocytes containing episomal HPV16 genomes were grown in undifferentiated monolayer culture (mono) or suspended in 1.6% methylcellulose (MC) to induce differentiation. Total RNAs were subjected to RT-qPCR using primers specific for Met. Values are the average of 4 independent experiments and bars represent ± one standard error of the mean. b. Representative western blot of total (top) or Y1234/1235 phosphorylated (bottom) Met levels in two HFK and two HPV16 containing cell lines grown in monolayer culture. c. Densitometric analysis of total Met and phospho-Met band intensities from six independent western blot experiments. *=p<0.05 as compared to HFK; ***=p<0.001.
Figure 2. HPV does not affect the subcellular localization of Met.

HFKs or HPV16-containing cells were treated with HGF (33 ng/ml) for 20 minutes or left untreated and stained using antibodies against Met (green) and the early endosomal marker EEA1 (red). The area in the white squares is shown magnified at the bottom of the figure. Arrows show sites of overlap between Met and EEA1.
Because HPV could increase Met levels, we used RT-qPCR to test whether other growth factor receptor kinases were also increased. Epidermal growth factor receptor (EGFR) and insulin-like growth factor-1 receptor (IGF1R) transcript levels and protein levels were increased in HPV-containing cells, under both differentiating and undifferentiated conditions (Figure 3a–c). In contrast, transforming growth factor-beta receptor II (TGFBR2) transcripts and protein were not changed in HPV-containing cells (Figure 3a and d), and fibroblast growth factor receptor-2 (FGFR2) transcripts were reduced (not shown), indicating the modification of growth factor receptor levels by HPV is relatively selective.
Figure 3. HPV increases levels of other cellular growth factor receptors.
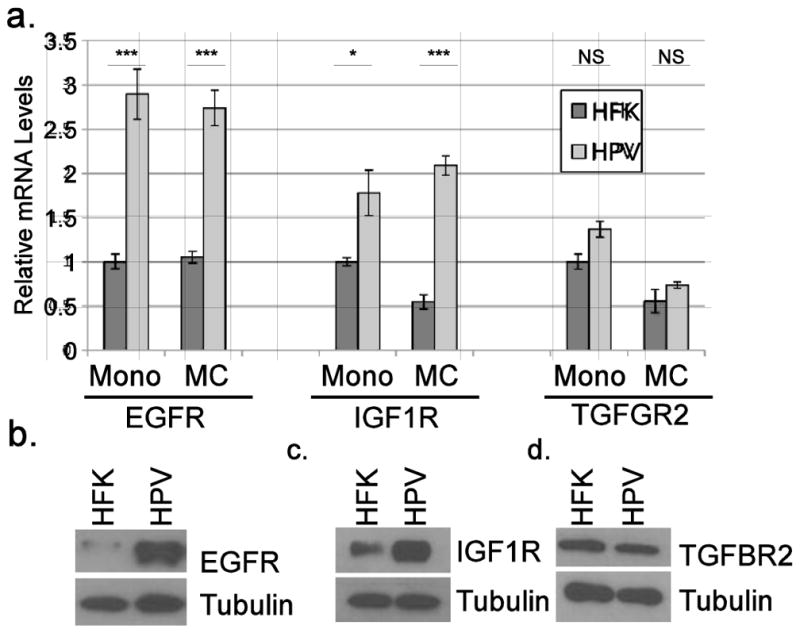
a. Total RNAs from HFKs or HPV16-containing keratinocytes were subjected to RT-qPCR using primers specific for EGFR, IGF1R, and TGFBR2. Values are the average of two independent donor backgrounds and bars represent ± one standard error of the mean. Representative western blots of total EGFR (b), IGF1R (c), and TGFBR2 (d) levels in the indicated cell lines. NS=not significant; *=p<0.05; ***=p<0.001.
E5 and E6 increase MET promoter activity
The levels of MET mRNA were higher in HPV-containing cells which led to the hypothesis that one or more HPV oncogenes are able to increase the activity of the MET promoter. To test this hypothesis, we employed a luciferase reporters containing the full length (3.1 kb) MET promoter (Hwang et al., 2011). Only low levels of reporter activity were observed when the reporters were transfected into HFK cells, but very high levels were seen in HPV-containing cells (Figure 4, note Y axis scale), confirming that HPV can increase transcription from the MET promoter. Next, we sought to identify the HPV gene product responsible for MET upregulation. Previous work has shown that MET transcription can be inhibited by p53 (Hwang et al., 2011). Because HPV16 encodes E6, which efficiently targets p53 for degradation (Howie et al., 2009), we hypothesized that the increase in MET transcripts was due to E6. MET reporters were transfected into cell lines stably expressing E6 and/or E7 from retroviral vectors. Cells expressing E6 with or without E7 activated the reporters (Figure 4), suggesting a role for E6 in increasing MET transcription. However, E6 was not sufficient to recapitulate the effect of the complete HPV16 genome, suggesting another viral product is necessary. Cells expressing E7 alone did not activate the promoter, and E7 seemed to dampen activation by E6.
Figure 4. E5 and E6 contribute to Met upregulation.

The indicated cell types were transfected with a Met promoter reporter construct (Hwang et al., 2011). 36 hours following transfection, luciferase activity was measured and normalized to Renilla luciferase internal control. Values are the average of at least 3 independent experiments and bars represent ± one standard error of the mean. ***=p<0.001 compared to HPV16 wild-type cells.
HPV16 encodes a third oncogene, E5. A possible role for E5 in MET transcription has been suggested in microarray data by others (Greco et al., 2011), but has not been further investigated. We employed cells stably maintaining the complete HPV16 genome harboring a translational stop mutation in the E5 open reading frame (E5 Stop cells). These mutant HPV genomes are maintained episomally and maintain E7 protein at levels similar to the wild-type HPV16 (Supplementary Figure 1a and b, see also (Genther et al., 2003)). Levels of p53 (as a surrogate for E6 expression) are not significantly altered, and mRNA levels of all three viral oncogenes are within 2 fold of wild type (Supplementary Figure 1 c and d). However, because of the stop mutation (confirmed by sequencing, not shown), E5 protein cannot be produced in E5 Stop cells. Upon transfection of MET promoter reporters into E5 Stop cells, we observed only moderate reporter activity, similar to cells containing E6 alone (p=0.31), but activity was much less than in cells containing the wild-type HPV genome, indicating that E5 is necessary for high level induction of Met by HPV16 (Figure 4). Repeated attempts to express E5 alone in keratinocytes were not successful. Altered levels of Met in E5 Stop cells cannot be attributed to differences in p53 levels because p53 levels do not differ significantly between E5 Stop cells and wild type HPV16-containing cells (Supplementary Figure 1b).
E5 is needed for induction of endogenous MET transcripts and protein by HPV16
To ensure that the results obtained using luciferase reporters accurately reflected endogenous MET promoter regulation, we used RT-qPCR to measure the levels of MET mRNA in cells containing various combinations of HPV16 oncogenes. As before, we noted that MET mRNA levels were significantly higher in cells containing the complete HPV16 genome than in uninfected HFKs (Figure 5a). Similar to the pattern observed in reporter assays, we found that E6/E7 expressed alone from a retroviral vector were not sufficient to recapitulate the high levels of MET transcripts seen in the cells containing the complete viral genome. Furthermore, HPV16 genomes lacking E5 expression were also unable to sustain elevated levels of MET mRNA, indicating that E5 is necessary to induce high levels of MET mRNA expression in HPV16-containing cells.
Figure 5. E5 is needed for increased of Met mRNA and protein.
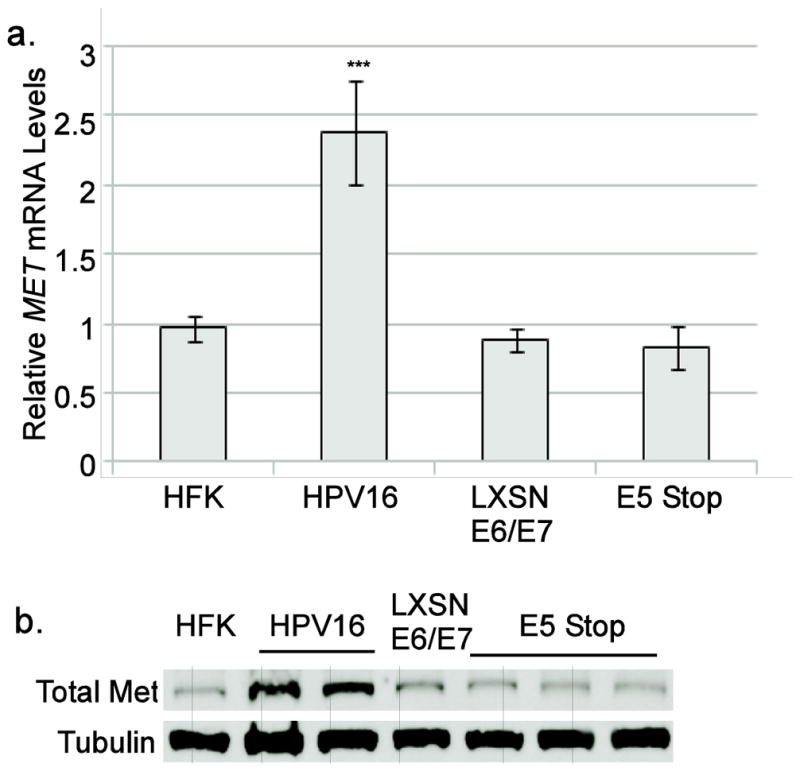
a. Total RNA from the indicated cell types were subjected to RT-qPCR using primers specific for Met. Values are the average of at least 3 independent experiments and bars represent ± one standard error of the mean. ***=p<0.001 vs. HFK. Other samples were not significant. b. Representative western blot of total Met levels in the indicated cell lines. Two independent wild-type HPV16 containing cell lines and three E5 Stop cell lines were included in this experiment.
To determine whether the ability of E5 to promote MET transcription would be reflected in higher Met protein levels, total protein lysates were harvested from cells containing HPV16 oncogenes. By immunoblotting, Met in cells containing the complete HPV16 genome was found to be higher than those seen in HFKs or E6/E7-expressing cells, confirming that E6 and E7 are not sufficient for the induction of high Met levels (Figure 5b). Met levels in E5 Stop cells were also reduced as compared to wild-type HPV16-containing cells, confirming that HPV16 E5 expression is necessary for HPV16-mediated upregulation of Met (Figure 5b). The requirement for E5 to increase both Met transcripts and protein levels was seen in the context of the complete viral genome and natural host cell type, giving strong support to the idea that Met is a physiological target of E5.
E5 increases MET transcripts through EGFR
E5 can potentiate activation of the EGFR, possibly by modifying vesicle trafficking and acidification, thereby prolonging receptor half-life and making EGFR more available for ligand binding(Dimaio and Petti, 2013). Activation of EGFR and downstream signaling targets can result in increased expression of Met (Bergstrom et al., 2000; Bonine-Summers et al., 2007; Seol et al., 2000; Velpula et al., 2012). These observations led us to hypothesize that E5 promotes the expression of Met by increasing EGFR levels and/or activation. First, to determine whether E5 expression could regulate the levels of EGFR in our cell system, levels of EGFR protein in HFKs and in cells containing either the wild-type or E5 Stop HPV16 genomes were examined. EGFR levels were notably higher in HPV-containing cells as compared to HFKs; however, cells containing the E5 Stop mutant did not show consistently increased levels of EGFR (Figure 6a). Using RT-qPCR, we found that the levels of EGFR mRNA were also higher in wild-type HPV16-containing cells than in HFKs and E5 Stop cells (Figure 6b), a pattern very similar to that seen for MET mRNA (Figure 5a). These results confirm that E5 can upregulate EGFR protein in the context of the complete viral genome and suggest that increased EGFR mRNA may contribute to this upregulation.
Figure 6. E5 enhances EGFR expression at both the protein and mRNA levels.

a. Representative western blot of total EGFR levels in the indicated cell lines. Two independent wild-type HPV16-containing cell lines and three E5 Stop cell lines were included in this experiment. b. Total RNA from the indicated cell types were subjected to RT-qPCR using primers specific for EGFR. Values are the average of six independent experiments and bars represent ± one standard error of the mean. *=p<0.05; ***=p<0.001.
To determine whether increased Met levels depend on EGFR, we transfected HFKs, HPV16-containing cells, or E5 Stop cells with the MET luciferase reporter plasmid and treated the cells with EGF or with the EGFR-specific inhibitor AG1478 for 24 hours. As before, cells lacking E5 expression displayed lower levels of MET reporter activity when treated with DMSO vehicle (p<0.01, Figure 7a). Treatment with EGF resulted in higher levels of MET promoter activity in cells with or without E5, but was only significant in cells containing wild-type HPV16, suggesting that the MET promoter is hypersensitive to EGF stimulation in the presence of E5. Importantly, AG1478 was able to reduce reporter expression in both basal and EGF-stimulated HPV16-containing cells, showing that the high basal level of MET transcripts seen in these cells requires EGFR activity. To corroborate these reporter data, we treated cells with EGF and/or AG1478 and examined Met levels in lysates by immunoblotting. EGF treatment trended toward increased levels of Met protein in cells containing wild-type HPV16, but not in HFK cells or cells containing E5 Stop mutant genomes. Treatment with AG1478 resulted in reduced Met protein levels in HPV containing cells regardless of the presence of E5 (Figure 7b and c). A corresponding increase in phosphorylated Met upon EGF treatment and decrease upon AG1478 treatment was also observed in wild type HPV16-containing cells but not HFKs or E5 Stop cells (Supp Figure 2a). Together these data show that E5-dependent upregulation of Met requires EGFR activity, and that E5 contributes to the ability of EGF to upregulate Met, reflecting an alteration of EGFR signaling by E5.
Figure 7. Met upregulation by E5 requires EGFR activity.

a. HFKs or keratinocytes containing wild-type or E5 Stop HPV16 genomes were transfected with the full-length MET reporter plasmid and treated with EGF or AG1478 for 24 hours. Luciferase activity was normalized to Renilla luciferase internal control. Values are the average of 3 independent experiments and bars represent ± one standard error of the mean. b. HFKs or keratinocytes containing wild-type or E5 Stop HPV16 genomes were treated with EGF or AG1478 for 24 hours and the levels of total Met were measured using western blotting. c. Densitometric analysis of 3 independent experiments. NS=not significant, *=p<0.05, **=p<0.01.
E5 promotes HGF-dependent motility
Met signaling controls many cellular processes, but the most distinctive is promoting cellular motility and invasion (Boccaccio and Comoglio, 2006; Gherardi et al., 2012; Trusolino et al., 2010). HPV oncogenes also have been shown to increase motility and invasiveness through several proposed mechanisms (Barbaresi et al., 2010; Charette and McCance, 2007; Liao et al., 2013). Because cells containing E5 Stop HPV16 genomes have reduced levels of Met as compared to wild-type, we predicted that these cells should show lower levels of Met-dependent motility. To determine whether the motility of keratinocytes containing HPV16 genomes is regulated by E5, live cell time-lapse video microscopy was used to observe the closure of a scratch wound in a cell monolayer. Cells harboring E5 Stop genomes closed experimental wounds more slowly than cells containing wild-type HPV with or without the addition of HGF (Figure 8), indicating that E5 promotes cell motility in the context of the complete viral genome, consistent with lower levels of Met, and thus a poor response to HGF, in these cells.
Figure 8. E5 promotes motility of HPV16-containing keratinocytes.
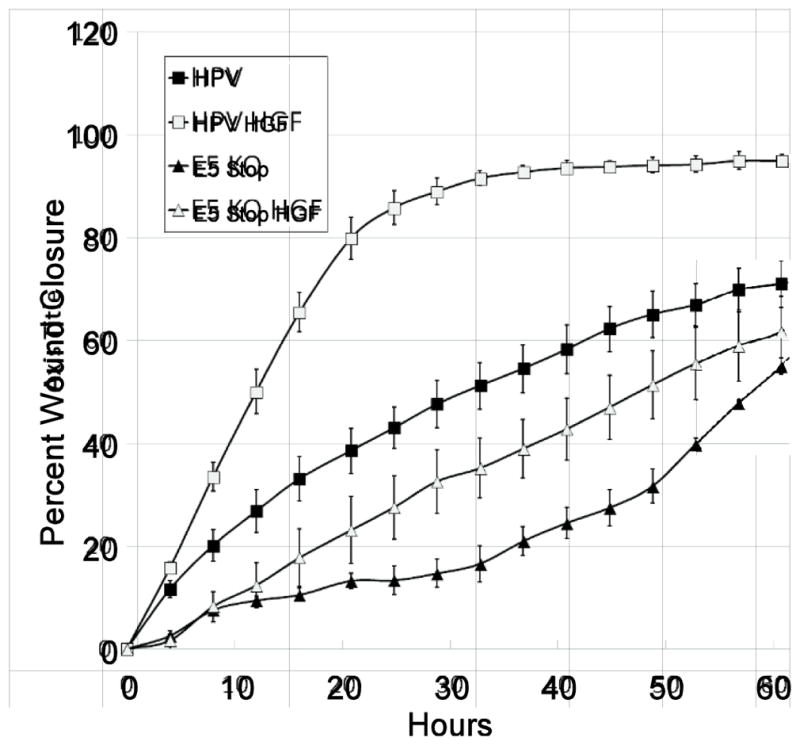
HFKs or keratinocytes containing wild-type or E5 Stop HPV16 genomes were plated into ImageLock 96 well plates and allowed to attach overnight. A wound was created by using the WoundMaker-96 and media was replaced with or without the 33 ng/ml HGF. Cells were returned to the IncuCyte ZOOM and subjected to live cell imaging, with images acquired every four hours. Wound closure over time during a representative experiment was calculated. Values represent the mean of four wells and bars represent ± one standard error of the mean.
Met supports HPV16 early and late transcript levels and episomal copy number
While these experiments have shown that HPV16 regulates Met expression in a manner dependent upon E5-induced EGFR activity and that Met levels influence cell motility, whether Met aids the viral life cycle was unclear. To determine if Met signaling can influence HPV16 gene expression, transcript levels from both the early and late viral promoters in HPV16-containing cells were examined. Cells were grown in mono or MC containing either HGF or the Met-specific inhibitor SU11274. We confirmed that HGF and SU11274 were both active on Met phosphorylation in HPV16 cells as expected (Supp Figure 2b). Interestingly, cells treated with HGF showed increased levels of both the early (E6-7) and late (E1^E4) transcripts in cells grown in methylcellulose (Figure 9a). Furthermore, cells treated with SU11274 showed reduced levels of early and late transcripts in monolayer cultures and a failure to activate viral transcription in methylcellulose cultures (Figure 9b), suggesting that Met signaling is needed for proper HPV16 gene expression. Because viral genomes are frequently found to be integrated in HPV-induced cancers (McBride and Warburton, 2017), we investigated whether the effects of the Met pathway would also be seen on transcription from integrated HPV16 genomes. Cells containing integrated HPV16 genomes were generated in the same manner as the episomal HPV16-containing cells, from transfection of primary human foreskin keratinocytes, with integration being confirmed by Southern blotting (not shown). Cells containing integrated (not shown) HPV16 were treated with HGF or SU11274. Similar to the effects seen in cells containing episomal HPV16, transcripts from integrated HPV16 genomes were increased upon treatment with HGF and decreased by SU11274 treatment (Figure 9c). These data indicate the Met pathway could be important for regulating HPV16 oncogene expression even following integration, and that increased levels of Met during cervical carcinogenesis (Baykal et al., 2003; Shimabukuro et al., 2001; Walker et al., 2003; Zhang et al., 2014) may act in part by regulating viral gene expression.
Figure 9. Met promotes HPV16 gene expression.

Keratinocytes containing either episomal (a,b) or integrated (c) HPV16 genomes were grown in either monolayer (Mono) or in 1.6% methylcellulose (MC) culture medium containing either 33 ng/ml HGF (a,c) or 10 μM SU11274 (b,c) for 24 hours. Total RNA was isolated and levels of early (E6-7) or late (E1^E4) transcripts were measured by RT-qPCR. *=p<0.05, **=p<0.01, ***p<0.001.
To confirm the effect of Met on HPV16 gene expression, Met was knocked down in HPV16-containing cells using lentivirally-expressed shRNA (Figure 10a). The number of HPV16 genome copies maintained in cells was reduced when Met was knocked down (Figure 10b), although these genomes remained present as episomes (Figure 10c). Cells with Met knocked down exhibited decreased baseline levels of transcripts derived from the early (E6/E7) and late (E1^E4) promoters in monolayer as well as a failure to upregulate transcripts upon methylcellulose-induced differentiation (Figure 10d). These results show that Met expression and activity are important for maintaining HPV16 genome copy number and transcription during both early and late stages of the viral life cycle.
Figure 10. Met knockdown affects HPV16 gene expression and viral copy number.
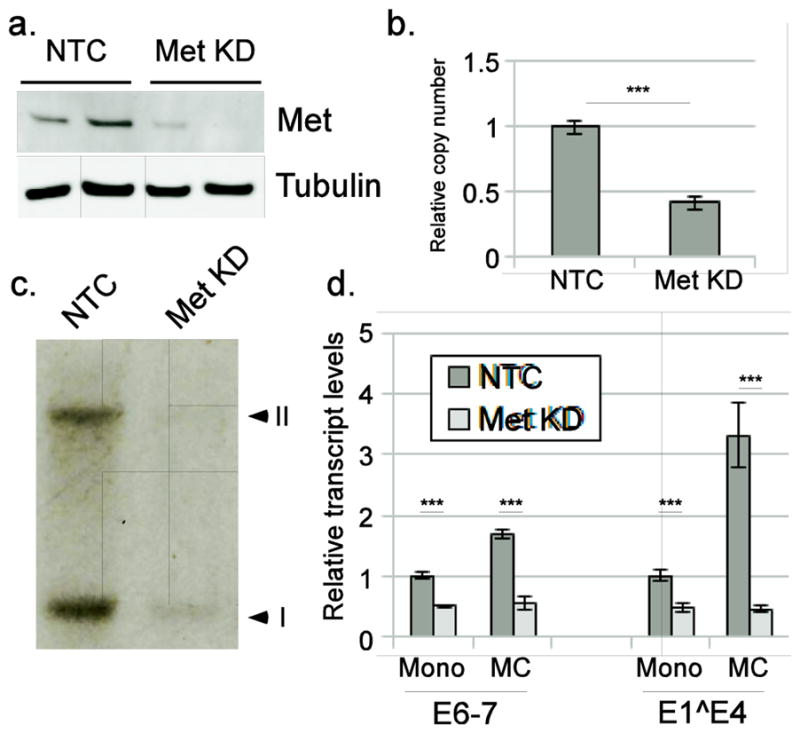
Keratinocytes containing the complete episomal HPV16 genome were infected with lentivirus expressing shRNA against Met (Met KD) or a non-target control (NTC), selected, and expanded in culture. a. Representative western blot (n=5) showing knockdown of Met in two independent backgrounds. b. qPCR showing the copy number of viral genomes. c. Southern Blot showing the banding pattern characteristic of episomal genome maintenance, with supercoiled (I) and nicked circular forms (II). d. Cells were cultured in monolayer (Mono) or 1.6% methylcellulose (MC) for 24 hours. Transcript levels for early (E6-7) and late (E1^E4) transcripts were measured using RT-qPCR. *** p<0.001.
Discussion
In most cases, infection by HPV causes only benign growth; progression to cancer is relatively rare, even for high risk viral types, but the sheer number of infections makes HPV-induced cancers very common (Parkin and Bray, 2006; zur Hausen, 2009). The HPV life cycle requires a balance between forced cell cycling on one hand (due to the activities of the viral oncogenes E5, E6, and E7) and maintenance of cellular differentiation on the other (Moody and Laimins, 2010; Munger et al., 2004). Alterations in this balance are thought to be the basis of HPV-induced cancer progression, but the specific events driven by high risk HPVs that lead to malignant progression from benign precursor lesions are only partially understood. Progression toward malignancy is detrimental to the HPV life cycle, in part because of lost differentiation potential to regulate viral late gene expression (Doorbar et al., 2012) and frequent integration of the viral genome into the host DNA (McBride and Warburton, 2017; Pett and Coleman, 2007). Furthermore, low risk HPV types use broadly similar life cycle strategies, promote unscheduled cell proliferation, replicate to high levels, and are evolutionarily successful in the absence of viral factors that foster malignant progression (Doorbar et al., 2012; Klingelhutz and Roman, 2012). Why cancer-promoting features are evolutionarily maintained in high risk HPV life cycles while low risk types are successful under similar conditions remains unclear. Therefore, it is necessary to better understand how cancer-associated factors induced by HPV oncogenes influence the evolved viral life cycle in the context of benign infection.
Here we report that levels of a host factor associated with advanced or metastatic cancers, Met, is increased by HPV in a model of early, benign HPV infection. Met-induced signaling promotes cellular proliferation, cell motility, and invasiveness (Boccaccio and Comoglio, 2006; Gherardi et al., 2012; Steffan et al., 2011; Trusolino et al., 2010) and is thus an important factor during later stages of cancer development (Boccaccio and Comoglio, 2006; Steffan et al., 2011). Our observations imply that E5-induced Met might be a factor promoting metastasis of HPV-containing malignancies, thus uncovering a potential new role for E5 in HPV pathogenesis. E5 has previously been found to promote motility and invasion (Barbaresi et al., 2010; Genther Williams et al., 2005; Liao et al., 2013; Maufort et al., 2010), and the presence of E5 in cancer is correlated with poor prognosis (Hsieh et al., 2000; Venuti et al., 2011). However, the role of E5 in the benign viral life cycle and the significance of E5 for cancer progression are unclear. During progression of HPV-associated cancers, viral integration into the host genome frequently results in the loss of the E2/E4/E5 region of the HPV DNA (Baker et al., 1987; Schwarz et al., 1985; zur Hausen, 1996), presumably resulting in the loss of E5 expression. However, E5 can be detected in some cervical cancer cell lines (Sahab et al., 2012). While E5 is not expressed in all HPV-induced cancers, E5-induced Met expression may contribute to cancer metastasis in cases in which E5 is present. In addition, a selective growth advantage provided by the ability of E5 to promote high levels of Met may explain why many cancers harboring HPV16 contain a high proportion of episomes rather than solely integrated HPV16 genomes (Cheung et al., 2013; Kristiansen et al., 1994; Woodman et al., 2007).
In our experiments, E6 and E7 expression were not sufficient to increase Met to the levels present in cells containing the complete HPV16 genome, although E6 was able to enhance Met promoter activity moderately, consistent with the ability of E6 to induce degradation of p53, which inhibits the Met promoter (Hwang et al., 2011). Whether the modest effect of E6 is mediated through p53 degradation remains to be seen. Because E6 and E7 are both needed for stable maintenance of the HPV16 genome, we are unable to test whether these proteins influence Met expression when expressed from the context of the genome as we were able to determine in the case of E5. Conversely, experiments using a variety of approaches to test whether E5 alone is sufficient to mediate upregulation of Met in keratinocytes were not successful. Nonetheless, we have found that upregulation of Met requires EGFR signaling, consistent with the known activities of E5. Whether E5 can bind to EGFR under physiological conditions is controversial, but E5-mediated protein stabilization is thought to be a primary mechanism for enhancement of EGFR signaling (Dimaio and Petti, 2013). Our studies have found that EGFR transcripts are also increased in HPV16-containing cells in an E5 dependent manner (Figure 6b). Studies using previously immortalized NIKS cells maintaining E5 mutant genomes suggested that E5 does not affect EGFR protein levels (Genther et al., 2003; Tummers et al., 2015). However, our cells were immortalized by the HPV16 genome itself, which may result in a different signaling environment than other forms of immortalization, but direct comparative experiments would be required to confirm this idea. Others have shown that E5 overexpression can increase EGFR levels in cells containing episomal genomes (Tummers et al., 2015). In any case, the mechanism by which E5 increases EGFR transcripts remains to be explored.
We observed that Met levels were enhanced by EGF treatment in cells containing E5, but only modestly in cells lacking E5. This finding suggests that E5 may cause a qualitative change in EGFR signaling, not just a quantitative change in EGFR levels (Figure 7). EGFR and Met also engage in extensive crosstalk and have overlapping effects on cell motility and proliferation (Accornero et al., 2012; Bergstrom et al., 2000; Bonine-Summers et al., 2007; Gherardi et al., 2012; Velpula et al., 2012). It is possible that some of the effects we have attributed to Met or EGFR may depend on a contribution from the other protein. Additionally, knowing whether E5, Met, and EGFR can form a complex in cells, how E5 alters the activity or regulation of these receptors with respect to one another, and whether the effects of E5 on motility and other cell behaviors is mediated through Met alone or also through EGFR would be important.
We found that Met influences transcription from both the early and late promoters of HPV16 and, probably as a consequence, is important for maintaining the normal viral copy number (Figure 9 and 10). We do not yet know the mechanism by which Met signaling regulates viral gene expression. However, the transcriptional effect of Met was most marked in in cells undergoing differentiation in methylcellulose, suggesting that Met may function in differentiated HPV-containing cells or that Met is needed to create conditions important for subsequent viral gene expression upon differentiation. Interestingly, we also observed that Met influences the transcription of integrated HPV16 genomes (Figure 9c). These findings indicate that in addition to promoting viral gene expression during the normal viral life cycle, Met may also promote oncogene expression in lesions with integrated genomes, thus helping to drive lesion progression and severity.
Met upregulation would presumably make HPV-containing cells more sensitive to HGF present in the cellular environment. Preliminary studies have found that HGF is not expressed in HPV-containing keratinocytes, but HGF is expressed in the feeder fibroblasts present in our culture system (MS and JB, unpublished data), consistent with the tumor stroma being a major source of HGF and other growth factors in vivo (Kankuri et al., 2005; Matsumoto and Nakamura, 2006; Steffan et al., 2011; Trusolino et al., 2010). Stromal factors are important for tumor growth, but a full picture of how factors derived from the stroma may affect the normal, benign life cycle of HPV is a critical area for future investigation.
Materials and Methods
Cell culture, Southern blotting, and reagents
Human foreskin keratinocytes (HFK) were isolated from neonatal foreskins by enzymatic disaggregation as previously described (Bodily et al., 2011a). HFKs and HFK-derived cell lines were cultured in E medium with 5% FBS on mitomycin C-treated NIH3T3 feeder fibroblast monolayers (Bodily et al., 2011a; Wilson and Laimins, 2005). Differentiation was induced by suspending cells in E medium containing 1.6% methylcellulose (MC) for 24 hours, followed by washing with PBS (Wilson and Laimins, 2005). Cell lines containing episomal HPV16 or E6/E7 oncogenes expressed from retroviral vectors were previously described (Bodily et al., 2013; Bodily et al., 2011a). HPV16 genomes containing a translational Stop codon at amino acid 11 in the E5 open reading frame (E5 Stop) were created by mutagenesis of wild-type pEGFP Ni HPV16 plasmid with the QuickChange II Site Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA) using the primers listed in Table 1. Keratinocytes containing the HPV16 E5 Stop genome were derived by transfection of HFKs followed by G418 selection using the protocol previously described (Bodily et al., 2011a). The presence of the mutation in E5 Stop cells was confirmed by sequencing. Episomal maintenance of the virus was confirmed by Southern analysis: total DNAs were isolated and digested with XhoI (which does not cut the HPV16 genome) before being analyzed by Southern analysis using the whole HPV16 genome as a probe as described previously (Bodily et al., 2011a). HFKs containing integrated HPV16 genomes were isolated when episomal HPV16 spontaneously integrated during routine cell culture. Integrated status of the HPV16 genome is confirmed using Southern blotting. Hepatocyte growth factor (HGF; EMD Millipore, Billerica, MA) was reconstituted in 0.1% BSA and used at 33 ng/ml. Epidermal growth factor (EGF; Sigma-Aldrich; St. Louis, MO) was reconstituted in 10mM acetic acid and used at 100 ng/ml. SU11274 and AG1478 (Sigma-Aldrich) were dissolved in DMSO and used at 10 μM and 1 μM, respectively (final).
Table 1.
Antibodies used in this study
| Name | Company | Catalog # |
|---|---|---|
| c-Met (western) | Invitrogen | 71-8000 |
| c-Met (IF) | R&D Systems | AF276 |
| EEA1 | BD Transduction | 610457 |
| p-Met (Tyr1234/1235) | Cell Signaling | 3126 |
| Anti-goat Dylight 594 | Jackson IR | 715-585-150 |
| Anti-mouse FITC | Jackson IR | 705-095-147 |
| Tubulin | Neo Markers | MS581P |
| EGFR | Cell Signaling | 2232 |
| HPV16 E7 | Valdospan, Gmbh | VS13004L |
shRNA-mediated Met knockdown
Keratinocytes containing the complete HPV16 genome were plated at a density of 500,000 cells per well in a 6-well plate for 24 hours. Culture media containing lentiviral particles encoding an shRNA against Met (TRC000019443, Sigma-Aldrich) were mixed with polybrene at a concentration of 8 μg/ml. This mixture was added to the cells for overnight infection. Following infection, infected cells were selected by the addition of 1 μg/ml puromycin for a period of two weeks. Following selection and expansion, DNA was harvested and subjected to Southern blotting to ensure the HPV16 genome remained episomal. RT-qPCR and immunoblotting were performed to ensure Met knockdown. Knockdown was repeated 5 times in two HFK donor backgrounds.
Immunoblotting and quantitative PCR
Protein lysates were prepared by adding 1x Cell Signaling Lysis Buffer (Cell Signaling, supplemented with 1 mM PMSF) to cells and incubating on ice for 5 minutes followed by scraping, brief sonication, and clarification by centrifugation. SDS-PAGE and Western blotting were performed as essentially as described previously (Nakamura et al., 2009), except SDS was excluded from the gels. Blocking and antibody dilution was performed using Li-Cor Odyssey® blocking buffer containing 0.1% tween-20 and images were acquired using a Li-Cor Odyssey® near infrared imaging system. Images included in the figures were unsaturated. Antibodies used in this study are shown in Table 1. RNA-STAT 60 (TelTest, Friendswood, TX) was used to isolate total RNAs which were subjected to reverse transcription using qScript™ cDNA SuperMix (Quanta Biosciences, Gaithersburg, MD). Quantitative PCR was performed using the PerfeCTa® SYBR® Green SuperMix ROX (Quanta) on an Applied Biosystems StepOne Plus™ real time PCR instrument. Values were normalized to Cyclophilin A as an internal control (Steele et al., 2002) and data are presented as a fold change in gene expression relative to the control within each experiment. qPCR to measure viral genome copy number used the E6-7 primer pair with total cellular DNAs as template, and values were normalized to GAPDH as an internal control. PCR primers used in this study are shown in Table 2. Statistical significance was calculated using Welch’s unequal variances t test.
Table 2.
Oligonucleotides used in this study
| Target | Primer Name | Sequence (5′-3′) |
|---|---|---|
| E5 Stop | 16 E5 Stop 5′ | TGATACTGCATCCACAACATaACTGGCGTGCTTTTTGCTT |
| 16 E5 Stop 3′ | AAGCAAAAAGCACGCCAGTtATGTTGTGGATGCAGTATCA | |
| Met | qcMet 5′ | GTGAGATGTCTCCAGCATTTTTACG |
| qcMet 3′ human | AGAGGACTTCGCTGAATTGACC | |
| EGFR | qEGFR 3′ | CATATTTCCTCTGATGATCTGCAGG |
| qEGFR 5′ | GAGGTGGTCCTTGGGAATTTG | |
| IGFR | qIGFR 3′ | CTTGGAGCATCTGAGCAGAAGTAA |
| qIGFR 5′ | GATCCACGACGGCGAGTG | |
| TGFβRII | qTGFRII 5′ | GAGCAACTGCAGCATCACCTC |
| qTGFRII 3′ | GGGGAGCTTGGGGTCATG | |
| FGFR2 | FGFR2 5′ | CCTCCCAGAGACCAACGTTC |
| FGFR2 3′ | TGAATACTGTTCGAGAGGTTGGC | |
| Cyclophilin A | qCyc 5′ | TGTACTATTAGCCATGGTCA |
| qCyc 3′ | CAGAGCACGAAAATTTTCTG | |
| 16 E6-7 | E6-7 5′ | GAACACGTAGAGAAACCCAGCTGTA |
| E6-7 3′ | GCTCATAACAGTAGAGATCAGTTGTCTCT | |
| 16 E1^E4 splice | E1^E4 5′ | CAGAAACCATAATCTACCATGGCTG |
| E1^E4 3′ | TGTGTTTCTTCGGTGCCCA | |
| GAPDH | GAPDH chip 5′ | CCAATGTGTCCGTCGTGGA |
| GAPDH chip 3′ | GTTGAAGTCGCAGGAGACACC |
Luciferase
MET luciferase reporters were constructed in Paolo Comoglio’s laboratory (Gambarotta et al., 1996) and were generously provided by Alexander Nikitin (Hwang et al., 2011). Cells were transfected with reporter plasmids using polyethyleneimine (PEI; Polysciences, Warrington, PA) as described (Bodily et al., 2011b). Cell lysates were harvested 36 hours after transfection and luciferase activity was measured using the Dual-Luciferase® Reporter Assay System (Promega, Madison, WI) according to manufacturer’s instructions with Renilla luciferase as an internal control.
Immunofluorescence
Cells were plated to approximately 50% confluency on glass coverslips in a 6-well plate overnight. Following treatment with HGF, cells were fixed in 4% paraformaldehyde, washed twice with PBS, and blocked for 30 minutes in 10% donkey serum in PBS with 0.1% saponin (DSP). Met and EEA1 primary antibodies were added simultaneously in DSP for 2 hours at room temperature (see Table 1). After washing three times with PBS, secondary antibodies conjugated to Dylight 594 and FITC were added in DSP for 2 hours at room temperature. Coverslips were washed again three times with PBS and then mounted with DAPI Slowfade (Life Technologies, Grand Island, NY). Images were taken on a Leica TCS SP5 Confocal Microscope at 60x magnification with oil immersion and enhanced using ImageJ software.
Motility assays
Feeder cells were removed and 20–30,000 cells were seeded in each well of a 96-well ImageLock plate (Essen BioScience, Ann Arbor, MI) and incubated overnight to allow cell attachment. Following wounding with the WoundMaker-96 (Essen BioScience), the plate was washed with PBS and E medium (containing HGF, if applicable) was added. The plate was placed into the IncuCyte™ ZOOM (Essen BioScience) and the IncuCyte Control Software (v 2013A) was programmed to scan the entire plate every 4 hours with phase imaging using a 10X objective. After data collection, integrated software was used to analyze collected images by creating a processing definition from representative images to train the software to identify the scratch wound and calculate the confluence of cells within each well. This definition was used to analyze the experiment for Relative Wound Density, which is the normalization of wound confluence to the starting confluence behind the scratch. Following analysis, data were exported and graphed.
Supplementary Material
Supplementary Figure 1. Characterization of E5 Stop cells. a. Southern blot of total cellular DNA digested with restriction enzymes that either do not cut the HPV16 genome (U) or cut it once (C). Supercoiled (I), nicked circular (II), linearized (III), or multimeric (multi) DNA forms are indicated. Western blots showing the levels of E7 (b) and p53 (c) in the designated cell type. (d) RT-qPCR measuring the levels of HPV16 transcripts from the E6, E7, and E7 ORFs. *=p<0.05; NS=not significant.
Supplementary Figure 2. Western Blot of (a) phosphorylated Met (p-Met) and tubulin in HFK cells, HPV16-containing cells and E5 Stop cells treated with DMSO (D), EGF (E), AG1478 (A) or both EGF and AG1478 (E/A) and (b) phosphorylated Met (p-Met), total Met, and tubulin as a loading control from HPV16-containing cells treated with DMSO (V), HGF, or SU11274 (SU).
Highlights.
Human papillomavirus type 16 increases the levels of Met in infected keratinocytes.
EGFR upregulation by oncoprotein E5 is primarily responsible for Met upregulation, with E6 playing a minor role.
E5 increases the motility of HPV-containing keratinocytes.
Met activity is needed for proper viral gene expression and genome copy number maintenance.
Acknowledgments
Funding
for this work was provided by the LSUHSC Research Council, the Feist-Weiller Cancer Center, the Biomedical Research Foundation of Northwest Louisiana, and grants from the National Institutes of Health (8 P20 GM103433, R01AI118904)
We thank Cynthia Rodriguez for technical assistance and Paolo Comoglio and Alexander Nikitin for the MET promoter reporter constructs. Support for this work was provided by the LSUHSC Research Council, the Feist-Weiller Cancer Center, the Biomedical Research Foundation of Northwest Louisiana, and grants from the National Institute of General Medical Sciences (P30-GM110703) and the National Institute of Allergy and Infectious Diseases (R01AI118904). The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Feist-Weiller Cancer Center.
Footnotes
Conflict of interest: The authors have no conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Accornero P, Miretti S, Bersani F, Quaglino E, Martignani E, Baratta M. Met receptor acts uniquely for survival and morphogenesis of EGFR-dependent normal mammary epithelial and cancer cells. PloS One. 2012;7:e44982. doi: 10.1371/journal.pone.0044982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CC, Phelps WC, Lindgren V, Braun MJ, Gonda MA, Howley PM. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. Journal of Virology. 1987;61:962–971. doi: 10.1128/jvi.61.4.962-971.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbaresi S, Cortese MS, Quinn J, Ashrafi GH, Graham SV, Campo MS. Effects of human papillomavirus type 16 E5 deletion mutants on epithelial morphology: functional characterization of each transmembrane domain. The Journal of General Virology. 2010;91:521–530. doi: 10.1099/vir.0.016295-0. [DOI] [PubMed] [Google Scholar]
- Baykal C, Ayhan A, Al A, Yuce K. Overexpression of the c-Met/HGF receptor and its prognostic significance in uterine cervix carcinomas. Gynecologic Oncology. 2003;88:123–129. doi: 10.1016/s0090-8258(02)00073-2. [DOI] [PubMed] [Google Scholar]
- Bergstrom JD, Westermark B, Heldin NE. Epidermal growth factor receptor signaling activates met in human anaplastic thyroid carcinoma cells. Experimental Cell Research. 2000;259:293–299. doi: 10.1006/excr.2000.4967. [DOI] [PubMed] [Google Scholar]
- Boccaccio C, Comoglio PM. Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nature Reviews Cancer. 2006;6:637–645. doi: 10.1038/nrc1912. [DOI] [PubMed] [Google Scholar]
- Bodily JM, Hennigan C, Wrobel GA, Rodriguez CM. Regulation of the human papillomavirus type 16 late promoter by E7 and the cell cycle. Virology. 2013;443:11–19. doi: 10.1016/j.virol.2013.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodily JM, Mehta KP, Cruz L, Meyers C, Laimins LA. The E7 Open Reading Frame Acts in cis and in trans To Mediate Differentiation-Dependent Activities in the Human Papillomavirus Type 16 Life Cycle. Journal of Virology. 2011a;85:8852–8862. doi: 10.1128/JVI.00664-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodily JM, Mehta KP, Laimins LA. Human papillomavirus E7 enhances hypoxia-inducible factor 1-mediated transcription by inhibiting binding of histone deacetylases. Cancer Research. 2011b;71:1187–1195. doi: 10.1158/0008-5472.CAN-10-2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonine-Summers AR, Aakre ME, Brown KA, Arteaga CL, Pietenpol JA, Moses HL, Cheng N. Epidermal growth factor receptor plays a significant role in hepatocyte growth factor mediated biological responses in mammary epithelial cells. Cancer Biology & Therapy. 2007;6:561–570. doi: 10.4161/cbt.6.4.3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caberg JH, Hubert PM, Begon DY, Herfs MF, Roncarati PJ, Boniver JJ, Delvenne PO. Silencing of E7 oncogene restores functional E-cadherin expression in human papillomavirus 16-transformed keratinocytes. Carcinogenesis. 2008;29:1441–1447. doi: 10.1093/carcin/bgn145. [DOI] [PubMed] [Google Scholar]
- Charette ST, McCance DJ. The E7 protein from human papillomavirus type 16 enhances keratinocyte migration in an Akt-dependent manner. Oncogene. 2007;26:7386–7390. doi: 10.1038/sj.onc.1210541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng YM, Chou CY, Hsu YC, Chen MJ, Wing LY. The role of human papillomavirus type 16 E6/E7 oncoproteins in cervical epithelial-mesenchymal transition and carcinogenesis. Oncology Letters. 2012;3:667–671. doi: 10.3892/ol.2011.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung JL, Cheung TH, Yu MY, Chan PK. Virological characteristics of cervical cancers carrying pure episomal form of HPV16 genome. Gynecologic Oncology. 2013;131:374–379. doi: 10.1016/j.ygyno.2013.08.026. [DOI] [PubMed] [Google Scholar]
- Chmielowiec J, Borowiak M, Morkel M, Stradal T, Munz B, Werner S, Wehland J, Birchmeier C, Birchmeier W. c-Met is essential for wound healing in the skin. The Journal of Cell Biology. 2007;177:151–162. doi: 10.1083/jcb.200701086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirri P, Chiarugi P. Cancer-associated-fibroblasts and tumour cells: a diabolic liaison driving cancer progression. Cancer Metastasis Reviews. 2012;31:195–208. doi: 10.1007/s10555-011-9340-x. [DOI] [PubMed] [Google Scholar]
- Craene BD, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nature Reviews Cancer. 2012;13:97–110. doi: 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- D’Costa ZJ, Jolly C, Androphy EJ, Mercer A, Matthews CM, Hibma MH. Transcriptional repression of E-cadherin by human papillomavirus type 16 E6. PloS One. 2012;7:e48954. doi: 10.1371/journal.pone.0048954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Boon JA, Pyeon D, Wang SS, Horswill M, Schiffman M, Sherman M, Zuna RE, Wang Z, Hewitt SM, Pearson R, Schott M, Chung L, He Q, Lambert P, Walker J, Newton MA, Wentzensen N, Ahlquist P. Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E3255–3264. doi: 10.1073/pnas.1509322112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimaio D, Petti LM. The E5 proteins. Virology. 2013;445:99–114. doi: 10.1016/j.virol.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorbar J, Egawa N, Griffin H, Kranjec C, Murakami I. Human papillomavirus molecular biology and disease association. Reviews in Medical Virology. 2015;25(Suppl 1):2–23. doi: 10.1002/rmv.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR, Stanley MA. The biology and life-cycle of human papillomaviruses. Vaccine. 2012;30(Suppl 5):F55–70. doi: 10.1016/j.vaccine.2012.06.083. [DOI] [PubMed] [Google Scholar]
- Drake LE, Macleod KF. Tumour suppressor gene function in carcinoma-associated fibroblasts: from tumour cells via EMT and back again? The Journal of Pathology. 2014;232:283–288. doi: 10.1002/path.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy CL, Phillips SL, Klingelhutz AJ. Microarray analysis identifies differentiation-associated genes regulated by human papillomavirus type 16 E6. Virology. 2003;314:196–205. doi: 10.1016/s0042-6822(03)00390-8. [DOI] [PubMed] [Google Scholar]
- Egawa N, Doorbar J. The low-risk papillomaviruses. Virus Research. 2017;231:119–127. doi: 10.1016/j.virusres.2016.12.017. [DOI] [PubMed] [Google Scholar]
- Faghihloo E, Akbari A, Adjaminezhad-Fard F, Mokhtari-Azad T. Transcriptional regulation of E-cadherin and oncoprotein E7 by valproic acid in HPV positive cell lines. Iranian Journal of Basic Medical Sciences. 2016;19:601–607. [PMC free article] [PubMed] [Google Scholar]
- Fehrmann F, Klumpp DJ, Laimins LA. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. Journal of Virology. 2003;77:2819–2831. doi: 10.1128/JVI.77.5.2819-2831.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambarotta G, Boccaccio C, Giordano S, Ando M, Stella MC, Comoglio PM. Ets up-regulates MET transcription. Oncogene. 1996;13:1911–1917. [PubMed] [Google Scholar]
- Geiger T, Sabanay H, Kravchenko-Balasha N, Geiger B, Levitzki A. Anomalous features of EMT during keratinocyte transformation. PloS One. 2008;3:e1574. doi: 10.1371/journal.pone.0001574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genther SM, Sterling S, Duensing S, Munger K, Sattler C, Lambert PF. Quantitative role of the human papillomavirus type 16 E5 gene during the productive stage of the viral life cycle. Journal of Virology. 2003;77:2832–2842. doi: 10.1128/JVI.77.5.2832-2842.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genther Williams SM, Disbrow GL, Schlegel R, Lee D, Threadgill DW, Lambert PF. Requirement of epidermal growth factor receptor for hyperplasia induced by E5, a high-risk human papillomavirus oncogene. Cancer Research. 2005;65:6534–6542. doi: 10.1158/0008-5472.CAN-05-0083. [DOI] [PubMed] [Google Scholar]
- Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nature Reviews Cancer. 2012;12:89–103. doi: 10.1038/nrc3205. [DOI] [PubMed] [Google Scholar]
- Gius D, Funk MC, Chuang EY, Feng S, Huettner PC, Nguyen L, Bradbury CM, Mishra M, Gao S, Buttin BM, Cohn DE, Powell MA, Horowitz NS, Whitcomb BP, Rader JS. Profiling microdissected epithelium and stroma to model genomic signatures for cervical carcinogenesis accommodating for covariates. Cancer Research. 2007;67:7113–7123. doi: 10.1158/0008-5472.CAN-07-0260. [DOI] [PubMed] [Google Scholar]
- Greco D, Kivi N, Qian K, Leivonen SK, Auvinen P, Auvinen E. Human papillomavirus 16 E5 modulates the expression of host microRNAs. PloS One. 2011;6:e21646. doi: 10.1371/journal.pone.0021646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hellner K, Mar J, Fang F, Quackenbush J, Munger K. HPV16 E7 oncogene expression in normal human epithelial cells causes molecular changes indicative of an epithelial to mesenchymal transition. Virology. 2009;391:57–63. doi: 10.1016/j.virol.2009.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie HL, Katzenellenbogen RA, Galloway DA. Papillomavirus E6 proteins. Virology. 2009;384:324–334. doi: 10.1016/j.virol.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CH, Tsao YP, Wang CH, Han CP, Chang JL, Lee JY, Chen SL. Sequence variants and functional analysis of human papillomavirus type 16 E5 gene in clinical specimens. Archives of Virology. 2000;145:2273–2284. doi: 10.1007/s007050070020. [DOI] [PubMed] [Google Scholar]
- Hwang CI, Matoso A, Corney DC, Flesken-Nikitin A, Korner S, Wang W, Boccaccio C, Thorgeirsson SS, Comoglio PM, Hermeking H, Nikitin AY. Wild-type p53 controls cell motility and invasion by dual regulation of MET expression. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:14240–14245. doi: 10.1073/pnas.1017536108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung YS, Kato I, Kim HR. A novel function of HPV16-E6/E7 in epithelial-mesenchymal transition. Biochemical and biophysical Research communications. 2013;435:339–344. doi: 10.1016/j.bbrc.2013.04.060. [DOI] [PubMed] [Google Scholar]
- Kankuri E, Cholujova D, Comajova M, Vaheri A, Bizik J. Induction of hepatocyte growth factor/scatter factor by fibroblast clustering directly promotes tumor cell invasiveness. Cancer Research. 2005;65:9914–9922. doi: 10.1158/0008-5472.CAN-05-1559. [DOI] [PubMed] [Google Scholar]
- Klingelhutz AJ, Roman A. Cellular transformation by human papillomaviruses: lessons learned by comparing high- and low-risk viruses. Virology. 2012;424:77–98. doi: 10.1016/j.virol.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen E, Jenkins A, Holm R. Coexistence of episomal and integrated HPV16 DNA in squamous cell carcinoma of the cervix. Journal of clinical pathology. 1994;47:253–256. doi: 10.1136/jcp.47.3.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorre IJ, Roh MH, Frese KK, Weiss RS, Margolis B, Javier RT. Viral oncoprotein-induced mislocalization of select PDZ proteins disrupts tight junctions and causes polarity defects in epithelial cells. Journal of cell science. 2005;118:4283–4293. doi: 10.1242/jcs.02560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MY, Chou CY, Tang MJ, Shen MR. Epithelial-mesenchymal transition in cervical cancer: correlation with tumor progression, epidermal growth factor receptor overexpression, and snail up-regulation. Clin Cancer Res. 2008;14:4743–4750. doi: 10.1158/1078-0432.CCR-08-0234. [DOI] [PubMed] [Google Scholar]
- Liao S, Deng D, Zhang W, Hu X, Wang W, Wang H, Lu Y, Wang S, Meng L, Ma D. Human papillomavirus 16/18 E5 promotes cervical cancer cell proliferation, migration and invasion in vitro and accelerates tumor growth in vivo. Oncology reports. 2013;29:95–102. doi: 10.3892/or.2012.2106. [DOI] [PubMed] [Google Scholar]
- Lopes-Bastos BM, Jiang WG, Cai J. Tumour-Endothelial Cell Communications: Important and Indispensable Mediators of Tumour Angiogenesis. Anticancer Research. 2016;36:1119–1126. [PubMed] [Google Scholar]
- Matsumoto K, Nakamura T. Hepatocyte growth factor and the Met system as a mediator of tumor-stromal interactions. International journal of cancer. 2006;119:477–483. doi: 10.1002/ijc.21808. [DOI] [PubMed] [Google Scholar]
- Maufort JP, Shai A, Pitot HC, Lambert PF. A role for HPV16 E5 in cervical carcinogenesis. Cancer Research. 2010;70:2924–2931. doi: 10.1158/0008-5472.CAN-09-3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride AA, Warburton A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS pathogens. 2017;13:e1006211. doi: 10.1371/journal.ppat.1006211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nature Reviews. 2010;10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- Munger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K. Mechanisms of human papillomavirus-induced oncogenesis. Journal of Virology. 2004;78:11451–11460. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myong NH. Loss of E-cadherin and Acquisition of Vimentin in Epithelial-Mesenchymal Transition are Notable Indicators of Uterine Cervix Cancer Progression. Korean Journal of Pathology. 2012;46:341–348. doi: 10.4132/KoreanJPathol.2012.46.4.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Bodily JM, Beglin M, Kyo S, Inoue M, Laimins LA. Hypoxia-specific stabilization of HIF-1alpha by human papillomaviruses. Virology. 2009;387:442–448. doi: 10.1016/j.virol.2009.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nees M, Geoghegan JM, Munson P, Prabhu V, Liu Y, Androphy E, Woodworth CD. Human papillomavirus type 16 E6 and E7 proteins inhibit differentiation-dependent expression of transforming growth factor-beta2 in cervical keratinocytes. Cancer Research. 2000;60:4289–4298. [PubMed] [Google Scholar]
- Parkin DM, Bray F. Chapter 2: The burden of HPV-related cancers. Vaccine. 2006;24(Suppl 3):S11–25. doi: 10.1016/j.vaccine.2006.05.111. [DOI] [PubMed] [Google Scholar]
- Patel D, Huang SM, Baglia LA, McCance DJ. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. The EMBO journal. 1999;18:5061–5072. doi: 10.1093/emboj/18.18.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pett M, Coleman N. Integration of high-risk human papillomavirus: a key event in cervical carcinogenesis? The Journal of Pathology. 2007;212:356–367. doi: 10.1002/path.2192. [DOI] [PubMed] [Google Scholar]
- Pim D, Thomas M, Banks L. Chimaeric HPV E6 proteins allow dissection of the proteolytic pathways regulating different E6 cellular target proteins. Oncogene. 2002;21:8140–8148. doi: 10.1038/sj.onc.1206026. [DOI] [PubMed] [Google Scholar]
- Polanska UM, Orimo A. Carcinoma-associated fibroblasts: non-neoplastic tumour-promoting mesenchymal cells. Journal of Cellular Physiology. 2013;228:1651–1657. doi: 10.1002/jcp.24347. [DOI] [PubMed] [Google Scholar]
- Roman A, Munger K. The papillomavirus E7 proteins. Virology. 2013;445:138–168. doi: 10.1016/j.virol.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahab Z, Sudarshan SR, Liu X, Zhang Y, Kirilyuk A, Kamonjoh CM, Simic V, Dai Y, Byers SW, Doorbar J, Suprynowicz FA, Schlegel R. Quantitative measurement of human papillomavirus type 16 E5 oncoprotein levels in epithelial cell lines by mass spectrometry. Journal of Virology. 2012;86:9465–9473. doi: 10.1128/JVI.01032-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz E, Freese UK, Gissmann L, Mayer W, Roggenbuck B, Stremlau A, zur Hausen H. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature. 1985;314:111–114. doi: 10.1038/314111a0. [DOI] [PubMed] [Google Scholar]
- Seol DW, Chen Q, Zarnegar R. Transcriptional activation of the hepatocyte growth factor receptor (c-met) gene by its ligand (hepatocyte growth factor) is mediated through AP-1. Oncogene. 2000;19:1132–1137. doi: 10.1038/sj.onc.1203404. [DOI] [PubMed] [Google Scholar]
- Shimabukuro K, Ichinose S, Koike R, Kubota T, Yamaguchi M, Miyasaka M, Aso T. Hepatocyte growth factor/scatter factor is implicated in the mode of stromal invasion of uterine squamous cervical cancer. Gynecologic Oncology. 2001;83:205–215. doi: 10.1006/gyno.2001.6347. [DOI] [PubMed] [Google Scholar]
- Spurgeon ME, Lambert PF. Human Papillomavirus and the Stroma: Bidirectional Crosstalk during the Virus Life Cycle and Carcinogenesis. Viruses. 2017:9. doi: 10.3390/v9080219. [DOI] [PMC free article] [PubMed]
- Steele BK, Meyers C, Ozbun MA. Variable expression of some “housekeeping” genes during human keratinocyte differentiation. Analytical Biochemistry. 2002;307:341–347. doi: 10.1016/s0003-2697(02)00045-3. [DOI] [PubMed] [Google Scholar]
- Steffan JJ, Coleman DT, Cardelli JA. The HGF-met signaling axis: emerging themes and targets of inhibition. Current protein & peptide science. 2011;12:12–22. doi: 10.2174/138920311795659425. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nature Reviews Molecular Cell Biology. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nature Reviews Molecular Cell Biology. 2010;11:834–848. doi: 10.1038/nrm3012. [DOI] [PubMed] [Google Scholar]
- Tummers B, Goedemans R, Pelascini LP, Jordanova ES, van Esch EM, Meyers C, Melief CJ, Boer JM, van der Burg SH. The interferon-related developmental regulator 1 is used by human papillomavirus to suppress NFkappaB activation. Nature Communications. 2015;6:6537. doi: 10.1038/ncomms7537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velpula KK, Dasari VR, Asuthkar S, Gorantla B, Tsung AJ. EGFR and c-Met Cross Talk in Glioblastoma and Its Regulation by Human Cord Blood Stem Cells. Translational Oncology. 2012;5:379–392. doi: 10.1593/tlo.12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venuti A, Paolini F, Nasir L, Corteggio A, Roperto S, Campo MS, Borzacchiello G. Papillomavirus E5: the smallest oncoprotein with many functions. Molecular Cancer. 2011;10:140. doi: 10.1186/1476-4598-10-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker F, Kermorgant S, Darai E, Madelenat P, Cremieux AC, Henin D, Lehy T. Hepatocyte growth factor and c-Met in cervical intraepithelial neoplasia: overexpression of proteins associated with oncogenic human papillomavirus and human immunodeficiency virus. Clin Cancer Res. 2003;9:273–284. [PubMed] [Google Scholar]
- Wilson R, Laimins LA. Differentiation of HPV-containing cells using organotypic “raft” culture or methylcellulose. Methods in molecular medicine. 2005;119:157–169. doi: 10.1385/1-59259-982-6:157. [DOI] [PubMed] [Google Scholar]
- Woodby B, Scott M, Bodily J. The Interaction Between Human Papillomaviruses and the Stromal Microenvironment. Progress in Molecular Biology and Translational Science. 2016;144:169–238. doi: 10.1016/bs.pmbts.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodman CB, Collins SI, Young LS. The natural history of cervical HPV infection: unresolved issues. Nature Reviews Cancer. 2007;7:11–22. doi: 10.1038/nrc2050. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wu JZ, Yang YQ, Ma R, Zhang JY, Feng JF. Expression of growthregulated oncogene1, hepatocyte growth factor, plateletderived growth factorAA and soluble Eselectin and their association with highrisk human papillomavirus infection in squamous cell carcinoma of the uterine cervix. Molecular Medicine Reports. 2014 doi: 10.3892/mmr.2014.2293. [DOI] [PubMed] [Google Scholar]
- zur Hausen H. Papillomavirus infections--a major cause of human cancers. Biochimica et Biophysica Acta. 1996;1288:F55–78. doi: 10.1016/0304-419x(96)00020-0. [DOI] [PubMed] [Google Scholar]
- zur Hausen H. Papillomaviruses in the causation of human cancers - a brief historical account. Virology. 2009;384:260–265. doi: 10.1016/j.virol.2008.11.046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Characterization of E5 Stop cells. a. Southern blot of total cellular DNA digested with restriction enzymes that either do not cut the HPV16 genome (U) or cut it once (C). Supercoiled (I), nicked circular (II), linearized (III), or multimeric (multi) DNA forms are indicated. Western blots showing the levels of E7 (b) and p53 (c) in the designated cell type. (d) RT-qPCR measuring the levels of HPV16 transcripts from the E6, E7, and E7 ORFs. *=p<0.05; NS=not significant.
Supplementary Figure 2. Western Blot of (a) phosphorylated Met (p-Met) and tubulin in HFK cells, HPV16-containing cells and E5 Stop cells treated with DMSO (D), EGF (E), AG1478 (A) or both EGF and AG1478 (E/A) and (b) phosphorylated Met (p-Met), total Met, and tubulin as a loading control from HPV16-containing cells treated with DMSO (V), HGF, or SU11274 (SU).