

Abstract
Natural products with diverse chemical scaffolds have been recognized as an invaluable source of compounds in drug discovery and development. However, systematic identification of drug targets for natural products at the human proteome level via various experimental assays is highly expensive and time-consuming. In this study, we proposed a systems pharmacology infrastructure to predict new drug targets and anticancer indications of natural products. Specifically, we reconstructed a global drug-target network with 7,314 interactions connecting 751 targets and 2,388 natural products and built predictive network models via a balanced substructure-drug-target network-based inference approach. A high area under receiver operating characteristic curve of 0.96 was yielded for predicting new targets of natural products during cross-validation. The newly predicted targets of natural products (e.g., resveratrol, genistein and kaempherol) with high scores were validated by various literatures. We further built the statistical network models for identification of new anticancer indications of natural products through integration of both experimentally validated and computationally predicted drug-target interactions of natural products with the known cancer proteins. We showed that the significantly predicted anticancer indications of multiple natural products (e.g., naringenin, disulfiram and metformin) with new mechanism-of-action were validated by various published experimental evidences. In summary, this study offers powerful computational systems pharmacology approaches and tools for development of novel targeted cancer therapies by exploiting the polypharmacology of natural products.
Graphical Abstract
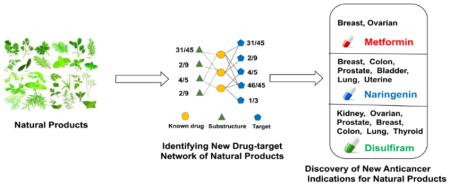
INTRODUCTION
Natural products and their derivatives, with diverse chemical structures, contribute greatly to the landscape of new chemical entities (NCEs) for drug discovery and development in the past several decades.1–3 Recent studies have suggested that Food and Drug Administration (FDA)-approved or clinically investigational natural products often target multiple proteins, called polypharmacology.4 However, polypharmacology of natural products are characterized with both therapeutic effects and unwanted adverse effects. Hence, systematic identification of drug-target interactions (DTIs) for natural products at the human proteome would provide unexpected opportunities for identification of new indications (e.g., drug repositioning) and reducing toxicity of natural products in de novo drug discovery and development.5–8
Traditionally experimental assays for identification of DTIs of natural products are highly expensive and time-consuming, which is not feasible for thousands of natural products available currently. Computational (in silico) approaches have been demonstrated as invaluable tools for prediction of DTIs of natural products, such as molecular docking,9 chemical similarity search,10, 11 and machine learning approaches.12, 13 Although remarkable advances of those computational approaches have been achieved, several potential limitations still have to be recognized. For example, most of the current approaches rely on the availability of three-dimensional (3D) structure of target protein (e.g., molecular docking) or chemical structures of ligands (e.g., chemical similarity and machine learning). However, the limited number of high-quality 3D structures of proteins and the structural complexity of natural products (e.g., multiple chiral atoms) restrict the application of current molecular docking and ligand 3D shape similarity approaches.14 In addition, most of machine learning approaches (except for k-nearest neighbor) require negative samples, while lack of high-quality negative samples further limit the accuracy of current machine learning-based models.6,15 It is urgently needed to develop new computational approaches for exploring the space of drug targets for natural products at the human proteome.
Recent advances in network-based approaches have provided useful tools for prediction of DTIs and assessment of drug safety profiles.16–24 These approaches have illustrated a great promise in drug discovery and development, since they do not rely on either 3D structures of target proteins or negative samples. For example, Cheng et al. proposed several network-based approaches, such as network-based inference (NBI)16 and the edge-weighted/node-weighted NBI17 for prediction of DTIs and drug repositioning. However, neither NBI nor the weighted NBIs can predict targets of NCEs whose targets are absent in the known drug-target network. To overcome this drawback, they further proposed a novel network-based approach, named substructure-drug-target network-based inference (SDTNBI),20 to identify potential targets for NCEs via integrating molecular fingerprints of drugs into drug-target network. As an improvement of SDTNBI, they further developed a balanced substructure-drug-target network-based inference (bSDTNBI) for identification of targets for both old drugs and NCEs by introducing three tunable parameters to balance the weights of each nodes and edges in networks. Via bSDTNBI, they identified several novel antagonistic or agonistic estrogen receptor alpha (ERα) ligands, and dual-effect ERα ligands by in vitro assays for the development of potential therapies in breast cancer or osteoporosis.21 Therefore, systematic identification of new target spaces of natural products via network-based approaches would provide unexpected opportunities for drug discovery and development by exploiting the pharmaceutical wealth of natural products.
In this study, we proposed a systems pharmacology infrastructure for in silico identification of drug targets and anticancer indications of natural products (Figure 1). Specifically, we re-constructed a global drug-target network of natural products and built the predictive network models via our previous network-based approach, named bSDTNBI.21 Then, we developed the statistical network models for identification of new anticancer indications of natural products through integration of both experimentally validated and computationally predicted DTIs with the known cancer proteins. We computationally identified multiple anticancer indications for several typical natural products with new mechanism-of-actions (MOA) across 13 cancer types. Altogether, this study offers a powerful systems pharmacology infrastructure for identification of new targets and anticancer indications of natural products.
Figure 1. Schematic diagram of a systems pharmacology infrastructure for identification of new targets and anticancer indications of natural products.

(A) Re-construction of drug-target network for natural products; (B) Building predictive network models via bSDTNBI for systematic prediction of new targets of natural products; (C) Performing network analyses for validating the new predicted drug-target interactions and for identifying testable hypothesis of new therapeutic effects of natural products; (D) Building the statistical network models for prioritizing new anticancer indication via integration of the computationally predicted (B) and known drug-target interaction network of natural products into the curated cancer-associated genes (proteins).
Materials and Methods
Constructing Drug-target Network of Natural Products
We firstly built a catalog of natural products with diverse chemical structures by integrating data from six publicly available natural product data sources: Traditional Chinese Medicine database (TCMDb),25 Chinese Natural Product database (CNPD),26 Traditional Chinese Medicine integrated database (TCMID),27 Traditional Chinese Medicine Systems Pharmacology (TCMSP),28 Traditional Chinese Medicine database@Taiwan (TCM@Taiwan),29 and Universal Natural Product Database (UNPD).30 For each data source, we changed its initial structure format (e.g., mol2) into unified SDF format using molecular operating environment (MOE) software.31 Subsequently, all chemical structures were stored in a single SDF file via merging six SDF files from the six data sources. InChIKey, a fixed length (25 character) condensed digital representation of the InChI, was calculated for each molecule using Open Babel (version 2.3.2).32 Finally, 259,547 unique natural products were collected after removing the duplicates according to InChIKey. The details are provided in a recent study.4
To construct a global drug-target network of natural products, we collected experimentally validated DTIs from two commonly used databases: ChEMBL (v21)33 and BindingDB (accessed in June 2016).34 All chemical structures were prepared and converted into canonical SMILES using Open Babel toolkit (version 2.3.2).32 In the preparation process, salt ions were removed and dative bonds were standardized. Herein, we only used the high-quality DTIs based on the following 5 criteria: (i) inhibitory constant (Ki), dissociation constant (Kd), half maximal inhibitory concentration (IC50) or half maximal effective concentration (EC50) ≤ 10 μM; (ii) the target is a human protein; (iii) the target can be represented by a unique UniProt accession number; (iv) each drug can be successfully represented by canonical SMILES format; and (v) each drug has more than one carbon atoms. Subsequently, we mapped 259,547 unique natural products into the abovementioned data items to extract experimentally validated DTIs using the ‘InChIKey’.
In order to focus on DTI networks of FDA-approved or investigational natural products, we extracted natural products via mapping global DTI network into 7,109 drugs in DrugBank (accessed in Sep 2016).35 In total, we reconstructed a local DTI network with 1,796 interactions connecting 276 natural products (including 146 FDA-approved drugs and 130 investigational drugs) and 453 targets.
To evaluate the model performance, we further reconstructed three independent DTI networks as external validation sets by integrating data from STITCH,36 Herbal Ingredients’ Targets Database (HIT),37 and the Comparative Toxicogenomics Database (CTD).38 For STITCH, the thickness of each interaction pair represented the confidence scores of DTIs. Only DTIs from Homo sapiens were downloaded, and high-confidence interactions (score ≥ 0.7) were retained in this study as the validation set A.36 We also extracted DTIs from HIT using a web crawler approach as the validation set B.37 Moreover, we constructed an additional DTI network as the validation set C by integrating data from CTD (accessed in June 2017).38 The duplicated DTIs between the global networks and three independent validation networks were excluded.
Description of Molecular Fingerprints
Here, we calculated substructure items of each compound using four types of molecular fingerprints from PaDEL-Descriptor (version 2.18),39 including Substructure (FP4), Klekota-Roth (KR), MACCS, and PubChem (PubChem).
Prediction of New Drug-target Interactions of Natural Products
In this study, we built predictive network models via bSDTNBI21 to predict targets of natural products. Specifically, bSDTNBI utilizes resource-diffusion processes in the substructure-drug (or NCE)-target network to prioritize potential targets for natural products. The substructure-drug (or NCE)-target network was constructed by integrating the known DTI network, drug-substructure associations and NCE-substructure associations (Figure 1B). Three tunable parameters were introduced into the resource-diffusion processes to improve the method performance.
To describe the resource-diffusion processes mathematically, assuming that the substructure-drug (or NCE)-target network used for prediction contains: (i) NC denotes NCEs without known targets, (ii) ND denotes drugs with known targets, (iii) NS denotes substructure items of molecular fingerprints, and (iv) NT denotes targets, we defined several matrices. For the NCE-substructure associations, we define a matrix: MCS(i, j) = 1 if NCE Ci contains substructure Sj (0 < i ≤ NC, 0 < j ≤ NS), otherwise = 0. For the drug-substructure associations, we define a matrix: MDS(i, j) = 1 if drug Di contains substructure Sj (0 < i ≤ ND, 0 < j ≤ NS), otherwise = 0. For the DTIs, we define a matrix: MDT(i, j) = 1 if drug Di has known interaction with target Tj (0 <i ≤ ND, 0 < j ≤ NT), otherwise = 0.
As shown in Figure 1B, the first parameter α ∈ [0,1) was introduced to balance the initial resource allocation of different node types (e.g., high vs. low connectivity). For each drug or NCE node, a total amount = α of initial resource was equally allocated to the substructure nodes linked with it, and a total amount = 1 − α of resource was equally allocated to the target nodes linked with it. By varying the value of α, we can adjust the importance of different node types (substructure nodes and target nodes) in initial resource allocation.
Mathematically, for NCE-substructure associations, an initial resource matrix can be defined as below (0 < i ≤ NC, 0 < j ≤ NS).
| (1) |
For drug-substructure associations, an initial resource matrix can be defined as below (0 < i ≤ ND, 0 < j ≤ NS).
| (2) |
For DTIs, an initial resource matrix can be defined as below (0 < i ≤ ND, 0 < ≤ j ≤ NT).
| (3) |
Using these matrices, the initial resource matrix for the substructure-drug (or NCE)-target network can be represented as:
| (4) |
Then, the second parameter β ∈ [0,1) was introduced to balance the weighted values of different edge types. Specifically, the weighted values of all drug-substructure associations were set to β, while the weighted values of all DTIs were set to 1 − β. By varying the value of β, we can adjust the importance of different edge types in resource-diffusion processes. In addition, the third parameter γ ∈(−∞,+∞) was imported to balance the influence of hub nodes in resource-diffusion processes. A positive/negative value of γ will strengthen/weaken the influence of hub nodes. Using the parameters β and γ, the aforementioned initial resource matrix A can be transformed into a final resource matrix as below.
Let B, C, and W be three square matrices, defined as below (0 < i ≤ NC + ND + NS + NT, 0 < j ≤ NC + ND + NS + NT):
| (5) |
| (6) |
| (7) |
The final resource matrix (score matrix) is calculated by the following equation:
| (8) |
where k is the number of resource-diffusion processes. The score of the interaction between NCE Ci and target Tj is the value of F(i, NC + ND + NS + j) (0 < i ≤ NC, 0 < j ≤ NT). The score of the interaction between drug Di and target Tj is F(NC + i, NC + ND + NS + j) (0 < i ≤ ND, 0 < j ≤ NT).
The parameters (α = β = 0.1, γ = −0.5, and k = 2) of bSDTNBI were used here based on our previous study.21
Performance Evaluation
In this study, both 10-fold cross validations and the external validation were used to evaluate the model performance. Four evaluation indications depending on the top prediction lists (e.g., L = 20), including precision (P), recall (R), precision enhancement (eP), and recall enhancement (eR) were calculated as below.
| (9) |
| (10) |
| (11) |
| (12) |
Where M and N are the number of drugs and targets participated in performance evaluation, Xi(L) is the number of the correctly predicted DTIs which were ranked in the top L places of Di’s newly predicted target list, Xi is the number of Di’s DTIs which were divided into test set, X is the total number of DTIs which were divided into test set.
Furthermore, another evaluation indicator independent of the L value, the area under the receiver operating characteristic curve (AUC), were calculated by computing the true positive rates and false positive rates under different L values. The details of these evaluation indicators were described in previous studies.16, 17, 20
We performed 10-fold cross validation via leaving out 10% of DTIs from the total links. For external validation, we only kept the novel DTIs not shared by the global DTI network that was used for model building. For each model, the processes of 10-fold cross validation were repeated 10 times, and the mean values and standard deviations (mean±SD) of each evaluation indicator were calculated to measure the model performance. Finally, three external test sets were employed to assess the generalization ability of in silico network models.
Curation of Cancer Proteins for 13 Cancer Types
We collected the products (proteins) of cancer-associated genes across 13 cancer types from 4 public available databases: the Online Mendelian Inheritance in Man (OMIM) database,40 HuGE Navigator,41 PharmGKB,42 and Comparative Toxicogenomics Database (CTD).38 The 13 major cancer types include leukemia, bladder cancer (bladder), breast cancer (breast), colon cancer (colon), glioblastoma multiforme (GBM), kidney cancer (kidney), lung cancer (lung), ovarian cancer (ovarian), prostate cancer (prostate), melanoma, stomach cancer (stomach), thyroid cancer (thyroid), and uterine cancer (uterine).The details are provided in Supporting Information, Table S1.
Prioritizing Anticancer Indications for Natural Products
Here, we developed the integrated statistical network models to prioritize new anticancer indications of natural products through incorporating both experimentally validated and the computationally predicted DTIs of natural products and the curated cancer proteins across 13 cancer types. The statistical network models assert that a natural product with multiple targets (named polypharmacological profiles) exhibits a high possibility to treat a particular cancer type if its targets are more likely to be cancer proteins in this specific cancer. A permutation testing was proposed to evaluate the statistical significance of a natural product to be prioritized for a specific cancer indication.
The null hypothesis supposes that the targets of a natural product randomly locate at cancer proteins in the human proteome, while the alternative hypothesis asserts that targets of a natural product are inclined to be cancer proteins than other proteins when this natural product shows potential treatment in this cancer type. The permutation testing was calculated as below:
| (13) |
A nominal P was computed for each natural product by counting the number of observed cancer proteins in a specific cancer type greater (Sm (p)) than the permutations (Sm). Herein, we performed 100,000 permutations by randomly selecting a set of proteins with the same number of cancer proteins in a cancer type from the genome-wide simulation (20,462 human protein-coding genes from the NCBI database, Supporting Information, Table S2). Then, the resulting P-values obtained from the permutation tests were corrected as adjusted P-values (q) using R based on Benjamini-Hochberg approach.43
In addition, a Z-score was also computed for each natural product across each specific cancer type during permutation testing in Equation 14, where x is the real number of cancer proteins targeted by a given natural product in a specific cancer type, μ is the mean number of cancer proteins targeted by a given natural product during 100,000 permutations, and σ is the standard
| (14) |
Network Visualization and Statistical Analysis
The statistical analysis was performed using the Python (v3.2, http://www.python.org/) and R platforms (v3.01, http://www.r-project.org/). Networks were visualized using Cytoscape (v3.2.0, http://www.cytoscape.org/).
Results
Reconstruction of Drug-target Network for Natural Products
In this study, we reconstructed a global drug-target network of natural products by integrating the high-quality experimental data (see Methods and Materials). As shown in Table 1, this network contains 7,314 interactions connecting 2,388 unique natural products and 751 targets. The average degree (i.e., connectivity) of a natural product is 3.06, while the sparsity value for the global network is 0.41%. The detailed DTI pairs of this network are provided in Supporting Information, Table S3.
Table 1.
The statistics of four independent drug-target interaction networks used for network model building and validation.
| Data set | ND | NT | NDTI | Sparsity (%) |
|---|---|---|---|---|
| Global network | 2,388 | 751 | 7,314 | 0.41 |
| Validation set A | 714 | 466 | 3,164 | 0.95 |
| Validation set B | 375 | 318 | 1,164 | 0.98 |
| Validation set C | 348 | 386 | 3,352 | 2.50 |
ND: the number of natural products, NT: the number of targets, NDTI: the number of drug-target interactions, Sparsity: the ratio of NDTI to the number of all possible drug-target interactions.
We examined the target coverage and chemical diversity of natural products in the global network. As presented in Figure 2A, the entire targets (n = 750) can be divided into five categories via mapping it to Therapeutic Target Database (TTD),44 including successful target (n = 131), clinical trial target (n = 113), research target (n = 142), discontinued target (n = 15) and others (n = 349), and the number of corresponding DTI pairs for the five categories was 1,631, 878, 1,446, 59 and 3,300, respectively (Figure 2B).
Figure 2. Analysis of target coverage and chemical diversity of natural products in the reconstructed global drug-target interaction network.

Classification of drug targets (A) and drug-target interactions (B) across five types of target proteins annotated in TTD.44 (C) Chemical structure clustering of 2,388 natural products based on FCFP_6 fingerprint. (D) The representative structures of 10 cluster centers.
Clustering analysis was performed to examine chemical scaffolds of natural products in the global network. The total 2,388 natural products are clustered into 10 groups based on FCFP_6 fingerprint using the Cluster ligands module in Discovery Studio 4.0,45 and each group has similar chemical features (Figure 2C). Among them, cluster 9 (Cluster center: chrysoeriol) shows the largest numbers of compounds (665 natural products), following by cluster 7 (Cluster center: 8-prenylnaringenin) with 307 natural products. The structures of each cluster center are shown in Figure 2D. Ten cluster centers are maslinic acid, 4-hydroxybenzoic acid, betaine, ZINC00083317, tetradecanoic acid, dronabinol, 8-prenylnaringenin, benzamide, chrysoeriol, and lactic acid, respectively. Other compounds in the same cluster share similar chemical scaffolds with the structure of cluster center. For instance, the structures in cluster 1 are mainly represented as steroids while the structures in cluster 4 are largely represented as unsaturated aliphatic hydrocarbon or unsaturated fatty acid. Taken together, current global DTI network covers wide FDA-approved or clinically investigational drug targets (Figure 2B) and diverse chemical scaffolds of various natural products (Figure 2D), offering a useful resource for natural product-focused drug discovery.
Prediction of New Targets for Natural Products
Here we built multiple predictive network models based on the above global DTI network for computational identification of new targets of natural products (Figure 1). To evaluate the performance of the predictive network models, 10-fold cross validation was used via implementing four different types of molecular fingerprints: FP4, KR, MACCS, and PubChem. Under the prior parameters of α = β = 0.1, γ = −0.5, and k = 2, the average AUC values of 0.955±0.005, 0.958±0.005, 0.954±0.005 and 0.953±0.005 were yielded for the models of bSDTNBI_FP4, bSDTNBI_KR, bSDTNBI_MACCS, and bSDTNBI_PubChem, respectively (Figure 3). The details of other evaluation indicators in 10-fold cross validation are given in Table 2. From these evaluation indicators, bSDTNBI_KR reveals the best performance with the highest value of P (0.049±0.001), R (0.754±0.016), eP (27.03±0.63), eR (27.30±0.64), and AUC (0.958±0.005).
Figure 3. Receiver operating characteristic (ROC) curves of four models in 10-fold cross validation.

For each model, the processes of 10-fold cross validation were repeated 10 times, and the mean values and standard deviation (mean±SD) of each evaluation indicator were calculated to measure the model performance. The shadow of each curve denotes the SD.
Table 2.
The performance of the predictive network models in 10-fold cross validation.
| Substructure | P (L=20) | R (L=20) | eP (L=20) | eR (L=20) | AUC |
|---|---|---|---|---|---|
| FP4 | 0.048±0.001 | 0.737±0.016 | 26.57±0.61 | 26.71±0.64 | 0.955±0.005 |
| KR | 0.049±0.001 | 0.754±0.016 | 27.03±0.63 | 27.30±0.64 | 0.958±0.005 |
| MACCS | 0.048±0.001 | 0.731±0.016 | 26.37±0.59 | 26.47±0.61 | 0.954±0.005 |
| PubChem | 0.047±0.001 | 0.716±0.017 | 25.94±0.63 | 25.93±0.65 | 0.953±0.005 |
P: precision, R: recall, eP: precision enhancement, eR: recall enhancement, AUC: area under the receiver operating characteristic curve. L: the length of the newly predicted target list for each natural product.
We further evaluated the generalization ability of the predictive network models using three external validations sets (Supporting Information, Table S3): (i) the external validation set A with 3,164 interactions connecting 714 natural products and 466 targets, (ii) the external validation set B with 1,164 interactions covering 375 natural products and 318 targets, and (iii) the external set C with 3,352 interactions connecting 348 natural products and 386 targets. As shown in Supporting Information, Figure S1, S2 and S3, the AUC value ranges from 0.661 to 0.671 for the external validation set A, 0.654 to 0.668 for the external validation set B, and 0.669 to 0.672 for the external validation set C. The detailed performance of the predictive network models on three independent external validation network sets are given in Table 3. As shown in Table 3, bSDTNBI_KR achieves the best performance with the highest value of P (0.023), R (0.152), eP (3.88), eR (5.71) for validation set A, P (0.014), R (0.128), eP (3.35), eR (4.82) for validation set B, as well as P (0.043), R (0.113), eP (3.37), eR (4.24) for validation set C. Figure 4 shows the precision-recall curves of four models evaluated by three independent drug-target networks, suggesting a reasonable accuracy for our predictive network models. Taken together, bSDTNBI_KR shows high performance during both cross-validation and reasonable accuracy in the external validation.
Table 3.
The performance of the predictive network models by three independent external validation network sets.
| Network sets | Substructure | P (L=20) | R (L=20) | eP (L=20) | eR (L=20) | AUC |
|---|---|---|---|---|---|---|
| Validation set A | FP4 | 0.021 | 0.126 | 3.62 | 4.73 | 0.671 |
| KR | 0.023 | 0.152 | 3.88 | 5.71 | 0.661 | |
| MACCS | 0.020 | 0.122 | 3.45 | 4.57 | 0.667 | |
| PubChem | 0.020 | 0.119 | 3.36 | 4.46 | 0.668 | |
| Validation set B | FP4 | 0.011 | 0.083 | 2.77 | 3.11 | 0.664 |
| KR | 0.014 | 0.128 | 3.35 | 4.82 | 0.654 | |
| MACCS | 0.010 | 0.069 | 2.35 | 2.60 | 0.659 | |
| PubChem | 0.010 | 0.071 | 2.45 | 2.68 | 0.668 | |
| Validation set C | FP4 | 0.043 | 0.119 | 3.37 | 4.45 | 0.669 |
| KR | 0.043 | 0.113 | 3.37 | 4.24 | 0.670 | |
| MACCS | 0.043 | 0.117 | 3.37 | 4.40 | 0.669 | |
| PubChem | 0.042 | 0.112 | 3.27 | 4.19 | 0.672 |
P: precision, R: recall, eP: precision enhancement, eR: recall enhancement, AUC: area under the receiver operating characteristic curve. L: the length of the newly predicted target list for each natural product.
Figure 4. Precision-recall curves of four models evaluated by three independent drug-target networks: validation set A (A), validation set B (B), and validation set C (C).
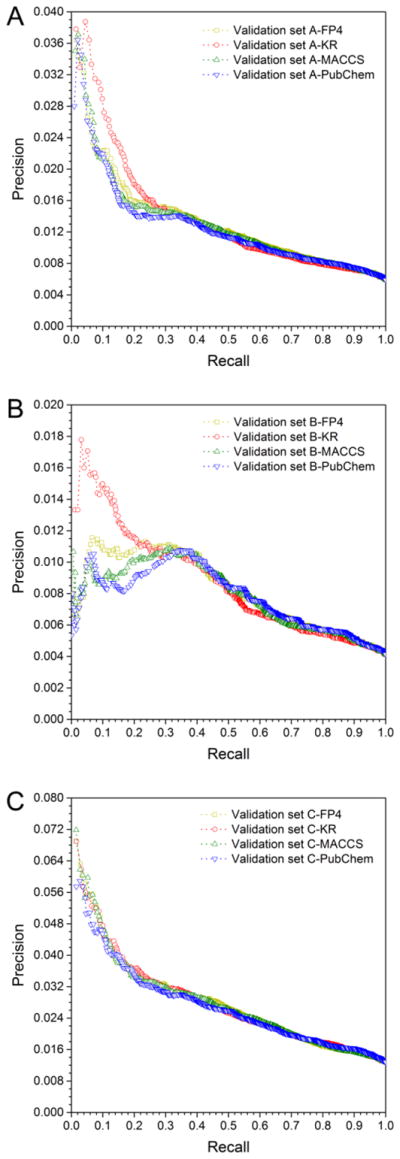
The detailed information of three independent validation drug-target networks is described in Table 1 and Table S3.
Discovery of New Targets for Natural Products
In total, we computationally predicted 42,225 new DTIs between 2,388 unique natural products and 680 targets using top 20 as a cut off (Supporting Information, Table S4). To examine how the network predictive model helps to identify new drug targets of natural products, we selected 3 typical FDA-approved or investigational natural products (kaempherol, resveratrol and genistein) as example drugs. In addition, we also investigated how the new prediction for these three natural products contributed to decipher the new mechanism-of-action (MOA) of natural products for treatment of various complex diseases, such as cancer.
As shown in Figure 5, the DTI bipartite network contains 124 DTIs (76 known DTI pairs and 48 predicted DTI pairs) connecting 3 natural products and 80 targets (48 cancer proteins and 32 non-cancer proteins). We systematically searched previously reported literatures in PubMed for the 48 predicted DTI pairs to kaempherol, resveratrol, and genistein. Among the 48 predicted DTI pairs, 19 (19/48, 39.6% success rate) had been experimentally validated in previously published data (Supporting Information, Table S5), suggesting a reasonable accuracy of our network model. The remaining 29 predicted DTIs are needed to be validated by experimental assays in the future.
Figure 5. Known and predicted drug-target network via the best model (bSDTNBI_KR) for 3 typical natural products (kaempherol, genistein and resveratrol).

This network includes 124 drug-target interactions connecting 3 natural products and 80 targets (48 cancer proteins and 32 non-cancer proteins [see Methods and Materials]).
Kaempherol, a natural flavonol, is mainly found in plants and plant-derived foods such as apples.46 Kaempherol possesses various anticancer effects in breast cancer47 and ovarian cancer.48 As shown in Figure 5, kaempherol interacts with 39 known (20) and predicted (19) targets (including 23 cancer proteins as well as 16 non-cancer proteins). For example, the two cancer proteins (MAPK10 and CBR1) were predicted as the targets of kaempherol. Recent studies have suggested that Kaempferol was a weak inhibitor of MAPK10 (19.1 μM)49 as well as a strong inhibitor of CBR1 (0.13 μM),50 indicating the potential MOAs of kaempferol in cancer.
Genistein, an isoflavonoid derived from soy products, has been reported to show the chemopreventive properties against several cancer types,51 including breast cancer (NCT00244933), bladder cancer (NCT00118040), and prostate cancer (NCT01325311). Figure 5 indicated that genistein was predicted to bind with 15 targets potentially. For example, nuclear factor-kappa B (NF-kB), playing crucial roles in tumor cell proliferation, survival, and angiogenesis,52 was predicted to be interacted by genistein. A recent study has suggested that genistein inhibited the proliferation of human multiple myeloma (MM) cells via suppressing the expression of NF-kB.53
Resveratrol, a non-flavonoid polyphenol derived from the skin of grapes, exerts a wide range of anticancer effects.54 Currently, approximate 20 clinical trials (http://clinicaltrials.gov/) are being conducted to treat various cancers by resveratrol, such as colon cancer (NCT00256334), and liver cancer (NCT02261844). The exact MOAs of anticancer activities by resveratrol are still unclear. In Figure 5, resveratrol interacts with 23 cancer proteins and 21 non-cancer proteins, consisting of 30 known and 14 predicted ones. Interestingly, 15-lipoxygenase (ALOX15) and estrogen receptor-β (ERβ encoded by ESR2) are the two predicted cancer proteins for resveratrol by our predictive network model. Recent studies have showed that resveratrol acted as a competitive weak inhibitor of ALOX15 with an IC50 value of 25 μM55 and inhibited ERβ to suppress cancer cell growth,56 indicating potential anticancer mechanism of resveratrol. A previous study has suggested that natural products often interacted with human proteins with moderate or weak binding affinities at micromolar level.57 A recent analysis on the binding affinities of natural products also suggests that therapeutic efficacy is not necessarily related to high binding affinity.58
Taken together, aforementioned examples show that our predictive network models provide useful computational tool to identify new potential drug targets that were involved in potential anticancer mechanisms of multiple natural products. Systems pharmacology-based integration of drug-target networks and the known cancer proteins would provide useful mechanism-based approaches to uncover new anticancer indications of natural products.
Discovery of New Anticancer Indications for Natural Products
We further built the statistical network models for identification of new anticancer indications of natural products annotated in the DrugBank35 (accessed in Sep 2016) by integrating DTIs of natural products into the known cancer proteins. Considering a lack of statistical power if the target number of a given natural product is 2 or 1, we excluded the natural products of which known target number was lower than 3. Based on this cutoff, we reconstructed an experimentally validated DTI network (named ExpNet) with 1,479 interactions connecting 163 natural products and 409 targets (Supporting Information, Table S6). To cover more potential targets of natural products, we further reconstructed the second network (named Exp&ComNet) by pooling both experimentally validated DTIs and the computationally predicted DTIs from the best predictive network model (bSDTNBI_KR). In order to increase the data quality of the computationally predicted DTIs, we only used the predicted targets ranked in top 5 candidates from the best network model (bSDTNBI_KR) described in our previous studies.20, 21 In total, Exp&ComNet contains 1,623 known and 1,259 predicted DTIs (Supporting Information, Table S7) connecting 275 natural products and 525 targets.
Table 4 illustrates two predicted drug-cancer indication networks of natural product drugs. Using the threshold of adjusted p-value (q) < 0.05, we computationally identified 635 anticancer indications of 124 natural products (Figure 6A) via the known DTIs (ExpNet). We further identified 993 anticancer indications (q < 0.05) of 196 natural products (Figure 6B) by pooling both the known DTIs and the computationally predicted DTIs (Exp&ComNet) via our predictive network model. The detailed predictions are provided in Supporting Information, Table S8. Among 196 natural product drugs having the significantly predicted anticancer indications (Figure 6B), 99 drugs cannot be predicted to have significant anticancer indication in any cancer type based on ExpNet only. We further compared natural products having at least 5 predicted anticancer indications in ExpNet and Exp&ComNet networks, resulting in 54 natural products (using ExpNet) and 85 natural drugs (using Exp&ComNet), respectively. We systematically searched previously reported data from PubMed for the 54 natural products against 13 cancer types. The detailed predictions are provided in Supporting Information, Table S9. Among 500 predicted anticancer indications, 232 (with a success rate of 46.4 % [232/500]) could be found to have the reported experimental data. This suggests a reliable accuracy of our proposed network-based model. The remaining 268 pairs without known experimental data need to be validated by various experimental assays in the future. Heat maps given in Figure 7 show Z-scores of the predicted indications using the experimentally validated DTIs only (Figure 7A) and the pooling data of the experimentally validated and computationally predicted DTIs (Figure 7B) against 13 cancer types. Compared with Figure 7A, Figure 7B shows the significantly predicted anticancer indications for 37 additional natural products based on Exp&ComNet. Collectively, we demonstrated that integration of the computationally predicted DTIs could help to uncover more new anticancer indications of natural products via overcoming the incompleteness of drug-target interactions
Table 4.
Summary of the newly predicted anticancer indications of natural products using the experimentally validated DTIs (ExpNet) only and the combination of the experimentally validated and network-based predicted DTIs (ExpNet&ComNet) respectively.
| Data source | # of DTIs (# targets, # drugs) | # SDCs (#drugs) (q<0.05) | # SDCs (# drugs) (q<1/10−5) |
|---|---|---|---|
| ExpNet | 1,479(409,163) | 635 (124) | 254 (66) |
| ExpNet&ComNet | 2,882 (525, 275) | 993 (196) | 519 (141) |
SDCs denote to significant drug-cancer indication pairs
Figure 6. Discovered drug-cancer indication networks.

(A) The predicted drug-cancer indication network based on the experimentally validated drug-target interaction (ExpNet) only, containing 635 significant drug-cancer indications pairs (SDCs) between 124 natural products and 13 cancer types. (B) The predicted drug-cancer indication network based on both experimentally validated and computationally predicted drug-target interactions (Exp&ComNet), containing 993 SDCs between 196 natural products and 13 cancer types. The 13 major cancer types are: leukemia, bladder, breast, colon, glioblastoma multiforme (GBM), kidney, lung, ovarian, prostate, melanoma, stomach, thyroid, and uterine cancers.
Figure 7. Heat maps show the predicted indications for FDA-approved or clinical investigational natural products against 13 cancer types.

(A) Predicted indications of 54 FDA-approved or clinical investigational natural products based on the experimentally validated drug-target interaction (ExpNet) only. (B) Predicted indications of 84 FDA-approved or clinical investigational natural products based on both experimentally validated and computationally prediction drug-target interactions (Exp&ComNet). The red asterisk in B reveals that a natural product does not show statistical significance based on ExpNet only (A). The area in gray represents the non-available value since no cancer proteins are overlapped with the known targets of a specific natural product. Color keys denote the predicted Z-scores. The area in red represents the natural product having the high Z-score across specific cancer indications. Abbreviates of 13 major cancer types are provided in the legend of Figure 6.
Uncovering New Anticancer Indications for Three Typical Natural Products
To further investigate the accuracy of the statistical network models, we selected 3 typical natural products (naringenin, disulfiram, and metformin) as case studies to illustrate their anticancer profiles with new MOAs.
Naringenin (DB03467), a flavanone, is mainly found in grapefruit, oranges, and tomatoes. It has been reported to possess various anticancer activities in breast, colon, pancreatic, lung, and prostate cancers.59 Figure 8 shows that naringenin is predicted to have potential indications for 6 cancer types: bladder (Z=12.12, q <1 ×10−5), lung (Z=9.59, q <1 × 10−5), uterine (Z=10.00, q <1 × 10−5), colon (Z=8.35, q <1 × 10−5), prostate (Z=7.83, q <1 × 10−5), and breast (Z=4.73, q =0.047). Among of them, breast, prostate and colon are the three new anticancer indications with non-significance based on ExpNet only. Previous studies have suggested that naringenin showed strong anticancer activities in three cancer types.60–63 For example, a previous study showed that naringenin prolonged the survival in breast cancer mice model.61 In addition, naringenin was reported to induce apoptotic cell death via regulating activation of PI3K/AKT and MAPK signaling pathways in prostate cancer cells.63
Figure 8. A discovered drug-target-disease network of 3 typical natural products.

The predicted indications for 3 typical natural products (naringenin, disulfiram, and metformin) against 13 cancer types and their corresponding targets are shown. The predicted anticancer indications are based on the pooling data of the experimentally validated and computationally predicted drug-target interactions. The thickness of red line and dotted red line is proportional to the predicted Z-score. Abbreviates of 13 major cancer types are provides in the legend of Figure 6.
Disulfiram (DB00822), a FDA-approved carbamate derivative for the treatment of chronic alcoholism, has showed potential in treating several cancer types.64 In this study, disulfiram was predicted to have potential indications for 8 cancer types. Five of them are the new anticancer indications using Exp&ComNet only, including breast (Z=6.10, q <1 × 10−5), colon (Z=6.51, q =0.0055), lung (Z=7.23, q <1 × 10−5), thyroid (Z=7.23, q =0.0026), and uterine (Z=6.39, q=0.0179). Our prediction is consistent with several previous in vitro or in vivo studies of disulfiram in cancer: (1) induce cell apoptosis in breast cancer cells through suppressing the activity of proteasome,65 and (2) elicit apoptosis and exclude cancer stem-like cells via the suppression of HER2/Akt signaling pathway in HER2-positive breast cancer.66–67
Metformin (DB00331) is a biguanide (mainly found in Galegaofficinalis) oral agent for treating type 2 diabetes. Nowadays, several clinical trials of metformin are being conducted to treat several cancer types, such as breast (NCT01266486), lung (NCT02109549), and ovarian (NCT01579812). In this work, none of cancer types was predicted using ExpNet only. Among top 5 predicted targets for metformin, and 3 targets were cancer proteins (TSHR, BLM, and ALDH1A1). Via Exp&ComNet, metformin was predicted to have potential in treating two cancer indications: breast (Z=6.41, q <1 × 10−5) and ovarian (Z=6.35, q=0.0106). Previous in vitro and clinical studies have suggested that metformin showed potential in treating breast cancer.68 In addition, metformin exerted anti-cancer effect in ovarian cancer cells via targeting AMPK/GSK3β signaling axis.69
Collectively, three case studies (naringenin, disulfiram, and metformin) demonstrated that our predictive network models showed promising to uncover new anticancer indications of natural products, despite the prediction are needed to be tested by experimental assays in the future. Interestingly, integration of both computationally predicted DTIs and known DTIs provide more comprehensive data for identifying new anticancer indications compared to using known DTIs alone. In addition, new predicted targets of natural products provide testable hypothesis to further investigate of MOAs of their anticancer activities.
Discussion
Quantitative and systems pharmacology is an emerging approach to combine computational and experimental methods for discovering new therapeutic agents and understanding of the therapeutic mechanisms of complex diseases.6, 70,71 In this study, we demonstrated a computational systems pharmacology framework for systematic identification of new drug targets and anticancer indications of natural products. Specifically, we developed predictive network models based on a global DTI network connecting natural products and their known targets. Compared with traditional molecular docking approaches or machine learning-based models, our network-based models have several advantages. For example, molecular docking-based methods always rely on high-quality 3D structures of proteins. Here, our network models is independent of 3D structures of targets, providing powerful approaches for targets without known 3D structures yet (e.g., membrane proteins). In addition, network-based models were built using the known DTIs (positive samples) only, whereas negative samples are not needed. The lack of high-quality negative samples often limits the accuracy and coverage of the machine learning models that are built using both positive and negative samples.15
Several potential limitations should be recognized in the current systems pharmacology framework. At first, potential data bias of the external validation sets as well as the incompleteness of known drug-target network of natural products may influence the model performance evaluation. In current study, external validation sets A and B were derived from STITCH36 and HIT,37 respectively. STITCH and HIT involved in a large number of DTI pairs of which experimental evidence (direct or indirect) were uncertain. Compared to validation sets A and B, a smooth recall vs. precision curve on validation set C (Figure 4C) was observed, suggesting a low quality of the external validation sets collected from STITCH and HIT. In the future, the independent test sets that only covered direct DTI pairs should be further evaluated. Secondly, the structure diversity of compounds in global DTI network may influence the performance of network-based models. In this study, the structure diversity of natural products is much less than that of synthesized compounds,21 which explains the marginal improvement of different molecular fingerprints in both cross-validations (Figure 3) and external validations (Figure 4). Thirdly, over 1,400 cancer proteins across 13 major cancer types were used in this study. The data quality and redundancy of the cancer proteins might directly influence our prediction results. For example, cancer proteins often have different biological effects, such as loss-of-function or gain-of-function. Inhibitors target cancer proteins with loss-of-function may cause adverse cancer effects. In the future, cancer proteins with specific biological functions should be considered in our updated systems pharmacology infrastructure. For example, integration of large-scale gene expression profiles of cancer genes in specific cancer types may provide reasonable biological effects of cancer proteins. Fourthly, in this study we focused on three well-known natural products because we can found more literature-reported data for validation. Further experimental assays should be performed to validate the predicted DTIs and anticancer effects for NCEs. Finally, integration of human protein protein interaction network, and drug microarray data such as the Connectivity Map (CMap)72 of natural products, may contribute to develop novel cancer targeted therapies in the future.73
Conclusions
In this study, we developed an in silico systems pharmacology framework for systematic prediction of drug-target interactions of natural products. Multiple predictive network models with high accuracy were built based on a global DTI network linking natural products to known target proteins. Network analyses provide testing hypothesis for exploring molecular mechanisms of therapeutic indications of natural products. Furthermore, we built the statistical network models to uncover new anticancer indications of natural products through integration of drug-target interaction network and well-known cancer proteins. We demonstrated that integration both computationally predicted drug-target interactions and experimentally validated ones could overcome the data incompleteness of current available experimental data. In summary, this study provides a powerful in silico systems pharmacology framework for the development of novel targeted cancer therapies by exploiting the polypharmacology of natural products in the post-genomics era.
Supplementary Material
Acknowledgments
This work was supported by the National Key Research and Development Program of China (Grant 2016YFA0502304), and the National Natural Science Foundation of China (Grants 81603318, 81603026, 81673356). This work was also supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Number K99HL138272 to FC. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The authors declare no competing financial interest.
Conflict of Interest: The authors declare no competing financial interest.
The AUC values of predictive network models for the external validation sets A (Figure S1), B (Figure S2), and C (Figure S3), the cancer-associated genes across 13 cancer types/subtypes (Table S1), 20,462 human protein-coding genes from the NCBI database (Table S2), the drug-target interaction (DTI) pairs for global network and three validation sets (Table S3), 42,225 predicted DTI pairs by bSDTNBI_KR (Table S4), the reported literatures by retrieving PubMed for 48 predicted DTI pairs (Table S5), the DTI network ExpNet (Table S6) and Exp&ComNet (Tables S7), the 635 and 993 significant drug-cancer indication pairs (SDCs) (Table S8), and the reported data in PubMed for 54 natural products having at least 5 predicted anticancer indications (Table S9). This material is available, free of charge, via the Internet at http://pubs.acs.org.
References
- 1.DeCorte BL. Underexplored Opportunities for Natural Products in Drug Discovery. J Med Chem. 2016;59:9295–9304. doi: 10.1021/acs.jmedchem.6b00473. [DOI] [PubMed] [Google Scholar]
- 2.Harvey AL, Edrada-Ebel R, Quinn RJ. The Re-emergence of Natural Products for Drug Discovery in the Genomics Era. Nat Rev Drug Discovery. 2015;14:111–129. doi: 10.1038/nrd4510. [DOI] [PubMed] [Google Scholar]
- 3.Li JW, Vederas JC. Drug Discovery and Natural Products: End of an Era or an Endless Frontier? Science. 2009;325:161–165. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 4.Fang J, Cai C, Wang Q, Lin P, Zhao Z, Cheng F. Systems Pharmacology-Based Discovery of Natural Products for Precision Oncology Through Targeting Cancer Mutated Genes. CPT Pharmacometrics Syst Pharmacol. 2017;6:177–187. doi: 10.1002/psp4.12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodrigues T, Reker D, Schneider P, Schneider G. Counting on Natural Products for Drug Design. Nat Chem. 2016;8:531–541. doi: 10.1038/nchem.2479. [DOI] [PubMed] [Google Scholar]
- 6.Fang J, Liu C, Wang Q, Lin P, Cheng F. In Silico Polypharmacology of Natural Products. Briefings Bioinf. 2017 doi: 10.1093/bib/bbx045. [DOI] [PubMed] [Google Scholar]
- 7.Luo H, Mattes W, Mendrick DL, Hong H. Molecular Docking for Identification of Potential Targets for Drug Repurposing. Curr Top Med Chem. 2016;16:3636–3645. doi: 10.2174/1568026616666160530181149. [DOI] [PubMed] [Google Scholar]
- 8.Ye H, Wei J, Tang K, Feuers R, Hong H. Drug Repositioning Through Network Pharmacology. Curr Top Med Chem. 2016;16:3646–3656. doi: 10.2174/1568026616666160530181328. [DOI] [PubMed] [Google Scholar]
- 9.Hsin KY, Matsuoka Y, Asai Y, Kamiyoshi K, Watanabe T, Kawaoka Y, Kitano H. SystemsDock: a Web Server for Network Pharmacology-based Prediction and Analysis. Nucleic Acids Res. 2016;44:W507–W513. doi: 10.1093/nar/gkw335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, Jensen NH, Kuijer MB, Matos RC, Tran TB, et al. Predicting New Molecular Targets for Known Drugs. Nature. 2009;462:175–181. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan X, Li J, Liu Z, Zheng M, Ge H, Xu J. Enhancing Molecular Shape Comparison by Weighted Gaussian Functions. J Chem Inf Model. 2013;53:1967–1978. doi: 10.1021/ci300601q. [DOI] [PubMed] [Google Scholar]
- 12.Cheng F, Zhao Z. Machine Learning-based Prediction of Drug-Drug Interactions by Integrating Drug Phenotypic, Therapeutic, Chemical, and Genomic Properties. J Am Med Inform Assoc. 2014;21:e278–286. doi: 10.1136/amiajnl-2013-002512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fang J, Yang R, Gao L, Zhou D, Yang S, Liu AL, Du GH. Predictions of BuChE Inhibitors Using Support Vector Machine and Naive Bayesian Classification Techniques in Drug Discovery. J Chem Inf Model. 2013;53:3009–3020. doi: 10.1021/ci400331p. [DOI] [PubMed] [Google Scholar]
- 14.Anighoro A, Bajorath J. Three-Dimensional Similarity in Molecular Docking: Prioritizing Ligand Poses on the Basis of Experimental Binding Modes. J Chem Inf Model. 2016;56:580–587. doi: 10.1021/acs.jcim.5b00745. [DOI] [PubMed] [Google Scholar]
- 15.Cheng F, Zhou Y, Li J, Li W, Liu G, Tang Y. Prediction of Chemical-protein Interactions: Multitarget-QSAR versus Computational Chemogenomic Methods. Mol Biosyst. 2012;8:2373–2384. doi: 10.1039/c2mb25110h. [DOI] [PubMed] [Google Scholar]
- 16.Cheng F, Liu C, Jiang J, Lu W, Li W, Liu G, Zhou W, Huang J, Tang Y. Prediction of Drug-Target Interactions and Drug Repositioning via Network-based Inference. PLoS Comput Biol. 2012;8:e1002503. doi: 10.1371/journal.pcbi.1002503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng F, Zhou Y, Li W, Liu G, Tang Y. Prediction of Chemical-protein Interactions Network with Weighted Network-based Inference Method. PloS One. 2012;7:e41064. doi: 10.1371/journal.pone.0041064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Lei K, Wu Z, Li W, Liu G, Liu J, Cheng F, Tang Y. Network-based Identification of MicroRNAs as Potential Pharmacogenomic Biomarkers for Anticancer Drugs. Oncotarget. 2016;7:45584–45596. doi: 10.18632/oncotarget.10052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J, Wu Z, Cheng F, Li W, Liu G, Tang Y. Computational Prediction of MicroRNA Networks Incorporating Environmental Toxicity and Disease Etiology. Sci Rep. 2014;4:5576. doi: 10.1038/srep05576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu Z, Cheng F, Li J, Li W, Liu G, Tang Y. SDTNBI: an Integrated Network and Chemoinformatics Tool for Systematic Prediction of Drug-target Interactions and Drug Repositioning. Briefings Bioinf. 2017;18:333–347. doi: 10.1093/bib/bbw012. [DOI] [PubMed] [Google Scholar]
- 21.Wu Z, Lu W, Wu D, Luo A, Bian H, Li J, Li W, Liu G, Huang J, Cheng F, et al. In silico Prediction of Chemical Mechanism of Action via an Improved Network-based Inference Method. Br J Pharmacol. 2016;173:3372–3385. doi: 10.1111/bph.13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng F, Li W, Wang X, Zhou Y, Wu Z, Shen J, Tang Y. Adverse Drug Events: Database Construction and In Silico Prediction. J Chem Inf Model. 2013;53:744–752. doi: 10.1021/ci4000079. [DOI] [PubMed] [Google Scholar]
- 23.Cheng F, Li W, Wu Z, Wang X, Zhang C, Li J, Liu G, Tang Y. Prediction of Polypharmacological Profiles of Drugs by the Integration of Chemical, Side Effect, and Therapeutic Space. J Chem Inf Model. 2013;53:753–762. doi: 10.1021/ci400010x. [DOI] [PubMed] [Google Scholar]
- 24.Lu W, Cheng F, Jiang J, Zhang C, Deng X, Xu Z, Zou S, Shen X, Tang Y, Huang J. FXR Antagonism of NSAIDs Contributes to Drug-induced Liver Injury Identified by Systems Pharmacology Approach. Sci Rep. 2015;5:8114. doi: 10.1038/srep08114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He M, Yan X, Zhou J, Xie G. Traditional Chinese Medicine Database and Application on the Web. J Chem Inf Comput Sci. 2001;41:273–277. doi: 10.1021/ci0003101. [DOI] [PubMed] [Google Scholar]
- 26.Shen J, Xu X, Cheng F, Liu H, Luo X, Shen J, Chen K, Zhao W, Shen X, Jiang H. Virtual Screening on Natural Products for Discovering Active Compounds and Target Information. Curr Med Chem. 2003;10:2327–2342. doi: 10.2174/0929867033456729. [DOI] [PubMed] [Google Scholar]
- 27.Xue R, Fang Z, Zhang M, Yi Z, Wen C, Shi T. TCMID: Traditional Chinese Medicine Integrative Database for Herb Molecular Mechanism Analysis. Nucleic Acids Res. 2013;41:D1089–D1095. doi: 10.1093/nar/gks1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ru J, Li P, Wang J, Zhou W, Li B, Huang C, Li P, Guo Z, Tao W, Yang Y, et al. TCMSP: a Database of Systems Pharmacology for Drug Discovery from Herbal Medicines. J Cheminform. 2014;6:13. doi: 10.1186/1758-2946-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen CY. TCM Database@Taiwan: the World's Largest Traditional Chinese Medicine Database for Drug Screening in Silico. PLoS One. 2011;6:e15939. doi: 10.1371/journal.pone.0015939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gu J, Gui Y, Chen L, Yuan G, Lu HZ, Xu X. Use of Natural Products as Chemical Library for Drug Discovery and Network Pharmacology. PloS one. 2013;8:e62839. doi: 10.1371/journal.pone.0062839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molecular Operating Environment (MOE), version 201010. Chemical Computing Group Inc; Montreal, Quebec, Canada: 2010. [Google Scholar]
- 32.O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an Open Chemical Toolbox. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bento AP, Gaulton A, Hersey A, Bellis LJ, Chambers J, Davies M, Kruger FA, Light Y, Mak L, McGlinchey S, et al. The ChEMBL Bioactivity Database: an Update. Nucleic Acids Res. 2014;42:D1083–1090. doi: 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gilson MK, Liu T, Baitaluk M, Nicola G, Hwang L, Chong J. BindingDB in 2015: a Public Database for Medicinal Chemistry, Computational Chemistry and Systems Pharmacology. Nucleic Acids Res. 2016;44:D1045–1053. doi: 10.1093/nar/gkv1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, Maciejewski A, Arndt D, Wilson M, Neveu V, et al. DrugBank 4.0: Shedding New Light on Drug Metabolism. Nucleic Acids Res. 2014;42:D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuhn M, Szklarczyk D, Pletscher-Frankild S, Blicher TH, von Mering C, Jensen LJ, Bork P. STITCH 4: Integration of Protein-chemical Interactions with User Data. Nucleic Acids Res. 2014;42:D401–D407. doi: 10.1093/nar/gkt1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye H, Ye L, Kang H, Zhang D, Tao L, Tang K, Liu X, Zhu R, Liu Q, Chen YZ, et al. HIT: Linking Herbal Active Ingredients to Targets. Nucleic Acids Res. 2011;39:D1055–D1059. doi: 10.1093/nar/gkq1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davis AP, Grondin CJ, Johnson RJ, Sciaky D, King BL, McMorran R, Wiegers J, Wiegers TC, Mattingly CJ. The Comparative Toxicogenomics Database: update 2017. Nucleic Acids Res. 2017;45:D972–D978. doi: 10.1093/nar/gkw838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yap CW. PaDEL-descriptor: an Open Source Software to Calculate Molecular Descriptors and Fingerprints. J Comput Chem. 2011;32:1466–1474. doi: 10.1002/jcc.21707. [DOI] [PubMed] [Google Scholar]
- 40.Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM(R)), an Online Catalog of Human Genes and Genetic Disorders. Nucleic Acids Res. 2015;43:D789–D798. doi: 10.1093/nar/gku1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu W, Gwinn M, Clyne M, Yesupriya A, Khoury MJ. A Navigator for Human Genome Epidemiology. Nat Genet. 2008;40:124–125. doi: 10.1038/ng0208-124. [DOI] [PubMed] [Google Scholar]
- 42.Whirl-Carrillo M, McDonagh EM, Hebert JM, Gong L, Sangkuhl K, Thorn CF, Altman RB, Klein TE. Pharmacogenomics Knowledge for Personalized Medicine. Clin Pharmacol Ther. 2012;92:414–417. doi: 10.1038/clpt.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benjamini Y, Hochberg Y. J R Stat Soc B. 1995. Controlling the False Discovery Rate: a Practical and Powerful Approach to Multiple Testing; pp. 289–300. [Google Scholar]
- 44.Yang H, Qin C, Li YH, Tao L, Zhou J, Yu CY, Xu F, Chen Z, Zhu F, Chen YZ. Therapeutic Target Database Update 2016: Enriched Resource for Bench to Clinical Drug Target and Targeted Pathway Information. Nucleic Acids Res. 2016;44:D1069–D1074. doi: 10.1093/nar/gkv1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Discovery Studio, version 4.0. Accelrys Inc; San Diego, CA: 2014. [Google Scholar]
- 46.Calderon-Montano JM, Burgos-Moron E, Perez-Guerrero C, Lopez-Lazaro M. A Review on the Dietary Flavonoid Kaempferol. Mini Rev Med Chem. 2011;11:298–344. doi: 10.2174/138955711795305335. [DOI] [PubMed] [Google Scholar]
- 47.Kim SH, Hwang KA, Choi KC. Treatment with Kaempferol Suppresses Breast Cancer Cell Growth Caused by Estrogen and Triclosan in Cellular and Xenograft Breast Cancer Models. J Nutr Biochem. 2016;28:70–82. doi: 10.1016/j.jnutbio.2015.09.027. [DOI] [PubMed] [Google Scholar]
- 48.Luo H, Rankin GO, Liu L, Daddysman MK, Jiang BH, Chen YC. Kaempferol Inhibits Angiogenesis and VEGF Expression Through both HIF Dependent and Independent Pathways in Human Ovarian Cancer Cells. Nutr Cancer. 2009;61:554–563. doi: 10.1080/01635580802666281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goettert M, Schattel V, Koch P, Merfort I, Laufer S. Biological Evaluation and Structural Determinants of p38alpha Mitogen-activated-protein Kinase and c-Jun-N-terminal Kinase 3 Inhibition by Flavonoids. Chembiochem. 2010;11:2579–2588. doi: 10.1002/cbic.201000487. [DOI] [PubMed] [Google Scholar]
- 50.Arai Y, Endo S, Miyagi N, Abe N, Miura T, Nishinaka T, Terada T, Oyama M, Goda H, El-Kabbani O, et al. Structure-activity Relationship of Flavonoids as Potent Inhibitors of Carbonyl Reductase 1 (CBR1) Fitoterapia. 2015;101:51–56. doi: 10.1016/j.fitote.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 51.Spagnuolo C, Russo GL, Orhan IE, Habtemariam S, Daglia M, Sureda A, Nabavi SF, Devi KP, Loizzo MR, Tundis R, et al. Genistein and Cancer: Current Status, Challenges, and Future Directions. Adv Nutr. 2015;6:408–419. doi: 10.3945/an.114.008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li F, Zhang J, Arfuso F, Chinnathambi A, Zayed ME, Alharbi SA, Kumar AP, Ahn KS, Sethi G. NF-kappaB in Cancer Therapy. Arch Toxicol. 2015;89:711–731. doi: 10.1007/s00204-015-1470-4. [DOI] [PubMed] [Google Scholar]
- 53.Xie J, Wang J, Zhu B. Genistein Inhibits the Proliferation of Human Multiple Myeloma Cells through Suppression of Nuclear Factor-kappaB and Upregulation of MicroRNA-29b. Mol Med Rep. 2016;13:1627–1632. doi: 10.3892/mmr.2015.4740. [DOI] [PubMed] [Google Scholar]
- 54.Varoni EM, Lo Faro AF, Sharifi-Rad J, Iriti M. Anticancer Molecular Mechanisms of Resveratrol. Front Nutr. 2016;3:8. doi: 10.3389/fnut.2016.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.MacCarrone M, Lorenzon T, Guerrieri P, Agro AF. Resveratrol Prevents Apoptosis in K562 Cells by Inhibiting Lipoxygenase and Cyclooxygenase Activity. Eur J Biochem. 1999;265:27–34. doi: 10.1046/j.1432-1327.1999.00630.x. [DOI] [PubMed] [Google Scholar]
- 56.Robb EL, Stuart JA. Resveratrol Interacts with Estrogen Receptor-beta to Inhibit Cell Replicative Growth and Enhance Stress Resistance by Upregulating Mitochondrial Superoxide Dismutase. Free Radic Biol Med. 2011;50:821–831. doi: 10.1016/j.freeradbiomed.2010.12.038. [DOI] [PubMed] [Google Scholar]
- 57.Wang J, Guo Z, Fu Y, Wu Z, Huang C, Zheng C, Shar PA, Wang Z, Xiao W, Wang Y. Weak-binding Molecules are not Drugs?-toward a Systematic Strategy for Finding Effective Weak-binding Drugs. Briefing Bioinf. 2017;18:321–332. doi: 10.1093/bib/bbw018. [DOI] [PubMed] [Google Scholar]
- 58.Mestres J, Gregori-Puigjane E. Conciliating Binding Ef ciency and Polypharmacology. Trends Pharmacol Sci. 2009;30:470–474. doi: 10.1016/j.tips.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 59.Mir IA, Tiku AB. Chemopreventive and Therapeutic Potential of "naringenin," a Flavanone Present in Citrus Fruits. Nutr Cancer. 2015;67:27–42. doi: 10.1080/01635581.2015.976320. [DOI] [PubMed] [Google Scholar]
- 60.Qin L, Jin L, Lu L, Lu X, Zhang C, Zhang F, Liang W. Naringenin Reduces Lung Metastasis in a Breast Cancer Resection Model. Protein Cell. 2011;2:507–516. doi: 10.1007/s13238-011-1056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang F, Dong W, Zeng W, Zhang L, Zhang C, Qiu Y, Wang L, Yin X, Zhang C, Liang W. Naringenin Prevents TGF-beta1 Secretion from Breast Cancer and Suppresses Pulmonary Metastasis by Inhibiting PKC Activation. Breast Cancer Res. 2016;18:38. doi: 10.1186/s13058-016-0698-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vanamala J, Leonardi T, Patil BS, Taddeo SS, Murphy ME, Pike LM, Chapkin RS, Lupton JR, Turner ND. Suppression of Colon Carcinogenesis by Bioactive Compounds in Grapefruit. Carcinogenesis. 2006;27:1257–1265. doi: 10.1093/carcin/bgi318. [DOI] [PubMed] [Google Scholar]
- 63.Lim W, Park S, Bazer FW, Song G. Naringenin-Induced Apoptotic Cell Death in Prostate Cancer Cells Is Mediated via the PI3K/AKT and MAPK Signaling Pathways. J Cell Biochem. 2017;118:1118–1131. doi: 10.1002/jcb.25729. [DOI] [PubMed] [Google Scholar]
- 64.Jiao Y, Hannafon BN, Ding WQ. Disulfiram's Anticancer Activity: Evidence and Mechanisms. Anticancer Agents Med Chem. 2016;16:1378–1384. doi: 10.2174/1871520615666160504095040. [DOI] [PubMed] [Google Scholar]
- 65.Chen D, Cui QC, Yang H, Dou QP. Disulfiram, a Clinically Used Anti-alcoholism Drug and Copper-binding Agent, Induces Apoptotic Cell Death in Breast Cancer Cultures and Xenografts via Inhibition of the Proteasome Activity. Cancer Res. 2006;66:10425–10433. doi: 10.1158/0008-5472.CAN-06-2126. [DOI] [PubMed] [Google Scholar]
- 66.Kim JY, Cho Y, Oh E, Lee N, An H, Sung D, Cho TM, Seo JH. Disulfiram Targets Cancer Stem-like Properties and the HER2/Akt Signaling Pathway in HER2-positive Breast Cancer. Cancer Lett. 2016;379:39–48. doi: 10.1016/j.canlet.2016.05.026. [DOI] [PubMed] [Google Scholar]
- 67.Liu X, Wang L, Cui W, Yuan X, Lin L, Cao Q, Wang N, Li Y, Guo W, Zhang X, et al. Targeting ALDH1A1 by Disulfiram/copper Complex Inhibits Non-small Cell Lung Cancer Recurrence Driven by ALDH-positive Cancer Stem Cells. Oncotarget. 2016;7:58516–58530. doi: 10.18632/oncotarget.11305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hatoum D, McGowan EM. Recent Advances in the Use of Metformin: Can Treating Diabetes Prevent Breast Cancer? Biomed Res Int. 2015;2015:548436. doi: 10.1155/2015/548436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gwak H, Kim Y, An H, Dhanasekaran DN, Song YS. Metformin Induces Degradation of Cyclin D1 via AMPK/GSK3beta Axis in Ovarian Cancer. Mol Carcinog. 2017;56:349–358. doi: 10.1002/mc.22498. [DOI] [PubMed] [Google Scholar]
- 70.Vicini P, van der Graaf PH. Systems Pharmacology for Drug Discovery and Development: Paradigm Shift or Flash in the Pan? Clin Pharmacol Ther. 2013;93:379–381. doi: 10.1038/clpt.2013.40. [DOI] [PubMed] [Google Scholar]
- 71.Visser SA, de Alwis DP, Kerbusch T, Stone JA, Allerheiligen SR. Implementation of Quantitative and Systems Pharmacology in Large Pharma. CPT Pharmacometrics Syst Pharmacol. 2014;3:e142. doi: 10.1038/psp.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et al. The Connectivity Map: Using Gene-expression Signatures to Connect Small Molecules, Genes, and Disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 73.Cheng F, Hong H, Yang S, Wei Y. Individualized Network-based Drug Repositioning Infrastructure for Precision Oncology in the Panomics Era. Briefings Bioinf. 2017;18:682–697. doi: 10.1093/bib/bbw051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.