

Abstract
Mutations in several lipid synthetic enzymes that block fatty acid and ceramide production produce autosomal recessive congenital ichthyoses (ARCIs) and associated abnormalities in permeability barrier homeostasis. However, the basis for the phenotype in patients with NIPAL4 (ichthyin) mutations (among the most prevalent ARCIs) remains unknown. Barrier function was abnormal in an index patient and in canines with homozygous NIPAL4 mutations, attributable to extensive membrane stripping, likely from detergent effects of nonesterified free fatty acid. Cytotoxicity compromised not only lamellar body secretion but also formation of the corneocyte lipid envelope (CLE) and attenuation of the cornified envelope (CE), consistent with a previously unrecognized, scaffold function of the CLE. Together, these abnormalities result in failure to form normal lamellar bilayers, accounting for the permeability barrier abnormality and clinical phenotype in NIPA-like domain–containing 4 (NIPAL4) deficiency. Thus, NIPAL4 deficiency represents another lipid synthetic ARCI that converges on the CLE (and CE), compromising their putative scaffold function. However, the clinical phenotype only partially improved after normalization of CLE and CE structure with topical ω-O-acylceramide because of ongoing accumulation of toxic metabolites, further evidence that proximal, cytotoxic metabolites contribute to disease pathogenesis.
The autosomal recessive congenital ichthyoses (ARCIs) include a group of nonsyndromic disorders1 associated with mutations in several genes responsible for the generation of a competent stratum corneum (SC). A substantial subgroup of these mutations, as well as some syndromic recessive ichthyosiform disorders, compromise enzymes that synthesize fatty acids and ceramides required for permeability barrier homeostasis2, 3, 4, 5 (Supplemental Figure S1). These disorders demonstrate a wide range of phenotypes, all of which to date have been accompanied by a defective permeability barrier.6 It is our view that the phenotypes in the ichthyoses reflect a best attempt by a metabolically compromised epidermis to generate a permeability barrier sufficient for survival in a desiccating terrestrial environment.6, 7 Per this paradigm, the structural and biochemical alterations in these disorders reflect not only pathomechanisms that compromise barrier function but also survival-enhancing compensatory responses (Supplemental Table S1).8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21
Several of the ARCIs and syndromic recessive disorders that affect lipid synthesis result in failure to form the corneocyte lipid envelope (CLE).3, 5, 20, 21, 22, 23, 24, 25 By compromising its putative scaffold function, loss of the CLE could in turn disrupt the formation or supramolecular organization of the extracellular lamellar bilayers that mediate the permeability barrier.26 Of these disorders, the pathogenesis of the ichthyosiform dermatosis in patients with mutations in NIPAL4, which account for a substantial proportion of the ARCIs,27, 28, 29, 30 remains poorly understood. A variety of missense, nonsense, and splice mutations in NIPAL4 have been identified in NIPA-like domain–containing 4 (NIPAL4)–deficient patients,30, 31, 32, 33 who have highly variable skin phenotypes and distinctive structural alterations in the stratum granulosum (SG).34
The function of the transmembrane protein encoded by NIPAL4 remains speculative. Because of its homology to G-protein–coupled receptors, it has been suggested that NIPAL4 could serve as a receptor for linoleic acid–derived ligands, such as trioxilins A3 and B3, which in turn could regulate epidermal homeostasis27, 35 (Supplemental Figure S2A), although these metabolites have not yet been identified in NIPAL4-deficient epidermis. Alternatively, one member of the NIPAL superfamily has been proposed to serve as a magnesium (Mg++) cation transporter.36 Hence, mutations in NIPAL4 could instead (or in addition) block provision of Mg++ ions necessary for activation of the acetyl coenzyme A synthetase moiety of fatty acid transporter 4 (FATP4) (Supplemental Figure S2B).37 The link between NIPAL4 and FATP4 is further supported by the colocalization of these two proteins within the SG37, 38 and shared, distinctive ultrastructural abnormalities.38
We report here that NIPAL4 mutations in an index patient provoke a modest permeability barrier abnormality that is mirrored by comparable functional defects in a recently described pedigree of American bulldogs.39, 40 These canines exhibit a homozygous, cytosine frameshift deletion in exon 6 of NIPAL4, which produces a truncated NIPAL4 protein of 248 amino acids versus the wild-type length of 404.40 Availability of this canine model allowed us to characterize certain cellular and metabolic abnormalities that likely account for the barrier abnormality and, therefore, for the putative, barrier-driven, clinical phenotype in patients with this ARCI. Loss of NIPAL4 results in evidence of extensive cytotoxicity that appeared to compromise lamellar body secretion and formation of the CLE, which has been proposed to serve as scaffold for the extracellular lamellar bilayers.26 Although replenishment with topical ω-O-acylceramide (acylCer) normalized the CLE [and reversed an unexpected, parallel abnormality in cornified envelopes (CEs)], it did not correct the clinical phenotype, indicating that cytotoxicity attributable to accumulation of free fatty acids (FFAs) and/or other proximal metabolites likely continue to drive the clinical phenotype.
Materials and Methods
Animals, Functional Assessments, and Experimental Protocols
Animal Models
The index patient was a 32-year–old man who presented with generalizedcongenital ichthyosiform erythroderma, fine scale, but an absence of pruritus. Sequencing revealed a homozygous mutation in NIPAL4 – c.527C>A; p.Ala176Asp. American bulldogs with ARCIs, donated to the University of Pennsylvania, School of Veterinary Medicine at 5 weeks of age, were studied and cared for according to the principles outlined in the NIH Guide for the Care and Use of Laboratory Animals.41 Age- and breed-matched wild-type dogs were used as controls. NIPAL4-associated ARCI was confirmed via gross and histologic morphologic analysis39 and DNA-mutation analysis.40 Seven age- and breed-matched wild-type dogs served as controls. Transepidermal water loss (TEWL) and SC hydration were measured18 over axilla and abdomen to minimize the interference by hair at a climate-controlled testing facility (cyberDERM, Inc., Broomhall, PA).
Functional Measurements
TEWL and SC hydration were measured as previously described.18 Assessed sites included axilla and abdomen to minimize the interference by hair. Dogs were sampled while laterally recumbent with minimal physical restraint. TEWL measurements were averaged over 30-second periods, with mean ambient temperatures and ambient humidities of 22.2°C and 25.5%, respectively. TEWL measurements were obtained on one 6-month–old female and two age- and breed-matched wild-type dogs at a climate controlled testing facility (cyberDERM, Inc.), and seven normal dogs also were subsequently tested at the same facility.
Lipid-Treatment Protocol
Sibling, 5-month–old female ARCI dogs (dogs 3 and 4) were used in the treatment trial. Before treatments, hair was clipped, and 0.2 mg of acylCer (acylCer/N-acyl = C32 + ω-esterified C18:2), synthesized as in the study by Opálka et al,42 and vehicle alone were applied to 2 cm2 sparsely haired regions on ventral abdominal skin twice daily for 42 days. The lipids were supplied by NeoPharm (Seoul, South Korea). TEWL was measured on days 0, 21, and 42, and skin biopsy specimens were obtained on day 42 for morphologic studies.
Morphologic Studies
Light Microscopy and Immunohistochemistry
Skin punch biopsy specimen measuring 6 μm were obtained with the animals under local anesthesia (1% lidocaine subcutaneously), fixed in 10% neutral-buffered formalin and routinely processed for hematoxylin and eosin staining. Paraffin-embedded or frozen sections were incubated with rabbit polyclonal anti-NIPAL4 (sc-133280; Santa Cruz Biotechnology, Santa Cruz, CA) or rabbit polyclonal FATP4 (ab199719; Abcam, Cambridge, UK) antibodies in a 1:50 dilution (0.865 mg/mL) or mouse monoclonal Ki-67 (M7240; Dako, Glostrup, Denmark) at a 1:50 dilution (46 mg/mL) on paraffin-embedded sections from affected and age-matched control canines. Negative controls omitted the primary antibody. For immunodetection of NIPAL4 and FATP4, a LSAB2 kit (Dako, Carpinteria, CA) was used, assisted by DAB and chromogen, 3,3′-diaminobenzidine, and hematoxylin counterstaining.
For immunofluorescence, skin samples were fixed in 4% paraformaldehyde, cryoprotected in 30% sucrose, and embedded in ornithine carbamoyltransferase. Cryosections 5 μm thick were air-dried, washed with 0.15 mol/L phosphate-buffered saline, and incubated for 18 hours with rabbit anti-NIPAL4 (sc-133280, Santa Cruz Biotechnology), diluted 1:300 in Antibody Diluent Reagent Solution (003118; Invitrogen, Carlsbad, CA), and incubated with a 1:500 dilution of AlexaFluor 568 Goat Anti-Rabbit IgG secondary antibody (A11036; Invitrogen). After phosphate-buffered saline washing, slides were mounted in Vectashield with DAPI (H-1200; Vector Labs, Burlingame, CA). The proliferation index was calculated as percentage of Ki-67–positive cells at ×400 magnification.
Ultrastructural Methods
Ultrastructure was analyzed in three biopsy specimens from affected and two wild-type American bulldogs, using i) reduced postfixation osmium and ruthenium tetroxide ,43 ii) absolute pyridine pretreatment,44 iii) lipase ultracytochemistry,45, 46 iv) calcium ion distribution by pyroantimonate precipitation,47 and/or v) vapor-fixed D-squame strippings.42 For pyridine, 2-mm punch biopsy specimens were flash frozen and thawed in pyridine for 2 hours at room temperature, rinsed in 0.1 mol/L cacodylate buffer, and postfixed with reduced osmium. CE thickness measurements were performed directly on the JEOL-100CX microscope (JEOL, Peabody, MA) or on randomly selected images taken of each sample micrograph taken with a Gatan Bioscan Camera model 792 (Gatan, Pleasanton, CA) at 29,000×, calibrated with crystalline catalase, and a diffraction grating replica. Fifteen measurements were made on the narrowest part of the CE, using a measuring tool and software on the Gatan Camera.
Assessment of SC Permeability by Lanthanum Perfusion
Lanthanum nitrate, a water soluble low-molecular-weight tracer, was used as a marker to assess not only the integrity but also the pathway of water egress across NIPAL4-deficient SC (normal SC excludes lanthanum). Two 2-mm or 4-mm punch biopsy specimens were obtained with the animals under local anesthesia from the glabrous skin of the abdomens of dogs 1, 3, and 4 for electron microscopy. The entire sample was then immersed in 4% colloidal LaNO3 solution for 1 hour before standard electron microscopy fixation and processing, as described above.
Lipase Ultrastructural Cytochemistry
To assess the delivery and secretion of lamellar body contents,45, 46 tissue samples were fixed in half-strength Karnovsky's solution, rinsed in 0.1 mol/L cacodylate buffer, transferred to the incubation medium (5% Tween 85; 0.2 mol/L HEPES buffer, pH 7.2), with 2.5% sodium taurocholate in 10% aqueous CaCl2; and microwaved for 30 seconds at 37°C, followed by further incubation (30 minutes) in a 37°C oven. The tissues then were immersed for 3 minutes in 2% EDTA in cacodylate buffer, rinsed in deionized water, and immersed in 0.1% lead nitrate for 10 minutes at room temperature and rinsed, followed by postfixation in reduced OsO4 for 1 hour in the dark. After postfixation, samples were rinsed, dehydrated, and embedded as above.
Epidermal Calcium Localization
Ion-capture ultrastructural cytochemistry was performed, as previously described.48 An aqueous solution that contained 0.8 g of paraformaldehyde in 10 mL of 2% glutaraldehyde was warmed to 60°C, and two drops of 1 N potassium hydroxide were added. The mixture was stirred and cooled to room temperature, followed by the addition of 0.579 g of potassium oxalate, 8 mL of 10% glutaraldehyde (1.6 of 50%), and 0.56 g of sucrose, and the volume adjusted to 40 mL with deionized water. A total of 30 mL was combined with 10 mL of 4% OsO4 and cooled on ice. Skin biopsy specimens were finely minced (<0.2 mm) and microwaved in the mixture on high power three times for 30 seconds in an ice bath, followed by further fixation for 1 hour, and transferred to ice-cold OsO4 in the dark for 2 hours, washed with deionized water at pH 10 on ice for 15 minutes, placed in 50% EtOH, followed by dehydration and embedding as above.
Lipid Extraction, Fractionation, and Quantitation of Epidermal Ceramides
Scale was acquired by surface skin scrapings from three affected and three unaffected canines. Scale samples were first homogenized in water (2 mg in 100 μL), and bound and unbound ceramides were extracted and quantitated by gas liquid chromatography coupled to electrospray ionization tandem mass spectrometry, as previously described.4 Ceramide species were monitored in the positive ion mode with their specific multiple reaction monitoring.49 Integrated peak areas of endogenous ceramide species were corrected against the peak area of the internal standard ceramides (d17:1/C18:0) and then normalized to the dry weight of the canine epidermal samples.
Results
Ichthyosiform Phenotype in NIPAL4-Deficient Humans and American Bulldogs
In addition to an index patient with a known NIPAL4 mutation (Materials and Methods), we examined a pedigree of four affected American bulldogs with a recently described mutation in NIPAL4 (Figure 1A).40 A prominent band of immunostaining for the NIPAL4 protein was present in the outermost nucleated layers of unaffected canines. NIPAL4 was not detectible in the epidermis of affected animals (Figure 1, B and C). All affected canines had ichthyosiform skin changes that were most apparent over less hairy skin sites, such as the abdomen (Figure 1D). On light microscopy, affected skin revealed minimal inflammation but marked compact orthohyperkeratosis and prominent epidermal hyperplasia (Figure 1, E and F). Accordingly, Ki-67 immunolabeling revealed a significant increase in nuclear staining, restricted to the basal layer (Supplemental Figure S3, A and B). The increase in epidermal proliferation was paralleled by a prominent decline in immunostaining for filaggrin (Supplemental Figure S3, C and D), whereas loricrin expression appeared largely unchanged (not shown). Thus, NIPAL4 deficiency provokes an ichthyosiform dermatosis in canines that mirrors the clinical and histologic features of NIPAL4 deficiency in humans.
Figure 1.

Consequences of NIPA-like domain–containing 4 (NIPAL4) deficiency in affected canines. A: Sequencing chromatograms of a region within exon 6 of canine NIPAL4. Comparison of the sequences from the normal dog versus a canine affected with NIPAL4 deficiency shows a missing cytosine residue (arrow) in the mutant canine. B: Immunohistochemistry for NIPAL4 protein (Ich). C: Note the absence of NIPAL4 immunostraining in the granular cell layer in an affected canine versus positive staining in the granular layer of a normal (N) canine (arrows). D: Adherent fine, silvery scale (arrows) with erythema and comedones on the abdominal skin of an affected canine. E: Epidermal hyperplasia, marked orthokeratotic hyperkeratosis, with minimal inflammation in an affected (Ich) canine (hematoxylin and eosin staining). F: Histologic analysis of skin of a normal (N) dog (hematoxylin and eosin staining). Scale bars = 50 μm (B, C, E, and F). Panel A has been modified from Casal et al,40 with permission from PLoS One.
Loss of NIPAL4 Provokes Abnormalities in Epidermal Function
Functional measurements in the genotyped, NIPAL4-deficient patient revealed a modest increase in TEWL levels (14 to 15 mg/cm2/hour), assessed over both the volar forearm and dorsal thigh skin compared with age- and sex-matched, historical controls (<10 mg/cm2/hour). Likewise, the four affected dogs had an approximately 2.8-fold increase in basal TEWL compared with normal canines (P < 0.0001) (Figure 2A), paralleled by a striking approximately fourfold reduction in SC hydration (P < 0.002) (Figure 2B).
Figure 2.

Functional abnormalities in NIPA-like domain–containing 4 (NIPAL4) deficiency reflect cytotoxicity, with impaired lipid secretion. A and B: Elevated transepidermal water loss (TEWL) and reduced stratum corneum (SC) hydration (by corneometry) in affected canines. C and F: Representative images show reduced lamellar contents at the stratum granulosum (SG)–SC interface (open arrows), with appearance of prominent nonlamellar domains (asterisks) (C, postfixation ruthenium tetroxide). D and E: Decreased or patchy Nile red lipid staining in frozen sections from NIPAL4-deficient (open arrows) versus normal canine (arrows). F and H: Stripped membranes, with positive acid lipase labeling (arrows) of cytosolic organelles in stratum spinosum (SS) and SG of affected [ichthyin (Ich)] canines and abnormal lamellar body structure (open arrows) (postfixation osmium tetroxide). G: Pyroantimonate cytochemistry identifies Ca++ ion deposits within stripped membranes (postfixation osmium tetroxide). Scale bars: 0.1 μm (C); 200 μm (D and E); 2 μm (F and H); 0.2 μm (G). AU, absolute units.
Barrier status in affected and normal canines was next assessed by an alternate method (ie, lanthanum perfusion, which can provide additional information about the subcellular pathway of excessive water movement).50 Lanthanum extended into the extracellular spaces of the SC in affected canines, whereas tracer rarely reached the SG-SC interface in normal canines (Supplemental Figure S4). Thus, NIPAL4 deficiency provokes a defect in epidermal permeability homeostasis, with the pathway for excessive TEWL occurring via the SC interstices.
Epidermal Cytotoxicity Likely Contributes to the Barrier Abnormality in NIPAL4 Deficiency
The basis for the permeability barrier abnormality in NIPAL4-deficient canines was next assessed (Figure 2, C–H). If NIPAL4 deficiency compromises FFA–coenzyme A esterification, a resultant increase in cytosolic, nonesterified FFAs could provoke detergent-like cytotoxicity in the form of stripped membranes, which are a prominent feature of excessive cytosolic FFA.21, 51, 52, 53 Accordingly, lipid biochemistry revealed a 1.75-fold increase in epidermal FFA content in NIPAL4-deficient canine epidermis (Table 1), which correlated with significant declines in the N-acyl fatty acids of all individual bound and unbound ceramide fractions (Supplemental Table S2). Possible, widespread detergent-induced cytotoxicity, consisting of stripped membranes along the margins of the Golgi apparatus, mitochondria, endoplasmic reticulum, and nuclear envelopes, was evident in all suprabasal layers of affected epidermis (Figure 2, F and H). With the use of a content marker for an endoplasmic reticulum–derived protein (ie, acidic lipase), the origin of these stripped membranes could be further ascertained—most of the membrane arrays were densely decorated with lipase (Supplemental Figure S5E and Supplemental Figure S6). Pertinently, these membrane structural abnormalities closely resemble the stripped membranes that occur in ichthyosis prematurity syndrome (IPS) and its transgenic murine analog52, 53, 54, 55, 56 (Supplemental Figure S5, A–D), as well as in Refsum disease, where branched chain FFAs of vegetable origin accumulate,20 and Sjögren-Larsson syndrome, where a blockade in fatty aldehyde metabolism results in FFA-induced cytotoxicity.57, 58 Consistent with compensatory up-regulation, FATP4 protein levels appeared much higher in NIPAL4-deficient than in normal canine epidermis (Supplemental Figure S6). Finally, frequent colocalization of Ca++ precipitates with stripped membranes was also noted; such Ca++-FFA soaps comprise further, putative evidence of FFA-induced cytotoxicity (Figure 2G).59
Table 1.
Lipid Biochemical Analysis of Ceramide Scale in Affected Canines (Pooled Samples from Two Animals Each)
| Ceramide | Normal, pM/mg dry scale | NIPAL4 deficient, pM/mg dry scale | Fold change vs normal |
|---|---|---|---|
| Unbound ceramide | |||
| ω-O-acylceramide | 0.196 | 0.189 | 0.97 |
| ω-O-acylceramide | 4.724 | 0.481 | 0.10 |
| Total ceramide | 438.0 | 443.3 | 1.01 |
| Bound ω-O-acylceramide | 1.266 | 0.197 | 0.16 |
NIPAL4, NIPA-like domain–containing 4.
A prominent downstream consequence of membrane stripping included abnormalities in the lamellar body secretory system34 (Figures 2C and 3A). Not only were the morphologic features of individual lamellar bodies abnormal, but these organelles also appeared to be present at lower densities, resulting in reduced deposition of organelle contents at the SG-SC interface (Figure 3A). Depletion of secreted lamellar body contents correlated in turn with a marked decrease in Nile red fluorescence for SC lipids (Figure 2, E and D), which was paralleled by a paucity of lamellar bilayer structures (ie, residual arrays of normal-appearing lamellar bilayers were interspersed with expanded, nonlamellar lacunae) (Figure 2C). Thus, the barrier abnormality in NIPAL4 deficiency likely can be attributed to epidermal cytotoxicity, with a resultant decrease in the generation, secretion, and organization of replete, extracellular lamellar bilayers.
Figure 3.

NIPA-like domain–containing 4 (NIPAL4) mutations result in loss of corneocyte lipid envelope (CLE) and attenuated cornified envelopes (CEs). A and B: Decreased secretion (open arrows, A), attenuated CEs (arrows), and absent CLEs (open arrows, B) in NIPAL4-deficient canines (postfixation osmium tetroxide). C: Normal CLEs (arrows) and CEs (open arrows) in normal (N) canines (postfixation ruthenium tetroxide). D: Significant reduction in thickness of CEs in NIPAL4-deficient canines. E: Topical ω-O-acylceramides restore CE dimensions in two NIPAL4-deficient canines. Data are expressed as means ± SEM. n = 8 normal (D); n = 7 affected (D). ∗∗∗∗P < 0.0001 versus normal or vehicle. Scale bars: 1 μm (A); 0.5 μm (B); 0.1 μm (C). Ich, affected; SC, stratum corneum; SG, stratum granulosum.
Failure to Form the CLE May also Contribute to the NIPAL4 Phenotype
Although exocytosis of lamellar bodies normally results in the deposition of extracellular lipids, it also is paralleled by formation of the CLE, a monolayer of ω-hydroxyceramides (ω-OH-Cers) that is covalently bound to the external face of the CE (Supplemental Figure S1B). Accordingly, an additional consequence of compromised lamellar body secretion was an apparent loss of CLEs in affected SC (Figure 3, A–C), a structural deficit that was paralleled by a near absence of covalently bound ω-OH-Cers in the SC of affected canines (>10-fold reduction) and an increase in ultra–long-chain FFAs (Table 1). Pertinently, the decrease in the N-acyl fatty acid content of bound ceramide was also paralleled by a decrease in N-acyl fatty acid content of unbound ceramide (Supplemental Table S2). As in NIPAL4 deficiency, CLEs also were absent in IPS (Supplemental Figure S5E), where similar lipid biochemical abnormalities have been reported,37, 54 further supporting a close association between these two disorders. Finally, not only CLEs but also CEs appeared substantially attenuated in NIPAL4-deficient canines (Figure 3, A and B), further confirmed by quantitative measurements (Figure 3, D and E). Thus, the barrier defect in NIPAL4 deficiency is associated with bidirectional abnormalities in the CE/CLE scaffold.
Lipid Repletion Largely Normalizes CLE/CE Structure but Fails to Correct the Clinical Phenotype in NIPAL4 Deficiency
Because the barrier abnormality appears to reflect insufficient delivery of the key pathway end product (ie, ω-OH-Cer) to the SC, it was next determined whether topical replenishment with acylCer, a lipid product distal to the mutation (Supplemental Figure S1), can bypass the site of metabolic blockade, thereby normalizing the phenotype and barrier abnormality in NIPAL4-deficient canines (Figures 4 and 5, A and B). One side of the abdominal skin of two affected canines was treated once daily for up to 6 weeks with a mixture of barrier lipids that contained the synthetic, skin-identical acylCer, whereas the opposite surface was treated with vehicle alone. The structure of CLE and CE is largely normalized in lipid- but not in vehicle-treated sites (Figure 4, C and D), which has been further confirmed by quantitative measurements that revealed a near normalization of CE dimensions (Figure 3E). Moreover, the outward passage of lanthanum tracer was largely but incompletely blocked in lipid- but not vehicle-treated skin sites (Figure 4, A and B). Finally, lamellar body contents, density, and secreted lamellar bilayers also largely normalized in lipid- versus vehicle-treated sites (Figure 5, C–G).
Figure 4.
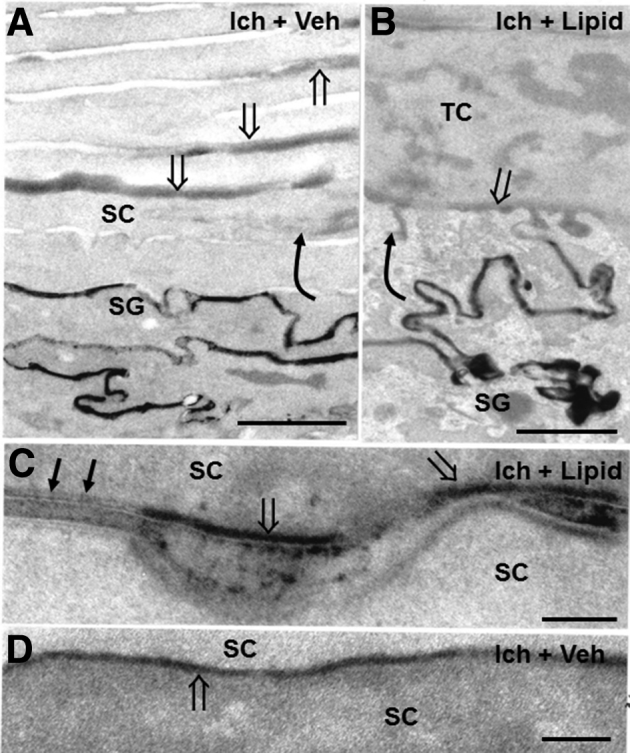
Topical ω-O-acylceramides (acylCers) restore corneocyte lipid envelope (CLE) and cornified envelope (CE), while partially normalizing barrier function in NIPA-like domain–containing 4 (NIPAL4)–deficient canines (postfixation osmium tetroxide). A: Lanthanum perfusion reveals free outward movement of tracer (curved arrow) through the stratum granulosum (SG)–stratum corneum (SC) interstices in vehicle-treated (Ich + Veh) NIPAL4-deficient canines (open arrows). B: Outward movement of tracer (curved arrow) is markedly reduced in acylCer-treated (Ich + Lipid) canine skin, reaching only the SG-SC interface (open arrow). C and D: Return of both CLEs (arrows) and CEs (open arrows in C) in acylCer- (Ich + Lipid) but not in vehicle-treated (Ich + Veh) canines (open arrows in D). Scale bars: 1 μm (A and B); 0.1 μm (C and D). TC, transitional cell.
Figure 5.
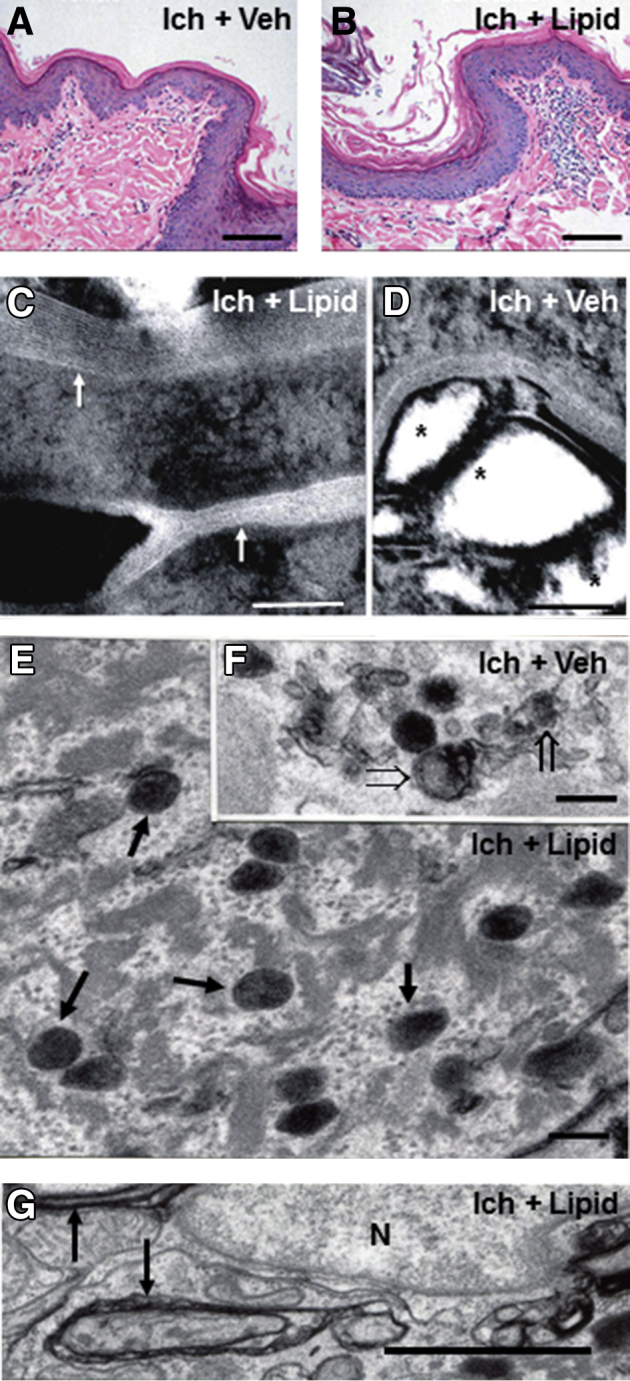
Although topical ω-O-acylceramides (acylCers) restore lamellar bilayers and lamellar body contents in NIPA-like domain–containing 4 (NIPAL4)–deficient canines, epidermal hyperplasia and cytotoxicity persist. A and B: Comparable epidermal hyperplasia and low-grade inflammation in lipid- (Ich + Lipid) versus vehicle-treated (Ich + Veh), NIPAL4 (Ich)–deficient skin (stained with hematoxylin and eosin). C and D: Return of lamellar bilayers in acylCer-treated, NIPAL4 (Ich)–deficient skin (arrows) and persistence of lamellar/nonlamellar phase separation in vehicle-treated skin (asterisks) (postfixation ruthenium tetroxide). E and F: Restoration of lamellar body contents in acylCer-treated (arrows) versus persistence of organelle abnormalities in vehicle-treated NIPAL4-deficient canines (open arrows) (postfixation osmium tetroxide). G: Evidence of ongoing membrane stripping despite lipid treatment of NIPAL4-deficient canines (arrows) (postfixation osmium tetroxide). Scale bars: 50 μm (A and B); 0.1 μm (C and D); 0.2 μm (E and F); 1 μm (G). N, nucleus.
However, despite an apparent improvement in the CE/CLE scaffold and the density, structure, and secretion of lamellar bodies, prominent macroscopic, histologic, and functional abnormalities persisted in lipid-treated sites (Figure 5, A and B), which correlated with the evidence of persistent cytotoxicity in these lipid-treated skin sites (Figure 5G). These results show that although lipid repletion largely corrected SC structure, it failed to normalize the clinical phenotype in affected canines, likely because of persistent cytotoxicity in lipid-treated skin sites. Because lipid repletion largely normalized not only the CLE but also the CE, the former structure appears to provide a scaffold necessary for the formation of replete subjacent CEs.
Discussion
Because all the ichthyoses examined to date have defects in permeability barrier function,5 the inability of a metabolically compromised epidermis to generate a competent permeability barrier likely shapes the clinical phenotype7, 50 (Supplemental Table S1). However, the cellular and metabolic basis for the barrier abnormality and subsequent phenotype often differs in different lipid synthetic ARCIs. For example, disease pathogenesis can reflect not only a failure to generate the pathway lipid product but also, in some cases, the accumulation of potentially toxic, metabolic intermediates. We found previously that such a dual pathogenic mechanism likely operates in a disorder of distal cholesterol metabolism, CHILD (congenital hemidysplasia with ichthyosiform erythroderma and limb defects) syndrome, where topical cotherapy with a statin (to reduce metabolite generation) and cholesterol (but not cholesterol alone) was required to correct the clinical phenotype.60 Likewise, our results suggest that not only a failure to generate ω-OH-Cers but also cytotoxicity from accumulated, excessive FFAs and/or other metabolic intermediates likely contributes to the pathogenesis of NIPAL4 deficiency.
If NIPAL4 serves as a membrane transporter that affects the bioavailability of the divalent cation, magnesium (Mg++), a cofactor required for activation of the acyl coenzyme synthase moiety in FATP4,37 nonesterified FFAs could (and did, as shown here) accumulate in the epidermis. The fact that the N-acyl, ultra–long-chain fatty acids in bound and unbound ceramides alone decreased further indicates that NIPAL4 deficiency compromises the synthesis and availability of ultra–long-chain fatty acids within the full spectrum of ceramide species found in normal SC. Pertinently, comparable abnormalities in the N-acyl acyl fatty acid content of bound and unbound ceramides occur in Fatp4-deficient mouse epidermis.54, 55, 56 Although the specific, responsible metabolite(s) was not identified, prominent membrane stripping and ultrastructural evidence of calcium soap formation were found, both likely reflecting detergent-like cytotoxicity attributable to the amphiphilic structure of these long-chain FFAs.21, 51, 52, 53, 59 Similar membrane stripping has been observed in IPS54 and Sjögren-Larsson syndrome,57, 58 where proximal accumulation of potentially toxic FFAs is known to occur. However, despite these similarities, the clinical phenotype in these two disorders differs greatly. These differences in clinical outcome can be explained by the known, compensatory postnatal up-regulation of other FATPs in epidermis.61, 62
The disruption of organelle membranes in NIPAL4-deficient epidermis in turn appeared to compromise the delivery of lamellar body contents to the SC interstices, thereby reducing the quantities and supramolecular organization of the extracellular lamellar bilayers. The lamellar bilayer abnormality in turn explains the permeability barrier abnormality, which in turn drives the clinical phenotype in NIPAL4 deficiency.6, 7 Because of the link between the proposed Mg++ transporter function of NIPAL4 and activation of the coenzyme A synthetase moiety in FATP4, which was expected, and indeed found, comparable ultrastructural alterations in patients with IPS, as well as in transgenic mice with deletion of this transporter (ie, curvilinear membrane sheets in patients with IPS and knockout mice, consistent again with FFA-induced cytotoxicity).54, 63
However, the phenotype in NIPAL4 deficiency likely reflects not only FFA-induced cytotoxicity but also a failure to generate the sphingolipid end products of this pathway. With the exception of mutations in transglutaminase 1 and ABCA12, all of the syndromic and nonsyndromic ARCIs compromise the synthesis or metabolism of epidermal ceramides (Supplemental Figure S1A). In several of these disorders, blockade in the proximal steps in this pathway decrease formation not only of N-acylCer, a key constituent of the extracellular lamellar bilayers, but also of the ω-OH-Cers that form the CLE.3, 22, 23, 24, 25, 64 This ω-OH-Cer–enriched structure forms a monolayer that is covalently bound to the external face of the CE,65, 66, 67 where it serves as a putative scaffold for the overlying lamellar bilayers.26 Indeed, we found biochemically and ultrastructurally that the CLE fails to form in NIPAL4-deficient epidermis. Thus, NIPAL4 deficiency can be added to the increasing list of mutations in fatty acid/ceramide metabolism that demonstrates a defective CLE, including patients or animal models with essential fatty acid deficiency,5 Refsum disease,21 the arachidonate lipoxygenase enzymes 12R-Lox and Aloxe3,25, 64 cgi-58 deficiency,23 ceramide synthase 3,3 and patatin-like phospholipase domain-containing protein 1 (Pnpla1) deficiency.22, 68, 69 The prominent abnormalities in barrier function and lamellar bilayer structure that accompany these mutations strongly suggest, but do not prove, that the CLE serves as a scaffold necessary for the formation and organization of the lamellar bilayers.70
Although we believe that the lack of a CLE scaffold, coupled with reduced delivery of lamellar body contents and the accumulation of proximal metabolites, can account for disease pathogenesis in these disorders, Fischer27 has championed an alternate pathogenic paradigm whereby NIPAL4 serves as a receptor for hepoxilin metabolites of FFAs. According to this view, failure of these intermediates to be internalized could in turn affect the activation of peroxisome proliferator-activated receptors (Supplemental Figure S2A), which is consistent with the known ability of exogenous peroxisome proliferator-activated receptor activators to improve barrier function in a variety of clinical and experimental settings.71 However, neither these metabolites nor their specific genetic signatures have been reported in NIPAL4-deficient patients. Moreover, defective hepoxilin ligand signaling likely does not account for the pathogenesis of NIPAL4 deficiency because the topical provision of the pathway end product, acylCer, improved barrier function by enhancing CLE/CE formation and the lamellar body secretory system. This observation supports the view that a lack of the CLE/CE scaffold contributes to the pathogenesis of NIPAL4 deficiency. Pertinently, Pnpla1-deficient mice have similar abnormalities in CLE structure and content,22, 68, 69 and both topical lipid applications and transfection of keratinocytes with wild-type Pnpla1 normalized the clinical phenotype by bypassing the site of metabolic blockade.22 In contrast, lipid replenishment alone reversed neither the epidermal hyperplasia nor the clinical phenotype in NIPAL4 deficiency. Thus, the CLE/CE defect alone cannot explain the phenotype—accumulation of toxic FFAs or other proximal metabolites likely also contribute to disease pathogenesis.
An entirely unanticipated outcome of these studies was the observation that reconstitution of the CLE after topical acylCer applications was paralleled by normalization of the underlying CE, suggesting that the CLE serves as a scaffold needed for the generation of a replete subjacent CE. It would be interesting to see whether absence of the CLE is accompanied by reduced CE formation in other ARCIs and whether correction of the CLE with topical pathway end products restores the CE in those disorders where the CLE is replenished. Pertinently, a normal CLE (and bound ω-OH-Cers) occur in TG1-deficient ARCIs (ie, lamellar ichthyosis) despite a markedly attenuated CE.18, 72 However, although lamellar bilayers are formed in this disorder, they are poorly organized,18 suggesting instead that the CE is required for the supramolecular organization of the extracellular lamellar bilayers.
Acknowledgments
We thank the patient with NIPAL4 deficiency and the Foundation for Ichthyosis and Related Skin Types.
Footnotes
Supported by the NIH National Institute of Arthritis, Musculoskeletal, and Skin grant AR061106, administered by the Northern California Institute for Research and Education, with additional resources provided by the Veterans Affairs Medical Center, San Francisco, CA, NIH grants P40 OD010939 and AR068392, the Commonwealth of Pennsylvania, and GAČR Czech Science Foundation grant 16-25687J.
Disclosures: None declared.
This content is solely the responsibility of the authors and does not necessarily represent the official views of either the National Institutes of Health or the Department of Veterans Affairs.
Supplemental material for this article can be found at https://doi.org/10.1016/j.ajpath.2018.02.008.
Supplemental Data
Supplemental Figure S1.

Fatty acid (FA) and ceramide synthetic pathway in autosomal recessive congenital ichthyosis converge on the corneocyte lipid envelope (CLE). A: Lipid metabolic pathway leading to formation of the CLE. Note close association of ichthyin and fatty acid transporter 4 (FATP4) in this pathway. B: Structure of the CLE and association to underlying cornified envelope (CE) (purple arrows). acylCer, ω-O-acylceramide; β-GlcCer'ase; β-glucocerebrosidase; FFA, free fatty acid; Glc, glucose; ω-acylGlcCer, ω-acyl-glucosylceramide; ω-OH-Cer, ω-hydroxyceramide; ω-OH-FA-CoA, ω-hydroxy-fatty acid-coenzyme A; ω-OH-FFA, ω-hydroxy-free fatty acid. Modified from Elias et al,5 with permission from Elsevier.
Supplemental Figure S2.

Basis for barrier abnormality in ichthyin deficiency and ichthyosis prematurity syndrome (IPS): alternate proposed pathogenic mechanisms. A: Representative role of ichthyin as a receptor for hepoxilin metabolites. B: Illustration of the paradigm proposed in this study. CLE, corneocyte lipid envelope; FATP4, fatty acid transporter 4; FFA, free fatty acid; LB, lamellar body; PPAR, peroxisome proliferator-activated receptor.
Supplemental Figure S3.

Basis for epidermal hyperplasia in affected canines: immunohistochemical staining. A and B: Ki-67 immunostaining in normal (A) versus affected (B) canines (arrows point to Ki-67–positive basal cells). C and D: Reduced filaggrin immunostaining (open arrows) in affected (Ich) versus normal (N) canines. Original magnification: ×10 (A and B); ×20 (C and D). Scale bar = 50 μm.
Supplemental Figure S4.

Lanthanum perfusion reveals partial correction of permeability barrier abnormality in lipid-treated, ichthyin-deficient canines. Lanthanum perfusion shows reduced outward movement (curved arrows) of tracer through stratum corneum (SC) and movement of tracer within extracellular spaces of SC (open arrows) in two affected (Ich) canines (A and B) but partial blockade of tracer egress beneath the stratum granulosum (SG) and SC interface in unaffected, normal (N) skin (C) (postfixation osmium tetroxide). SC1, first layer of stratum corneum; SC2, second layer of stratum corneum. Scale bars: 0.5 μm (A); 1 μm (B and C).
Supplemental Figure S5.
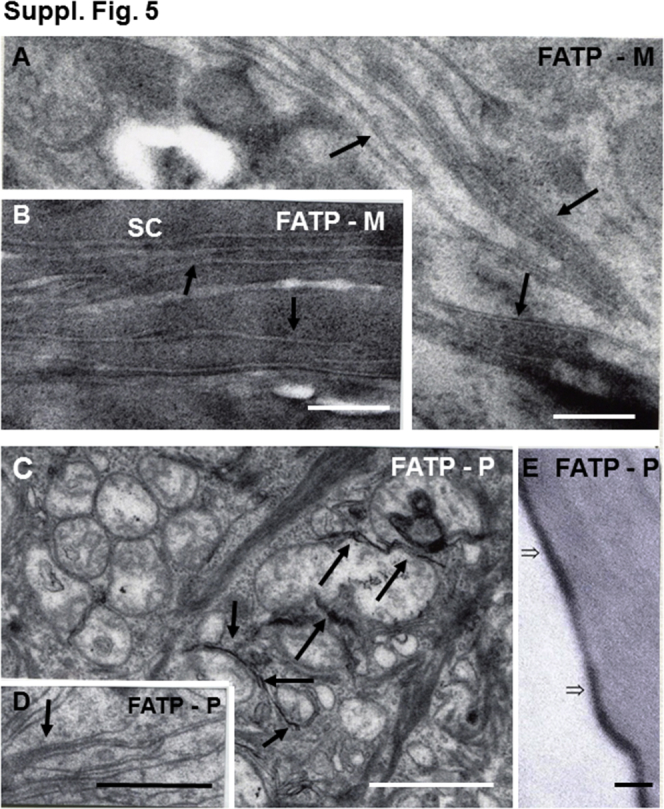
Evidence of cytotoxicity in fatty acid transporter 4 (FATP4)–deficient patients (P) and murine (M) epidermis: similarities to ichthyin deficiency. A–D: Note membrane stripping (arrows) and mitochondrial degeneration (postfixation osmium tetroxide). E: Attenuation of cornified envelopes and absence of the corneocyte lipid envelope (open arrows) in a sample from a patient with ichthyosis prematurity syndrome. Scale bars: 0.2 μm (A and B); 1 μm (C and D); 50 μm (E). FATP-M, transgenic knockout mice with loss of FATP4; FATP-P, human patient with FATP4 mutation; SC, stratum corneum.
Supplemental Figure S6.

Fatty acid transporter 4 (FATP4) immunostaining in ichthyin (Ich)- and patatin-like phospholipase domain-containing protein 1 (Pnpla1)–deficient versus normal canine epidermis. Scale bar = 50 μm.
References
- 1.Oji V., Tadini G., Akiyama M., Blanchet Bardon C., Bodemer C., Bourrat E. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Soreze 2009. J Am Acad Dermatol. 2010;63:607–641. doi: 10.1016/j.jaad.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 2.Holleran W.M., Man M.Q., Gao W.N., Menon G.K., Elias P.M., Feingold K.R. Sphingolipids are required for mammalian epidermal barrier function. Inhibition of sphingolipid synthesis delays barrier recovery after acute perturbation. J Clin Invest. 1991;88:1338–1345. doi: 10.1172/JCI115439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jennemann R., Rabionet M., Gorgas K., Epstein S., Dalpke A., Rothermel U., Bayerle A., van der Hoeven F., Imgrund S., Kirsch J., Nickel W., Willecke K., Riezman H., Grone H.J., Sandhoff R. Loss of ceramide synthase 3 causes lethal skin barrier disruption. Hum Mol Genet. 2012;21:586–608. doi: 10.1093/hmg/ddr494. [DOI] [PubMed] [Google Scholar]
- 4.Uchida Y., Cho Y., Moradian S., Kim J., Nakajima K., Crumrine D., Park K., Ujihara M., Akiyama M., Shimizu H., Holleran W.M., Sano S., Elias P.M. Neutral lipid storage leads to acylceramide deficiency, likely contributing to the pathogenesis of Dorfman-Chanarin syndrome. J Invest Dermatol. 2010;130:2497–2499. doi: 10.1038/jid.2010.145. [DOI] [PubMed] [Google Scholar]
- 5.Elias P.M., Gruber R., Crumrine D., Menon G., Williams M.L., Wakefield J.S., Holleran W.M., Uchida Y. Formation and functions of the corneocyte lipid envelope (CLE) Biochim Biophys Acta. 2014;1841:314–318. doi: 10.1016/j.bbalip.2013.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elias P.M., Williams M.L., Crumrine D., Schmuth M. S. Kargar AG; Basel: 2010. Ichthyoses - Clinical, Biochemical, Pathogenic, and Diagnostic Assessment. [Google Scholar]
- 7.Williams M.L., Elias P.M. From basketweave to barrier: unifying concepts for the pathogenesis of the disorders of cornification. Arch Dermatol. 1993;129:626–629. doi: 10.1001/archderm.129.5.626. [DOI] [PubMed] [Google Scholar]
- 8.Elias P.M., Williams M.L., Choi E.H., Feingold K.R. Role of cholesterol sulfate in epidermal structure and function: lessons from X-linked ichthyosis. Biochim Biophys Acta. 2014;1841:353–361. doi: 10.1016/j.bbalip.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rizzo W.B., S'Aulis D., Jennings M.A., Crumrine D.A., Williams M.L., Elias P.M. Ichthyosis in Sjogren-Larsson syndrome reflects defective barrier function due to abnormal lamellar body structure and secretion. Arch Dermatol Res. 2010;302:443–451. doi: 10.1007/s00403-009-1022-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vasireddy V., Uchida Y., Salem N., Jr., Kim S.Y., Mandal M.N., Reddy G.B., Bodepudi R., Alderson N.L., Brown J.C., Hama H., Dlugosz A., Elias P.M., Holleran W.M., Ayyagari R. Loss of functional ELOVL4 depletes very long-chain fatty acids (> or =C28) and the unique omega-O-acylceramides in skin leading to neonatal death. Hum Mol Genet. 2007;16:471–482. doi: 10.1093/hmg/ddl480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmuth M., Yosipovitch G., Williams M.L., Weber F., Hintner H., Ortiz-Urda S., Rappersberger K., Crumrine D., Feingold K.R., Elias P.M. Pathogenesis of the permeability barrier abnormality in epidermolytic hyperkeratosis. J Invest Dermatol. 2001;117:837–847. doi: 10.1046/j.0022-202x.2001.01471.x. [DOI] [PubMed] [Google Scholar]
- 12.Chan A., Holleran W.M., Ferguson T., Crumrine D., Goker-Alpan O., Schiffmann R., Tayebi N., Ginns E.I., Elias P.M., Sidransky E. Skin ultrastructural findings in type 2 Gaucher disease: diagnostic implications. Mol Genet Metab. 2011;104:631–636. doi: 10.1016/j.ymgme.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holleran W.M., Ziegler S.G., Goker-Alpan O., Eblan M.J., Elias P.M., Schiffmann R., Sidransky E. Skin abnormalities as an early predictor of neurologic outcome in Gaucher disease. Clin Genet. 2006;69:355–357. doi: 10.1111/j.1399-0004.2006.00589.x. [DOI] [PubMed] [Google Scholar]
- 14.Zhang L., Ferreyros M., Feng W., Hupe M., Crumrine D.A., Chen J., Elias P.M., Holleran W.M., Niswander L., Hohl D., Williams T., Torchia E.C., Roop D.R. Defects in stratum corneum desquamation are the predominant effect of impaired Abca12 function in a novel mouse model of Harlequin ichthyosis. PLoS One. 2016;11:e0161465. doi: 10.1371/journal.pone.0161465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choate K.A., Lu Y., Zhou J., Choi M., Elias P.M., Farhi A., Nelson-Williams C., Crumrine D., Williams M.L., Nopper A.J., Bree A., Milstone L.M., Lifton R.P. Mitotic recombination in patients with ichthyosis causes reversion of dominant mutations in KRT10. Science. 2010;330:94–97. doi: 10.1126/science.1192280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choate K.A., Lu Y., Zhou J., Elias P.M., Zaidi S., Paller A.S., Farhi A., Nelson-Williams C., Crumrine D., Milstone L.M., Lifton R.P. Frequent somatic reversion of KRT1 mutations in ichthyosis with confetti. J Clin Invest. 2015;125:1703–1707. doi: 10.1172/JCI64415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gruber R., Elias P.M., Crumrine D., Lin T.K., Brandner J.M., Hachem J.P., Presland R.B., Fleckman P., Janecke A.R., Sandilands A., McLean W.H., Fritsch P.O., Mildner M., Tschachler E., Schmuth M. Filaggrin genotype in ichthyosis vulgaris predicts abnormalities in epidermal structure and function. Am J Pathol. 2011;178:2252–2263. doi: 10.1016/j.ajpath.2011.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elias P.M., Schmuth M., Uchida Y., Rice R.H., Behne M., Crumrine D., Feingold K.R., Holleran W.M. Basis for the permeability barrier abnormality in lamellar ichthyosis. Exp Dermatol. 2002;11:248–256. doi: 10.1034/j.1600-0625.2001.110308.x. [DOI] [PubMed] [Google Scholar]
- 19.Schmuth M., Fluhr J.W., Crumrine D.C., Uchida Y., Hachem J.P., Behne M., Moskowitz D.G., Christiano A.M., Feingold K.R., Elias P.M. Structural and functional consequences of loricrin mutations in human loricrin keratoderma (Vohwinkel syndrome with ichthyosis) J Invest Dermatol. 2004;122:909–922. doi: 10.1111/j.0022-202X.2004.22431.x. [DOI] [PubMed] [Google Scholar]
- 20.Hachem J.P., Wagberg F., Schmuth M., Crumrine D., Lissens W., Jayakumar A., Houben E., Mauro T.M., Leonardsson G., Brattsand M., Egelrud T., Roseeuw D., Clayman G.L., Feingold K.R., Williams M.L., Elias P.M. Serine protease activity and residual LEKTI expression determine phenotype in Netherton syndrome. J Invest Dermatol. 2006;126:1609–1621. doi: 10.1038/sj.jid.5700288. [DOI] [PubMed] [Google Scholar]
- 21.Menon G.K., Orso E., Aslanidis C., Crumrine D., Schmitz G., Elias P.M. Ultrastructure of skin from Refsum disease with emphasis on epidermal lamellar bodies and stratum corneum barrier lipid organization. Arch Dermatol Res. 2014;306:731–737. doi: 10.1007/s00403-014-1478-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grond S., Eichmann T.O., Dubrac S., Kolb D., Schmuth M., Fischer J., Crumrine D., Elias P.M., Haemmerle G., Zechner R., Lass A., Radner F.P. PNPLA1 deficiency in mice and humans leads to a defect in the synthesis of omega-O-acylceramides. J Invest Dermatol. 2017;137:394–402. doi: 10.1016/j.jid.2016.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grond S., Radner F.P., Eichmann T.O., Kolb D., Grabner G.F., Wolinski H., Gruber R., Hofer P., Heier C., Schauer S., Rulicke T., Hoefler G., Schmuth M., Elias P.M., Lass A., Zechner R., Haemmerle G. Skin barrier development depends on CGI-58 protein expression during late-stage keratinocyte differentiation. J Invest Dermatol. 2017;137:403–413. doi: 10.1016/j.jid.2016.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uchida Y., Holleran W.M. Omega-O-acylceramide, a lipid essential for mammalian survival. J Dermatol Sci. 2008;51:77–87. doi: 10.1016/j.jdermsci.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 25.Krieg P., Rosenberger S., de Juanes S., Latzko S., Hou J., Dick A., Kloz U., van der Hoeven F., Hausser I., Esposito I., Rauh M., Schneider H. Aloxe3 knockout mice reveal a function of epidermal lipoxygenase-3 as hepoxilin synthase and its pivotal role in barrier formation. J Invest Dermatol. 2013;133:172–180. doi: 10.1038/jid.2012.250. [DOI] [PubMed] [Google Scholar]
- 26.Behne M., Uchida Y., Seki T., de Montellano P.O., Elias P.M., Holleran W.M. Omega-hydroxyceramides are required for corneocyte lipid envelope (CLE) formation and normal epidermal permeability barrier function. J Invest Dermatol. 2000;114:185–192. doi: 10.1046/j.1523-1747.2000.00846.x. [DOI] [PubMed] [Google Scholar]
- 27.Fischer J. Autosomal recessive congenital ichthyosis. J Invest Dermatol. 2009;129:1319–1321. doi: 10.1038/jid.2009.57. [DOI] [PubMed] [Google Scholar]
- 28.Sugiura K., Akiyama M. Update on autosomal recessive congenital ichthyosis: mRNA analysis using hair samples is a powerful tool for genetic diagnosis. J Dermatol Sci. 2015;79:4–9. doi: 10.1016/j.jdermsci.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 29.Akiyama M. Updated molecular genetics and pathogenesis of ichthiyoses. Nagoya J Med Sci. 2011;73:79–90. [PMC free article] [PubMed] [Google Scholar]
- 30.Pigg M.H., Bygum A., Ganemo A., Virtanen M., Brandrup F., Zimmer A.D., Hotz A., Vahlquist A., Fischer J. Spectrum of autosomal recessive congenital ichthyosis in Scandinavia: clinical characteristics and novel and recurrent mutations in 132 patients. Acta Derm Venereol. 2016;96:932–937. doi: 10.2340/00015555-2418. [DOI] [PubMed] [Google Scholar]
- 31.Lefevre C., Bouadjar B., Karaduman A., Jobard F., Saker S., Ozguc M., Lathrop M., Prud'homme J.F., Fischer J. Mutations in ichthyin a new gene on chromosome 5q33 in a new form of autosomal recessive congenital ichthyosis. Hum Mol Genet. 2004;13:2473–2482. doi: 10.1093/hmg/ddh263. [DOI] [PubMed] [Google Scholar]
- 32.Wajid M., Kurban M., Shimomura Y., Christiano A.M. NIPAL4/ichthyin is expressed in the granular layer of human epidermis and mutated in two Pakistani families with autosomal recessive ichthyosis. Dermatology. 2010;220:8–14. doi: 10.1159/000265757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kusakabe M., Nagai M., Nakano E., Jitsukawa O., Nishigori C., Yamanishi K. A Japanese case of ichthyosiform erythroderma with a novel mutation in NIPAL4/ichthyin. Acta Derm Venereol. 2017;97:397–398. doi: 10.2340/00015555-2550. [DOI] [PubMed] [Google Scholar]
- 34.Dahlqvist J., Klar J., Hausser I., Anton-Lamprecht I., Pigg M.H., Gedde-Dahl T., Jr., Ganemo A., Vahlquist A., Dahl N. Congenital ichthyosis: mutations in ichthyin are associated with specific structural abnormalities in the granular layer of epidermis. J Med Genet. 2007;44:615–620. doi: 10.1136/jmg.2007.050542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brash A.R., Yu Z., Boeglin W.E., Schneider C. The hepoxilin connection in the epidermis. FEBS J. 2007;274:3494–3502. doi: 10.1111/j.1742-4658.2007.05909.x. [DOI] [PubMed] [Google Scholar]
- 36.Goytain A., Hines R.M., Quamme G.A. Functional characterization of NIPA2, a selective Mg2+ transporter. Am J Physiol Cell Physiol. 2008;295:C944–C953. doi: 10.1152/ajpcell.00091.2008. [DOI] [PubMed] [Google Scholar]
- 37.Li H., Vahlquist A., Torma H. Interactions between FATP4 and ichthyin in epidermal lipid processing may provide clues to the pathogenesis of autosomal recessive congenital ichthyosis. J Dermatol Sci. 2013;69:195–201. doi: 10.1016/j.jdermsci.2012.11.593. [DOI] [PubMed] [Google Scholar]
- 38.Dahlqvist J., Westermark G.T., Vahlquist A., Dahl N. Ichthyin/NIPAL4 localizes to keratins and desmosomes in epidermis and Ichthyin mutations affect epidermal lipid metabolism. Arch Dermatol Res. 2012;304:377–386. doi: 10.1007/s00403-012-1207-7. [DOI] [PubMed] [Google Scholar]
- 39.Mauldin E.A., Wang P., Evans E., Cantner C.A., Ferracone J.D., Credille K.M., Casal M.L. Autosomal recessive congenital ichthyosis in American bulldogs is associated with NIPAL4 (ICHTHYIN) deficiency. Vet Pathol. 2015;52:654–662. doi: 10.1177/0300985814551425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Casal M.L., Wang P., Mauldin E.A., Lin G., Henthorn P.S. A defect in NIPAL4 is associated with autosomal recessive congenital ichthyosis in American bulldogs. PLoS One. 2017;12:e0170708. doi: 10.1371/journal.pone.0170708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Committee for the Update of the Guide for the Care and Use of Laboratory Animals. National Research Council . National Academies Press; Washington, DC: 2011. Guide for the Care and Use of Laboratory Animals: Eighth Edition. [Google Scholar]
- 42.Opálka L., Kovacik A., Sochorova M., Roh J., Kunes J., Lenco J., Vavrova K. Scalable synthesis of human ultralong chain ceramides. Org Lett. 2015;17:5456–5459. doi: 10.1021/acs.orglett.5b02816. [DOI] [PubMed] [Google Scholar]
- 43.Hou S.Y., Mitra A.K., White S.H., Menon G.K., Ghadially R., Elias P.M. Membrane structures in normal and essential fatty acid-deficient stratum corneum: characterization by ruthenium tetroxide staining and x-ray diffraction. J Invest Dermatol. 1991;96:215–223. doi: 10.1111/1523-1747.ep12461361. [DOI] [PubMed] [Google Scholar]
- 44.Lin T.K., Crumrine D., Ackerman L.D., Santiago J.L., Roelandt T., Uchida Y., Hupe M., Fabrias G., Abad J.L., Rice R.H., Elias P.M. Cellular changes that accompany shedding of human corneocytes. J Invest Dermatol. 2012;132:2430–2439. doi: 10.1038/jid.2012.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Menon G.K., Grayson S., Elias P.M. Cytochemical and biochemical localization of lipase and sphingomyelinase activity in mammalian epidermis. J Invest Dermatol. 1986;86:591–597. doi: 10.1111/1523-1747.ep12355263. [DOI] [PubMed] [Google Scholar]
- 46.Menon G.K., Ghadially R., Williams M.L., Elias P.M. Lamellar bodies as delivery systems of hydrolytic enzymes: implications for normal and abnormal desquamation. Br J Dermatol. 1992;126:337–345. doi: 10.1111/j.1365-2133.1992.tb00675.x. [DOI] [PubMed] [Google Scholar]
- 47.Menon G.K., Elias P.M. Ultrastructural localization of calcium in psoriatic and normal human epidermis. Arch Dermatol. 1991;127:57–63. [PubMed] [Google Scholar]
- 48.Menon G.K., Elias P.M., Lee S.H., Feingold K.R. Localization of calcium in murine epidermis following disruption and repair of the permeability barrier. Cell Tissue Res. 1992;270:503–512. doi: 10.1007/BF00645052. [DOI] [PubMed] [Google Scholar]
- 49.Eckl K.M., Tidhar R., Thiele H., Oji V., Hausser I., Brodesser S., Preil M.L., Onal-Akan A., Stock F., Muller D., Becker K., Casper R., Nurnberg G., Altmuller J., Nurnberg P., Traupe H., Futerman A.H., Hennies H.C. Impaired epidermal ceramide synthesis causes autosomal recessive congenital ichthyosis and reveals the importance of ceramide acyl chain length. J Invest Dermatol. 2013;133:2202–2211. doi: 10.1038/jid.2013.153. [DOI] [PubMed] [Google Scholar]
- 50.Elias P.M., Williams M.L., Feingold K.R. Abnormal barrier function in the pathogenesis of ichthyosis: therapeutic implications for lipid metabolic disorders. Clin Dermatol. 2012;30:311–322. doi: 10.1016/j.clindermatol.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iwig M., Glaesser D., Fass U., Struck H.G. Fatty acid cytotoxicity to human lens epithelial cells. Exp Eye Res. 2004;79:689–704. doi: 10.1016/j.exer.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 52.Glaesser D., Fass U., Gruner M., Thust O., Iwig M., Spindler M. Low concentrations of cis-linoleic acid induce cell damage in epithelial cells from bovine lenses. Eur J Cell Biol. 1996;71:286–292. [PubMed] [Google Scholar]
- 53.Lieberthal W., Sheridan A.M., Schwartz J.H. Fatty acid-induced cytotoxicity: differences in susceptibility between MDCK cells and primary cultures of proximal tubular cells. J Lab Clin Med. 1997;129:260–265. doi: 10.1016/s0022-2143(97)90148-7. [DOI] [PubMed] [Google Scholar]
- 54.Moulson C.L., Lin M.H., White J.M., Newberry E.P., Davidson N.O., Miner J.H. Keratinocyte-specific expression of fatty acid transport protein 4 rescues the wrinkle-free phenotype in Slc27a4/Fatp4 mutant mice. J Biol Chem. 2007;282:15912–15920. doi: 10.1074/jbc.M701779200. [DOI] [PubMed] [Google Scholar]
- 55.Khnykin D., Ronnevig J., Johnsson M., Sitek J.C., Blaas H.G., Hausser I., Johansen F.E., Jahnsen F.L. Ichthyosis prematurity syndrome: clinical evaluation of 17 families with a rare disorder of lipid metabolism. J Am Acad Dermatol. 2012;66:606–616. doi: 10.1016/j.jaad.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 56.Herrmann T., van der Hoeven F., Grone H.J., Stewart A.F., Langbein L., Kaiser I., Liebisch G., Gosch I., Buchkremer F., Drobnik W., Schmitz G., Stremmel W. Mice with targeted disruption of the fatty acid transport protein 4 (Fatp 4, Slc27a4) gene show features of lethal restrictive dermopathy. J Cell Biol. 2003;161:1105–1115. doi: 10.1083/jcb.200207080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ito M., Oguro K., Sato Y. Ultrastructural study of the skin in Sjogren-Larsson syndrome. Arch Dermatol Res. 1991;283:141–148. doi: 10.1007/BF00372053. [DOI] [PubMed] [Google Scholar]
- 58.Matsuoka L.Y., Kousseff B.G., Hashimoto K. Studies of the skin in Sjogren-Larsson syndrome by electron microscopy. Am J Dermatopathol. 1982;4:295–301. doi: 10.1097/00000372-198208000-00002. [DOI] [PubMed] [Google Scholar]
- 59.Palmquist D.L., Jenkins T.C., Joyner A.E., Jr. Effect of dietary fat and calcium source on insoluble soap formation in the rumen. J Dairy Sci. 1986;69:1020–1025. doi: 10.3168/jds.S0022-0302(86)80497-0. [DOI] [PubMed] [Google Scholar]
- 60.Paller A.S., van Steensel M.A., Rodriguez-Martin M., Sorrell J., Heath C., Crumrine D., van Geel M., Cabrera A.N., Elias P.M. Pathogenesis-based therapy reverses cutaneous abnormalities in an inherited disorder of distal cholesterol metabolism. J Invest Dermatol. 2011;131:2242–2248. doi: 10.1038/jid.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmuth M., Ortegon A.M., Mao-Qiang M., Elias P.M., Feingold K.R., Stahl A. Differential expression of fatty acid transport proteins in epidermis and skin appendages. J Invest Dermatol. 2005;125:1174–1181. doi: 10.1111/j.0022-202X.2005.23934.x. [DOI] [PubMed] [Google Scholar]
- 62.Lin M.H., Miner J.H. Fatty acid transport protein 1 can compensate for fatty acid transport protein 4 in the developing mouse epidermis. J Invest Dermatol. 2015;135:462–470. doi: 10.1038/jid.2014.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin M.H., Khnykin D. Fatty acid transporters in skin development, function and disease. Biochim Biophys Acta. 2014;1841:362–368. doi: 10.1016/j.bbalip.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zheng Y., Yin H., Boeglin W.E., Elias P.M., Crumrine D., Beier D.R., Brash A.R. Lipoxygenases mediate the effect of essential fatty acid in skin barrier formation: a proposed role in releasing omega-hydroxyceramide for construction of the corneocyte lipid envelope. J Biol Chem. 2011;286:24046–24056. doi: 10.1074/jbc.M111.251496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wertz P.W., Swartzendruber D.C., Kitko D.J., Madison K.C., Downing D.T. The role of the corneocyte lipid envelope in cohesion of the stratum corneum. J Invest Dermatol. 1989;93:169–172. doi: 10.1111/1523-1747.ep12277394. [DOI] [PubMed] [Google Scholar]
- 66.Sandhoff R. Very long chain sphingolipids: tissue expression, function and synthesis. FEBS Lett. 2010;584:1907–1913. doi: 10.1016/j.febslet.2009.12.032. [DOI] [PubMed] [Google Scholar]
- 67.Kihara A. Synthesis and degradation pathways, functions, and pathology of ceramides and epidermal acylceramides. Prog Lipid Res. 2016;63:50–69. doi: 10.1016/j.plipres.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 68.Hirabayashi T., Anjo T., Kaneko A., Senoo Y., Shibata A., Takama H., Yokoyama K., Nishito Y., Ono T., Taya C., Muramatsu K., Fukami K., Munoz-Garcia A., Brash A.R., Ikeda K., Arita M., Akiyama M., Murakami M. PNPLA1 has a crucial role in skin barrier function by directing acylceramide biosynthesis. Nat Commun. 2017;8:14609. doi: 10.1038/ncomms14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ohno Y., Kamiyama N., Nakamichi S., Kihara A. PNPLA1 is a transacylase essential for the generation of the skin barrier lipid omega-O-acylceramide. Nat Commun. 2017;8:14610. doi: 10.1038/ncomms14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Akiyama M. Corneocyte lipid envelope (CLE), the key structure for skin barrier function and ichthyosis pathogenesis. J Dermatol Sci. 2017;88:3–9. doi: 10.1016/j.jdermsci.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 71.Schmuth M., Jiang Y.J., Dubrac S., Elias P.M., Feingold K.R. Thematic review series: skin lipids: peroxisome proliferator-activated receptors and liver X receptors in epidermal biology. J Lipid Res. 2008;49:499–509. doi: 10.1194/jlr.R800001-JLR200. [DOI] [PubMed] [Google Scholar]
- 72.Paige D.G., Morse-Fisher N., Harper J.I. Quantification of stratum corneum ceramides and lipid envelope ceramides in the hereditary ichthyoses. Br J Dermatol. 1994;131:23–27. doi: 10.1111/j.1365-2133.1994.tb08452.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.