

Abstract
The transcription factor Hes family basic helix-loop-helix transcription factor 1 (Hes1) is a downstream effector of Notch signaling and plays a crucial role in orchestrating developmental processes during the embryonic stage. However, its aberrant signaling in adulthood is linked to the pathogenesis of cancer. In the present study, we report the discovery of small organic molecules (JI051 and JI130) that impair the ability of Hes1 to repress transcription. Hes1 interacts with the transcriptional corepressor transducing-like enhancer of split 1 (TLE1) via an interaction domain comprising two tryptophan residues, prompting us to search a chemical library of 1,800 small molecules enriched for indole-like π-electron–rich pharmacophores for a compound that blocks Hes1-mediated transcriptional repression. This screening identified a lead compound whose extensive chemical modification to improve potency yielded JI051, which inhibited HEK293 cell proliferation with an EC50 of 0.3 μm. Unexpectedly, using immunomagnetic isolation and nanoscale LC-MS/MS, we found that JI051 does not bind TLE1 but instead interacts with prohibitin 2 (PHB2), a cancer-associated protein chaperone. We also found that JI051 stabilizes PHB2's interaction with Hes1 outside the nucleus, inducing G2/M cell-cycle arrest. Of note, JI051 dose-dependently reduced cell growth of the human pancreatic cancer cell line MIA PaCa-2, and JI130 treatment significantly reduced tumor volume in a murine pancreatic tumor xenograft model. These results suggest a previously unrecognized role for PHB2 in the regulation of Hes1 and may inform potential strategies for managing pancreatic cancer.
Keywords: chemical biology, small molecule, Notch pathway, basic helix-loop-helix transcription factor, cell proliferation, chaperone, pancreatic cancer, Prohibitin 2
Introduction
The basic helix-loop-helix transcription factor Hes1 is a downstream effector of Notch signaling that is required for the maintenance of progenitor cells in the nervous and digestive systems (1). A number of studies also suggest that activating mutations in Notch receptors (2) and the up-regulation of Notch pathway components are associated with tumorigenesis (3–5), whereas Notch receptors are down-regulated under normal physiological conditions (6).
Notch signaling is conveyed from cell to cell when the Notch receptor protein interacts with its ligand, leading to receptor cleavage by γ-secretase. Following its cleavage, Notch intracellular domain (NICD)4 translocates to the nucleus and associates with RBP-J to recruit coactivators, leading to the activation of downstream target genes, including Hes1 (7). Hes1 down-regulates its own expression by binding to N-box sequences (CACNAG) within its promoter region (8), leading to the recruitment of the transcriptional corepressor TLE1 (9). The interaction between Hes1 and TLE1 is mediated by the WRPW (Trp-Arg-Pro-Trp) motif of Hes1 (10) and the WD-repeat domain of TLE1 (11).
Over the past decade, γ-secretase inhibitors have been investigated for their potential to prevent Notch receptor activation in cancer cells (12). However, because γ-secretase has over 100 identified and postulated substrates, such cancer therapies have untoward side effects (13). Targeting Notch signaling more directly with Hes1 inhibitors may therefore have the advantage of limiting off-target effects. Although small-molecule Hes1 dimer inhibitors (14, 15) and Hes1 modulators with unknown mode of action (16) have previously been reported, Hes1 inhibitors to slow down cancer progression are still highly needed.
In this study, we searched a focused chemical library for small-molecule Hes1 inhibitors that competitively block TLE1 binding to Hes1. Extensive structural optimization of a hit molecule led to the discovery of JI051, which reverted Hes1-mediated transcriptional repression and interfered with cell proliferation. In a surprising twist, JI051 did not bind to TLE1 but instead interacted with an unexpected player, PHB2.
Results
Small-molecule screening for Hes1 inhibitors
Because the WRPW Hes1–TLE1 interaction domain is composed of two tryptophan residues each comprising a side-chain indole (17), we searched a chemical library enriched with indole-like π-electron–rich pharmacophores for a small molecule that would mimic the protein–protein interface to block Hes1-mediated transcriptional repression. A total of 1,800 compounds were screened with a luciferase reporter assay to study the activity of the Hes1 promoter (Fig. 1A). As shown in Fig. S1A, exogenous Hes1 expression decreased the expression of the reporter gene to 56.5 ± 4.1% compared with cells transfected with an empty vector (control), due to negative feedback regulation on the Hes1 promoter. The ability of each small molecule to revert the Hes1-mediated luciferase gene repression was reported as an increase in the luminescence signal compared with cells treated with DMSO. Of the compounds tested, 17 increased the level of luciferase expression to more than 80% of control levels.
Figure 1.
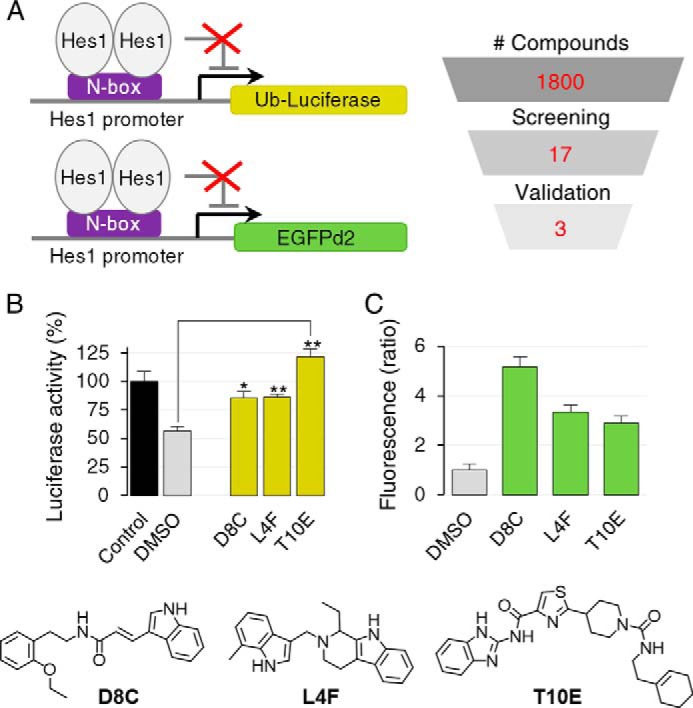
Isolation of Hes1 inhibitors. A, procedure for the selection and validation of compounds from the chemical library. HEK293 cells were transfected with the indicated reporter genes expressed under the control of the Hes1 promoter, together with Hes1 under the control of a constitutive promoter (pCMV-Hes1) to repress transcription. B, effect of validated compounds (2.5 μm) on the Hes1-mediated repression of luciferase gene expression. Data are displayed as means ± S.D. Two-tailed Student's t test was used for statistical analysis (*, p ≤ 0.01, and **, p ≤ 0.001, compared with DMSO control). C, effect of validated compounds (10 μm) on the Hes1-mediated repression of EGFPd2 gene expression. Data are mean ± S.D. from a minimum of nine individual fields representing at least 366 cells per condition.
As benzothiazoles, benzimidazoles, and benzoxazoles have been previously reported to act as firefly luciferase stabilizers (18), we performed a validation screening by confocal microscopy using a Hes1 promoter-driven EGFPd2 reporter assay to limit false-positive compounds (Fig. 1A). Of the compounds that initially passed the screening with the luciferase reporter gene, three of them (D8C, L4F, and T10E) produced a fluorescence signal 3-fold higher compared with DMSO in cells overexpressing Hes1 (Fig. S1B). Among these validated compounds, D8C, an indolylacrylamide molecule, induced the most significant response, with a 5.2-fold increase in fluorescence signal. Based on these results, D8C was chosen as the lead compound for further analysis. Fig. 1, B and C, highlights the effects of the validated compound on Hes1-mediated transcriptional repression on luciferase and the EGFPd2 reporter gene, respectively.
Effect of D8C derivatives of cell proliferation
Hes1 is essential for cell proliferation as progenitor cells with inactivated Hes1 choose to exit the cell cycle (19). To examine whether D8C influences cell growth, a colorimetric cell proliferation assay was carried out using the tetrazolium salt WST-8. As shown in Fig. 2A, D8C decreased the cell proliferation to 69.9 ± 2.4% of the levels observed in cells treated with DMSO after an incubation period of 24 h. The inhibition of cell growth was time-dependent and reached a plateau after 72 h (37.8 ± 1.3% compared with DMSO).
Figure 2.

D8C inhibits cell proliferation. A, HEK293 cells were incubated with 2.5 μm D8C before cell proliferation analysis with the tetrazolium salt WST-8. B, dose–response curve of the effect of D8C derivatives on cell proliferation following a 24-h incubation. C, effect of Hes1 gene knockdown on the JI051-mediated response. Cells were treated with 1 μm JI051 for 24 h. Data are presented as mean ± S.D. Bottom, chemical structures of D8C derivatives and corresponding EC50 values (n/a = not applicable).
To improve the potency of D8C, we synthesized a series of 130 compounds derived from its chemical structure, including modifications of the ethoxyphenetyl, amide, alkene, and indole moieties (Table S1 and supporting Chemical Synthesis). Structure–activity relationship studies revealed that the ethoxy group needs to be located in the ortho-position of the phenyl ring, as the meta- and para-positions resulted in a significant loss of activity. Introduction of longer alkyl groups or substitution with halogen atoms also resulted in a loss of activity. Other modifications around the amide bond were not tolerated for activity, and deletion of the double bond or the indole ring resulted in activity loss. In contrast, modifications including the introduction of a methoxy group (JI051) or a methyl group (JI021) at position 7 of the indole ring resulted in improved EC50 values compared with D8C (Fig. 2B).
JI051 induces G2/M cell-cycle arrest
To determine whether the decrease in proliferation could be attributed to cell-cycle arrest, HEK293 cells were transfected with the fluorescent ubiquitination-based cell-cycle indicators (Fucci) (20). These probes exploit the cell cycle-dependent proteolysis of Cdt1 and geminin by E3 ligases, enabling the visualization of G1 and G2/M phases in red and green, respectively. As shown in Fig. 3A, treatment with JI051 induced a dose-dependent increase in the geminin/Cdt1 fluorescence ratio, suggesting an increase in the cell population in G2/M. A similar increase was also observed with the microtubule-depolarizing agent nocodazole, which is known to arrest cell cycle progression in G2/M (21). As the Fucci probes do not enable the distinction between G2 and M phases of the cell cycle, we examined the effect of JI051 on chromatin condensation to evaluate the percentage of cells undergoing mitosis. As shown in Fig. 3B, the proportion of cells with visible chromosomes increased from 3.6 to 37.9% following treatment with JI051. Fluorescence microscopy analysis also showed incomplete chromosome congression at the metaphase plate, suggesting that the cells are blocked in mitotic prometaphase.
Figure 3.
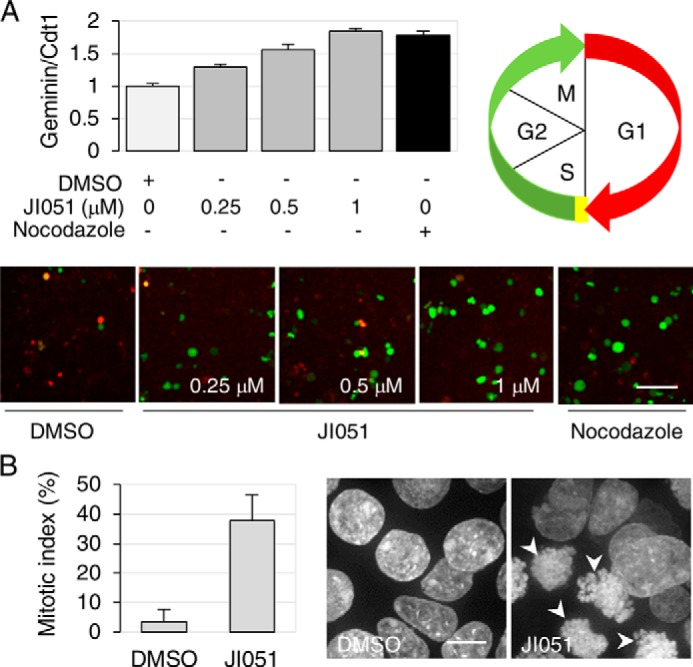
JI051 induces cell-cycle arrest in G2/M. A, HEK293 cells expressing the Fucci fluorescent probes mCherry-hCdt1 (30/120) (red) and AmCyan-hGeminin (green) were treated with 1 μm JI051 for 24 h prior to confocal microscopy imaging in live cells. An increase in geminin/Cdt1 ratio indicates an increase in the cell population in the G2/M phase of the cell cycle. Nocodazole (1000 ng/ml) was used as a positive control. Data are mean ± S.D. from triplicates (nine fields each) representing a minimum of 762 cells per condition. Scale bar, 50 μm. B, calculation of the mitotic index (% of cells with condensed chromatin) following a 24-h incubation with 1 μm JI051 or DMSO. Nuclei were stained with Hoechst 33342. Arrowheads indicate condensed chromatin. Data represent mean ± S.D. from a minimum of 22 individual fields representing at least 373 cells per condition. Scale bar, 10 μm.
Hes1 is important for JI051 response
To confirm whether the JI051 effect on cell proliferation is mediated through Hes1, we carried out a series of experiments in mouse embryonic fibroblasts (MEF) derived from Hes1–KO mice (Fig. S2). As shown in Fig. 2C, Hes1 gene depletion induced a 40.1 ± 8.6% decrease in cell growth as compared with control cells. This reduction was equivalent to that observed in control cells treated with JI051 (35.2 ± 6.5%). However, incubation with JI051 did not further reduce cell growth (42.2 ± 2.7%) in Hes1 KO cells, suggesting that Hes1 is required for the JI051 response.
We also tested the effect of Hes1 overexpression on G2/M cell-cycle arrest. For this experiment, HEK293 cells were transfected with the Fucci probes together with increasing amounts of pC1–Hes1 cDNA. As shown in Fig. S3, overexpression of Hes1 blocked JI051-mediated G2/M cell-cycle arrest in a dose-dependent manner, reaching levels similar to those observed in the absence of the compound. Taken together, these results indicate that JI051 mediates cell-cycle arrest by interfering with Hes1 function.
Target identification for JI051
As our initial strategy was to mimic the WRPW motif of Hes1 for blocking Hes1–TLE1 interaction, we attempted to determine whether JI051 interacts with TLE1. For this purpose, we synthesized a JI051 photoreactive probe (JITV10) equipped with a diazirine group for covalent binding upon UV irradiation, and a biotin moiety for pulldown with NeutrAvidin-agarose beads (Fig. S4A). Although TLE1 was efficiently pulled down in HEK293 cells overexpressing FLAG–TLE1, a 5-fold excess of JI051 failed to compete with JITV10 and comparable labeling was obtained with JIN04 (a negative control lacking JI051 moiety), showing that TLE1 labeling is not specific for JI051 (Fig. S4B).
To identify the true protein target for JI051, we carried out immunomagnetic isolation of target protein(s) with streptavidin-coupled Dynabeads for optimal washing efficiency. We also used a biotinylated version of JI051 (JITV14) without the diazirine moiety to reduce nonspecific binding. Analysis of bound proteins by SDS-PAGE (Fig. 4A) showed a distinct band pattern for JITV14 compared with JIN05 (negative control without the JI051 moiety). The protein bands that were efficiently displaced by a 10-fold excess of JI130 (a JI051 derivative with improved solubility; Table S1) were isolated and digested with trypsin, and the resulting peptides were analyzed by nanoscale LC/tandem MS (nano LC-MS/MS). MS/MS ion search (22) identified three potential targets: Prohibitin 2 (PHB2); glyceraldehyde 3-phosphate dehydrogenase; and β-actin. Because the last two proteins represent nonspecific abundant proteins frequently occurring during target identification, we therefore focused on PHB2, a cancer-related protein chaperone, which showed 12 matching peptides covering 51% of the entire protein sequence (Fig. S5).
Figure 4.

Prohibitin 2 is JI051 target. A, target identification by microsequencing analysis. Silver staining of proteins isolated by streptavidin beads upon incubation with a biotinylated version of JI051 (JITV14) before or after displacement with a 10-fold excess of JI130. DMSO and JIN05 were used as negative controls. Bands indicated by arrows were analyzed by microsequencing. B, validation of PHB2 as a target for JI051 by pulldown with a photoreactive probe. FLAG–PHB2-expressing HEK293 cells were incubated with NeutrAvidin beads together with JIN04 (negative control) or JITV10 (diazirine photoreactive moiety), with or without a 2.5-fold excess of JI051, prior to Western blotting with anti-FLAG antibody. Input represents the supernatant fraction of cell lysates. C, KD determination of JITV10. Human recombinant GST-tagged PHB2 protein was incubated with JITV10 before UV exposure and Western blotting with HRP-conjugated streptavidin or PHB2 antibody. The densitometry analysis of HRP–streptavidin signal was normalized to PHB2.
We then performed a series of pulldown studies in HEK293 cells overexpressing FLAG–PHB2 to confirm that PHB2 is a target for JI051. Western blotting revealed that PHB2 was photoreacted and pulled down with JITV10, but not with JIN04 (negative control without the JI051 moiety), and displaced with a 2.5-fold excess of JI051 (bound fraction, Fig. 4B). To determine whether JITV10 binds directly to PHB2 or needs additional proteins found in cell lysates, a series of photoaffinity studies with human recombinant PHB2 protein was carried out, using streptavidin conjugated with horseradish peroxidase (HRP) for detection. As shown in Fig. S6A, HRP–streptavidin labeling confirmed the biotinylation of PHB2 following incubation with 25 μm JITV10, suggesting that JITV10 binds directly to PHB2. The signal was displaced with increasing amounts of JI051, and UV irradiation was required for JITV10/PHB2 cross-linking. Analysis of PHB2 recombinant protein incubated with JITV10 also revealed a dose-dependent biotinylation of PHB2 with a KD of 3.7 μm (Fig. S6B and Fig. 4C), indicating that JITV10 binds directly to PHB2. In addition, labeling of JITV10 with a streptavidin–Alexa 568 conjugate displayed a substantial overlap with PHB2 immunostaining in HEK293 cells (Fig. S7). Taken together, these results suggest that PHB2 is a bona fide target for JI051.
Effect of PHB2 depletion on JI051 response
We next carried out a series of siRNA experiments to determine whether PHB2 is important for JI051 response. PHB2 siRNAs decreased PHB2 protein levels by at least 50% (Fig. S8A) and induced a substantial decrease in cell proliferation compared with cells treated with scrambled (control) siRNAs (77.4 ± 4.7% and 26.6 ± 0.6%, respectively; Fig. S8, B and C). This reduction in cell growth is consistent with previous studies in PHB2-depleted cells (23, 24). However, condensed chromatin was not observed in cells treated with PHB2 siRNAs alone (Fig. S8, D and E), suggesting that PHB2-depleted cells are not arrested in mitosis. Next, we investigated the effect of PHB2 knockdown on JI051 response. As shown in Fig. S9, JI051-induced condensed chromatin was significantly reduced in cells treated with PHB2 siRNAs as compared with control siRNAs (10.3 ± 3.2 and 27.4%± 6.5%, respectively), indicating that PHB2 is critical for JI051 response.
Effect of JI051 on Hes1–PHB2 interaction
We then tested the possibility that PHB2 regulates Hes1 signaling by direct interaction through a series of coimmunoprecipitation studies in HEK293 cell lysates. Western blotting with a Hes1 antibody revealed that Hes1 is specifically coimmunoprecipitated with PHB2 (Fig. 5A) in cells transfected with FLAG–PHB2 and Hes1, suggesting that the two proteins interact with each other. The amount of Hes1 coimmunoprecipitated with PHB2 strongly increased upon incubation with JI051, reaching saturation around 1 μm. These results not only indicate that Hes1 interacts directly with PHB2 but also suggest that JI051 serves as a stabilizer for the Hes1–PHB2 interaction.
Figure 5.
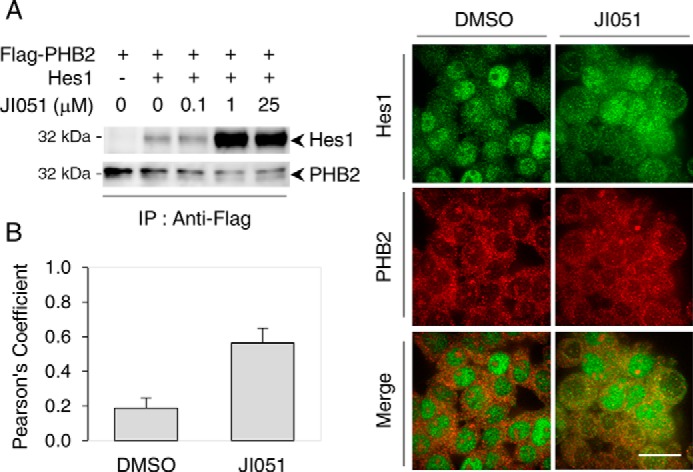
PHB2 is interacting with Hes1. A, lysates from HEK293 cells expressing FLAG–PHB2 alone or together with Hes1 were incubated with JI051 at the indicated concentrations together with anti-FLAG antibody, prior to pulldown with protein A-Sepharose beads. Western blot analysis was carried out with either Hes1 or PHB2 antibodies. IP, immunoprecipitation. Blots are representative of three individual experiments. B, Pearson correlation coefficient analysis showing the colocalization of Hes1 and PHB2 following a 24-h incubation with 1 μm JI051 or DMSO before dual immunocytochemistry with Hes1 and PHB2 antibodies. Data are mean ± S.D. from a minimum of 22 individual fields representing at least 399 cells per condition. Scale bar, 25 μm.
To determine whether the compound increases the colocalization of Hes1 and PHB2 in intact cells, double immunostaining was carried out in HEK293 cells. As shown in Fig. 5B, treatment with 1 μm JI051 for 24 h induced a 3-fold increase in Pearson's correlation coefficient, indicating that the compound enhances spatial overlap between Hes1 and PHB2. Confocal microscopy imaging showed that colocalization was mainly found in the cytoplasm. Also, cells without Hes1 nuclear labeling were observed, suggesting that Hes1 nuclear trafficking may be disrupted.
Effect of JI051 and its derivatives on pancreatic cancer cells
As Notch signaling pathway appears to be activated in human pancreatic cancer, we tested the effects of our Hes1 inhibitors on a pancreatic ductal adenocarcinoma cell line. As shown in Fig. 6A and Fig. S10A, treatment of MIA PaCa-2 cells with JI051 and JI130 resulted in a dose-dependent reduction in cell growth as compared with cells treated with DMSO. Dose-response studies revealed an EC50 of 49 nm for JI130, comparable with 59 nm for the pyrimidine antimetabolite gemcitabine, a chemotherapy drug recommended as a first-line treatment in pancreatic cancer in combination with nab-paclitaxel (Fig. 6B) (25). Fluorescence imaging following staining with Hoechst 33342 not only indicated the presence of condensed chromatin (Fig. 6C) but also revealed additional morphological alterations such as micronucleation and multinucleation. These features are prominent characteristics of mitotic catastrophe, a mechanism of delayed cell death resulting from aberrant mitosis (26). In addition to MIA PaCa-2 cells, JI130 was able to suppress cell growth in several pancreatic cancer cell lines, including CFPAC-1, PK9, and KP4-1 (Fig. S10B).
Figure 6.

JI130 inhibits MIA PaCa-2 cell growth. A, cells were treated for the indicated periods of time with 1 μm JI130 or DMSO prior to cell proliferation analysis with the tetrazolium salt 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt. Data represent mean ± S.D. B, dose-response curve of the effect of JI130 and the chemotherapy drug gemcitabine on cell proliferation following a 72-h incubation. Data are expressed as mean ± S.D. C, chemical structures of JI130 and corresponding EC50 value. D, effect of a 24-h incubation with JI130 (100 nm and 1 μm) or DMSO on nucleus appearance. Nuclei were stained with Hoechst 33342. Arrowheads indicate cells with micronuclei. DIC, differential interference contrast. Scale bar, 25 μm.
Finally, we tested the effects of JI130 on tumor growth in a pancreatic tumor xenograft model created with implanted MIA PaCa-2 cells. Mice were treated according to the protocol detailed in Fig. 7A. As shown in Fig. 7B, treatment with JI130 induced a gradual decrease in the tumor volume compared with DMSO, reaching a 48.2% reduction after 21 days. A decrease in tumor weight was also observed after treatment with JI130 (Fig. 7, C and D) without any noticeable change in body weight (Fig. 7E). To examine whether our Hes1 inhibitor also induced cell-cycle arrest in vivo, immunohistochemical staining on MIA PaCa-2 xenograft tumors with the proliferation marker Ki-67 was carried out. As shown in Fig. S11, treatment with JI130 induced a significant reduction in the proportion of Ki-67 positive cells compared with DMSO (26.8 ± 3.3 and 51.2 ± 4.5%, respectively), suggesting that our Hes1 inhibitor is decreasing tumor volume/weight by interfering with cell proliferation.
Figure 7.

JI130 inhibits tumor growth in xenograft model. A, MIA PaCa-2 cells were implanted in 28-day-old nude mice. Mice were treated with JI130 at a concentration of 50 mg/kg body weight 5 days/week for a total of 3 weeks. B, tumor volume of the mice treated with JI130 or DMSO. Data are mean ± S.E. from seven mice in each group. Two-tailed Student's t test was used for statistical analysis (*, p ≤ 0.01m and **, p ≤ 0.002 compared with DMSO). C, boxed plot illustrating the tumor weight following treatment with JI130 or DMSO. Data are mean ± S.D. from seven mice in each group. D, representative pictures of mice treated with JI130 or DMSO. E, transition of body weight of the mice treated with JI130 or DMSO. Data are mean ± S.D. from seven mice in each group.
Discussion
This study reports the discovery of a novel Hes1 inhibitor that highlights PHB2 as a new player in Hes1 signaling. Most importantly, our results indicate that JI051 stabilizes the interaction between Hes1 and PHB2 and induces cell-cycle arrest by inhibiting the Notch downstream effector gene Hes1.
Few small molecules that act as protein–protein interaction stabilizers have been reported in the literature (27–34), and JI051 represents the first small molecule that stabilizes the interaction of PHB2 with its client protein(s). Because PHB2 is mainly a cytoplasmic/mitochondrial protein (35), our small molecule may be interfering with Hes1 function by confining it outside of the nucleus through its interaction with PHB2.
Regulation of transcriptional activity by nuclear exclusion has been previously reported for other transcription factors, including Forkhead box protein family members (36, 37), NFAT1 (38), TCF11 (39), and Tbx5 (40). Proposed mechanisms include retention in the cytoplasm due to interaction with 14-3-3 scaffolding proteins (36) and prevention of the recognition of nuclear-localization signal (NLS) by the nuclear import machinery (37). Prediction of putative NLS within the Hes1 sequence (NLS Mapper: http://nls-mapper.iab.keio.ac.jp5 (54–56)) highlighted a bipartite motif between amino acids 42 and 69. Nevertheless, it remains unclear whether the PHB2 interaction with Hes1 hides/prevents the recognition of critical NLS.
In addition to inhibiting Hes1-mediated transcription, our small molecule also induced cell-cycle arrest in the G2/M phase of the cell cycle. Our experiments showed that both PHB2 and Hes1 are critical for the effect of JI051 on cell proliferation, as gene depletion of either PHB2 or Hes1 was sufficient to suppress the response. Because the depletion of PHB2 alone did not induce chromatin condensation, inhibition of Hes1 through PHB2 is likely responsible for aberrant mitosis. We also showed that our Hes1 inhibitor has an anti-tumor effect on pancreatic cancer in vitro and in vivo, which is consistent with the pro-tumorigenic role of Notch signaling in pancreatic ductal adenocarcinoma (41, 42). Previous studies have shown that Hes1 regulates the cell cycle progression by repressing the expression of cyclin-dependent kinase inhibitors (CDKI), including p21, p27, and p57, of the kinase inhibitor protein family (18, 44), and that depletion of Hes1 results in their up-regulation and concomitant cell cycle exit (44, 45). Whether these proteins or other cell cycle–related proteins are up-regulated following Hes1 inhibition by JI051 remains to be investigated.
Although Prohibitin was initially identified as a negative controller of cell proliferation (46), PHB1 and PHB2 play crucial roles in cancer development and progression through the regulation of Ras/MAPK, TGF-β, and estrogen receptor signaling (47–49). As overexpression of PHB1 and PHB2 is also commonly found in a wide range of tumors, including pancreatic cancer (50–52), PHB2 may prove to serve as a new target for modulating Hes1 in future cancer therapies.
Experimental procedures
Cell culture and transfection
HEK293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 5% fetal bovine serum (FBS; HyClone) and 100 units/ml penicillin and streptomycin (PS; Invitrogen). Cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2. Cells were transfected with FuGENE HD (Promega) according to the manufacturer's instructions. Human pancreatic cancer cell lines (MIA PaCa-2, CFPAC-1, PK9, and KP4-1) were maintained at 37 °C and 5% CO2 in DMEM or Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco) supplemented with 10% FBS (Gibco) and 100 units/ml PS. Immortalized MEF from the Hes1–KO mice (53) were generated as below. Primary culture of the E14.5 Hes1–KO and its littermate WT control embryo were maintained for 1 month with DMEM with 10% FBS and 100 units/ml PS. Spontaneously immortalized colonies were picked up and expanded. Genotyping of the Hes1 locus was confirmed by PCR using the genomic DNA of MEFs.
Plasmid cDNA
pC1–Hes1, pHes1–EGFPd2, pHes1–Ub–luciferase, and pEF–NICD were provided by Prof. Ryoichiro Kageyama (Kyoto University, Japan). pCMV3–FLAG–PHB2 and pcDNA3.1 were purchased from Sino Biological, Inc., and Thermo Fisher Scientific, respectively. pMXs–mCherry–hCdh1 (30:120) and pCSII–AmCyan–hGeminin (1:110) were provided by Prof. Atsushi Miyawaki (RIKEN BSI, Japan). pCMV–Hes1–DsRed was generated by amplifying Hes1 (from the pC1–Hes1 plasmid) with sense and antisense primers comprising an NheI and an AgeI site, respectively, before insertion into pDsRed Monomer Golgi vector (Clontech) predigested with NheI and AgeI to remove the Golgi-targeting signal peptide. pCMV2–FLAG–Gro/TLE1 was provided by Prof. Stefano Stifani (McGill University, Canada).
Compound screening
A chemical library of 1,800 small organic molecules containing indole moieties (Tripos Receptor Research) was screened in HEK293 cells expressing pHes1–Ub–luciferase with or without pC1–Hes1 following a 2.5 μm treatment for 48 h. Luminescence was amplified using the Luciferase Assay System (Promega) and measured with a microplate reader (MTP-880 Corona). To validate hit compounds, HEK293 cells expressing pHes1–EGFPd2 together with pCMV–Hes1–DsRed were treated with 10 μm compounds for 48 h, prior to confocal microscopy imaging with CellVoyagerTM CV1000 (Yokogawa). Image data corresponding to fluorescence intensities were quantified using ImageJ software. The fluorescence intensity of the EGFPd2 signal was divided by the intensity of the DsRed signal for normalization of transfection efficacy.
Cell proliferation
Cell proliferation was evaluated using WST-8 assay kit (Dojindo Molecular Technologies) or CellTiter 96 Aqueous One Solution (Promega) for HEK293 and pancreatic cancer cells, respectively. Absorbance was measured using a microplate reader. For mitotic index determination, DNA was stained with 20 μm Hoechst 33342 (Thermo Fisher Scientific) for 10 min at room temperature before confocal microscopy imaging. The percentage of cells displaying condensed chromatin was determined following cell counting using the cell counter plugin in ImageJ.
Target identification
HEK293 cells were sonicated with an ultrasonic processor (Astrason W-385; Heat System-ultrasonics) in PBS with protease inhibitors (Complete Mini; Roche Applied Science). Resulting lysates were incubated with compounds together with Dynabeads M-280 streptavidin (Thermo Fisher Scientific) overnight at 4 °C. Samples were boiled at 100 °C for 5 min before separation onto Mini II polyacrylamide 4/20 gels (Cosmo Bio Co., Ltd.), followed by silver staining (Silver Stain MS Kit; Wako Pure Chemical Industries, Ltd.). Gel pieces were excised and destained according to the manufacturer's instructions. Samples were digested with trypsin, and resulting peptides were analyzed by nanoscale LC-tandem MS (nano LC-MS/MS). Target proteins were identified following MS/MS ion search using Mascot server software from Matrix Science.
Target validation
HEK293 cells expressing pCMV2–FLAG–Gro/TLE1 or pCMV3–FLAG–PHB2 were lysed in 150 mm Tris-HCl (pH 7.4), 150 mm NaCl, and 1% Nonidet P-40 substitute (Amresco) with protease inhibitors before centrifugation. Cell lysates were incubated with the indicated compounds for 1 h at 4 °C and exposed to UV light for 30 min at 4 °C (Bio-link Cross-linker BLX-365; Cosmo Bio). Lysates were then incubated with NeutrAvidin-agarose beads (Thermo Fisher Scientific) for 2 h at 4 °C. Resin-bound complexes were washed three times with lysis buffer and boiled at 100 °C for 10 min in reducing 2× Laemmli sample buffer (Bio-Rad) for elution. Human recombinant PHB2 protein with an N-terminal GSH–S-transferase (GST) tag (Abnova) was incubated with increasing concentrations of JITV10 overnight at 4 °C and exposed to UV light for 30 min at 4 °C. Samples were then boiled at 100 °C for 10 min in reducing 2× Laemmli sample buffer and resolved by SDS-PAGE. Biotinylated compounds were detected with a high sensitivity streptavidin–HRP conjugate (Thermo Fisher Scientific).
Western blotting
Samples were resolved on 10% mini-protean TGX gels (Bio-Rad) before transfer onto nitrocellulose blotting membranes (GE Healthcare). Membranes were incubated overnight with primary antibodies, followed by a 1-h incubation at room temperature with ECL–peroxidase-labeled anti-mouse (GE Healthcare) or horseradish peroxidase (HRP)-linked anti-rabbit (Cell Signaling Technology) in immunoreaction enhancer solution (Can Get Signal; Toyobo Co., Ltd.). Immunoreactive proteins were visualized using Amersham Biosciences ECL Prime Western blotting detection reagent (GE Healthcare) and an ImageQuant LAS 500 imaging system (GE Healthcare).
Co-immunoprecipitation studies
HEK293 cells expressing FLAG–PHB2 alone or together with Hes1 were lysed in 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, and 1% Nonidet P-40 substitute with protease inhibitors. Cell lysates were precleared with 2 mg of protein A-Sepharose (Sigma) for 45 min at 4 °C and incubated for 48 h at 4 °C with FLAG M2 mouse mAb, together with JI051 at the concentration indicated. Labeled proteins were immunoprecipitated with 6 mg of protein A-Sepharose for 2 h at 4 °C. Complexes were washed three times with lysis buffer and dissolved in reducing 2× Laemmli sample buffer before a 10-min incubation at 100 °C.
siRNA experiments
siRNA oligonucleotide duplexes targeting human PHB2 were purchased from OriGene (PHB2B, 5′-GUGAUUUCCUACAGUGUUGUUCCCT-3′, and PHB2C, 5′-UCUAUCUCACAGCUGACAACCUUGT-3′). HEK293 cells were transfected with siRNAs using Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific). Western blotting and confocal imaging were performed 72 h after transfection.
Fluorescence imaging
For immunocytochemistry, cells were fixed with a 4% paraformaldehyde solution (PFA; Muto Pure Chemicals Co. Ltd.), prior to blocking with 5% Blocking One-P (Nacalai Tesque Inc.) and 0.1% Triton X-100 (Sigma) in PBS. Incubation with primary antibodies was carried out in antibody dilution buffer (2.5% Blocking One-P and 0.05% Triton X-100 in PBS) overnight at 4 °C. Cells were then incubated with Alexa Fluor 488- and Alexa Fluor 568-conjugated secondary antibodies (Thermo Fisher Scientific) for 1 h at room temperature and imaged by confocal microscopy. To evaluate colocalization between PHB2 and Hes1, Pearson correlation coefficient was determined using the JACoP plugin (43) from ImageJ. For JITV10 imaging, cells were incubated with JITV10 (20 μm) for 45 min at 37 °C. Cells were fixed with 4% PFA and incubated with a Streptavidin-Alexa Fluor 568 conjugate (Thermo Fisher Scientific) for 1 h at room temperature before confocal microscopy imaging.
Aqueous solubility
Aqueous solubility was evaluated using a high-throughput turbidimetric assay. Compounds were diluted in PBS (pH 7.4) and 1% DMSO at the following concentrations: 1, 3, 10, 30, and 100 μm and incubated for 2 h at 37 °C, prior to absorbance measurements at 620 nm.
Animals and xenograft model
Immune–incompetent nude (Nu/Nu) mice were purchased from SLC (Japan). All animal care and experiments were conducted following the guidelines for Japan's Act on Welfare and Management of Animals and approved by Kyoto University Graduate School. All procedures were performed when mice were anesthetized with chloral hydrate, isoflurane, or diethyl ether, and all efforts were made to minimize the number of animals used and their suffering. 1 × 106 MIA PaCa-2 pancreatic cancer cells, suspended with 50 μl of DMEM without FBS and antibiotics, were injected subcutaneously into the right flank of 4-week-old female immune–incompetent nude (Nu/Nu) mice. Tumors were measured with a digital caliper in two dimensions, and volume was calculated (π/6 × length × width2). When the tumor volume had reached 100 mm3, mice were assigned to the control group (DMSO) or the treatment group (JI130). JI130 dissolved in DMSO (100 mg/ml) was administered intraperitoneally at 50 mg/kg body weight 5 days a week for 3 weeks. Tumor volume was calculated three times a week, and body weight was measured at the same time. After 3 weeks treatment, mice were sacrificed, and tumor weight was measured.
Author contributions
A. P., R. K., Y. K., and M. U. conceptualization; A. P., Y. N., J. I., H. S., J. T., and K. K. data curation; A. P., Y. N., H. S., and K. K. formal analysis; A. P., Y. K., and M. U. supervision; A. P., Y. K., and M. U. funding acquisition; A. P. and Y. N. validation; A. P., Y. N., J. I., H. S., J. T., K. K., I. I., N. M. M., and M. T. investigation; A. P., Y. N., J. I., and I. I. methodology; A. P. writing-original draft; A. P. project administration; M. U. writing-review and editing.
Supplementary Material
Acknowledgments
This research was inspired by the international and interdisciplinary environments of the iCeMS and JSPS Asian CORE Program, “Asian Chemical Biology Initiative.”
This work was supported by a start-up grant from Kyoto University (to A. P.), AMED Project for Development for Innovative Research on Cancer Therapeutics (P-DIRECT), and MEXT (JSPS) KAKENHI Grants-in-aid for Scientific Research 25870365 (to A. P.) and 26220206 (to M. U.). The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as one of our Editors' Picks.
This article contains Figs. S1–S11, Table S1, and supporting Chemical Synthesis.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- NICD
- Notch intracellular domain
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- PFA
- paraformaldehyde
- MEF
- mouse embryonic fibroblast
- NLS
- nuclear-localization signal
- HRP
- horseradish peroxidase
- PS
- penicillin and streptomycin.
References
- 1. Kageyama R., Shimojo H., and Imayoshi I. (2015) Dynamic expression and roles of Hes factors in neural development. Cell Tissue Res. 359, 125–133 10.1007/s00441-014-1888-7 [DOI] [PubMed] [Google Scholar]
- 2. Weng A. P., Ferrando A. A., Lee W., Morris J. P. 4th., Silverman L. B., Sanchez-Irizarry C., Blacklow S. C., Look A. T., and Aster J. C. (2004) Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269–271 10.1126/science.1102160 [DOI] [PubMed] [Google Scholar]
- 3. Miyamoto Y., Maitra A., Ghosh B., Zechner U., Argani P., Iacobuzio-Donahue C. A., Sriuranpong V., Iso T., Meszoely I. M., Wolfe M. S., Hruban R. H., Ball D. W., Schmid R. M., and Leach S. D. (2003) Notch mediates TGF α-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 3, 565–576 10.1016/S1535-6108(03)00140-5 [DOI] [PubMed] [Google Scholar]
- 4. Sjölund J., Johansson M., Manna S., Norin C., Pietras A., Beckman S., Nilsson E., Ljungberg B., and Axelson H. (2008) Suppression of renal cell carcinoma growth by inhibition of Notch signaling in vitro and in vivo. J. Clin. Invest. 118, 217–228 10.1172/JCI32086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Santagata S., Demichelis F., Riva A., Varambally S., Hofer M. D., Kutok J. L., Kim R., Tang J., Montie J. E., Chinnaiyan A. M., Rubin M. A., and Aster J. C. (2004) JAGGED1 expression is associated with prostate cancer metastasis and recurrence. Cancer Res. 64, 6854–6857 10.1158/0008-5472.CAN-04-2500 [DOI] [PubMed] [Google Scholar]
- 6. Jensen J. N., Cameron E., Garay M. V., Starkey T. W., Gianani R., and Jensen J. (2005) Recapitulation of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology 128, 728–741 10.1053/j.gastro.2004.12.008 [DOI] [PubMed] [Google Scholar]
- 7. Jarriault S., Brou C., Logeat F., Schroeter E. H., Kopan R., and Israel A. (1995) Signalling downstream of activated mammalian Notch. Nature 377, 355–358 10.1038/377355a0 [DOI] [PubMed] [Google Scholar]
- 8. Takebayashi K., Sasai Y., Sakai Y., Watanabe T., Nakanishi S., and Kageyama R. (1994) Structure, chromosomal locus, and promoter analysis of the gene encoding the mouse helix-loop-helix factor HES-1. Negative autoregulation through the multiple N box elements. J. Biol. Chem. 269, 5150–5156 [PubMed] [Google Scholar]
- 9. Buscarlet M., Perin A., Laing A., Brickman J. M., and Stifani S. (2008) Inhibition of cortical neuron differentiation by Groucho/TLE1 requires interaction with WRPW, but not Eh1, repressor peptides. J. Biol. Chem. 283, 24881–24888 10.1074/jbc.M800722200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grbavec D., and Stifani S. (1996) Molecular interaction between TLE1 and the carboxyl-terminal domain of HES-1 containing the WRPW motif. Biochem. Biophys. Res. Commun. 223, 701–705 10.1006/bbrc.1996.0959 [DOI] [PubMed] [Google Scholar]
- 11. Jennings B. H., Pickles L. M., Wainwright S. M., Roe S. M., Pearl L. H., and Ish-Horowicz D. (2006) Molecular recognition of transcriptional repressor motifs by the WD domain of the Groucho/TLE corepressor. Mol. Cell 22, 645–655 10.1016/j.molcel.2006.04.024 [DOI] [PubMed] [Google Scholar]
- 12. Olsauskas-Kuprys R., Zlobin A., and Osipo C. (2013) γ-Secretase inhibitors of Notch signaling. Onco. Targets Ther. 6, 943–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barten D. M., Meredith J. E. Jr., Zaczek R., Houston J. G., and Albright C. F. (2006) γ-Secretase inhibitors for Alzheimer's disease: balancing efficacy and toxicity. Drugs R D 7, 87–97 10.2165/00126839-200607020-00003 [DOI] [PubMed] [Google Scholar]
- 14. Arai M. A., Masada A., Ohtsuka T., Kageyama R., and Ishibashi M. (2009) The first Hes1 dimer inhibitors from natural products. Bioorg. Med. Chem. Lett. 19, 5778–5781 10.1016/j.bmcl.2009.07.146 [DOI] [PubMed] [Google Scholar]
- 15. Arai M. A., Ishikawa N., Tanaka M., Uemura K., Sugimitsu N., Suganami A., Tamura Y., Koyano T., Kowithayakorn T., and Ishibashi M. (2016) Hes1 inhibitor isolated by target protein oriented natural products isolation (TPO-NAPI) of differentiation activators of neural stem cells. Chem. Sci. 7, 1514–1520 10.1039/C5SC03540F [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sail V., and Hadden M. K. (2013) Identification of small molecule Hes1 modulators as potential anticancer chemotherapeutics. Chem. Biol. Drug Des. 81, 334–342 10.1111/cbdd.12059 [DOI] [PubMed] [Google Scholar]
- 17. Fisher A. L., Ohsako S., and Caudy M. (1996) The WRPW motif of the hairy-related basic helix-loop-helix repressor proteins acts as a 4-amino-acid transcription repression and protein–protein interaction domain. Mol. Cell. Biol. 16, 2670–2677 10.1128/MCB.16.6.2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Georgia S., Soliz R., Li M., Zhang P., and Bhushan A. (2006) P57 and Hes 1 coordinate cell cycle exit with self-renewal of pancreatic progenitors. Dev. Biol. 298, 22–31 10.1016/j.ydbio.2006.05.036 [DOI] [PubMed] [Google Scholar]
- 19. Sakaue-Sawano A., Kurokawa H., Morimura T., Hanyu A., Hama H., Osawa H., Kashiwagi S., Fukami K., Miyata T., Miyoshi H., Imamura T., Ogawa M., Masai H., and Miyawaki A. (2008) Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498 10.1016/j.cell.2007.12.033 [DOI] [PubMed] [Google Scholar]
- 20. Choi H. J., Fukui M., and Zhu B. T. (2011) Role of cyclin B1/Cdc2 up-regulation in the development of mitotic prometaphase arrest in human breast cancer cells treated with nocodazole. PLoS One 6, e24312 10.1371/journal.pone.0024312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thorne N., Inglese J., and Auld D. S. (2010) Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem. Biol. 17, 646–657 10.1016/j.chembiol.2010.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perkins D. N., Pappin D. J., Creasy D. M., and Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 10.1002/(SICI)1522-2683(19991201)20:18%3C3551::AID-ELPS3551%3E3.0.CO%3B2-2 [DOI] [PubMed] [Google Scholar]
- 23. Merkwirth C., Dargazanli S., Tatsuta T., Geimer S., Löwer B., Wunderlich F. T., von Kleist-Retzow J. C., Waisman A., Westermann B., and Langer T. (2008) Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Gene Dev. 22, 476–488 10.1101/gad.460708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Von Hoff D. D., Ervin T., Arena F. P., Chiorean E. G., Infante J., Moore M., Seay T., Tjulandin S. A., Ma W. W., Saleh M. N., Harris M., Reni M., Dowden S., Laheru D., Bahary N., et al. (2013) Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 369, 1691–1703 10.1056/NEJMoa1304369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mc Gee M. M. (2015) Targeting the mitotic catastrophe signaling pathway in cancer. Mediators Inflamm. 2015, 146282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peyroche A., Antonny B., Robineau S., Acker J., Cherfils J., and Jackson C. L. (1999) Brefeldin A acts to stabilize an abortive ARF-GDP-Sec7 domain protein complex: involvement of specific residues of the Sec7 domain. Mol. Cell 3, 275–285 10.1016/S1097-2765(00)80455-4 [DOI] [PubMed] [Google Scholar]
- 27. Sunahara R. K., Dessauer C. W., Whisnant R. E., Kleuss C., and Gilman A. G. (1997) Interaction of Gsα with the cytosolic domains of mammalian adenylyl cyclase. J. Biol. Chem. 272, 22265–22271 10.1074/jbc.272.35.22265 [DOI] [PubMed] [Google Scholar]
- 28. Tan X., Calderon-Villalobos L. I., Sharon M., Zheng C., Robinson C. V., Estelle M., and Zheng N. (2007) Mechanism of auxin perception by the TIR1 ubiquitin ligase. Nature 446, 640–645 10.1038/nature05731 [DOI] [PubMed] [Google Scholar]
- 29. Nogales E., Wolf S. G., Khan I. A., Ludueña R. F., and Downing K. H. (1995) Structure of tubulin at 6.5 A and location of the taxol-binding site. Nature 375, 424–427 10.1038/375424a0 [DOI] [PubMed] [Google Scholar]
- 30. Liu J., Farmer J. D. Jr., Lane W. S., Friedman J., Weissman I., and Schreiber S. L. (1991) Calcineurin is a common target of cyclophilin-cyclosporine-A and Fkbp-Fk506 complexes. Cell 66, 807–815 10.1016/0092-8674(91)90124-H [DOI] [PubMed] [Google Scholar]
- 31. Würtele M., Jelich-Ottmann C., Wittinghofer A., and Oecking C. (2003) Structural view of a fungal toxin acting on a 14-3-3 regulatory complex. EMBO J. 22, 987–994 10.1093/emboj/cdg104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Viaud J., Zeghouf M., Barelli H., Zeeh J. C., Padilla A., Guibert B., Chardin P., Royer C. A., Cherfils J., and Chavanieu A. (2007) Structure-based discovery of an inhibitor of Arf activation by Sec7 domains through targeting of protein–protein complexes. Proc. Natl. Acad. Sci. U.S.A. 104, 10370–10375 10.1073/pnas.0700773104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kita M., Hirayama Y., Yoneda K., Yamagishi K., Chinen T., Usui T., Sumiya E., Uesugi M., and Kigoshi H. (2013) Inhibition of microtubule assembly by a complex of actin and antitumor macrolide aplyronine A. J. Am. Chem. Soc. 135, 18089–18095 10.1021/ja406580w [DOI] [PubMed] [Google Scholar]
- 34. Mishra S., Murphy L. C., and Murphy L. J. (2006) The prohibitins: emerging roles in diverse functions. J. Cell. Mol. Med. 10, 353–363 10.1111/j.1582-4934.2006.tb00404.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brunet A., Bonni A., Zigmond M. J., Lin M. Z., Juo P., Hu L. S., Anderson M. J., Arden K. C., Blenis J., and Greenberg M. E. (1999) Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 96, 857–868 10.1016/S0092-8674(00)80595-4 [DOI] [PubMed] [Google Scholar]
- 36. Wolfrum C., Besser D., Luca E., and Stoffel M. (2003) Insulin regulates the activity of forkhead transcription factor Hnf-3β/Foxa-2 by Akt-mediated phosphorylation and nuclear/cytosolic localization. Proc. Natl. Acad. Sci. U.S.A. 100, 11624–11629 10.1073/pnas.1931483100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shaw K. T., Ho A. M., Raghavan A., Kim J., Jain J., Park J., Sharma S., Rao A., and Hogan P. G. (1995) Immunosuppressive drugs prevent a rapid dephosphorylation of transcription factor Nfat1 in stimulated immune cells. Proc. Natl. Acad. Sci. U.S.A. 92, 11205–11209 10.1073/pnas.92.24.11205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Husberg C., Murphy P., Bjørgo E., Kalland K. H., and Kolstø A. B. (2003) Cellular localisation and nuclear export of the human bZIP transcription factor TCF11. Biochim. Biophys. Acta 1640, 143–151 10.1016/S0167-4889(03)00041-7 [DOI] [PubMed] [Google Scholar]
- 39. Kulisz A., and Simon H. G. (2008) An evolutionarily conserved nuclear export signal facilitates cytoplasmic localization of the Tbx5 transcription factor. Mol. Cell. Biol. 28, 1553–1564 10.1128/MCB.00935-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Plentz R., Park J. S., Rhim A. D., Abravanel D., Hezel A. F., Sharma S. V., Gurumurthy S., Deshpande V., Kenific C., Settleman J., Majumder P. K., Stanger B. Z., and Bardeesy N. (2009) Inhibition of γ-secretase activity inhibits tumor progression in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology 136, 1741–1749 10.1053/j.gastro.2009.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. De La O J. P., Emerson L. L., Goodman J. L., Froebe S. C., Illum B. E., Curtis A. B., and Murtaugh L. C. (2008) Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc. Natl. Acad. Sci. U.S.A. 105, 18907–18912 10.1073/pnas.0810111105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Murata K., Hattori M., Hirai N., Shinozuka Y., Hirata H., Kageyama R., Sakai T., and Minato N. (2005) Hes1 directly controls cell proliferation through the transcriptional repression of p27(Kip1). Mol. Cell. Biol. 25, 4262–4271 10.1128/MCB.25.10.4262-4271.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sievers C., Billig G., Gottschalk K., and Rudel T. (2010) Prohibitins are required for cancer cell proliferation and adhesion. PLoS One 5, e12735 10.1371/journal.pone.0012735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Monahan P., Rybak S., and Raetzman L. T. (2009) The Notch target gene Hes1 regulates cell cycle inhibitor expression in the developing pituitary. Endocrinology 150, 4386–4394 10.1210/en.2009-0206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McClung J. K., Danner D. B., Stewart D. A., Smith J. R., Schneider E. L., Lumpkin C. K., Dell'Orco R. T., and Nuell M. J. (1989) Isolation of a cDNA that hybrid selects antiproliferative mRNA from rat liver. Biochem. Biophys. Res. Commun. 164, 1316–1322 10.1016/0006-291X(89)91813-5 [DOI] [PubMed] [Google Scholar]
- 46. Rajalingam K., Wunder C., Brinkmann V., Churin Y., Hekman M., Sievers C., Rapp U. R., and Rudel T. (2005) Prohibitin is required for Ras-induced Raf-MEK-ERK activation and epithelial cell migration. Nat. Cell Biol. 7, 837–843 10.1038/ncb1283 [DOI] [PubMed] [Google Scholar]
- 47. Zhu B., Zhai J., Zhu H., and Kyprianou N. (2010) Prohibitin regulates TGF-β induced apoptosis as a downstream effector of Smad-dependent and -independent signaling. Prostate 70, 17–26 10.1002/pros.21033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kasashima K., Ohta E., Kagawa Y., and Endo H. (2006) Mitochondrial functions and estrogen receptor-dependent nuclear translocation of pleiotropic human prohibitin 2. J. Biol. Chem. 281, 36401–36410 10.1074/jbc.M605260200 [DOI] [PubMed] [Google Scholar]
- 49. Asamoto M., and Cohen S. M. (1994) Prohibitin gene is overexpressed but not mutated in rat bladder carcinomas and cell-lines. Cancer Lett. 83, 201–207 10.1016/0304-3835(94)90320-4 [DOI] [PubMed] [Google Scholar]
- 50. Cao Y., Liang H., Zhang F., Luan Z., Zhao S., Wang X. A., Liu S., Bao R., Shu Y., Ma Q., Zhu J., and Liu Y. (2016) Prohibitin overexpression predicts poor prognosis and promotes cell proliferation and invasion through ERK pathway activation in gallbladder cancer. J. Exp. Clin. Cancer Res. 35, 68 10.1186/s13046-016-0346-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Luan Z., He Y., Alattar M., Chen Z., and He F. (2014) Targeting the prohibitin scaffold-CRAF kinase interaction in RAS-ERK-driven pancreatic ductal adenocarcinoma. Mol. Cancer 13, 38 10.1186/1476-4598-13-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ishibashi M., Ang S. L., Shiota K., Nakanishi S., Kageyama R., and Guillemot F. (1995) Targeted disruption of mammalian hairy and Enhancer of split homolog-1 (HES-1) leads to up-regulation of neural helix-loop-helix factors, premature neurogenesis, and severe neural tube defects. Gene Dev. 9, 3136–3148 10.1101/gad.9.24.3136 [DOI] [PubMed] [Google Scholar]
- 53. Bolte S., and Cordelières F. P. (2006) A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 10.1111/j.1365-2818.2006.01706.x [DOI] [PubMed] [Google Scholar]
- 54. Kosugi S., Hasebe M., Tomita M., and Yanagawa H. (2009) Systematic identification of yeast cell cycle-dependent nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc. Natl. Acad. Sci. U.S.A. 106, 10171–10176 10.1073/pnas.0900604106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kosugi S., Hasebe M., Matsumura N., Takashima H., Miyamoto-Sato E., Tomita M., and Yanagawa H. (2009) Six classes of nuclear localization signals specific to different binding grooves of importin α. J. Biol. Chem. 284, 478–485 10.1074/jbc.M807017200 [DOI] [PubMed] [Google Scholar]
- 56. Kosugi S., Hasebe M., Entani T., Takayama S., Tomita M., and Yanagawa H. (2008) Design of peptide inhibitors for the importin α/β nuclear import pathway by activity-based profiling. Chem. Biol. 15, 940–949 10.1016/j.chembiol.2008.07.019 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.