

Abstract
In aortic vascular smooth muscle (VSM), the canonical Wnt receptor LRP6 inhibits protein arginine (Arg) methylation, a new component of noncanonical Wnt signaling that stimulates nuclear factor of activated T cells (viz. NFATc4). To better understand how methylation mediates these actions, MS was performed on VSM cell extracts from control and LRP6-deficient mice. LRP6-dependent Arg methylation was regulated on >500 proteins; only 21 exhibited increased monomethylation (MMA) with concomitant reductions in dimethylation. G3BP1, a known regulator of arteriosclerosis, exhibited a >30-fold increase in MMA in its C-terminal domain. Co-transfection studies confirm that G3BP1 (G3BP is Ras-GAP SH3 domain–binding protein) methylation is inhibited by LRP6 and that G3BP1 stimulates NFATc4 transcription. NFATc4 association with VSM osteopontin (OPN) and alkaline phosphatase (TNAP) chromatin was increased with LRP6 deficiency and reduced with G3BP1 deficiency. G3BP1 activation of NFATc4 mapped to G3BP1 domains supporting interactions with RIG-I (retinoic acid inducible gene I), a stimulus for mitochondrial antiviral signaling (MAVS) that drives cardiovascular calcification in humans when mutated in Singleton-Merten syndrome (SGMRT2). Gain-of-function SGMRT2/RIG-I mutants increased G3BP1 methylation and synergized with osteogenic transcription factors (Runx2 and NFATc4). A chemical antagonist of G3BP, C108 (C108 is 2-hydroxybenzoic acid, 2-[1-(2-hydroxyphenyl)ethylidene]hydrazide CAS 15533-09-2), down-regulated RIG-I–stimulated G3BP1 methylation, Wnt/NFAT signaling, VSM TNAP activity, and calcification. G3BP1 deficiency reduced RIG-I protein levels and VSM osteogenic programs. Like G3BP1 and RIG-I deficiency, MAVS deficiency reduced VSM osteogenic signals, including TNAP activity and Wnt5-dependent nuclear NFATc4 levels. Aortic calcium accumulation is decreased in MAVS-deficient LDLR−/− mice fed arteriosclerotic diets. The G3BP1/RIG-I/MAVS relay is a component of Wnt signaling. Targeting this relay may help mitigate arteriosclerosis.
Keywords: Wnt signaling, calcification, atherosclerosis, protein methylation, cardiovascular disease, vascular biology, vascular smooth muscle cells, Type 2 diabetes, metabolic syndrome, arteriosclerosis
Introduction
The metabolic insults of hyperglycemia, hyperlipidemia, and hyperphosphatemia, either with or without uremia, accelerate conduit vessel aging (1). In concert with hypertension, these common dysmetabolic states induce arteriosclerotic calcification and conduit vessel stiffening (2). This reduction in vessel compliance impairs Windkessel physiology, the rubbery elasticity of conduit arteries necessary for smooth distal tissue perfusion throughout the cardiac cycle (3). Wall thickening, fibrosis, elastin fragmentation, medial and atherosclerotic calcification, matrix “browning,” and endothelial dysfunction are all contributors to sclerotic aging of the conduit arteries (1). Work from numerous laboratories, including our own, has identified that osteogenic morphogens of the Wnt and BMP gene families and their inhibitors regulate neointima formation, matrix deposition and remodeling, and vascular calcification in arteriosclerotic disease (4–12). It was Mani et al. (13) who first discovered that a nonsynonymous private mutation in the Wnt receptor LRP6 (LRP6 is the low-density lipoprotein receptor-related protein 6) predisposes to precocious aging phenotypes in humans, viz. atherosclerotic heart disease and osteoporosis. Recently, Mani and co-workers (14) and our laboratory (9) outlined the cell-autonomous actions of LRP6 in vascular smooth muscle (VSM)3 as relevant to the pathobiology of arteriosclerosis. In addition to conveying canonical Wnt signals, LRP6 restrains noncanonical signaling programs in VSM that promote cardiovascular calcification (9), arteriosclerotic stiffening (9), and neointima formation (14) in mice. Similar noncanonical Wnt signals participate in aortic valve calcification as well (15, 16). Thus, human genetics and murine molecular genetics have converged to firmly place Wnt/LRP6 signaling as a key component of cardiometabolic disease, including arteriosclerosis.
In our studies of LRP6 actions, we identified VSM protein arginine methylation as being directly regulated by LRP6 (9). Asymmetric dimethylarginine (ADMA) modification of histone H3 Arg-17, and a number of unknown cellular proteins, was increased with LRP6 deficiency (9). LRP6-dependent reductions in the broad-specificity demethylase Jmjd6 (Jumonji domain containing 6 arginine demethylase) were responsible in part for these changes, and Jmjd6 expression inhibited key features of noncanonical Wnt signaling, including USF1 (upstream stimulatory factor 1) transactivation (9).
To better understand the roles and targets of arginine methylation in LRP6 actions, we performed a whole-cell Arg methylome analysis (17) from LRP6-replete and LRP6-deficient primary aortic VSM (9). Major increases were noted in proteins undergoing arginine monomethylation (MMA) with LRP6 deficiency, with one-third of these being RNA-binding proteins. One highly regulated protein, G3BP1, was recently identified, via visceral (thoracic) tissue eQTL bioinformatics, to be a global regulator of cardiovascular disease severity in humans (18). We demonstrate that G3BP1 promotes pro-sclerotic noncanonical Wnt signaling through NFATc4 nuclear localization. This is mediated in part via functional and physical interactions with RIG-I, an activator of mitochondrial antiviral signaling protein (MAVS, also known as CARDIF, IPS1, VISA) (19), and dependent upon the C-terminal G3BP1 arginine methylation domain.
Results
LRP6 regulates the MMA/SDMA modification of G3BP1, a genetic determinant of cardiovascular disease severity
We recently identified that conditional deletion of LRP6 increases the accumulation of VSM proteins undergoing asymmetric dimethylation arginine (ADMA) modification (9). To better understand the landscape of arginine methylation in primary VSM cells and its regulation by LRP6, we implemented commercially available MethylScan immunoaffinity MS (17) to interrogate the cellular protein arginine methylome as modified by LRP6 deficiency. Because perturbations in activity of one protein arginine methyltransferase (PRMT) result in compensatory “substrate scavenging” by related PRMT family members (20), we focused upon proteins where reciprocal changes were observed between MMA, ADMA), and SDMA in response to LRP6 deficiency. The LDLR−/− background was used because murine LDLR deficiency provides susceptibility to Western diet-induced insulin-resistant hyperglycemia, dyslipidemia, and arteriosclerotic calcification in vivo- recapitulating key features of human type 2 diabetes and the metabolic syndrome (2, 9, 21). Using a 2.5-fold change threshold in duplicate samples, over 500 proteins modified by arginine methylation were differentially expressed in SM22-Cre;LRP6(fl/fl);LDLR−/− VSM versus LRP6(fl/fl);LDLR−/− controls (Fig. 1A), but only 21 specific proteins exhibited increases in MMA or ADMA modification with reductions in SDMA accumulation (supporting data S1). The plurality of the regulated Arg-methylated proteins contain RNA-binding domains (Fig. 1A) (22). Two of these proteins, G3BP1 (Fig. 1B) and G3BP2, immediately commanded attention, because (a) Bikkavilli and Malbon (23) had previously shown canonical Wnt-dependent Arg methylation of G3BP1 and G3BP2 (24); and (b) expression of G3BP1 was recently identified by expressed quantitative trait locus (eQTL) analysis as a master regulator of angiographically-adjudicated cardiovascular disease severity in humans (18). Because of the relationship between expression and cardiovascular disease (18), we focused upon G3BP1 (Fig. 1B). With LRP6 deficiency, three Arg residues (Arg-443, Arg-445, and Arg-458; NCBI accession no. NP_038744) in the mouse G3BP1 C-terminal domain (exhibited > = 10-fold increase in MMA, with a concomitant 5-fold reduction in SDMA modification of Arg-458 (Fig. 1B and supporting data S1 and S2). Residue 458 in murine G3BP1 is equivalent to human G3BP1 Arg-460 (NCBI accession no. NP_005745; Fig. 1B). The reciprocal change in the MMA/SDMA ratio was accompanied by a 75% increase (p = 0.004) in total G3BP1 protein levels in LRP6-deficient VSMs as determined by Western blotting (Fig. 1C). G3BP1 mRNA levels were unaltered by LRP6 deficiency (supporting data S3), and Western blot analysis again confirmed (9) reductions in LRP6 protein levels with SM22-Cre expression in LRP6(fl/fl) (where LRP6(fl/fl) is LRP6 gene floxed, e.g. flanked by lox P) primary VSM cultures (supporting data S4). Immunofluorescence confirmed the expression of G3BP1 in both medial and adventitial compartments of the thoracic aorta (Fig. 1, E and D). The latter largely overlapped the adiponectin-positive cell populations in the fibrofatty arterial adventitia (Fig. 1D, supporting data S5), and the former co-localized to the α-actin–positive cells (Fig. 1E; note co-staining with FITC-conjugated VSM α-actin also known as Acta2 (25)). No background signal was observed in the absence of primary antibodies (supporting data S6).
Figure 1.

LRP6 deficiency alters the C-terminal Arg methylation of G3BP1, an RNA-binding protein genetically implicated in human cardiovascular disease. A, arginine methylome analysis reveals a plurality of RNA-binding proteins are regulated by LRP6 deficiency in primary aortic VSM cells. Applying a 2.5-fold threshold for change (2.5-fold increase, 60% decrease) in Arg methylation, 673 VSM proteins were altered with LRP6 deficiency. B, domain structure and C-terminal sequence of murine and corresponding human G3BP1. Residues differentially methylated in VSM with LRP6 deficiency are indicated by asterisks. See also supporting data S1 and S2. C, cytosolic levels of G3BP1 protein are significantly increased in LRP6-deficient VSM cells. D, consistent with results from CARDIoGRAM (18), G3BP1 protein is detected in arterial smooth muscle (arrows) and adjacent fibrofatty adventitia of LDLR-deficient mice fed atherogenic diets (yellow overlays in Merge). Note that although adiponectin is not expressed in endothelial cells, adiponectin protein is taken up by macrovascular endothelial cells (106) and can be immunologically detected (arrowheads). E, confocal co-localization with Acta2 confirms G3BP1 expression in the VSM cells of the tunica media. Note that the fibrofatty adventitial cell G3BP1 expression cell population is Acta2-negative (E) but is adiponectin-positive (D).
To confirm and extend these results, we examined the Arg methylation modification of human G3BP1 in transient transfection assays of HEK293T cells followed by Western blotting using Arg methylation-specific immunoreagents (9, 17). In transient co-transfection, both PRMT1 and PRMT5, but not PRMT4, increased G3BP1 MMA as revealed by Western blotting (Fig. 2A). Moreover, MMA modification was reduced by co-expression of LRP6 (Fig. 2B) as consistent with the methylome analyses from LRP6-deficient VSM (supporting data S1 and S2). Mass spectrometry performed on immunoprecipitates of FLAG-tagged G3BP1 confirmed regulation of Arg-methylated G3BP1 (data not shown). Thus, LRP6 negatively regulates the Arg methylation of G3BP1 in HEK293T cells (Fig. 2, A and B) as well as in VSM cells (supporting data S1 and S2).
Figure 2.

PRMT1 and PRMT5 increase, whereas LRP6 inhibits, G3BP1 MMA. A, expression vectors for human G3BP1, PRMT4, PRMT1, and PRMT5 were co-transfected as indicated; G3BP1 was immunoprecipitated, and Western blottings were performed assessing total Arg monomethylation (upper panel) and G3BP1 levels (lower panel). B, expression vectors for human G3BP1 (Myc- and FLAG- tagged) and LRP6 (untagged) were co-transfected as indicated. Following G3BP1 immunoprecipitated with anti-FLAG, western blots were performed assessing G3BP1 expression (anti-Myc, upper panel) and Arg monomethylation (lower panel). Note that although PRMT1 and PRMT5 increase G3BP1 MMA, LRP6 reduces G3BP1 Arg MMA levels.
LRP6 inhibits and G3BP1 supports NFATc4 localization to osteogenic chromatin targets and NFAT transcriptional activation
We previously demonstrated that LRP6 inhibits noncanonical Wnt signaling, including NFAT activation, as a key component of its anti-sclerotic actions in the VSM (9). Because LRP6 regulated G3BP1 Arg methylation, we examined the impact of G3BP1 expression upon NFAT expression and transactivation, a feature of the calcium-dependent noncanonical Wnt signaling program. LRP6 deficiency increased the relative gene expression of NFATc4 mRNA in VSM (Fig. 3A) along with nuclear NFATc4 protein (Fig. 3B; and supporting data S7), so we focused upon NFATc4 activity. Chromatin immunoprecipitation (ChIP) assays confirmed increased accumulation of NFATc4 on endogenous osteogenic genomic targets (26–28) OPN and TNAP in LRP6-deficient VSM (Fig. 3C), and RNAi targeting NFATc4 reduced numerous osteogenic mRNAs, including OPN, TNAP, Msx2, Sox2, and Wnt4 but not Runx2 (Fig. 3D). NFATc1 was unaltered, but NFATc3 was reduced by 50%. As shown in Fig. 4, co-expression of G3BP1 enhanced NFATc4-dependent transactivation. OPN promoter activation by NFATc4 in MOVAS VSM (29, 30) cultures was synergistically enhanced by G3BP1 co-expression (Fig. 4A); synergy was not noted between G3BP1 and NFATc2. Similarly, transcriptional activation of the artificial NFATLUC reporter in HEK293T cells was enhanced by co-expression of human G3BP1 with either NFATc4 or NFATc3, with little impact upon NFATc1 activity (Fig. 4B). Up-regulation of NFATLUC activity by activation of noncanonical Wnt signaling with Fzd5 and the ligand Wnt7b was also enhanced by G3BP1 co-expression (Fig. 4C). Interestingly, human G3BP2 was inactive in this assay (Fig. 4, C and D). Structure–function studies demonstrated that both the N-terminal NTF2 (31) and C-terminal methylation domains of G3BP1 (Fig. 1B) were required to augment noncanonical Wnt/NFAT signaling (Fig. 4D; and supporting data S8). Mutation of Arg-460 to a Gln residue in human G3BP1, corresponding to mouse G3BP1 residue Arg-458 that is differentially methylated with LRP6 deficiency (Fig. 1B and supporting data S1 and S2), significantly reduced but did not abrogate G3BP1-dependent enhancement of NFAT activity by noncanonical Wnt signaling (Fig. 4E). RNAi confirmed the importance of G3BP1 in the maintenance of osteogenic gene expression in primary VSM, with activity overlapping yet distinct from that of G3BP2 (Fig. 4F). In addition to reducing OPN mRNA, G3BP1 “knockdown” reduced the accumulation of Msx2 and LEF1 genes, components of Wnt signaling important to both craniofacial (32, 33) and ectopic arterial (6, 34) mineralization. Western blot analysis confirmed selective reductions in G3BP1 protein accumulation with G3BP1 siRNA (Fig. 4G).
Figure 3.

Conditional deletion of LRP6 in VSM increases NFATc4 mRNA, NFATc4 nuclear accumulation, and NFATc4 association with osteogenic genomic targets OPN and TNAP. A, primary aortic VSM cultures were prepared as detailed previously (6, 9) from LRP6-deficient SM22-Cre-LRP6(fl/fl);LDLR−/− mice and LRP6(fl/fl);LDLR−/− controls, and RNA was extracted for quantitation of NFATc messages as indicated. NFATc4 was significantly up-regulated with LRP6 deficiency. B, nuclear NFATc4 protein is also increased in primary aortic VSM cultures lacking LRP6. C, ChIP assays demonstrate that NFATc4 association with osteogenic OPN and TNAP promoters is increased with LRP6 deficiency. Associated with the TNF (tumor necrosis factor) control promoter was unaltered with LRP6 deficiency. D, RNAi targeting NFATc4 reduces osteogenic mRNA accumulation (OPN, TNAP, Msx2, Sox2, etc.) in cultures of aortic VSM cells. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001 versus corresponding control.
Figure 4.

G3BP1 enhances NFATc4-dependent transcription and supports OPN expression. A, MOVAS murine aortic VSM cells were transiently co-transfected with the 2-kb murine OPN promoter-luciferase reporter (OPNLUC) (96), with or without the pCMV-based expression vectors for NFATc4, NFATc2, and G3BP1 as indicated. TK Renilla was included as an internal control for transfection efficiency. Note that NFATc4 and G3BP1 synergistically increase OPN promoter activity. *, **, p ≤ 0.05 and p ≤ 0.01 versus control. #, p ≤ 0.01 versus G3BP1 + NFATc4. B, HEK293T cells were transiently transfected with a concatemerized NFAT cognate-driven luciferase reporter derived from the interleukin 2 promoter (NFATLUC) and the indicated expression vectors. Note that G3BP1 enhances both NFATc3- and NFATc4-dependent transcription. **, p ≤ 0.01 versus corresponding control. C, HEK293T cells were transiently transfected NFATLUC and pCMV-based expression vector for Wnt7b, Fzd5, G3BP1, and G3BP2 as indicated. Note that although G3BP1 enhances NFATLUC activation by the noncanonical Wnt7b/Fzd5 cascade, G3BP2 is inactive in this assay *, ***, p ≤ 0.05 and p ≤ 0.001 versus Wnt7b/Fzd5 with pCMV; #, p ≤ 0.001 versus G3BP1 with Wnt7b/Fzd5. D, both the NTF2 and C-terminal Arg methylation domains of G3BP1 are required to enhance Wnt/NFAT signaling. **, p ≤ 0.01 versus all others; #, p ≤ 0.05 versus Wnt7b/Fzd5 with pCMV, and p ≤ 0.01 versus Wnt7b/Fzd5 with G3BP1. E, G3BP1(R460Q) substitution in human G3BP1 reduces G3BP1-dependent enhancement of noncanonical Wnt/NFAT signaling. **, p ≤ 0.01 versus all others. F, RNAi targeting G3BP1 down-regulates OPN mRNA accumulation in primary aortic VSM. G3BP2 siRNA does not inhibit OPN expression, even though it significantly reduces Msx2, LEF1, and Myh11 (*, p ≤ 0.05 versus control siRNA). G, Western blotting analyses demonstrate efficient and selective reductions in G3BP1 and G3BP2 proteins by RNAi in aortic VSM cultures.
To confirm and extend these results, we implemented CRISPR/Cas9 (35, 36) to reduce G3BP1 expression in primary VSM (pools of clones). G3BP1 protein levels were reduced by ∼60% in cell cultures transduced and selected for expression of both Cas9 and G3BP1 guide RNA (gRNA). Concomitantly, nuclear NFATc4 protein levels (Fig. 5A) and NFATc4 binding to osteogenic chromatin in ChIP assays (OPN, TNAP; Fig. 5B) were reduced with G3BP1 deficiency. Thus, G3BP1 supports and LRP6 inhibits NFATc4 nuclear accumulation, osteogenic chromatin association, and noncanonical Wnt/NFAT signaling. The LRP6-inhibited Arg methylation target G3BP1 augments osteochondrogenic gene expression in part via enhanced NFATc4 transcriptional regulation.
Figure 5.
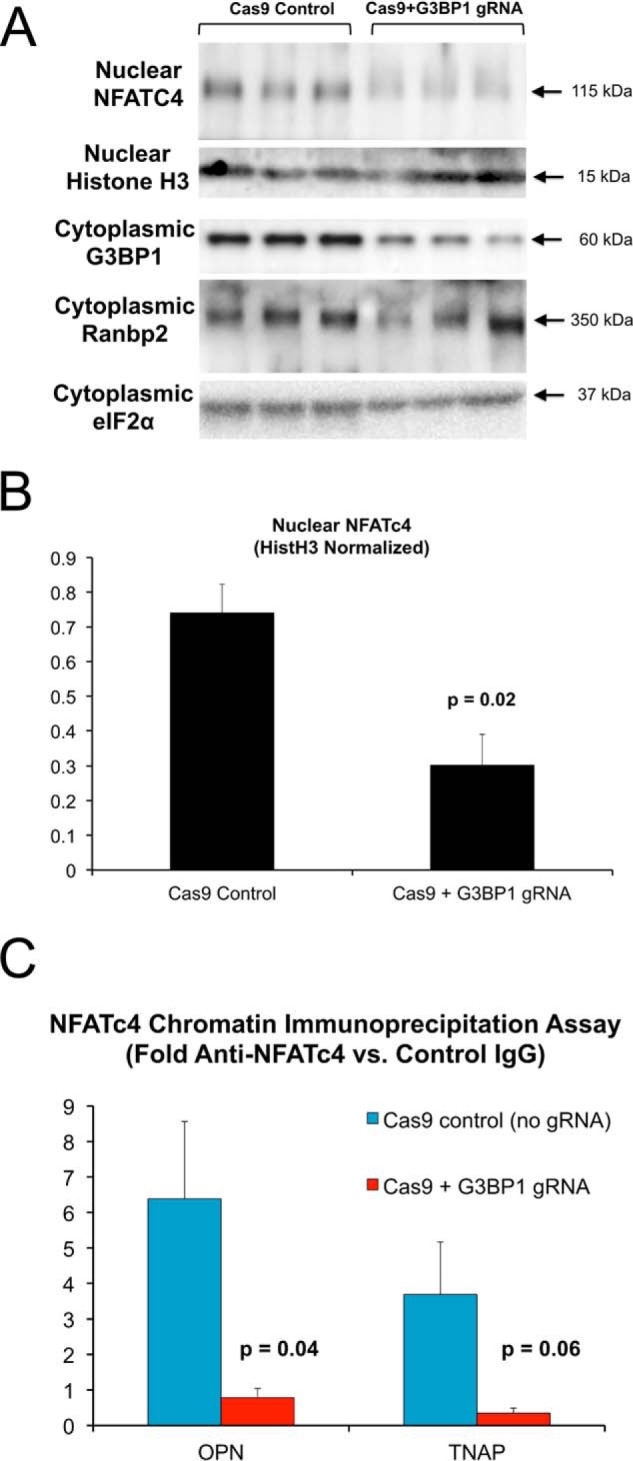
CRISPR/Cas9 down-regulation of G3BP1 in aortic VSM cell cultures reduces nuclear NFATc4 protein accumulation and NFATc4 association with OPN and TNAP promoters. Aortic VSM were transduced with antibiotic-selectable Cas9-expressing lentivirus. Cas9-expression pooled clones were then transduced with lentivirus expressing a G3BP1 guide RNA (gRNA), and NFATc4 and G3BP1 protein levels were assessed by Western blot analysis (A and B) or cultures were cross-linked and processed for ChIP assay (C). A, nuclear NFATc4 levels were down-regulated by G3BP1-directed gRNA. B, quantitative image analysis of relative NFATc4 expression in A. C, ChIP assay demonstrated reduced NFATc4 association with OPN and TNAP promoters in G3BP1-depleted VSM cells.
G3BP1 regulation of NFAT-dependent transcription is conveyed via G3BP1 interactions with RIG-I-like receptor activators of MAVS
To better understand the mechanisms whereby G3BP1 participates in noncanonical Wnt signaling, we carefully reviewed a recently published comprehensive G3BP1 interactome (37) and performed our own independent studies to identify interaction partners. Importantly, G3BP1 interactions with the RIG-l-like receptor (RLR) IFIH1 emerged from the systematic interactome (37). This immediately caught our attention because hereditary syndromes associated with arteriosclerotic aortic and coronary calcification have been newly mapped to gain-of-function mutations in the RLR family (38, 39). Thus, using transient co-transfection studies in HEK293T cells, we confirmed that the two RLRs IFIH1 (also known as MDA5; Fig. 6A) and RIG-I (retinoic acid inducible- I; also known as Ddx58; Fig. 6B) co-precipitate with G3BP1. Moreover, whereas deletion of the N-terminal domain had little effect, loss of the G3BP1 C-terminal domain reduced RIG-I co-immunoprecipitation with FLAG-tagged G3BP1 (Fig. 6C).
Figure 6.

G3BP1 and the RIG-I like receptors IFIH1 and RIG-I/Ddx58 co-precipitate in HEK293T cells. A, FLAG-tagged G3BP1 and IFIH1 were co-expressed in HEK293T cells as indicated. Cell extracts were prepared, immunoprecipitated with anti-G3BP1 antibody, and FLAG epitope-tagged proteins identified by Western blotting. Note that IFIH1 co-precipitates with G3BP1 consistent with recent comprehensive interactome studies (Ref. 36; see text). B, Myc-tagged RIG-I was co-expressed with untagged G3BP1, and cell extracts were prepared and immunoprecipitated with anti-Myc antibody, and RIG-I (anti-Myc; 100 kDa) and G3BP1 (anti-G3BP1; 60 kDa) were identified by Western blotting. Note that G3BP1 co-precipitates with RIG-I. Total levels of RIG-I protein were also increased by the co-expression of G3BP1. C, whereas deletion of the N-terminal domain had little effect, loss of the G3BP1 C-terminal domain reduced RIG-I co-immunoprecipitation with FLAG-tagged G3BP1. Arrows, co-precipitated proteins; filled circle, immunoglobulin IgG heavy chain background (50 kDa).
We reasoned that if one or both RLRs participate in the noncanonical Wnt programs held in check by LRP6, the steady-state levels of these proteins might change with LRP6 deficiency. As shown in Fig. 7A, levels of RIG-I/Ddx58 but not IFIH1 were significantly increased in LRP6-deficient VSM. Conversely, RIG-I levels, but not IFIH1, were reciprocally regulated with G3BP1 deficiency, reduced by ∼50% in primary VSM from G3BP1+/− mice (Fig. 7B), with concomitant reductions in RIG-I mRNA and the expression of other osteogenic gene programs (Fig. 7C). Intriguingly, the expression of Wnt antagonists (40, 41) secreted frizzled related protein 2 (SFRP2) and sclerostin (SOST) were concomitantly increased with G3BP1 deficiency (Fig. 7C). Confocal immunofluorescence confirmed the partial co-localization of endogenous RIG-I with G3BP1 in VSM (Fig. 7D), but not with the RIG-I effector, MAVS (19). RIG-I and G3BP1 co-localization is most apparent in the perinuclear region of VSM cells (Fig. 7E).
Figure 7.

VSM cell RIG-I/Ddx58 protein levels are increased with LRP6 deficiency and decreased with G3BP1 deficiency. A, cell extracts were prepared from SM22-Cre;LRP6(fl/fl);LDLR−/− VSM and LRP6(fl/fl);LDLR−/− controls as described previously, and Western blotting analyses were performed for RIG-I and IFIH1 as indicated. Note that RIG-I protein levels are increased with LRP6 deficiency, a setting where both G3BP1 methylation and protein levels are also increased (Fig. 1, B and C, and supporting data S1 and S2). B, VSM cells were prepared from control G3BP1+/+ and heterozygous G3BP1+/− mice, and cellular extracts were analyzed for G3BP1, RIG-I, IFIH1, and controls as indicated. Note the selective down-regulation of Ddx58 paralleling changes in G3BP1 protein levels. C, osteogenic mRNAs are also reduced in heterozygous G3BP1+/− VSM cell cultures as compared with G3BP1+/+ controls. D, RIG-I and G3BP1 partially co-localize in primary VSM cells (left panel), whereas G3BP1 and MAVS do not (right panel). E, RIG-I and G3BP1 co-localization is most apparent in the perinuclear region of VSM cells (yellow arrows). *, p ≤ 0.05 versus G3BP1+/+ control; **, p ≤ 0.01 versus G3BP1+/+ control. NS, not significant.
We continued to focus upon RIG-I because its protein levels were reciprocally regulated by LRP6 (increased) and G3BP1 (decreased) deficiencies. As a component of noncanonical Wnt signaling, we examined the impact on G3BP1-dependent NFAT transcription. In MOVAS VSM cells (29, 30), NFATc4-driven transcription was synergistically enhanced by G3BP1 and RIG-I co-expression (Fig. 8A). Synergy depended upon the G3BP1 C-terminal Arg methylation domain and could not be achieved by co-expression of G3BP2 (Fig. 8A). Likewise, activation of the TNAP promoter by NFATc4 and G3BP1 was augmented by Ddx58 co-expression (Fig. 8B). Furthermore, RNAi targeting RIG-I reduced osteogenic mRNA accumulation in primary VSM (Fig. 8C).
Figure 8.
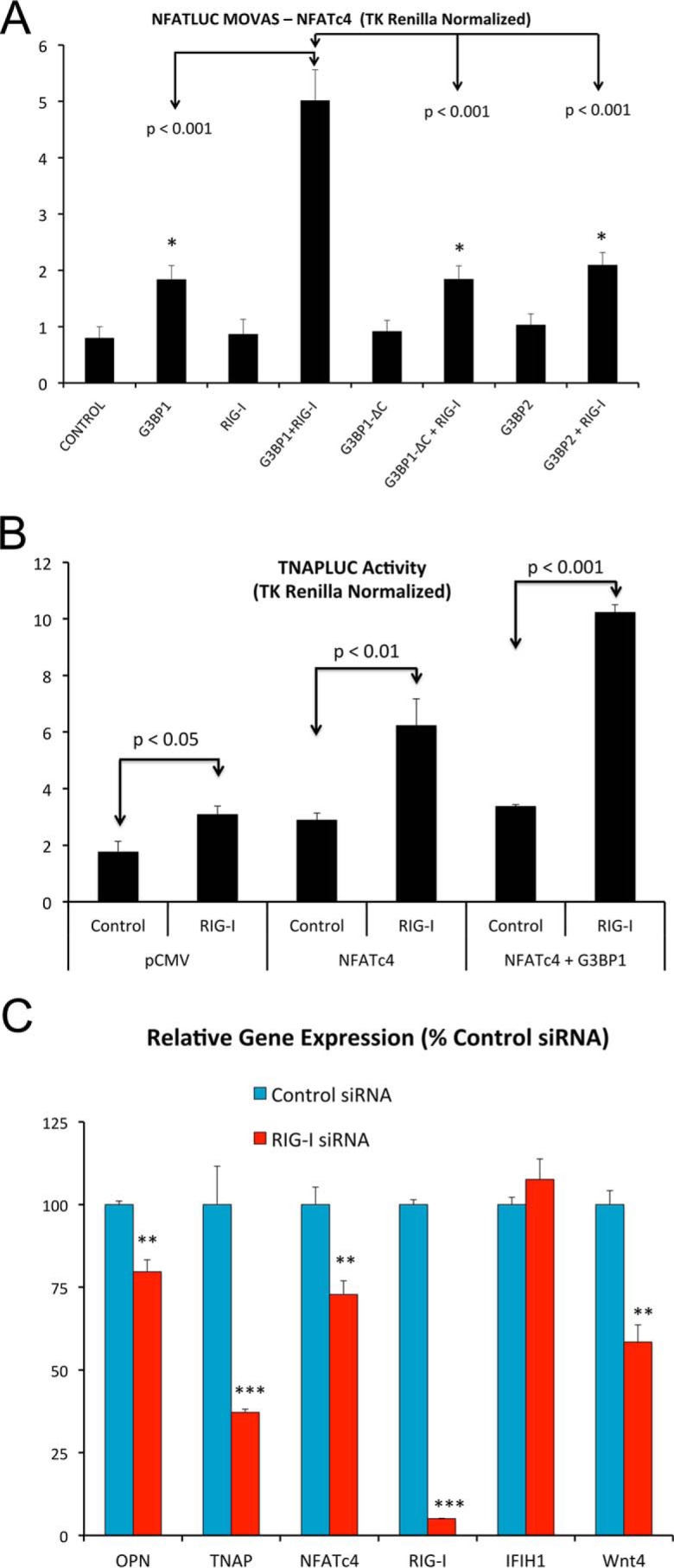
Ddx58/RIG-I expression enhances G3BP1-stimulated NFATc4 transactivation and supports osteogenic gene expression in VSM. A, co-expression of RIG-I in MOVAS VSM cells enhances G3BP1-stimulated NFATLUC activity (NFATc4 supported). Stimulation required the C-terminal Arg methylation domain of G3BP1. B, RIG-I also enhances G3BP1-stimulated NFATc4 transactivation of the TNAP promoter. C, RNAi targeting RIG-I/Ddx58 down-regulates osteogenic gene expression in aortic VSM cells. See Fig. 4G for efficiency of RIG-I protein down-regulation by RNAi. *, **, ***, p ≤ 0.05, ≤ 0.01, and ≤ 0.001 versus corresponding controls.
To confirm and extend the role for RIG-I in VSM arteriosclerotic responses, we implemented CRISPR-Cas9 methodology to target the RIG-I/Ddx58 gene; this strategy reduced VSM RIG-I protein levels by ∼75% (Fig. 9A). Once again, the expression of osteosclerotic mRNAs (OPN, TNAP, Osx, and Wnt4) and NFATc4 were reduced with RIG-I deficiency (Fig. 9B). Importantly, bone alkaline phosphatase enzyme activity, a key ectoenzyme in osteochondronic mineralization encoded by the TNAP gene (26, 27), was also reduced with RIG-I down-regulation (Fig. 9C). Thus, the G3BP1-interacting protein RIG-I supports osteogenic programs in aortic VSM cells.
Figure 9.

CRISPR/Cas9 down-regulation of RIG-I/Ddx58 reduces osteogenic differentiation of aortic VSM cells. Aortic VSM were transduced with antibiotic-selectable Cas9-expressing lentivirus. Cas9-expression pooled clones were then transduced with lentiviruses expressing RIG-I/Ddx58 gRNA, and RIG-I protein levels were assessed by Western blot analysis (A). In parallel, cultures were grown under mineralizing conditions, and osteogenic re-programming assessed by RT-qPCR (B) or alkaline phosphatase enzyme activity (C). Note that down-regulation of VSM RIG-I (A) is again associated with reductions in the osteogenic potential of aortic VSM (B and C). *, **, ***, p ≤ 0.05, ≤ 0.01, and ≤ 0.001 versus corresponding controls.
Arteriosclerotic gain-of-function RIG-I variant enhances NFATc4- and Runx2-dependent transcription with G3BP1
Very recently, gain-of-function mutations in RIG-I (also known as Ddx58) and IFIH1 (also known as MDA5) have been demonstrated to cause Singleton-Merten syndrome, a disorder characterized by precocious coronary, aortic valve, and aortic calcification with glaucoma, bone loss, and psoriasis (38, 39). The gain-of-function variants of RIG-I result in aortic and coronary calcification with less severe skeletal involvement (39). We therefore examined the impact of RIG-I(E373A), one of these constitutively active RIG-I (cRIG-I) variants (39), on NFATc4-dependent transcription. As shown in Fig. 10A, RIG-I(E373A), henceforth cRIG-I, synergistically enhances NFATc4 transcription with G3BP1. Activation depended upon the intact G3BP1 N- and C-terminal domains (Fig. 10A) and was inhibited by LRP6 (Fig. 10B). Of note, deletion of the G3BP1 N-terminal domain encompassing NTF2 generated a variant that functioned as a RIG-I-dependent dominant-negative inhibitor of NFAT signaling (Fig. 10A). Furthermore, introduction of the point mutation R460Q in human G3BP1, corresponding to the residue exhibiting reciprocal MMA/SDMA modification with LRP6 deficiency (Fig. 1B and supporting data S1 and S2), reduced cRIG-I activation of NFATc4-driven transcription (Fig. 10C).
Figure 10.

Constitutively active, pro-arteriosclerotic variants of Ddx58/RIG-I enhance G3BP1-stimulated transcription. The RIG-I/Ddx58 variants Ddx58(E373A) and Ddx58(C268F) have recently been identified to cause Singleton-Merten syndrome and characterized by precocious aortic, aortic valve, and coronary calcification (39). A, G3BP1 + RIG-I(E373A) activation of NFAT transcription (NFATc4) requires intact G3BP1 NTF2 and C-terminal Arg methylation domains. RIG-I(E373A) is hence denoted as cRIG-I. **, p ≤ 0.01 versus control lacking any expression vector (far left, control); NS, not significant. B, cRIG-I activation of NFATc4 is inhibited by LRP6 expression. ***, p ≤ 0.001 versus control. #, p ≤ 0.001 versus cRIG-I. C, as compared with WT G3BP1, G3BP1(R460Q) is not capable of synergizing with cRIG-I to activate NFAT signaling (NFATc4 co-expression). ***, p ≤ 0.001 versus all others. D, both RIG-I/Ddx58 variants RIG-I (E373A) and RIG-I (C268F) exhibit gain-of-function synergy with Runx2 activation of the OPN promoter. ***, p ≤ 0.001 versus RIG-I and all others. ****, p < 0.0001 versus G3BP1 + Runx2 + RIG-I. E, Runx2 but not Runx1 is active in this assay. ***, p ≤ 0.001 versus Runx2 + G3BP1, and #, p ≤ 0.01 versus pCMV control. F, cRIG-I activation of the OPN promoter requires the G3BP1 C-terminal Arg methylation domain. #, p ≤ 0.001 versus all others, and ***, p ≤ 0.001 versus G3BP1 + Runx2 with cRIG-I.
Because the osteochondrogenic transcription factor Runx2 also supports OPN gene expression (42) and arteriosclerotic responses in the vessel wall (43–45), we assessed whether the novel G3BP1/RIG-I cascade participated in Runx2 activity. As shown in Fig. 10D, WT RIG-I and gain-of-function (39) RIG-I(E373A) and RIG-I(C268F) variants enhanced Runx2 + G3BP1 activation of OPN promoter activity; however, the Singleton-Merten syndrome variants RIG-I(E373A) and RIG-I(C268F) (39) exhibited much greater capacity to activate OPN in this assay (Fig. 10D). No synergy was observed with either Runx1 (Fig. 10E) or when G3BP1 was replaced by G3BP2 (data not shown). As with NFAT activation, the C-terminal G3BP1 methylation domain was critical to Runx2 transactivation (Fig. 10F). Runx2 structure–function studies mapped activation by cRIG-I to the Runx2 N-terminal activation domain AD1 (supporting data S9). Thus, the arteriosclerotic Singleton-Merten syndrome RIG-I/Ddx58 variants (39) greatly enhance osteogenic transcriptional responses in transient transfection assays, dependent upon the G3BP1 C-terminal Arg methylation domain.
Arteriosclerotic RIG-I(E373A) variant increases G3BP1 Arg monomethylation
Because the arteriosclerotic variant RIG-I(E373A) (39) enhances G3BP1 functions, and G3BP1 activity in LRP6-deficient VSM tracked increased G3BP1 Arg MMA, we reasoned that cRIG-I might control G3BP1 MMA. To test this notion, we co-transfected FLAG-tagged G3BP1 either with an empty vector or a vector driving the expression of cRIG-I (also FLAG-tagged) in HEK293T cells. We then immunoprecipitated the FLAG-tagged proteins and blotted with either FLAG or MMA. As shown in Fig. 11A, co-expression of cRIG-I with G3BP1 increased levels of G3BP1 protein. More importantly, cRIG-I co-expression significantly enhanced G3BP1 MMA modification (Fig. 11, B and C). Mass spectrometry of G3BP1 immunoprecipitates confirmed that cRIG-I co-expression promoted human G3BP1 Arg methylation including residues Arg-435, Arg-447, and Arg-460 (data not shown). Thus, the arteriosclerotic cRIG-I variant that enhances osteogenic G3BP1-NFATc4 and G3BP1-Runx2 activity increases G3BP1 Arg monomethylation.
Figure 11.

Constitutively active RIG-I/Ddx58 stimulates G3BP1 MMA. HEK293T cells were co-transfected with FLAG-tagged G3BP1 and constitutively active RIG-I (cRIG-I) as indicated; cell extracts were prepared, proteins isolated by anti-FLAG immunoprecipitation, and analyzed by Western blot analysis. A, anti-FLAG analyses of FLAG-tagged proteins in the immunoprecipitate. B, anti-MMA modification in the immunoprecipitated proteins. Note increased MMA modification of G3BP1 with cRIG-I co-expression. C, image analysis quantification a significant 6-fold increase in G3BP1 MMA signal with cRIG-I expression. ***, p ≤ 0.001 versus all others.
G3BP antagonist C108 inhibits NFAT signaling, G3BP1 Arg monomethylation, and osteogenic differentiation of primary VSM
Using a high-throughput screening strategy to identify compounds that induce breast cancer cell death without harming nonmalignant cells, the benzohydrazide compound C108 (also known as 2-hydroxy-N′-[1-(2 hydroxyphenyl)ethylidene] benzohydrazide) was recently identified (Fig. 12A) (46). Affinity purification revealed that both G3BP1 and G3BP2 were key targets of C108 inhibition (46), with G3BP2 inhibition mediating beneficial reductions in tumor growth. Given our above data, we reasoned that this G3BP inhibitor might impact pro-sclerotic programs in primary VSM. Hence, we contracted with Cayman Chemical (Ann Arbor, MI) for custom synthesis of C108. C108 indeed inhibits cRIG-I activation of NFATc4 signaling (Fig. 12B) in transfected HEK293T cells. By contrast, two commercially available benzohydrazide derivatives of C108, R699497 (N′-(phenylethylidene)benzohydrazide) and R700061 (4-hydroxy-N′-(4-hydroxy-3-methoxybenzylidene)benzohydrazide) did not inhibit noncanonical Wnt/NFAT signaling (Fig. 12C). Of note, C108 did not exhibit direct inhibition of Runx2 transactivation function in one-hybrid assays (supporting data S10). Consistent with the inhibitory actions of C108 on noncanonical NFATc4 signaling, C108 inhibited the up-regulation of both OPN and TNAP in LRP6-deficient VSM cultures (Fig. 12, D and E). Moreover, alkaline phosphatase enzyme activity was also significantly reduced (Fig. 12F) with concomitant reductions in Alizarin red staining (Fig. 12G). Spautin, and inhibitor of the G3BP1-associated protein USP10 (47), also reduced mineralization (Fig. 12G).
Figure 12.

G3BP inhibitor C108 antagonizes NFAT transactivation stimulated by Wnt/Fzd or by G3BP1/RIG-I signaling. A, chemical structure of the compound C108, recently identified to bind G3BP1 and G3BP2 and to inhibit G3BP2-dependent tumor initiation (45). B, NFATLUC activation (NFATc4 co-expression) by either Wnt7b/Fzd5 or by G3BP1/cRIG-I is inhibited by C108 treatment. C, chemically similar benzohydrazides R699497 and R700061 are inactive as compared with C108. ***, p ≤ 0.001 versus DMSO and all other treatments in Wnt7b(Fzd5)-supported NFATLUC activity. #, p ≤ 0.001 versus all samples lacking Wnt7b(Fzd5). D and E, C108 antagonizes the up-regulation of OPN and TNAP in LRP6-deficient VSM cells. F, C108 inhibits alkaline phosphatase activity in VSM cells cultured under mineralizing conditions. G, calcium deposition assessed by Alizarin red staining was also reduced by C108 treatment. Comparison is made with spautin, an inhibitor of the G3BP1-binding protein USP10 (47), and metformin. *, **, ***, p ≤ 0.05, ≤ 0.01, and ≤ 0.001 versus corresponding controls.
Because G3BP1 Arg methylation tracks noncanonical osteogenic signaling with LRP6 deficiency (9), and C108 inhibits these programs, we reasoned that C108 might modulate G3BP1 Arg methylation. Therefore, we co-transfected HEK293T cells with FLAG-tagged G3BP1 and cRIG-I in the presence of either DMSO or 3 or 10 μm C108. Following immunoprecipitation with anti-FLAG, Western blottings were performed with anti-FLAG, anti-MMA, or anti-SDMA as indication. As shown in Fig. 13A, levels of G3BP1 MMA were significantly reduced by C108 treatment. By contrast, SDMA modification of G3BP1 was unaltered (Fig. 13B) as were total G3BP1 protein levels (Fig. 13C). Thus, the small molecule G3BP inhibitor C108 (46) mitigates the arteriosclerotic gene expression, enzyme activity, and mineralization programs of cultured aortic VSM cells.
Figure 13.

C108 selectively inhibits G3BP1 MMA but not G3BP1 Arg SDMA. HEK293T cells were transfected with either empty vector (lanes 1–3) or with FLAG-tagged G3BP1 + cRIG-I (lanes 4–14), and treated with either DMSO vehicle (lanes 1–6) or C108 (lanes 7–14) as indicated. Following immunoprecipitation with anti-FLAG antibody, aliquots of immunoprecipitates were analyzed by Western blotting for either MMA (A), SDMA (B), or FLAG-tagged proteins (C). Note that C108 in the 3–10 μm range inhibited G3BP1 MMA (A) without altering SDMA accumulation (B) or total protein accumulation (C).
Deficiency in MAVS, the RIG-I mitochondrial signaling platform, reduces osteochondrogenic differentiation and arteriosclerotic calcification
RIG-I/Ddx58 and IFIH1/MDA5 function as components of viral innate immunity (48). Upon binding and sensing foreign viral RNAs, these RLRs activate downstream NF-κB and interferon cascades via MAVS, the mitochondrial antiviral signaling platform protein (19, 49). Because of the role for RIG-I in G3BP1 activity and methylation in noncanonical Wnt osteosclerotic programs, we reasoned that MAVS might play a critical role in this cascade as well. To examine the role for MAVS in VSM arteriosclerotic responses, we first quantified the osteochondrogenic differentiation programs (50) elaborated by primary VSM from MAVS−/− mice when cultured under mineralizing conditions. As shown in Fig. 14A, MAVS−/− VSM cells exhibit a 40% reduction in the expression of OPN protein. Moreover, Wnt5b-dependent up-regulation of nuclear NFATc4 was reduced in MAVS−/− primary VSM (Fig. 14B), to confirm the role of MAVS in noncanonical Wnt signaling. Of note, Runx2 nuclear protein accumulation was unaltered. Furthermore, RT-qPCR analyses revealed the down-regulation of multiple arteriosclerotic genes in MAVS−/− VSM cultures, including OPN, TNAP, Msx2, Msx1, Wnt4, Sox2, and Osx (Fig. 14C). Again, the Wnt inhibitor Sost was up-regulated, along with preservation of VSM phenotypic markers myocardin (Myocd), myosin heavy chain 11 (Myh11), and elastin. Although Rankl, a prosclerotic inflammatory regulator (51–54), was down-regulated in MAVS−/− VSM, its autocrine antagonist osteoprotegerin (Opg) was increased (Fig. 14C). Alkaline phosphatase (TNAP) enzyme activity was also down-regulated with MAVS deficiency (Fig. 14D), with concomitant reductions in calcium deposition as visualized by Alizarin red staining (Fig. 14E).
Figure 14.

RIG-I effector MAVS supports osteogenic differentiation and noncanonical Wnt/NFAT signaling in aortic VSM. Primary aortic VSM cells were prepared from WT and MAVS−/− mice and analyzed for protein expression, osteogenic differentiation, and mineralization as indicated. A, Western blot analysis confirms absence of MAVS, with concomitant down-regulation of OPN expression. B, although Wnt5b treatment increases nuclear NFATc4 protein accumulation in WT cells, stimulation is markedly reduced in MAVS−/− VSM cells. Runx2 protein accumulation is unaltered. NFATc4 is significantly reduced in MAVS−/− nuclei (p < 0.05 all comparisons). C, osteogenic mRNA accumulation is reduced in cultures of MAVS−/− aortic VSM cells. D, alkaline phosphatase enzyme activity is reduced in MAVS−/− VSM cultured under mineralizing conditions. E, Alizarin red staining with digital image analysis (94) confirms significant reduction in VSM calcification with MAVS deficiency. F, aortic calcium accrual is significantly reduced in male MAVS−/−;LDLR−/− mice fed arteriosclerotic high-fat diets for 12 weeks as compared with male LDLR−/− controls. Systemic inflammation as indicated by serum haptoglobin (supporting data S12) and aortic sinus atheroma lesion areas (supporting data S13) were not reduced. See text for details. *, **, #, p ≤ 0.05, ≤ 0.01, and ≤ 0.001 versus corresponding controls.
When fed a high-fat diet with a composition resembling that of Western societies (42% of calories from fat), male LDLR−/− mice develop obesity, insulin-resistant diabetes, and arteriosclerotic calcification. Therefore, to assess the contribution of the MAVS effector to aortic calcification in vivo, male MAVS−/−; LDLR−/− mice (n = 6) were challenged for 3 months with the high-fat diabetogenic Western diet, and aortic calcium content was biochemically compared with that of MAVS-replete LDLR−/− controls (n = 18). As shown in Fig. 14F, aortic calcium was reduced by ∼40% in MAVS−/−;LDLR−/− mice as compared LDLR−/− controls (p = 0.03), consistent with primary VSM cell culture results (Fig. 14, A–F). Histological staining for calcium in sections of the aortic sinus and ascending aorta, implementing an Alexa647-labeled amino-bisphosphonate (55), confirmed that calcium was accruing in both atherosclerotic lesions and the tunica media (supporting data S11) as seen previously (7). At necropsy, no differences were exhibited between groups with respect to fasting hypercholesterolemia (LDLR−/− versus MAVS−/−;LDLR−/− and 975 ± 34 versus 1012 ± 37 mg/dl; p = NS), hypertriglyceridemia (296 ± 25 versus 316 ± 40 mg/dl; p = NS), weight (34.0 ± 2.1 versus 33.7 ± 1.0 g; p = NS), or diabetes in the homeostatic model assessment of insulin resistance (56) (HOMA-IR; 17 ± 5 versus 16 ± 7; p = NS). MAVS deficiency did not globally reduce diet-induced increases in the inflammation associated with insulin resistance (57, 58) as indicated by the circulating biomarker haptoglobin (7, 57) (supporting data S12). Atheroma lesion area, assessed by Mac2 staining at the level of the aortic sinus, was unaltered (p = 0.88; supporting S13). Thus, MAVS, a key component of the arteriosclerotic G3BP1/RIG-I signaling relay, supports noncanonical Wnt signaling and arteriosclerotic relays in VSM cells (Fig. 15).
Figure 15.

Arteriosclerotic G3BP1/RIG-I signaling in aortic VSM, a working model. We (9) and others (14) previously identified that LRP6 expression in VSM restrains noncanonical Wnt signals that (a) promote phenotype modulation and neointima formation and (b) enhance arteriosclerotic mineralization. We demonstrated that protein Arg methylation is profoundly altered with LRP6 deficiency and have identified G3BP1 as a key MMA target that is negatively regulated by LRP6. We further show that RIG-I, also known as Ddx58, functionally and physically interacts with G3BP1 to enhance G3BP1 Arg methylation and the noncanonical Wnt/NFAT pathway. This is mediated in part via NFATc4 nuclear translocation and association with osteogenic genomic targets such as the OPN and TNAP promoters. The mechanisms whereby nuclear Runx2 transactivation is enhanced by the G3BP1/cRIG-I cascade have yet to be determined (indicated by the ?). Not shown are the contributions of nuclear USF1 and Sp1 recently identified as also held in check by VSM LRP6 signaling (9, 14). See text for details.
Discussion
Cardiovascular disease remains a leading cause of morbidity and mortality (59). Remarkable therapeutic advances have successfully mitigated cardiovascular disease by targeting key metabolic stressors (cholesterol, glucose, phosphate, and oxylipids) and the neuroendocrine contributors to hypertension, vascular remodeling and inflammation, and myocardial pump function (60, 61). However, the clinical burden of unmet need, particularly in the settings of diabetes and declining renal function, continues to challenge patients and clinicians (62). Human molecular genetics has identified a number of novel contributors to cardiovascular disease biology (18), but these discoveries do not immediately illuminate the molecular mechanisms altering cardiovascular health.
G3BP1 was recently identified by eQTL analysis of visceral (thoracic) adventitial fat as a master determinant of the extent of cardiovascular disease as adjudicated by angiography (18). The biochemical and cellular roles of G3BP1 are only beginning to be understood, and multiple functions are emerging. The single Drosophila G3BP gene, Rasputin, was discovered as a component of the planar cell polarity controlling compound eye development (63, 64). In vertebrates, studies of G3BP1, and its homolog G3BP2, have emphasized roles in the formation of RNA stress granules in interferon-mediated and translation-modulated antiviral responses (65, 66). Recent data have demonstrated that Arg methylation of G3BP1 inhibits RNA binding and stress granule formation (67). However, G3BP1 has roles beyond RNA stress granule formation, including regulation of c-Myc RNA turnover (68), inhibition of miR-1 biogenesis (69), intron splicing (70), and signal transduction (23, 71, 72).
The Bikkavilli and Malbon (23, 24) have best characterized the roles of G3BP1 and G3BP2 in signal transduction. They first identified that G3BPs associate with the Wnt signaling platform protein Disheveled and that Wnt agonists promote Arg methylation of both G3BP1 and G3BP2 (23, 24). This is consistent with data from others suggesting that Wnt signals inhibit stress granule formation (73). Our data have newly revealed that the up-regulation of noncanonical Wnt signals, because of loss of VSM LRP6, also enhances G3BP1 Arg monomethylation, with concomitant activation of osteogenic transcriptional programs mediated in part by NFATc4 and Runx2. Both the N-terminal nuclear transport factor 2 (NTF2) (31) and C-terminal Arg methylation domains of G3BP1 are important in activating these transcriptional responses. Because RRM domains encode nuclear localization functions that become evident in the absence of RNA binding (22) and G3BP protein Arg methylation inhibits RRM binding to RNA cognates (67), it is probable that Arg methylation of G3BP1 controls key nuclear transport functions, including NFATc4 localization. Our unpublished data4 point to an important role for specific G3BP1-interacting peripheral nucleoporins (74), dependent upon the G3BP1 NTF2 domain and RIG-I. Importantly, we show that loss of LRP6 and loss of G3BP1 reciprocally regulate the nuclear localization of NFATc4 as well as NFATc4 association with the osteogenic chromatin targets OPN and TNAP. Thus, in this model, the G3BP1 either promotes nuclear transport or prevents nuclear egress of NFATc4 (Fig. 15). The G3BP1 NTF2 domain does indeed interact with the FXFG motifs of the nucleoporins (31), and the crystal structure has been solved (31). G3BP1 residue Phe-33 forms part of the NTF2-binding pocket for the FXFG cognate (31). Like G3BP1-ΔN and G3BP1-ΔC, G3BP1(F33W) fails to activate NFATc4 signaling (data not shown). However, this domain and residue are also required for G3BP1 interactions with caprin and USP10 (66), interactions that either promote (former) or inhibit (latter) RNA stress granule assembly (66). Because 1) G3BP1 methylation is up-regulated with noncanonical Wnt/NFAT signaling; 2) G3BP1 and G3BP2 methylation inhibits RNA stress granule formation (67); 3) both G3BP1 and G3BP2 support RNA stress granule formation (66); yet 4) G3BP1 but not G3BP2 supports the noncanonical Wnt/NFAT cascade; and 5) knockdown of G3BP1 and G3BP2 in VSM cells differentially regulate gene expression, we surmise that noncanonical Wnt activation by G3BP1 does not proceed via classical RNA stress granule assembly. Rather, we speculate that nuclear transport regulatory functions of G3BP1 are critical for this Wnt response. Future studies characterizing stress granule assembly and cytoplasmic–nuclear transport in G3BP1-null versus G3BP2-null VSM will directly test this notion.
Our data also reveal new regulatory components of noncanonical Wnt signaling in VSM, viz. the contributions of the G3BP1/MAVS relay. In systematic analyses of complex interactomes (37), the RIG-I like receptors (RLRs) have emerged as novel G3BP-binding proteins as our data independently confirm. Although RLRs are traditionally studied as part of the antiviral response (75), human molecular genetics have revealed the roles of the RLRs RIG-I/Ddx58 and IFIH1/MDA5 in arteriosclerotic vascular calcification (38, 39). As introduced above, autosomal dominant gain-of-function point mutations in RIG-I/Ddx58 and IFIH1/MDA5 are associated with familial Singleton-Merten syndrome, a disorder characterized by precocious (under age 20) cardiovascular calcification (38, 39). Our data functionally confirm and extend these data from human studies to identify that RIG-I promotes G3BP1 Arg methylation and activation of downstream osteogenic mineralization responses in VSM cultures. This mechanism is further supported by our data demonstrating that the prototypic RLR effector MAVS is necessary for the robust osteogenic differentiation, mineralization of aortic VSM, and aortic calcium accrual in male LDLR−/− mice fed high-fat diets. Of note, in the Dallas Heart Study I cohort, we recently identified an uncommon variant of RIG-I that is associated with reduced risk for coronary calcification; this variant is hypomorphic in assays of NFAT and NF-κB transactivation.4 Demer and Tintut (76) were among the first to highlight that arterial calcification in metabolic disorders is likely a consequence of innate immune programs that drive calcification to wall off pathogens in the lung or dermis. Aging and irradiation alter the integrity of the nuclear envelope in ways that lead to aberrant cytosolic accumulation of nuclear RNAs capable of activating RIG-I (77), thereby eliciting innate inflammatory responses optimally directly toward RNA virus pathogens. Therefore, we speculate that some features of cardiovascular sclerosis arising with aging (78) and irradiation (79) may reflect “intracrine” activation of the pro-sclerotic G3BP1/RIG-I/MAVS program. This remains to be tested.
In our immunofluorescence studies of G3BP1 and RIG-I in VSM, there appeared to be co-localization in juxtanuclear membranes in addition to the nuclear periphery. RIG-I has been identified in mitochondrion-associated membranes of the endoplasmic reticulum (80, 81). Indeed, RNA degradation associated with the endoplasmic reticulum stress response may activate the RIG-I pathway (81), which is relevant to vascular calcification as highlighted by Miyazaki and co-workers (82). Our Western blotting studies also identified G3BP1 protein accumulation in the total cellular membrane fraction. Future studies will address the potential differences in subcellular targeting of G3BP1–RIG-I complexes in arteriosclerotic signal transduction.
There are, of course, limitations to our study. We have focused on G3BP1 actions in the aortic VSM cell. However, G3BP1 expression is widespread, including within the myeloid and adipocyte lineages (18). We have yet to characterize the roles and regulation of G3BP1 in these cellular backgrounds. Indeed, Foroughi Asl et al. (18) suggested that G3BP1 inhibits cholesterol ester accumulation in the THP1 cell culture model of foam cell formation. Although G3BP1 is readily detected in both medial and fibrofatty adventitial compartments, roles served by G3BP1 may differ between these cell types. Future studies will examine the cell type as well as the integrated global impact of G3BP1 deficiency in murine cardiometabolic disease models. Although G3BP1 enhances NFATc4 nuclear accumulation and transcription, the precise nucleoporin transporters mediating these responses have yet to be determined. Furthermore, although we have used both gain- and loss-of-function approaches to confirm that endogenous G3BP1, RIG-I, and MAVS protein levels regulate VSM osteogenic programs, structure–function analyses by co-transfection have limitations; thus, future studies will analyze mice possessing “knockin” alleles for RIG-I gain-of-function to extend these results and provide a preclinical model for the Singleton-Merten syndrome. G3BP1 Arg methylation inhibits stress granule assembly; hence, the roles for specific G3BP1–RNA complexes in this regulatory cascade are still unknown. It is interesting to note that Jmjd6, the broad specificity demethylase we identified as regulating osteogenic histone Arg methylation downstream of LRP6 (9), has been recently show to demethylate G3BP1 as well (83). Precisely how active RIG-I enhances while LRP6 and C108 inhibit G3BP1 Arg monomethylation remains to be determined, but it may involve Jmjd6 activity (9, 83). Because co-expression of RIG-I and G3BP1 appears to increase not only G3BP1 protein Arg methylation but also G3BP1 protein levels, RIG-I may control of G3BP1 protein turnover via ubiquitination or sumoylation or may emerge as an important co-regulatory feature for nuclear transport (84). Indeed, G3BP1 does interact with certain ubiquitin- specific proteases (85). Moreover, we have yet to determine how other regulators of vascular calcification, including AMP kinases (86) and O-GlcNacylation (87), intersect with noncanonical Wnt programs. Our previous data (9) revealed cdc42/Rac1 promotes Arg methylation downstream of noncanonical Wnt/Fzd activation held in check by LRP6 (Fig. 15). Whether RIG-I also requires intact cdc42/Rac1 activity is unknown, but studies of RIG-I–deficient macrophages suggest that this may be the case (88), because microvesicles arising from the macrophage lineage can also nucleate calcification (89). Finally, because Runx2 is constitutively nuclear in its localization (90), the mechanisms whereby G3BP1 augments Runx2 (but not Runx1)-dependent transcription are unknown. Activation by RIG-I and G3BP1 maps to the N terminus of Runx2, a p38 MAPK and core binding factor β-regulated transactivation function (91). Future studies will examine whether a MAPK or another modifier of Runx2 function shuttles to the nucleus in response to activation of the G3BP1/RIG-I relay. Nevertheless, our data indicate that both pharmacological and molecular targeting of the noncanonical Wnt/G3BP1/RIG-I relay is feasible, and it may offer a novel therapeutic strategy to limit the extent and severity of arteriosclerotic disease.
Experimental procedures
Biochemical and tissue culture supplies and reagents
Tissue culture reagents were purchased from Thermo Fisher Scientific. Molecular biology reagents, enzymes, biochemicals, and synthetic oligodeoxynucleotides were purchased from Thermo Fisher Scientific, Sigma, Cayman, or ACROS as indicated. Luciferase enzyme assay reagents were purchased from Promega. Custom synthesis and purification of C108 (also known as 2-hydroxy-N′-[1-(2 hydroxyphenyl)ethylidene] benzohydrazide; CAS no. 15533-09-2) was performed by Cayman Chemical; this is now available as item no. 9002951. The structurally similar molecules N′-(1-phenylethylidene)benzohydrazide (R699497) and 4-hydroxy-N′-(4-hydroxy-3-methoxybenzylidene)benzohydrazide (R700061) were purchased from Sigma. Commercially available antibodies, TaqMan gene expression assays (Life Technologies, Inc.), siRNA/RNAi reagents (GE Dharmacon), and lentiviruses used for CRISP-Cas9 targeted gene disruption (GE Dharmacon) in primary VSM are listed in the supporting data S14, along with primers used for ChIP assay analyses. Plasmids generated by the Towler laboratory, provided as gifts via Addgene or obtained from commercial sources, are also presented in supporting data S14.
Transgenic mice, primary aortic VSM culture, and Arg methylation proteomics
All procedures for handling mice were approved by the University of Texas Southwestern Institutional Animal Care and Use Committees. SM22-Cre;LRP6(fl/fl);LDLR−/− mice and LRP6(fl/fl);LDLR−/− controls were generated on the C57Bl/6 as described previously (9, 92). MAVS-deficient mice were generated as described previously (19) and bred onto the C57Bl/6 background for 10 generations. G3BP1+/− mice on the C57Bl/6 background (G3bp1tm1a(KOMP)Wtsi) from knockout-first clone EPD0551_4_H12) were obtained from the University of California at Davis KOMP Repository. All experimental animals and controls used to generate primary aortic VSM cells were male sibling cohorts. Aortic primary cells were isolated from genetically engineered mice as described previously (6) using timed digest with type 1 collagenase (Worthington, catalog no. LS004149, 1 mg/ml), DNase I (Sigma, D5025, 60 units/ml), and hyaluronidase (Sigma, H3506, 0.5 mg/ml) in DMEM with 2× penicillin/streptomycin fungizone supplementation (2.5 μg/ml). For routine experiments, pools of cells isolated from 8 to 10 male animals per genotype were expanded with passage on collagen in growth media as described previously (6, 9). Results were confirmed in at least two independent primary culture preparations (range 2–6), with 3–4 replicates per condition as indicated. For arginine methylome analysis (17), primary VSM cultures from a larger set of SM22-Cre;LRP6(fl/fl);LDLR−/− mice and LRP6(fl/fl);LDLR−/− control cohorts (10–15 animals per genotype) were prepared, expanded for seven passages on collagen-coated plates, and then plated at 150,000 cells/cm2 into 20 150-mm diameter tissue culture plastic plates per genotype. Two days later, at confluence, the monolayers were rinsed twice with ice-cold PBS; proteins were extracted by scraping into 6 m urea, combined into 2 aliquots representing the SM22-Cre;LRP6(fl/fl);LDLR−/− mice and LRP6(fl/fl);LDLR−/− controls, frozen on dry ice, and shipped to Cell Signaling Technologies for fee-for-service MethylScan methylation proteomics analysis (17). This immunoaffinity LC-tandem MS technique (IA-LC/MS/MS) has been detailed previously (17). Briefly, following trypsin proteolysis of extracted cellular proteins, Arg-methylated peptides were affinity-captured by antibodies to MMA (Cell Signaling reagents 8015/8711); ADMA (Cell Signaling reagent 13522); or SDMA (Cell Signaling reagent 13222). For LC-MS/MS analysis, captured peptides were eluted, loaded directly onto a 10-cm × 75-μm PicoFrit capillary column packed with Magic C18 AQ reversed-phase resin. The column was developed with a 120-min linear gradient of acetonitrile in 0.125% formic acid delivered at 280 nl/min and characterized by MS/MS using an LTQ-Orbitrap-Velos (settings: MS run time of 126 min; MS1 scan range (300.0–1500.00); Top 20 MS/MS (minimum signal 500); isolation width 2.0; normalized collision energy 35.0; activation-Q 0.250; activation time 20.0; lock mass 371.101237; charge state rejection enabled; charge state 1+ rejected; dynamic exclusion enabled; repeat count 1; repeat duration 35.0; exclusion list size 500; exclusion duration 40.0; exclusion mass width relative to mass; exclusion mass width 10 ppm). MS/MS spectra were evaluated using SEQUEST (93). Searches were performed against the most recent update of the NCBI Mus musculus database with mass accuracy of ±50 ppm for precursor ions and 1 Da for product ions. Results were filtered with mass accuracy of ±5 ppm on precursor ions and presence of the intended motif. IA-LC/MS/MS analyses were performed in independent duplicates for both genotypes and each Arg methylation modification. Label-free quantitation from methylated peptide spectral counts was used to identify Arg methylation proteins that were changed at least 2.5-fold (increased by 250% or decreased by 60%) in SM22-Cre;LRP6(fl/fl);LDLR−/− VSM cultures versus LRP6(fl/fl);LDLR−/− controls. Data for the 21 proteins that exhibited reciprocal increases in MMA and decreases in SDMA are presented in supporting data S1. Other features of the VSM Arg methylome will be presented elsewhere when functionally validated. Accession numbers for protein sequences are as follows: murine G3BP1 NCBI accession no. NP_038744; human G3BP1 NCBI accession no. NP_938405; murine RIG-I NCBI accession no. NP_766277; and human RIG-I NCBI accession no. NP_055129.
Mineralization analysis of aortic primary cell cultures
Aortic primary VSM cells were plated onto type I collagen-coated 12-well plates (100,000 cells/well, 3 wells/genotype). Two days after seeding, cells were treated with β-glycerophosphate (3 mm) and ascorbic acid (50 μg/ml) and refed ever 2–3 days for 3 weeks. For experiments involving treatment with inhibitors, cells were treated either with vehicle (0.1%; DMSO) or C108 (15 μm), spautin (15 μm), or metformin (200 μm) in minimal essential growth medium every 2–3 days for 11 days. Cells were washed with 1× TBS (20 mm Tris-HCl, 1.5 m NaCl, pH 7.5) three times followed by fixation with 4% paraformaldehyde in 1× TBS for 4 min and washed again three times with 1× TBS. Mineralized matrix was detected by Alizarin red S staining, as described previously (94), and digitally captured for presentation and image analysis quantification (n = 3 to 4 per condition), as detailed previously (94).
Alkaline phosphatase enzyme activity
Assays for bone alkaline phosphatase enzyme activity were carried out as detailed previously (9). Briefly, aortic primary VSM were plated onto type I collagen-coated 12-well cluster dishes and treated with either vehicle (DMSO) or indicated reagents. Following the end of culture and treatment periods, cells were washed twice with 1× TBS and extracted with 10 mm Tris-HCl, pH 8.0, containing 0.5 mm MgCl2 and 0.1% Triton X-100 (0.5 ml/well). Extracts and cell residue were scraped into sonication tubes, processed by three cycles of 20-s sonication and 90-s settling at 4 °C with a Diagenode Bioruptor Pico Sonication System. After 20 s of 12,000 rpm centrifugation to remove debris, the alkaline phosphatase activity in the cleared sonicates were measured using p-nitrophenyl phosphate as substrate, as described previously (9). Protein concentration was measured using Pierce BCA protein assay kit (Thermo Fisher Scientific, product no. 23227). Alkaline phosphatase activity in each cell extract was expressed as nanomoles of p-nitrophenol produced per min/mg cellular protein.
RNA analysis
Total RNA of aortic primary cells were isolated using RNeasy kit (Qiagen), whereas total RNA of whole aorta was isolated using RNeasy Lipid Tissue Mini kit after homogenization with Rotorstator in QIAzol Lysis Reagent and chloroform extraction as described previously (6, 95). To quantify mRNA levels, real-time fluorescence RT-PCR was performed, as described previously (6, 9), using commercially available, inventoried TaqMan probes that provided optimal coverage (Applied Biosystems, Foster City, CA). Relative mRNA accumulation was normalized to 18S rRNA levels.
Plasmids, siRNA, and transfection
Our murine OPNLUC (osteopontin promoter–luciferase reporter; −1976 to +78) construct has been previously detailed (96). NFATLUC reporter was obtained from Stratagene/Agilent (Path Detect plasmid 219088-51). TK-Renilla (E2241), and CMV-Renilla (E2261) were obtained from Promega. Site-directed modifications of parental plasmids listed in supporting data S14 were generated by using In-Fusion HD cloning kit (catalog no. 639692) according to the protocol provided by Clontech (PT5162-1). The remaining plasmids were purchased either from Origene (TrueORF Gold) or from Addgene as indicated. For plasmid transfection, HEK293T cells (ATCC; CRL-3216) or MOVAS mouse aortic smooth muscle cells (ATCC; CRL-2797) (29, 30) were seeded the day before at 65,000 cells/well in 12-well culture plates, and transient transfections and reporter assays were carried out as detailed previously (97), but using Qiagen Superfect. All transfections were performed in triplicates and repeated at least once. For siRNA transfection, aortic primary cells were seeded at 100,000 cells/well in type I collagen-coated 12-well culture plates the day before transfection. Control or siRNA (listed in supporting data S14) at 60 nm was incubated with Lipofectamine RNAiMAX reagent (3 μl/well, Invitrogen, catalog no. 13778-150) in Opti-MEM for 20 min. During incubation, cells were washed once with DMEM, and fresh DMEM growth medium containing 10% FBS and penicillin/streptomycin was added (0.9 ml/well). At the end of incubation, siRNA mixture (103 μl/well) was added in triplicate to cells. Cells were harvested for RNA isolation 3 days later.
Western blotting analyses
Western blotting analyses were performed using either whole-cell extracts or extracts subjected to subcellular fractionation as indicated. For whole-cell extracts, cell layers were rinsed twice with 1× TBS and extracted with modified RIPA buffer containing 10 mm Tris-HCl, pH 8.0, 1 mm EDTA, 140 mm NaCl, 1% Triton X-100, and 0.5% Nonidet P-40 plus cocktails of protease inhibitors and phosphatase inhibitors (Sigma). For subcellular fractions, confluent cells in 10-cm culture dishes were processed using a subcellular protein fractionation kit for cultured cells (Pierce catalog no. 78840) according to the manufacturer's protocol. Extracts and fractions containing equal amounts of protein were subjected to SDS-PAGE, and Western blotting was performed as described previously, with immune complexes visualized by enhanced chemiluminescence using alkaline phosphatase-conjugated secondary antibodies and Bio-Rad ChemiDoc Touch Imaging System (linear signal response over 3–4 orders of magnitude). The integrated band signal minus background was quantified and compared as described previously (95, 98) using Fiji ImageJ for Mac OS X (99). Antibodies utilized for Western blot analysis are listed in supporting data S14.
Co-immunoprecipitation studies
To analyze the co-localization of G3BP1 and the RLRs, HEK293T cells in 6-well plates were transfected with the indicated C-terminally tagged or untagged proteins as indicated in each figure. Seventy two hours after transfection, cells were washed with cold PBS, incubated in 1× TBS containing protease and phosphatase inhibitor cocktails (Sigma) (9), and then harvested into microcentrifuge tubes, and cell pellets were obtained by centrifugation at 5000 rpm for 1 min. Cell pellets were then extracted with 500 μl of modified RIPA buffer/pellet as described above for 80 min at 4 °C. After centrifugation at 12,500 rpm for 10 min, supernatants were harvested and stored frozen at −80 °C until immunoprecipitation. For immunoprecipitation, cell extracts were thawed on ice. Protein G Dynabeads (Thermo Fisher Scientific) were resuspended by vortexing, and 50 μl of the slurry was transferred to a 1.7-ml microcentrifuge tube. The tubes were placed in a magnetic holder and liquid-aspirated. After washing once with PBS-T (PBS with 0.02% Tween 20, 100 μl/tube), 200 μl of rabbit anti-FLAG antibody (10 μg antibody) in PBS-T was added to each tube. Antibody was allowed to bind to protein G Dynabeads by rotating the tubes on a Nutator at room temperature for 20 min. At the end of incubation, the beads were washed once with PBS-T. All but 40 μl (reserved for input) of modified RIPA buffer cell extract was added to each tube. The solution was gently mixed via pipetting up and down a few times. Antigen–antibody interaction was allowed to proceed on a Nutator for 40 min at room temperature. After removal of the supernatant, the Dynabeads were washed three times with 200 μl of PBS-T/tube. Protein G Dynabead-bound antigen was then extracted with 20 μl of elution buffer (50 mm glycine, pH 2.8). Ten microliters of 5× Laemmli buffer containing β-mercaptoethanol (3%) and protein and phosphatase inhibitors and 20 μl of 100 mm DTT were also added. Twenty microliters of Elution buffer, 15 μl of 5× Laemmli buffer with β-mercaptoethanol and inhibitors, and 20 μl of 100 mm DTT were also added to each input. Both immunoprecipitates (IP) and inputs were placed in a 95 °C heating block for 10 min for denaturation and reduction. After cooling, IP and inputs were applied to 15 × 1.5-mm 4–12% acrylamide gradient Tris-glycine gels (Thermo Fisher Scientific-Novex; 15 μl/lane). Western blot analysis was performed using anti-FLAG, anti-Myc, anti-G3BP1, or anti-MMA/SDMA/ADMA antibodies as indicated (see supporting data S14 for immunoreagents).
Immunohistochemistry and immunofluorescence imaging
Immunofluorescence and immunohistochemistry were carried out essentially as we have described previously (9, 95, 100), but with the following specifications. Confocal immunolocalization of aortic G3BP1 was carried out on 5-μm fresh frozen tissue sections, and co-localization studies in cultured cells were performed using primary VSM cultured on collagen-coated 4-well chamber slides (Nunc Lab Tek; Thermo Fisher Scientific). Sections were washed with 1× TBS twice for 5 min each followed by fixation with 4% paraformaldehyde in 1× TBS for 8 min at room temperature. Sections were further washed with 1× TBS three times and incubated in TBST (TBS with 0.1% Tween 20) for 15 min at room temperature to permeabilize the cells. After washing three times with 1× TBS, sections were blocked for 1 h with 5% normal goat serum in 1× TBST. When goat primary antibodies were used, normal donkey serum was used as the blocking agent. Incubation with primary antibodies (e.g. rabbit anti-G3BP1 Proteintech, 13057-2-AP 1:250) was carried out at 4 °C overnight. All antibodies were made up in TBST with 5% blocking serum. After incubation, sections were washed with 1× TBS three times followed by incubation with the secondary antibody (Alexa Fluor conjugated, Life Technologies, Inc., 1:200; see supporting data S1) for 1 h. Sections were washed again with 1× TBS and mounted with ProLong Gold Antifade Reagent (Invitrogen P-36931) containing 4′,6-diamidino-2-phenylindole for nuclei staining. Mac2-positive atheroma area was performed precisely as described previously (9), except that an Alexa647-conjugated secondary antibody was implemented for immune complex visualization. Staining of aortic calcium deposition in frozen sections using fluor-conjugated amino-bisphosphonate was implemented using the techniques of New et al. (101), but implementing Alexa647-conjugated zoledronic acid (BioVinc AF647-ZOL) (55) (supporting data S14), an amino-bisphosphonate with a very high binding affinity for calcium phosphate mineralized matrix (102), in lieu of fluor-conjugated pamidronic acid (101). Fluorescent images were captured using a Leica true confocal scanning SP8 microscope outfitted with an acousto-optical beam splitter and HyD spectral detection. Leica LAS X co-localization and 3D visualization software were used to create images for export and publication. When performed, image overlay contrast editing was carried out using Fiji ImageJ for Mac OS X (99).
ChIP assays
ChIP was carried out following our modification of the previously reported fast ChIP method of Nelson et al. (103), as we have extensively detailed (9, 104). Input and precipitated DNA was quantified using SYBR Green, as described previously (9, 104), with an Applied Biosystems 7300 Real Time PCR instrument. One pg of mouse genomic DNA was assessed in parallel aliquots as a standard to normalize quantitation between each plate and experiment. ChIP antibodies and primer pairs are presented in supporting data S14.
Diet-induced diabetes and arteriosclerotic calcification in LDLR−/− and MAVS−/−;LDLR−/− mice
The induction and characterization of arteriosclerotic calcification and insulin-resistant hyperglycemia in the male LDLR−/− mouse was carried out as detailed previously (6) and reviewed (2). Briefly, beginning at 8–10 weeks of age, male MAVS−/−;LDLR−/− mice (n = 6; bred 10 generations on the C57BL6/J background as above) and male LDLR−/− controls (n = 18, also C57BL6/J) were fed with the Western diet TD88137 (42% of calories from fat, 34% sucrose by weight, 0.2% cholesterol; Envigo). After 12 weeks of dietary challenge, mice were fasted for 5 h and then euthanized by exsanguination (cardiac puncture under tribromoethanol general anesthesia). At necropsy, aortic tissue (ascending aorta, arch, and thoracic aorta) was harvested, dried, and extracted with formic acid overnight with heating (6). Following alkalinization, calcium was measured in the aortic extract by spectrophotometry using the cresolphthalein complexone assay (Point Scientific, C7503) (6). Data are presented as micrograms of calcium/mg of aortic weight. Plasma insulin, glucose, cholesterol, and triglycerides were measured, and the HOMA-IR index of insulin resistance was calculated precisely as described previously (9). Plasma haptoglobin was measured as we described previously (7) using a commercially available ELISA from Life Diagnostics, Inc. (catalog no. HAPT-1).
Statistical analyses
All statistical analyses were performed with 3–6 independent replicates per group as indicated. Statistical analyses were performed using GraphPad Prism 7.0a software for Mac OS X, implementing two-tailed standard parametric methods after Shapiro-Wilk normality testing, or with nonparametric methods when indicated. Graphic data are presented as mean ± S.E. When one-way analysis of variance was applied and significant differences were detected between two or more groups (p < 0.05), Holm-Sidak post hoc analysis was implemented to correct for multiple comparisons during the pairwise testing for significant differences between each group (105).
Author contributions
B. R., J. N. S., S.-L. C., A. S. B., A. G., L. L., M. M., J. K., A. L., and H. M. data curation; B. R., J. N. S., S.-L. C., A. S. B., A. G., L. L., M. M., J. K., A. L., H. M., Z. C., and D. A. T. formal analysis; B. R., J. N. S., S.-L. C., A. G., L. L., M. M., J. K., A. L., H. M., Z. C., and D. A. T. investigation; B. R., J. N. S., S.-L. C., A. S. B., A. G., L. L., M. M., J. K., A. L., H. M., Z. C., and D. A. T. methodology; B. R., J. N. S., S.-L. C., A. S. B., A. G., L. L., J. K., A. L., and D. A. T. writing-review and editing; A. S. B. and D. A. T. project administration; J. K., A. L., and H. M. resources; A. L., H. M., Z. C., and D. A. T. supervision; D. A. T. conceptualization; D. A. T. funding acquisition; D. A. T. writing-original draft.
Supplementary Material
Acknowledgments
We thank Dr. Bart O. Williams (Van Andel Research Institute) for generously providing the LRP6-flox mice, Dr. Joseph Ready (University of Texas Southwestern) for helpful discussions, and the University of Texas Southwestern Proteomics Core Facility.
This work was supported by National Institutes of Health Grants HL114806 (to D. A. T.) and HL069229 (to D. A. T.), the J. D. and Maggie E. Wilson Endowed Chair in Biomedical Research (to D. A. T.), the Louis V. Avioli Professorship in Mineral Metabolism Research (to D. A. T.), and the Howard Hughes Medical Institute (to Z. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supporting data S1–S14.
B. Ramachandran, A. S. Behrmann, A. Gay, J. Kozlitina, A. Lemoff, H. Mirzaei, and D. A. Towler, unpublished data.
- VSM
- vascular smooth muscle
- ADMA
- asymmetric dimethylarginine
- CMV
- cytomegalovirus promoter/enhancer vector
- CRISPR
- Clustered Regularly Interspaced Short Palindromic Repeats
- eQTL
- expressed quantitative trait locus
- IP
- immunoprecipitation
- Fzd
- frizzled
- HEK
- human embryonic kidney cell line
- LDLR
- low density lipoprotein receptor
- LRP
- LDL receptor-related protein
- LUC
- luciferase reporter
- MAVS
- mitochondrial antiviral signaling protein
- NFAT
- nuclear factor of activated T cell
- OPN
- osteopontin
- Osx
- osterix
- PRMT
- protein arginine N-methyltransferase
- Rankl
- receptor activator of nuclear factor κB ligand
- RLR
- RIG-I like receptor
- RNAi
- RNA interference
- SDMA
- symmetric dimethylation
- siRNA
- small interfering RNA
- TK
- HSV thymidine kinase promoter
- TNAP
- tissue nonspecific alkaline phosphatase; upstream stimulatory factor
- Wnt
- Wingless/int-1 family member
- gRNA
- guide RNA
- HOMA-IR
- homeostatic model assessment of insulin resistance
- DMEM
- Dulbecco's modified Eagle's medium
- qPCR
- quantitative PCR
- NS
- not significant.
References
- 1. Greenwald S. E. (2007) Ageing of the conduit arteries. J. Pathol. 211, 157–172 10.1002/path.2101 [DOI] [PubMed] [Google Scholar]
- 2. Stabley J. N., and Towler D. A. (2017) Arterial calcification in diabetes mellitus: preclinical models and translational implications. Arterioscler. Thromb. Vasc. Biol. 37, 205–217 10.1161/ATVBAHA.116.306258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Westerhof N., Lankhaar J. W., and Westerhof B. E. (2009) The arterial Windkessel. Med. Biol. Eng. Comput. 47, 131–141 10.1007/s11517-008-0359-2 [DOI] [PubMed] [Google Scholar]
- 4. Bostrom K., and Demer L. L. (2000) Regulatory mechanisms in vascular calcification. Crit. Rev. Eukaryot Gene Expr. 10, 151–158 [PubMed] [Google Scholar]
- 5. Boström K., Watson K. E., Horn S., Wortham C., Herman I. M., and Demer L. L. (1993) Bone morphogenetic protein expression in human atherosclerotic lesions. J. Clin. Invest. 91, 1800–1809 10.1172/JCI116391 10.1172/JCI116391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cheng S. L., Behrmann A., Shao J. S., Ramachandran B., Krchma K., Bello Arredondo Y., Kovacs A., Mead M., Maxson R., and Towler D. A. (2014) Targeted reduction of vascular Msx1 and Msx2 mitigates arteriosclerotic calcification and aortic stiffness in LDLR-deficient mice fed diabetogenic diets. Diabetes 63, 4326–4337 10.2337/db14-0326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Al-Aly Z., Shao J. S., Lai C. F., Huang E., Cai J., Behrmann A., Cheng S. L., and Towler D. A. (2007) Aortic Msx2-Wnt calcification cascade is regulated by TNF-α–dependent signals in diabetic Ldlr−/− mice. Arterioscler. Thromb. Vasc. Biol. 27, 2589–2596 10.1161/ATVBAHA.107.153668 [DOI] [PubMed] [Google Scholar]
- 8. Shao J. S., Cheng S. L., Pingsterhaus J. M., Charlton-Kachigian N., Loewy A. P., and Towler D. A. (2005) Msx2 promotes cardiovascular calcification by activating paracrine Wnt signals. J. Clin. Invest. 115, 1210–1220 10.1172/JCI24140 10.1172/JCI24140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cheng S. L., Ramachandran B., Behrmann A., Shao J. S., Mead M., Smith C., Krchma K., Bello Arredondo Y., Kovacs A., Kapoor K., Brill L. M., Perera R., Williams B. O., and Towler D. A. (2015) Vascular smooth muscle LRP6 limits arteriosclerotic calcification in diabetic LDLR−/− mice by restraining noncanonical Wnt signals. Circ. Res. 117, 142–156 10.1161/CIRCRESAHA.117.306712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Williams H., Mill C. A., Monk B. A., Hulin-Curtis S., Johnson J. L., and George S. J. (2016) Wnt2 and WISP-1/CCN4 induce intimal thickening via Promotion of smooth muscle cell migration. Arterioscler. Thromb. Vasc. Biol. 36, 1417–1424 10.1161/ATVBAHA.116.307626 [DOI] [PubMed] [Google Scholar]
- 11. Tsaousi A., Williams H., Lyon C. A., Taylor V., Swain A., Johnson J. L., and George S. J. (2011) Wnt4/β-catenin signaling induces VSMC proliferation and is associated with intimal thickening. Circ. Res. 108, 427–436 10.1161/CIRCRESAHA.110.233999 [DOI] [PubMed] [Google Scholar]
- 12. Woldt E., Terrand J., Mlih M., Matz R. L., Bruban V., Coudane F., Foppolo S., El Asmar Z., Chollet M. E., Ninio E., Bednarczyk A., Thiersé D., Schaeffer C., Van Dorsselaer A., Boudier C., Wahli W., Chambon P., Metzger D., Herz J., and Boucher P. (2012) The nuclear hormone receptor PPARgamma counteracts vascular calcification by inhibiting Wnt5a signalling in vascular smooth muscle cells. Nat. Commun 3, 1077 10.1038/ncomms2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mani A., Radhakrishnan J., Wang H., Mani A., Mani M. A., Nelson-Williams C., Carew K. S., Mane S., Najmabadi H., Wu D., and Lifton R. P. (2007) LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science 315, 1278–1282 10.1126/science.1136370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Srivastava R., Zhang J., Go G. W., Narayanan A., Nottoli T. P., and Mani A. (2015) Impaired LRP6-TCF7L2 Activity Enhances Smooth Muscle Cell Plasticity and Causes Coronary Artery Disease. Cell Rep. 13, 746–759 10.1016/j.celrep.2015.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Siddique A., Yu B., Khan K., Buyting R., Al-Kindi H., Alaws H., Rhéaume E., Tardif J. C., Cecere R., and Schwertani A. (2018) Expression of the frizzled receptors and their co-receptors in calcified human aortic valves. Can. J. Physiol Pharmacol. 96, 208–214 [DOI] [PubMed] [Google Scholar]
- 16. Albanese I., Yu B., Al-Kindi H., Barratt B., Ott L., Al-Refai M., de Varennes B., Shum-Tim D., Cerruti M., Gourgas O., Rhéaume E., Tardif J. C., and Schwertani A. (2017) Role of noncanonical Wnt signaling pathway in human aortic valve calcification. Arterioscler. Thromb. Vasc. Biol. 37, 543–552 10.1161/ATVBAHA.116.308394 [DOI] [PubMed] [Google Scholar]
- 17. Guo A., Gu H., Zhou J., Mulhern D., Wang Y., Lee K. A., Yang V., Aguiar M., Kornhauser J., Jia X., Ren J., Beausoleil S. A., Silva J. C., Vemulapalli V., Bedford M. T., and Comb M. J. (2014) Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell. Proteomics 13, 372–387 10.1074/mcp.O113.027870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Foroughi Asl H., Talukdar H. A., Kindt A. S., Jain R. K., Ermel R., Ruusalepp A., Nguyen K. D., Dobrin R., Reilly D. F., Schunkert H., Samani N. J., Braenne I., Erdmann J., Melander O., Qi J., et al. (2015) Expression quantitative trait Loci acting across multiple tissues are enriched in inherited risk for coronary artery disease. Circ. Cardiovasc. Genet. 8, 305–315 10.1161/CIRCGENETICS.114.000640 [DOI] [PubMed] [Google Scholar]
- 19. Seth R. B., Sun L., Ea C. K., and Chen Z. J. (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 122, 669–682 10.1016/j.cell.2005.08.012 [DOI] [PubMed] [Google Scholar]
- 20. Dhar S., Vemulapalli V., Patananan A. N., Huang G. L., Di Lorenzo A., Richard S., Comb M. J., Guo A., Clarke S. G., and Bedford M. T. (2013) Loss of the major type I arginine methyltransferase PRMT1 causes substrate scavenging by other PRMTs. Sci. Rep. 3, 1311 10.1038/srep01311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shao J. S., Cheng S. L., Sadhu J., and Towler D. A. (2010) Inflammation and the osteogenic regulation of vascular calcification: a review and perspective. Hypertension 55, 579–592 10.1161/HYPERTENSIONAHA.109.134205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cassola A., Noé G., and Frasch A. C. (2010) RNA recognition motifs involved in nuclear import of RNA-binding proteins. RNA Biol. 7, 339–344 10.4161/rna.7.3.12087 [DOI] [PubMed] [Google Scholar]
- 23. Bikkavilli R. K., and Malbon C. C. (2011) Arginine methylation of G3BP1 in response to Wnt3a regulates β-catenin mRNA. J. Cell Sci. 124, 2310–2320 10.1242/jcs.084046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bikkavilli R. K., and Malbon C. C. (2012) Wnt3a-stimulated LRP6 phosphorylation is dependent upon arginine methylation of G3BP2. J. Cell Sci. 125, 2446–2456 10.1242/jcs.100933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Milewicz D. M., Trybus K. M., Guo D. C., Sweeney H. L., Regalado E., Kamm K., and Stull J. T. (2017) Altered smooth muscle cell force generation as a driver of thoracic aortic aneurysms and dissections. Arterioscler. Thromb. Vasc. Biol. 37, 26–34 10.1161/ATVBAHA.116.303229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Narisawa S., Yadav M. C., and Millán J. L. (2013) In vivo overexpression of tissue-nonspecific alkaline phosphatase increases skeletal mineralization and affects the phosphorylation status of osteopontin. J. Bone Miner. Res. 28, 1587–1598 10.1002/jbmr.1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harmey D., Hessle L., Narisawa S., Johnson K. A., Terkeltaub R., and Millán J. L. (2004) Concerted regulation of inorganic pyrophosphate and osteopontin by akp2, enpp1, and ank: an integrated model of the pathogenesis of mineralization disorders. Am. J. Pathol. 164, 1199–1209 10.1016/S0002-9440(10)63208-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fakhry M., Roszkowska M., Briolay A., Bougault C., Guignandon A., Diaz-Hernandez J. I., Diaz-Hernandez M., Pikula S., Buchet R., Hamade E., Badran B., Bessueille L., and Magne D. (2017) TNAP stimulates vascular smooth muscle cell trans-differentiation into chondrocytes through calcium deposition and BMP-2 activation: Possible implication in atherosclerotic plaque stability. Biochim. Biophys. Acta 1863, 643–653 10.1016/j.bbadis.2016.12.003 [DOI] [PubMed] [Google Scholar]
- 29. Kelynack K. J., and Holt S. G. (2016) An in vitro murine model of vascular smooth muscle cell mineralization. Methods Mol. Biol. 1397, 209–220 10.1007/978-1-4939-3353-2_14 [DOI] [PubMed] [Google Scholar]
- 30. Mackenzie N. C., Zhu D., Longley L., Patterson C. S., Kommareddy S., and MacRae V. E. (2011) MOVAS-1 cell line: a new in vitro model of vascular calcification. Int. J. Mol. Med. 27, 663–668 [DOI] [PubMed] [Google Scholar]
- 31. Vognsen T., Møller I. R., and Kristensen O. (2013) Crystal structures of the human G3BP1 NTF2-like domain visualize FxFG Nup repeat specificity. PLoS ONE 8, e80947 10.1371/journal.pone.0080947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Satokata I., Ma L., Ohshima H., Bei M., Woo I., Nishizawa K., Maeda T., Takano Y., Uchiyama M., Heaney S., Peters H., Tang Z., Maxson R., and Maas R. (2000) Msx2 deficiency in mice causes pleiotropic defects in bone growth and ectodermal organ formation. Nat. Genet. 24, 391–395 10.1038/74231 [DOI] [PubMed] [Google Scholar]
- 33. Wu X., Li Y., Wang F., Hu L., Li Y., Wang J., Zhang C., and Wang S. (2017) Spatiotemporal expression of Wnt/β-catenin signaling during morphogenesis and odontogenesis of deciduous molar in miniature pig. Int. J. Biol. Sci. 13, 1082–1091 10.7150/ijbs.20905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Saito Y., Nakamura K., Miura D., Yunoki K., Miyoshi T., Yoshida M., Kawakita N., Kimura T., Kondo M., Sarashina T., Akagi S., Watanabe A., Nishii N., Morita H., and Ito H. (2017) Suppression of Wnt signaling and osteogenic changes in vascular smooth muscle cells by eicosapentaenoic acid. Nutrients 9, E858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mali P., Yang L., Esvelt K. M., Aach J., Guell M., DiCarlo J. E., Norville J. E., and Church G. M. (2013) RNA-guided human genome engineering via Cas9. Science 339, 823–826 10.1126/science.1232033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., and Zhang F. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hein M. Y., Hubner N. C., Poser I., Cox J., Nagaraj N., Toyoda Y., Gak I. A., Weisswange I., Mansfeld J., Buchholz F., Hyman A. A., and Mann M. (2015) A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163, 712–723 10.1016/j.cell.2015.09.053 [DOI] [PubMed] [Google Scholar]
- 38. Rutsch F., MacDougall M., Lu C., Buers I., Mamaeva O., Nitschke Y., Rice G. I., Erlandsen H., Kehl H. G., Thiele H., Nürnberg P., Höhne W., Crow Y. J., Feigenbaum A., and Hennekam R. C. (2015) A specific IFIH1 gain-of-function mutation causes Singleton-Merten syndrome. Am. J. Hum. Genet. 96, 275–282 10.1016/j.ajhg.2014.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jang M. A., Kim E. K., Now H., Nguyen N. T., Kim W. J., Yoo J. Y., Lee J., Jeong Y. M., Kim C. H., Kim O. H., Sohn S., Nam S. H., Hong Y., Lee Y. S., Chang S. A., et al. (2015) Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome. Am. J. Hum. Genet. 96, 266–274 10.1016/j.ajhg.2014.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Duan P., and Bonewald L. F. (2016) The role of the wnt/β-catenin signaling pathway in formation and maintenance of bone and teeth. Int. J. Biochem. Cell Biol. 77, 23–29 10.1016/j.biocel.2016.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gay A., and Towler D. A. (2017) Wnt signaling in cardiovascular disease: opportunities and challenges. Curr. Opin. Lipidol. 28, 387–396 10.1097/MOL.0000000000000445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shen Q., and Christakos S. (2005) The vitamin D receptor, Runx2, and the Notch signaling pathway cooperate in the transcriptional regulation of osteopontin. J. Biol. Chem. 280, 40589–40598 10.1074/jbc.M504166200 [DOI] [PubMed] [Google Scholar]
- 43. Sun Y., Byon C. H., Yuan K., Chen J., Mao X., Heath J. M., Javed A., Zhang K., Anderson P. G., and Chen Y. (2012) Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ. Res. 111, 543–552 10.1161/CIRCRESAHA.112.267237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Raaz U., Schellinger I. N., Chernogubova E., Warnecke C., Kayama Y., Penov K., Hennigs J. K., Salomons F., Eken S., Emrich F. C., Zheng W. H., Adam M., Jagger A., Nakagami F., Toh R., et al. (2015) Transcription factor Runx2 promotes aortic fibrosis and stiffness in type 2 diabetes mellitus. Circ. Res. 117, 513–524 10.1161/CIRCRESAHA.115.306341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lin M. E., Chen T. M., Wallingford M. C., Nguyen N. B., Yamada S., Sawangmake C., Zhang J., Speer M. Y., and Giachelli C. M. (2016) Runx2 deletion in smooth muscle cells inhibits vascular osteochondrogenesis and calcification but not atherosclerotic lesion formation. Cardiovasc. Res. 2016, cvw205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gupta N., Badeaux M., Liu Y., Naxerova K., Sgroi D., Munn L. L., Jain R. K., and Garkavtsev I. (2017) Stress granule-associated protein G3BP2 regulates breast tumor initiation. Proc. Natl. Acad. Sci. U.S.A. 114, 1033–1038 10.1073/pnas.1525387114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sowa M. E., Bennett E. J., Gygi S. P., and Harper J. W. (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403 10.1016/j.cell.2009.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu F., and Gu J. (2011) Retinoic acid inducible gene-I, more than a virus sensor. Protein Cell 2, 351–357 10.1007/s13238-011-1045-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sun Q., Sun L., Liu H. H., Chen X., Seth R. B., Forman J., and Chen Z. J. (2006) The specific and essential role of MAVS in antiviral innate immune responses. Immunity 24, 633–642 10.1016/j.immuni.2006.04.004 [DOI] [PubMed] [Google Scholar]
- 50. Cohen M. M., Jr. (2006) The new bone biology: pathologic, molecular, and clinical correlates. Am. J. Med. Genet. A. 140, 2646–2706 [DOI] [PubMed] [Google Scholar]
- 51. Morony S., Sage A. P., Corbin T., Lu J., Tintut Y., and Demer L. L. (2012) Enhanced mineralization potential of vascular cells from SM22α-Rankl (tg) mice. Calcif. Tissue Int. 91, 379–386 10.1007/s00223-012-9655-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Byon C. H., Sun Y., Chen J., Yuan K., Mao X., Heath J. M., Anderson P. G., Tintut Y., Demer L. L., Wang D., and Chen Y. (2011) Runx2-up-regulated receptor activator of nuclear factor κB ligand in calcifying smooth muscle cells promotes migration and osteoclastic differentiation of macrophages. Arterioscler. Thromb. Vasc. Biol. 31, 1387–1396 10.1161/ATVBAHA.110.222547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tintut Y., and Demer L. (2006) Role of osteoprotegerin and its ligands and competing receptors in atherosclerotic calcification. J. Investig. Med. 54, 395–401 10.2310/6650.2006.06019 [DOI] [PubMed] [Google Scholar]
- 54. Morony S., Tintut Y., Zhang Z., Cattley R. C., Van G., Dwyer D., Stolina M., Kostenuik P. J., and Demer L. L. (2008) Osteoprotegerin inhibits vascular calcification without affecting atherosclerosis in ldlr(−/−) mice. Circulation 117, 411–420 10.1161/CIRCULATIONAHA.107.707380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sun S., Blażewska K. M., Kadina A. P., Kashemirov B. A., Duan X., Triffitt J. T., Dunford J. E., Russell R. G., Ebetino F. H., Roelofs A. J., Coxon F. P., Lundy M. W., and McKenna C. E. (2016) Fluorescent bisphosphonate and carboxyphosphonate probes: a versatile imaging toolkit for applications in bone biology and biomedicine. Bioconjug. Chem. 27, 329–340 10.1021/acs.bioconjchem.5b00369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Berglund E. D., Li C. Y., Poffenberger G., Ayala J. E., Fueger P. T., Willis S. E., Jewell M. M., Powers A. C., and Wasserman D. H. (2008) Glucose metabolism in vivo in four commonly used inbred mouse strains. Diabetes 57, 1790–1799 10.2337/db07-1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Can U., Buyukinan M., Guzelant A., Ugur A., Karaibrahimoglu A., and Yabancıun S. (2016) Investigation of the inflammatory biomarkers of metabolic syndrome in adolescents. J. Pediatr. Endocrinol. Metab. 29, 1277–1283 [DOI] [PubMed] [Google Scholar]
- 58. Muhammad I. F., Borné Y., Östling G., Kennbäck C., Gottsäter M., Persson M., Nilsson P. M., and Engström G. (2017) Acute phase proteins as prospective risk markers for arterial stiffness: the Malmo diet and cancer cohort. PLoS ONE 12, e0181718 10.1371/journal.pone.0181718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Benjamin E. J., Blaha M. J., Chiuve S. E., Cushman M., Das S. R., Deo R., de Ferranti S. D., Floyd J., Fornage M., Gillespie C., Isasi C. R., Jiménez M. C., Jordan L. C., Judd S. E., Lackland D., Lichtman J. H., et al. (2017) Heart Disease and Stroke Statistics–2017 Update: a report from the American Heart Association. Circulation 135, e146 10.1161/CIR.0000000000000485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ritchey M. D., Loustalot F., Wall H. K., Steiner C. A., Gillespie C., George M. G., and Wright J. S. (2017) Million hearts: description of the national surveillance and modeling methodology used to monitor the number of cardiovascular events prevented during 2012–2016. J. Am. Heart Assoc 6, e006021 10.1161/JAHA.117.006021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Karmali K. N., Lloyd-Jones D. M., Berendsen M. A., Goff D. C. Jr, Sanghavi D. M., Brown N. C., Korenovska L., and Huffman M. D. (2016) Drugs for primary prevention of atherosclerotic cardiovascular disease: an overview of systematic reviews. JAMA Cardiol. 1, 341–349 10.1001/jamacardio.2016.0218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sidney S., Quesenberry C. P. Jr, Jaffe M. G., Sorel M., Nguyen-Huynh M. N., Kushi L. H., Go A. S., and Rana J. S. (2016) Recent trends in cardiovascular mortality in the United States and Public Health Goals. JAMA Cardiol. 1, 594–599 10.1001/jamacardio.2016.1326 [DOI] [PubMed] [Google Scholar]
- 63. Pazman C., Mayes C. A., Fanto M., Haynes S. R., and Mlodzik M. (2000) Rasputin, the Drosophila homologue of the RasGAP SH3 binding protein, functions in ras- and Rho-mediated signaling. Development 127, 1715–1725 [DOI] [PubMed] [Google Scholar]
- 64. Irvine K., Stirling R., Hume D., and Kennedy D. (2004) Rasputin, more promiscuous than ever: a review of G3BP. Int. J. Dev. Biol. 48, 1065–1077 10.1387/ijdb.041893ki [DOI] [PubMed] [Google Scholar]
- 65. Valiente-Echeverría F., Melnychuk L., Vyboh K., Ajamian L., Gallouzi I. E., Bernard N., and Mouland A. J. (2014) eEF2 and Ras-GAP SH3 domain-binding protein (G3BP1) modulate stress granule assembly during HIV-1 infection. Nat. Commun. 5, 4819 10.1038/ncomms5819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kedersha N., Panas M. D., Achorn C. A., Lyons S., Tisdale S., Hickman T., Thomas M., Lieberman J., McInerney G. M., Ivanov P., and Anderson P. (2016) G3BP–Caprin1–USP10 complexes mediate stress granule condensation and associate with 40S subunits. J. Cell Biol. 212, 845–860 10.1083/jcb.201508028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tsai W. C., Gayatri S., Reineke L. C., Sbardella G., Bedford M. T., and Lloyd R. E. (2016) Arginine demethylation of G3BP1 promotes stress granule assembly. J. Biol. Chem. 291, 22671–22685 10.1074/jbc.M116.739573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tourrière H., Gallouzi I. E., Chebli K., Capony J. P., Mouaikel J., van der Geer P., and Tazi J. (2001) RasGAP-associated endoribonuclease G3Bp: selective RNA degradation and phosphorylation-dependent localization. Mol. Cell. Biol. 21, 7747–7760 10.1128/MCB.21.22.7747-7760.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. He M., Yang Z., Abdellatif M., and Sayed D. (2015) GTPase activating protein (Sh3 domain) binding protein 1 regulates the processing of MicroRNA-1 during cardiac hypertrophy. PLoS ONE 10, e0145112 10.1371/journal.pone.0145112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Martin S., Bellora N., González-Vallinas J., Irimia M., Chebli K., de Toledo M., Raabe M., Eyras E., Urlaub H., Blencowe B. J., and Tazi J. (2016) Preferential binding of a stable G3BP ribonucleoprotein complex to intron-retaining transcripts in mouse brain and modulation of their expression in the cerebellum. J. Neurochem. 139, 349–368 10.1111/jnc.13768 [DOI] [PubMed] [Google Scholar]
- 71. Zhang H., Ma Y., Zhang S., Liu H., He H., Li N., Gong Y., Zhao S., Jiang J. D., and Shao R. G. (2015) Involvement of Ras GTPase-activating protein SH3 domain-binding protein 1 in the epithelial-to-mesenchymal transition-induced metastasis of breast cancer cells via the Smad signaling pathway. Oncotarget 6, 17039–17053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhang H., Zhang S. H., He H. W., Zhang C. X., Yu D. K., and Shao R. G. (2013) Downregulation of G3BPs inhibits the growth, migration and invasion of human lung carcinoma H1299 cells by suppressing the Src/FAK-associated signaling pathway. Cancer Gene Ther. 20, 622–629 10.1038/cgt.2013.62 [DOI] [PubMed] [Google Scholar]
- 73. Sahoo P. K., Murawala P., Sawale P. T., Sahoo M. R., Tripathi M. M., Gaikwad S. R., Seshadri V., and Joseph J. (2012) Wnt signalling antagonizes stress granule assembly through a Dishevelled-dependent mechanism. Biol. Open 1, 109–119 10.1242/bio.2011023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ibarra A., and Hetzer M. W. (2015) Nuclear pore proteins and the control of genome functions. Genes Dev. 29, 337–349 10.1101/gad.256495.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cai X., Xu H., and Chen Z. J. (2017) Prion-like polymerization in immunity and inflammation. Cold Spring Harb. Perspect. Biol. 9, a023580 10.1101/cshperspect.a023580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Demer L. L., and Tintut Y. (2014) Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler. Thromb. Vasc. Biol. 34, 715–723 10.1161/ATVBAHA.113.302070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ranoa D. R., Parekh A. D., Pitroda S. P., Huang X., Darga T., Wong A. C., Huang L., Andrade J., Staley J. P., Satoh T., Akira S., Weichselbaum R. R., and Khodarev N. N. (2016) Cancer therapies activate RIG-I-like receptor pathway through endogenous non-coding RNAs. Oncotarget 7, 26496–26515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Whelton S. P., Silverman M. G., McEvoy J. W., Budoff M. J., Blankstein R., Eng J., Blumenthal R. S., Szklo M., Nasir K., and Blaha M. J. (2015) Predictors of long-term healthy arterial aging: coronary artery calcium nondevelopment in the MESA study. JACC Cardiovasc. Imaging 8, 1393–1400 10.1016/j.jcmg.2015.06.019 [DOI] [PubMed] [Google Scholar]
- 79. Rademaker J., Schöder H., Ariaratnam N. S., Strauss H. W., Yahalom J., Steingart R., and Oeffinger K. C. (2008) Coronary artery disease after radiation therapy for Hodgkin's lymphoma: coronary CT angiography findings and calcium scores in nine asymptomatic patients. AJR Am. J. Roentgenol 191, 32–37 10.2214/AJR.07.3112 [DOI] [PubMed] [Google Scholar]
- 80. Horner S. M., Wilkins C., Badil S., Iskarpatyoti J., and Gale M. Jr. (2015) Proteomic analysis of mitochondrial-associated ER membranes (MAM) during RNA virus infection reveals dynamic changes in protein and organelle trafficking. PLoS ONE 10, e0117963 10.1371/journal.pone.0117963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Janssens S., Pulendran B., and Lambrecht B. N. (2014) Emerging functions of the unfolded protein response in immunity. Nat. Immunol. 15, 910–919 10.1038/ni.2991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Masuda M., Miyazaki-Anzai S., Levi M., Ting T. C., and Miyazaki M. (2013) PERK-eIF2α-ATF4-CHOP signaling contributes to TNFα-induced vascular calcification. J. Am. Heart Assoc. 2, e000238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tsai W. C., Reineke L. C., Jain A., Jung S. Y., and Lloyd R. E. (2017) Histone arginine demethylase JMJD6 is linked to stress granule assembly through demethylation of the stress granule nucleating protein G3BP1. J. Biol. Chem. 292, 18886–18896 10.1074/jbc.M117.800706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ptak C., and Wozniak R. W. (2017) SUMO and nucleocytoplasmic transport. Adv. Exp. Med. Biol. 963, 111–126 10.1007/978-3-319-50044-7_7 [DOI] [PubMed] [Google Scholar]
- 85. Soncini C., Berdo I., and Draetta G. (2001) Ras-GAP SH3 domain binding protein (G3BP) is a modulator of USP10, a novel human ubiquitin specific protease. Oncogene 20, 3869–3879 10.1038/sj.onc.1204553 [DOI] [PubMed] [Google Scholar]
- 86. Cai Z., Ding Y., Zhang M., Lu Q., Wu S., Zhu H., Song P., and Zou M. H. (2016) Ablation of adenosine monophosphate-activated protein kinase α1 in vascular smooth muscle cells promotes diet-induced atherosclerotic calcification in vivo. Circ. Res. 119, 422–433 10.1161/CIRCRESAHA.116.308301 [DOI] [PubMed] [Google Scholar]
- 87. Heath J. M., Sun Y., Yuan K., Bradley W. E., Litovsky S., Dell'Italia L. J., Chatham J. C., Wu H., and Chen Y. (2014) Activation of AKT by O-linked N-acetylglucosamine induces vascular calcification in diabetes mellitus. Circ. Res. 114, 1094–1102 10.1161/CIRCRESAHA.114.302968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kong L., Sun L., Zhang H., Liu Q., Liu Y., Qin L., Shi G., Hu J. H., Xu A., Sun Y. P., Li D., Shi Y. F., Zang J. W., Zhu J., Chen Z., Wang Z. G., and Ge B. X. (2009) An essential role for RIG-I in toll-like receptor-stimulated phagocytosis. Cell Host Microbe 6, 150–161 10.1016/j.chom.2009.06.008 [DOI] [PubMed] [Google Scholar]
- 89. New S. E., and Aikawa E. (2013) Role of extracellular vesicles in de novo mineralization: an additional novel mechanism of cardiovascular calcification. Arterioscler. Thromb. Vasc. Biol. 33, 1753–1758 10.1161/ATVBAHA.112.300128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Pockwinse S. M., Kota K. P., Quaresma A. J., Imbalzano A. N., Lian J. B., van Wijnen A. J., Stein J. L., Stein G. S., and Nickerson J. A. (2011) Live cell imaging of the cancer-related transcription factor RUNX2 during mitotic progression. J. Cell. Physiol. 226, 1383–1389 10.1002/jcp.22465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Greenblatt M. B., Shim J. H., and Glimcher L. H. (2013) Mitogen-activated protein kinase pathways in osteoblasts. Annu. Rev. Cell Dev. Biol. 29, 63–79 10.1146/annurev-cellbio-101512-122347 [DOI] [PubMed] [Google Scholar]
- 92. Zhong Z., Baker J. J., Zylstra-Diegel C. R., and Williams B. O. (2012) Lrp5 and Lrp6 play compensatory roles in mouse intestinal development. J. Cell. Biochem. 113, 31–38 10.1002/jcb.23324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lundgren D. H., Martinez H., Wright M. E., and Han D. K. (2009) Protein identification using Sorcerer 2 and SEQUEST. Curr. Protoc. Bioinformatics Chapter 13, Unit 13.3 10.1002/0471250953.bi1303s28 [DOI] [PubMed] [Google Scholar]
- 94. Lai C. F., Shao J. S., Behrmann A., Krchma K., Cheng S. L., and Towler D. A. (2012) TNFR1-activated reactive oxidative species signals up-regulate osteogenic Msx2 programs in aortic myofibroblasts. Endocrinology 153, 3897–3910 10.1210/en.2012-1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Shao J. S., Sierra O. L., Cohen R., Mecham R. P., Kovacs A., Wang J., Distelhorst K., Behrmann A., Halstead L. R., and Towler D. A. (2011) Vascular calcification and aortic fibrosis: a bifunctional role for osteopontin in diabetic arteriosclerosis. Arterioscler. Thromb. Vasc. Biol. 31, 1821–1833 10.1161/ATVBAHA.111.230011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bidder M., Shao J. S., Charlton-Kachigian N., Loewy A. P., Semenkovich C. F., and Towler D. A. (2002) Osteopontin transcription in aortic vascular smooth muscle cells is controlled by glucose-regulated upstream stimulatory factor and activator protein-1 activities. J. Biol. Chem. 277, 44485–44496 10.1074/jbc.M206235200 [DOI] [PubMed] [Google Scholar]
- 97. Sierra O. L., and Towler D. A. (2010) Runx2 trans-activation mediated by the MSX2-interacting nuclear target requires homeodomain interacting protein kinase-3. Mol. Endocrinol. 24, 1478–1497 10.1210/me.2010-0029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Cheng S. L., Shao J. S., Halstead L. R., Distelhorst K., Sierra O., and Towler D. A. (2010) Activation of vascular smooth muscle parathyroid hormone receptor inhibits Wnt/β-catenin signaling and aortic fibrosis in diabetic arteriosclerosis. Circ. Res. 107, 271–282 10.1161/CIRCRESAHA.110.219899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., and Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sierra O. L., Cheng S. L., Loewy A. P., Charlton-Kachigian N., and Towler D. A. (2004) MINT, the Msx2 interacting nuclear matrix target, enhances Runx2-dependent activation of the osteocalcin fibroblast growth factor response element. J. Biol. Chem. 279, 32913–32923 10.1074/jbc.M314098200 [DOI] [PubMed] [Google Scholar]
- 101. New S. E., Goettsch C., Aikawa M., Marchini J. F., Shibasaki M., Yabusaki K., Libby P., Shanahan C. M., Croce K., and Aikawa E. (2013) Macrophage-derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ. Res. 113, 72–77 10.1161/CIRCRESAHA.113.301036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lawson M. A., Xia Z., Barnett B. L., Triffitt J. T., Phipps R. J., Dunford J. E., Locklin R. M., Ebetino F. H., and Russell R. G. (2010) Differences between bisphosphonates in binding affinities for hydroxyapatite. J. Biomed. Mater. Res. B. Appl. Biomater. 92, 149–155 [DOI] [PubMed] [Google Scholar]
- 103. Nelson J. D., Denisenko O., and Bomsztyk K. (2006) Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat. Protoc. 1, 179–185 10.1038/nprot.2006.27 [DOI] [PubMed] [Google Scholar]
- 104. Shafer S. L., and Towler D. A. (2009) Transcriptional regulation of SM22α by Wnt3a: convergence with TGFβ(1)/Smad signaling at a novel regulatory element. J. Mol. Cell. Cardiol. 46, 621–635 10.1016/j.yjmcc.2009.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Glantz S. A. (2005) in Primer of Biostatistics (Diedrich C., and Pancotti R., eds) 6th Ed., pp. 64–67, McGraw-Hill Medical Publishers, New York [Google Scholar]
- 106. Komura N., Maeda N., Mori T., Kihara S., Nakatsuji H., Hirata A., Tochino Y., Funahashi T., and Shimomura I. (2013) Adiponectin protein exists in aortic endothelial cells. PLoS ONE 8, e71271 10.1371/journal.pone.0071271 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.