

Abstract
PH domain leucine-rich repeat protein phosphatase (PHLPP) is a serine/threonine phosphatase that has been shown to regulate cell growth and survival through dephosphorylation of several members of the AGC family of kinases. G-protein–coupled receptor kinase 5 (GRK5) is an AGC kinase that regulates phenylephrine (PE)-induced cardiac hypertrophy through its noncanonical function of directly targeting proteins to the nucleus to regulate transcription. Here we investigated the possibility that the PHLPP2 isoform can regulate GRK5-induced cardiomyocyte hypertrophy in neonatal rat ventricular myocytes (NRVMs). We show that removal of PHLPP2 by siRNA induces hypertrophic growth of NRVMs as measured by cell size changes at baseline, potentiated PE-induced cell size changes, and re-expression of fetal genes atrial natriuretic factor and brain natriuretic peptide. Endogenous GRK5 and PHLPP2 were found to interact in NRVMs, and PE-induced nuclear accumulation of GRK5 was enhanced upon down-regulation of PHLPP2. Conversely, overexpression of PHLPP2 blocked PE-induced hypertrophic growth, re-expression of fetal genes, and nuclear accumulation of GRK5, which depended on its phosphatase activity. Finally, using siRNA against GRK5, we found that GRK5 was necessary for the hypertrophic response induced by PHLPP2 knockdown. Our findings demonstrate for the first time a novel regulation of GRK5 by the phosphatase PHLPP2, which modulates hypertrophic growth. Understanding the signaling pathways affected by PHLPP2 has potential for new therapeutic targets in the treatment of cardiac hypertrophy and failure.
Keywords: cardiac hypertrophy, cardiomyocyte, cell growth, cell signaling, phosphatase, GRK5, PHLPP2
Introduction
The balance between protein phosphorylation and dephosphorylation represents an important regulatory step in maintaining cellular homeostasis. The precise control between protein phosphatases and kinases is crucial for cellular decisions that lead to cell growth, metabolism, proliferation, and hypertrophy (1–3). Activation of G-protein–coupled receptors (GPCRs)2 by hypertrophic agonists such as PE engages a number of intracellular signaling pathways that are important transducers of the hypertrophic response. These pathways include calcineurin-nuclear factor of activated T cells (NFAT) (4), Ca2+/calmodulin-dependent kinase II (5, 6), mitogen-activated protein kinases (7, 8), PKC (9), GRK5 (10), and the Akt-mechanistic target of rapamycin pathway (11, 12) among many others.
The PHLPP family consists of two members of serine/threonine phosphatases (PHLPP1 and PHLPP2) with identical domain structures in which a PH domain is followed by a region of leucine-rich repeats, a PP2C phosphatase domain, and a C-terminal PDZ ligand domain (13). In addition, PHLPP1β and PHLPP2 contain a Ras-association domain preceding the PH domain (14). PHLPP has been shown to directly target several members of the AGC family of kinases, including Akt and PKC (14–18). PHLPP1 and PHLPP2 are ubiquitously expressed (16), and dysregulation of PHLPP has been associated with several disease pathologies including cancer, diabetes, and cardiovascular disease (19–21).
Loss of either PHLPP1 or PHLPP2 in various cancer cell lines was found to be accompanied by increased phosphorylation and activity of Akt (22, 23). Genomic analysis of prostate cancers revealed that both PHLPP1 and PHLPP2 genes are deleted at high frequency, and this correlates with increased proliferation rate caused by changes in Akt activity (23).
In the heart, Akt is a nodal kinase crucial for balancing cell survival and growth of cardiomyocytes (12, 24, 25). We previously found in vitro that knockdown of PHLPP1 increased Akt activity and protected cardiomyocytes from oxidative damage (18). In vivo studies of the mouse heart revealed that removal of PHLPP1 increased Akt activity basally without exhibiting hypertrophic growth, whereas induction of pathological hypertrophy in vivo by pressure overload was attenuated in the PHLPP1 knockout mouse compared with their WT counterpart (26).
On the other hand, the role of the PHLPP2 isoform on hypertrophic growth of cardiomyocytes remains largely unknown. Whereas both isoforms of PHLPP are shown to dephosphorylate Akt at Ser473 and inhibit its activity in cancer cells (14, 23, 27), recent studies in primary astrocytes and cardiomyocytes suggest that PHLPP2 does not target Akt and PKC, indicating possible cell type specificity (18, 28).
GRK5 belongs to the GRK family of membrane-bound kinases that is known to phosphorylate GPCRs and lead to their desensitization (10, 29). Unlike other GRKs, GRK5 translocates to the nucleus in a calmodulin-dependent manner in response to selective activation of Gq-coupled α-adrenergic and angiotensin receptor stimulation to regulate cardiac hypertrophy (30–32). GRK5 interacts with several nuclear targets including HDAC5 and can bind DNA directly, which in turn regulates transcription of genes involved in cardiomyocyte hypertrophy (30, 33–35). The targets of PHLPP2 in cardiomyocytes are relatively unknown; accordingly we investigated whether PHLPP2 affects the AGC kinase GRK5 to modulate hypertrophy. We found that removal of PHLPP2 in cardiomyocytes basally increased hypertrophic growth as measured by changes in fetal gene expression and cell area. PE-induced hypertrophic growth and fetal gene expression were also accentuated in cardiomyocytes lacking PHLPP2 compared with control. Removal of PHLPP2 was also shown to increase PE-induced nuclear accumulation of GRK5 in a PHLPP2 phosphatase–dependent manner. Lastly, we determined that GRK5 was necessary for cardiomyocyte hypertrophy induced by PHLPP2 knockdown. Overall, our data revealed for the first time that the phosphatase PHLPP2 plays a novel role in regulating PE-induced cardiac hypertrophy via a GRK5-dependent pathway.
Results
PHLPP2 removal potentiates PE-induced hypertrophic growth and fetal gene expression in cardiomyocytes
We have previously demonstrated that removal of PHLPP1 increases Akt activity and elicits a more “physiological” response to pathological hypertrophy in vivo. Here we extended our studies to determine the effect of PHLPP2 removal on cardiomyocyte growth in vitro. NRVMs were transfected with siRNA for PHLPP1, PHLPP2, or control and treated with PE for 48 h to induce hypertrophic growth. The efficiency of knockdown for PHLPP1 (∼70–80%) or PHLPP2 (∼50–60%) protein and mRNA (∼80–90%) relative to control are represented in the Figs. S1 (A–D) and S2 (A and B). Removal of PHLPP2 significantly increased the basal expression of mRNA for the fetal genes ANF and BNP, markers of hypertrophy (Fig. 1, A and B). Following stimulation with the hypertrophic agonist PE, ANF, and BNP mRNA expression was significantly increased in control, and this response was not significantly altered by the removal of PHLPP1 (Fig. 1, A and B). Treatment with AngII significantly increased ANF and BNP mRNA expression but to a lesser extent than PE (Fig. 2, A and B). Surprisingly, removal of PHLPP2 significantly increased PE (Fig. 1, A and B) and AngII-induced (Fig. 2, A and B) ANF and BNP mRNA expression compared with treated control. Removal of PHLPP2 also significantly increased hypertrophic growth, assessed by examination of cell size basally (Fig. 1, C, panel a versus panel e, and D) and in response to PE (Fig. 1, C, panel b versus panel f, and D) compared with control. There was no significant difference between control and PHLPP1 knockdown cells following PE stimulation (Fig. 1, C, panel b versus panel d, and D). These findings suggest that PHLPP2 regulates cardiomyocyte growth.
Figure 1.
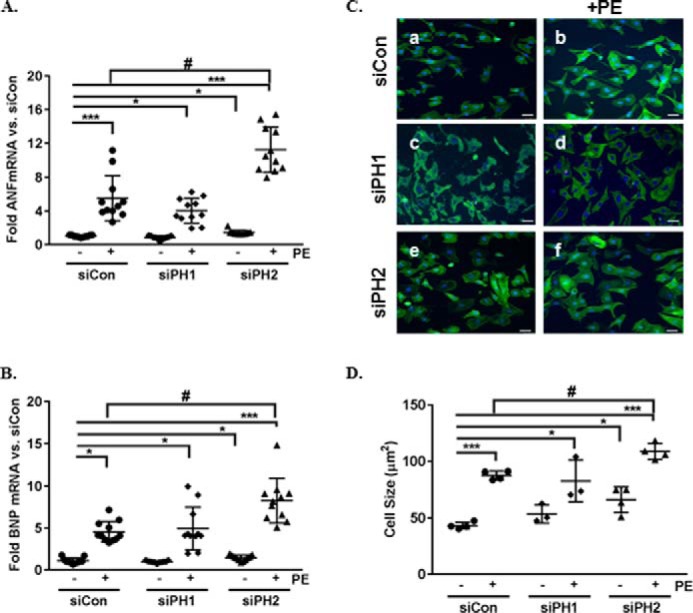
PHLPP2 knockdown induces hypertrophic growth of cardiomyocytes. NRVMs were transfected with siRNA (2 μm) containing control (siCon), PHLPP1 (siPH1), or PHLPP2 (siPH2) and treated with the hypertrophic agonist PE (50 μm) for 48 h. A and B, the mRNA expression of the genes ANF (A) and BNP (B) were measured by RT-PCR. The graph represents the fold change in ANF and BNP mRNA versus nontreated siCon. ***, p < 0.001 versus siCon; *, p < 0.05 versus siCon; #, <0.05 versus siCon+PE (n = 11 independent experiments, means ± S.D.). C, representative images of Immunocytochemistry on NRVMs to visualize cell morphology, phalloidin (green), and DAPI (blue) (20× magnification). The scale bars represent 25 μm. D, cell size was quantified using National Institutes of Health ImageJ software. ***, p < 0.001 versus siCon; *, p < 0.05 versus siCon; #, p < 0.05 versus siCon+PE (n = 4 (siCon and siPH2) and n = 3 independent experiments (siPH1), 400 cells/experiment, means ± S.D.).
Figure 2.

PHLPP2 knockdown potentiates angiotensin II–induced hypertrophic gene expression. A and B, NRVMs were transfected with siRNA (2 μm) containing control (siCon) or PHLPP2 (siPH2) and treated with the hypertrophic agonist AngII (10 nm) for 48 h. The mRNA expression of the genes ANF and BNP were measured by RT-PCR. The graph represents the fold change in ANF and BNP mRNA versus nontreated siCon. *, p < 0.05 versus siCon; #, p < 0.05 versus siCon+AngII (n = 4/5 independent experiments with biological replicates, means ± S.D.). C and D, NRVMs were infected with adenoviruses expressing GFP (adGFP) or full-length PHLPP2 (adPH2) (50 MOI) and treated with AngII (10 nm) for 48 h. The mRNA expression of the hypertrophic genes ANF and BNP were measured by RT-PCR. The graph represents fold change versus nontreated GFP. *, p < 0.05 versus adGFP (n = 5 independent experiments with biological replicates, means ± S.D.).
PHLPP2 activity is necessary for inhibition of hypertrophic growth
To examine the ability of PHLPP2 to block hypertrophy, NRVMs were infected with adenoviruses expressing GFP or PHLPP2. Following 24 h of expression, cardiomyocytes were stimulated with PE for 48 h, and fetal gene expression, as well as growth, was examined. Overexpression of PHLPP2 attenuated PE-induced re-expression of the hypertrophic genes ANF and BNP compared with GFP-expressing cells (Fig. 3, A and B). Also, overexpression of PHLPP2 attenuated AngII-induced ANF and BNP mRNA expression (Fig. 2, C and D). Inhibition of the hypertrophic response was also evidenced by a reduction in cell size in PE-treated PHLPP2 cells compared with GFP treated (Fig. 3, C and D). To determine whether the phosphatase activity of PHLPP2 is necessary for inhibition of myocyte growth, a mutant construct of PHLPP2 with the phosphatase domain deleted (ΔPP2C) was examined. Overexpression of the PHLPP2 phosphatase dead mutant acted as a dominant negative of PHLPP2 and caused an elevated fetal gene expression at baseline (Fig. 3, A and B), but gene expression was not further elevated in response to PE stimulation (Fig. 3, A and B). Similarly, PHLPP2 mutant–overexpressing cells displayed significant hypertrophic growth at baseline (Fig. 3, C and D) comparable with PE-treated GFP cells. The cell size of the PHLPP2 mutant expressing cells was not further increased by PE treatment. Together these findings suggest that PHLPP2 negatively regulates hypertrophic growth and that this depends upon its phosphatase activity.
Figure 3.
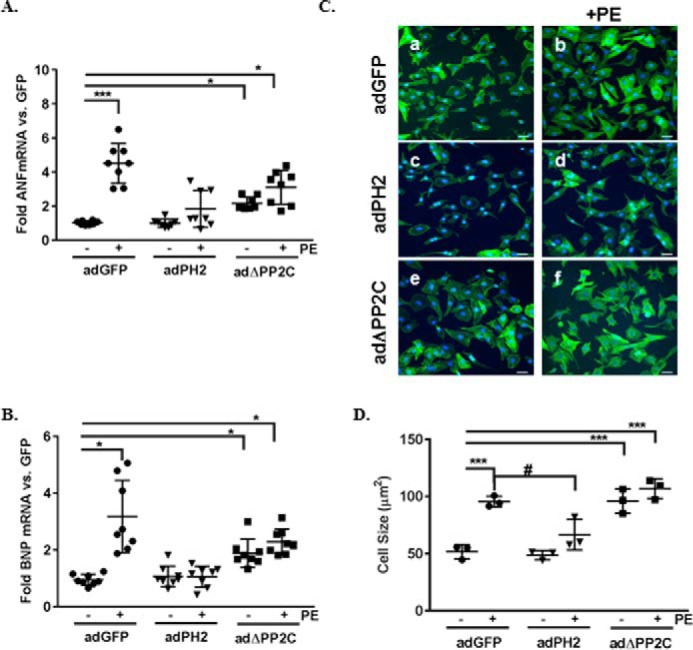
PHLPP2 activity blocks hypertrophic growth induced by PE. NRVMs were infected with adenoviruses expressing GFP, full-length PHLPP2 (PH2), and a phosphatase dead mutant construct (ΔPP2C) (50 MOI) and treated with PE (50 μm) for 48 h. A and B, the mRNA expression of the hypertrophic genes ANF (A) and BNP (B) were measured by RT-PCR. The graph represents fold change versus nontreated GFP. *, p < 0.05; ***, p < 0.001 versus GFP; #, p < 0.05 versus GFP+PE (n = 8 independent experiments, means ± S.D.) C, representative images of immunocytochemistry on NRVMs to visualize cell morphology: phalloidin (green) and DAPI (blue) (20× magnification). The scale bars represent 25 μm. D, cell size was quantified using National Institutes of Health ImageJ software. ***, p < 0.001 versus GFP; #, p < 0.05 versus GFP+PE (n = 3 independent experiments, 400 cells/experiment, means ± S.D.).
PHLPP2 binds GRK5 in cardiomyocytes
Numerous studies have shown that activation of Akt leads to hypertrophic growth of cardiomyocytes in culture (12, 36–38). Because removal of PHLPP2 in cardiomyocytes has no effect on Akt phosphorylation or activity in contrast to what occurs in several other cell types (14, 16, 18, 26, 28), we postulated that PHLPP2 regulates myocyte hypertrophy through another AGC kinase. GRK5 is an AGC kinase that has been demonstrated to regulate myocyte hypertrophy both in vitro and in vivo (30, 32, 39). To determine whether PHLPP2 and GRK5 interact in NRVMs, we performed a co-immunoprecipitation experiment. Adenoviral overexpression of PHLPP2 and GRK5 in cardiomyocytes demonstrated that the proteins interacted (data not shown). To confirm that the interaction between PHLPP2 and GRK5 was present basally and not due to overexpression, we analyzed endogenous binding of PHLPP2 and GRK5. Our immunoprecipitation demonstrated that endogenous PHLPP2 and GRK5 bind in cardiomyocytes (Fig. 4A). This is the first evidence that PHLPP2 and GRK5 interact in cardiomyocytes.
Figure 4.

PHLPP2 and GRK5 interact in NRVMs. A, IP of NRVM extracts (150 μg) treated with or without PE (50 μm) for 30 min with antibodies for IgG or GRK5. PHLPP2 (150 kDa) was blotted (IB) for binding and GRK5 (68 kDa) for IP control. The endogenous expression of PHLPP2 was blotted as control for input (bottom panel). B, quantitation of bound PHLPP2 to GRK5 with and without PE stimulation (∼30% decrease versus Con (−)). The graph represents n = 3 independent experiments with biological replicates. **, p < 0.01 versus nontreated (means ± S.D.).
PHLPP2 modulates GRK5 translocation in cardiomyocytes
It is well-established that GRK5, acting through a noncanonical pathway downstream of Gαq, can regulate hypertrophic growth by its localization in the nucleus (10, 32). In the nucleus, GRK5 acts as an HDAC kinase and leads to derepression of fetal gene transcription and increased hypertrophy (10, 32, 40). Following PE stimulation, the interaction of PHLPP2 and GRK5 are decreased significantly (Fig. 4B). Thus, PHLPP2 may act as a negative regulator of GRK5 translocation. Therefore we examined whether removal of PHLPP2 altered nuclear accumulation of GRK5 induced by PE stimulation. As previously demonstrated, GRK5 accumulates in the nucleus following 30 min of PE stimulation (Fig. 5, A and B). Knockdown of PHLPP2 with siRNA increased basal nuclear accumulation of GRK5, and this accumulation was significantly potentiated following PE stimulation versus the siControl-treated group (Fig. 5, A and B). In accordance with the previously demonstrated effect of nuclear GRK5 on HDAC5, PE-induced nuclear accumulation of GRK5 led to a decrease of HDAC5 in the nucleus (Fig. 5, A and C). Removal of PHLPP2 significantly decreased the basal levels of nuclear HDAC5, but there was no further effect of PE treatment (Fig. 5, A and C).
Figure 5.

Knockdown of PHLPP2 potentiates GRK5 nuclear translocation. NRVMs were transfected (2 μm) with either siControl (siCon) or siPHLPP2 (siPh2). After 24 h of transfection, the cells were infected with adenoviruses (50 MOI) expressing GRK5 (adGRK5) or a N-terminal calmodulin-binding mutant of GRK5 (adW30A) and cultured overnight. At 48 h post-transfection, the cells were treated with PE (50 μm) for 30 min and fractionated. A, representative Western blotting of cytosolic (Cyto) and nuclear (Nuc) fractions probed for GRK5 (68 kDa) and HDAC5 (122 kDa). Purity of the fraction was determined by blotting for lamin A/C (nuclear marker, 70 kDa) and Rho GDI (cytosol marker, 26 kDa). B and C, the amounts of GRK5 (B; #, p < 0.05 versus siCon+PE; *, p < 0.05 versus siCon; **, p < 0.01 versus siPH2; n = 6 independent experiments, means ± S.D.) and HDAC5 in the nucleus were quantified and normalized to lamin A/C and reported as fold change versus siCon (C; *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus siCon; n = 6 independent experiments, means ± S.D.). D, NRVMs were infected with adenoviruses expressing GFP (adGFP), PHLPP2 (adPH2), or a phosphatase dead mutant of PHLPP2 (adΔPP2C) with GRK5 (adGRK5) overnight. The following day, the cells were treated with PE (50 μm) and for 30 min and fractionated. The amount of GRK5 in the nucleus was quantified and normalized to lamin A/C and reported as fold change versus nontreated virus. *, p < 0.05 versus GFP; #, p < 0.05 versus GFP+PE; n.s., not significant (n = 6 independent experiments, means ± S.D.).
Because nuclear localization of GRK5 in response to PE requires calmodulin (CaM) binding to the GRK5 N terminus (32), we investigated whether the effect of knocking down PHLPP2 on GRK5 translocation required CaM binding. We infected NRVMs with a previously characterized GRK5 mutant construct in which the N-terminal CaM domain was mutated (W30A and K312Q) to inhibit PE-induced translocation (Fig. S3A) (32). The CaM-binding mutant GRK5 W30A failed to accumulate in the nucleus following PHLPP2 knockdown or following PE stimulation (Fig. 5B). We also determined that overexpression of PHLPP2 blocked PE-induced nuclear accumulation of GRK5 (Fig. 5D), and this inhibitory ability depended on its phosphatase activity because removal of the PP2C domain led to increased GRK5 nuclear accumulation (Fig. 5D). These findings suggest that PHLPP2 affects GRK5 nuclear accumulation through its phosphatase activity and actions at the site known to bind calmodulin.
GRK5 is required for the hypertrophic response induced by PHLPP2 removal in NRVMs
PHLPP2 interacts with GRK5 and regulates GRK5 nuclear translocation. To determine whether GRK5 mediates the hypertrophic response induced by PHLPP2 removal, we used siRNA to knockdown both GRK5 and PHLPP2 in NRVMs (∼70–80% protein knockdown; Figs. S1, E and F, and S2, A and B). As previously demonstrated in Fig. 1 (A–D) and restated here, knockdown of PHLPP2 significantly accentuated PE-induced hypertrophic growth (Fig. 6, A, panel b versus panel d, and B–D). In contrast, knockdown of GRK5 attenuated PE-induced hypertrophic growth compared with siControl-treated (Fig. 6, A, panel b versus panel f, and B–D). The ability of PHLPP2 knockdown to accentuate PE-induced hypertrophy was prevented by removal of GRK5 (Fig. 6, A, panel e versus panel h, and B–D). These findings support the necessity of GRK5 for the heightened hypertrophic response to PE following PHLPP2 knockdown.
Figure 6.

GRK5 is necessary for hypertrophy induced by PHLPP2 knockdown. NRVMs were transfected with siControl (siCon), siPHLPP2 (siPH2), siGRK5 (siG5), or siGRK5+siPHLPP2 (siG5/PH2, 2 μm). Following knockdown, the cells were treated with PE (50 μm) for 48 h. A, representative images of immunocytochemistry on NRVMs to visualize cell morphology, phalloidin (green) and DAPI (blue) (20× magnification). The scale bars represent 25 μm. B, cell size was quantified using National Institutes of Health ImageJ software. ***, p < 0.001 versus siCon; #, p < 0.05 versus siCon+PE; ##, p < 0.05 versus siPH2+PE (n = 4 independent experiments, 400 cells/experiment, means ± S.D.). C and D, expression of ANF (C) and BNP (D) mRNA were determined following PE stimulation and represented as fold change over siControl. *, p < 0.05; ***, p < 0.01 versus siCon; #, p < 0.05 versus siCon+PE; ##, p < 0.05 versus siPH2+PE; n.s., not significant (n = 11 independent experiments for siCon and siPH2; n = 6 independent experiments for siG5 and siG5/PH2, means ± S.D.).
Discussion
PHLPP1 and PHLPP2 are a family of serine/threonine protein phosphatases that target multiple AGC kinases including Akt and PKC (2, 16, 27). Activation of the Akt signaling pathway in many cell types including cardiomyocytes leads to increased protein synthesis, growth, and survival (12, 41). Based on our previous studies, removal of PHLPP1 in cardiomyocytes and in the heart resulted in increased Akt activity (18), and under pathological stress PHLPP1 knockout mice presented with an attenuated hypertrophic response caused by increased angiogenesis (26). Although PHLPP1 has been demonstrated to regulate Akt in cardiomyocytes (18, 26, 28), the target of PHLPP2 is largely unknown. Here we demonstrate for the first time that PHLPP2 is both necessary and sufficient to inhibit PE-induced hypertrophic growth of cardiomyocytes.
Removal of PHLPP2 increases proliferation rate and survival usually through up-regulation of the Akt signaling pathway in many cancers including colorectal, breast, and ovarian (19, 42, 43). We have demonstrated, however, in cardiomyocytes that removal of PHLPP2 does not alter Akt signaling (18). We found that PHLPP2 and GRK5 can complex in cardiomyocytes and that following hypertrophic stimulation this interaction decreased, suggesting that PHLPP2 may be an important modulator of GRK5. During this study we wanted to determine whether PHLPP2 regulated hypertrophic growth through the AGC kinase GRK5.
GRK5 is one of a family of serine/threonine kinases that phosphorylates and desensitizes agonist occupied GPCRs (10). It is known that GRKs are regulated by a group of calcium sensor proteins (44), among which is the ubiquitously expressed calcium-binding protein CaM. CaM inhibits GRK5 activity by promoting its autophosphorylation and decreases its association at its site of action, the plasma membrane (45). GRK5 is distinctive from other family members in that it contains a nuclear localization and export signal that is important for subcellular targeting and protein interaction (40). In cardiomyocytes, α-adrenergic stimulation with PE causes CaM-dependent nuclear translocation of GRK5, which regulates cardiomyocyte hypertrophy through its ability to phosphorylate and de-repress HDAC5 (30, 32). In the current study we demonstrate that removal of PHLPP2 in cardiomyocytes increases basal and PE-induced nuclear translocation of GRK5 and concomitant export of HDAC5. Using a CaM-binding mutant of GRK5, we established that in the presence of PHLPP2 knockdown, GRK5 requires CaM binding for nuclear translocation. Given that PHLPP2 and GRK5 interact, and PHLPP2 phosphatase activity is required to block GRK5 translocation in cardiomyocytes, we hypothesize that PHLPP2 may directly or indirectly affect the phosphorylation state of GRK5 to mediate CaM binding and nuclear translocation.
Little is known about the effect of phosphorylation and dephosphorylation on the function of GRK5 and its role in GRK5 regulation of cardiomyocyte hypertrophy. CaM binding induces GRK5 autophosphorylation and decreased membrane binding (45). The only data regarding regulation of GRK5 activity by phosphorylation comes from studies in the Benovic laboratory (46). They demonstrated that PKC can phosphorylate GRK5 in its C-terminal domain and that this blocks its ability to desensitize receptors. Because activation of adrenergic receptors stimulate PKC isoforms downstream of phospholipase C to induce cardiomyocyte hypertrophy in vitro (42, 47), whether PKC activity alters GRK5-induced hypertrophy in cardiomyocytes is unknown. Although PHLPP1 and PHLPP2 have the ability to target PKC isoforms in several cell systems causing its destabilization at the membrane (13, 15), removal of either isoform does not alter the levels of PKCα and PKCβ in cardiomyocytes (18). Preliminary findings suggest that removal of PHLPP2 alters the phosphorylation status of GRK5 following PE stimulation (data not shown), and our data demonstrate that PHLPP2 phosphatase activity is required for inhibition of GRK5 nuclear localization. Taken together, our findings suggest that GRK5 phosphorylation may be important for its ability to regulate myocyte hypertrophy. Because there are no commercial antibodies available against putative phosphorylation sites on GRK5, mapping the sites on GRK5 that are altered by PHLPP2 is out of the scope of this paper but will be examined in the future.
In light of our finding that overexpression of PHLPP2 blocked hypertrophy induced by both agonists PE and AngII, as well as the discovery that endogenous levels of PHLPP2 are decreased following hypertrophic stimulation (Figs. S1B and S2A), our data suggest that PHLPP2 is a novel regulator of cardiomyocyte hypertrophy. Although we found that GRK5 is necessary for the growth induced by PHLPP2 knockdown, we cannot rule out the possibility that there are other hypertrophic signaling pathways affected by PHLPP2 removal. GRK2, a member of the G-protein–coupled receptor kinase family, is also an important regulator of cardiomyocyte physiology (48) and can bind PHLPP2 (data not shown). The relevance of this finding and its consequence to GRK2-regulated signaling in cardiomyocytes are unknown. Further studies are needed to determine the importance of this interaction in vitro.
Lastly, we find that removal of the phosphatase activity of PHLPP2 increases cardiomyocyte hypertrophy basally, and this is not further potentiated in the presence of hypertrophic stimulation. Because we have demonstrated that binding of CaM is required for the GRK5-mediated cardiac hypertrophy as discussed above, it is possible that the removal of PHLPP2 activity alters Ca2+/calmodulin-dependent activation of calcineurin/NFAT and Ca2+/calmodulin-dependent kinase II, which also regulate cardiomyocyte hypertrophy (4, 49). NFAT proteins are a family of calcium level–regulated transcriptional factors that activate a wide range of genes including fetal genes (49, 50). Because GRK5 has been shown to enhance NFAT activity in cardiomyocytes (51), it is possible that NFAT might also be regulating the induced fetal gene expression seen with the increased accumulation of GRK5 in nucleus. Even though we saw increased export of HDAC5 from the nucleus, multiple signaling pathways may regulate the hypertrophic response following PHLPP2 knockdown. Future studies will define the contribution of other signaling pathways to the hypertrophic growth induced by PHLPP2 removal in vitro.
Our lab is the first to demonstrate in cardiomyocytes an interaction between the phosphatase PHLPP2 and GRK5 and an in vitro role for this in modulating cardiomyocyte growth. Understanding the biological function and signaling pathways altered by PHLPP2 in cardiomyocytes may help delineate therapeutic targets for cardiac hypertrophy.
Experimental procedures
Reagents
Phenylephrine ((R)-(−)-phenylephrine hydrochloride, PE) was purchased from Sigma–Aldrich and used at 50 μm. AngII was purchased from Bachem Americas, Inc. (Vista, CA) and used at 10 nm.
Animals
All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of University of California San Diego. Sprague–Dawley rats (1–2 days old) were used for cell isolation experiments.
Isolation of neonatal rat ventricular myocytes
Neonatal rat ventricular myocytes were isolated from 1–2-day-old Sprague–Dawley rat pups (Harlan, Indianapolis, IN). Myocytes were isolated using the Neonatal Cardiomyocyte Isolation System (Worthington, Lakewood, NJ) and plated at density of 3.5 × 104/cm2 in Dulbecco's modified Eagle's medium (DMEM) with 15% fetal bovine serum and antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin) at 37 °C in 5% CO2 as previously described (18). After overnight culture, the cells were placed in serum-free DMEM and transfected or infected as described below. DMEM, fetal bovine serum, and antibiotics were purchased from Thermo Fisher Scientific (Waltham, MA).
Transfection and adenoviral infection
NRVMs were transfected with siRNA as previously described (18). Briefly, predesigned siRNA (2 μm) for PHLPP2 (ON-TARGET plus rat PHLPP2, J-104590-07; Dharmacon, Lafayette, CO; targeting sequence UAGUCUGAGUCUUCGGAAA), PHLPP1 (ON-TARGET plus rat PHLPP1, J-094929-09; Dharmacon; targeting sequence GAAUGUACAAUGUCCGAAA), GRK5 (rat GRK5, SI01518650; Qiagen; targeting sequence GCAUGUAUUUUGACCGUUU), or control siRNA (rat, 1027281, Qiagen; targeting sequence AATTCTCCGAACGTGTCACGT) was transfected into NRVMs using DharmaFECT-1 (Dharmacon) transfection reagent in a 1:3 ratio, respectively. Following 48 h of transfection, the cells were treated with or without PE for various time points as described in the figure legends. The mRNA (Fig. S1, A, C, and E) and protein levels (Fig. S1, B, D, and F, and S2, A and B) following siRNA knockdown of PHLPP1, PHLPP2, and GRK5 are represented in the supporting information.
For adenoviral infection, adenoviral vectors at 50 MOI were added to cells 24 h after transfection or after plating as previously described (52). Adenoviruses used expressed the following genes: full-length GRK5, GFP, V5-tagged full-length mouse PHLPP2, phosphatase dead mutant of PHLPP2, V5-tagged ΔPP2C in which amino acids 782–1030 were removed, and a GRK5 mutant (W30A/K31Q) that inhibits N-terminal CaM binding (32). Expression of the adenoviruses used with or without siRNA is represented in Fig. S3 (A and B).
Immunostaining
Following 48 h of PE treatment, NRVMs were fixed with 4% paraformaldehyde. The cells were visualized using Phalloidin; Dylight 488-conjugated antibody at 1:200 dilution (21833, lot RI2262261, Thermo Scientific); and Vectashield anti-fade mounting medium with DAPI (Vector Lab, Burlingame, CA) for nuclear staining. Images were acquired at 20× magnification using a Leica DMi8 fluorescence microscope and DFC450C camera (Leica Microsystems, Wetzlar, Germany). Cell area was quantified using ImageJ software (National Institutes of Health, Bethesda, MD), and the area across cells with central nuclei was measured. For each condition at least 400 cells per experiment were measured for cell size analysis.
Quantitative PCR
For quantitative PCR, following 48 h of PE treatment, total RNA was isolated using a micro-RNA isolation kit (Invitrogen) and cDNA synthesized using the Verso cDNA synthesis kit (Thermo Scientific) based on the manufacturer's instructions as previously described (26). Hypertrophic gene expression was analyzed using probe sets from Applied Biosystems for ANF (NPPA (natriuretic peptide A)), BNP (NPPB (natriuretic peptide B)), and GAPDH as internal control. Relative quantification was analyzed using the comparative threshold cycle (Ct) method normalized to GAPDH as previously described (26).
Protein isolation
For protein analysis, whole cell extracts were isolated from NRVMs using radioimmune precipitation assay buffer as previously described (18). For fractionation experiments, cytosolic and nuclear fractions were isolated by differential centrifugation as previously described (18). Protein concentration was measured using a micro BCA protein assay kit (Thermo Scientific). For fractionation, 5 μg of protein was run for the nuclear and cytosolic fractions. For immunoprecipitation (IP) of GRK5 for binding experiments, whole cell lysates (150 μg) were incubated with 50 μl of protein A/G-agarose beads (Santa Cruz Biotechnology) in a 50% slurry along with specific antibodies for GRK5 (1 μg/100 μg, rabbit polyclonal, sc-565, lot E2915) from Santa Cruz Biotechnology (Santa Cruz, CA) or rabbit IgG (Santa Cruz Biotechnology) as control at 4 °C overnight. Immuocomplexes were washed and protein eluted in SDS followed by Western blotting analysis.
Western blotting
Electrophoresis and Western blotting were performed as previously described (18, 26). Primary antibodies used are as follows: PHLPP2 (1:2000 rabbit polyclonal, A300-661A, lot A300-661A-1) and PHLPP1 (1:2000 rabbit polyclonal, A300-660A, lot A300-660A-1) from Bethyl Laboratories (Montgomery, TX). Lamin A/C (1:1000 rabbit polyclonal, 2032S, lot 5), RhoGDI (1:1000 rabbit polyclonal, 2564S, lot 1), HDAC5 (1:1000 rabbit polyclonal, 2082BC, lot 2), GAPDH (1:1000 rabbit polyclonal, 5174, lot 6) actinin (1:1000 rabbit polyclonal, 6487, lot 1), and V5 (1:1000 rabbit polyclonal, 13202, lot 2) antibodies were from Cell Signaling Technology. The GRK5 antibody (1:500 rabbit polyclonal, sc-565, lot E2915) were from Santa Cruz Biotechnology. All antibodies were diluted in 5% BSA/TBS/Tween-20. Secondary anti-rabbit antibody (Sigma, A6154, lot SLBP3451V) was used at 1:8000, and anti-mouse antibody (Sigma, A5278, lot SLBK2640V) was used at 1:2000 dilution in 5% milk/TBS-Tween-20. Antibodies were validated by either knockdown of the protein using siRNA or overexpression with adenoviruses when possible.
Statistical analysis
Researchers were blinded to the treatment group during analyses. The data are represented as means ± S.D. Differences are considered statistically significant (p < 0.05) assessed using unpaired Student's t test (for two groups) and analysis of variance (for multiple comparisons involving two variables) with post hoc Tukey analysis using the GraphPad Prism software (GraphPad, La Jolla, CA).
Author contributions
N. H. P. and W. J. K. contributed to the experimental design of the work. S.-T. Y., C. M. Z., W. J. K., and N. H. P. contributed to the acquisition, analysis, and interpretation of data. N. H. P. and S. Y. wrote the paper. All authors approved the manuscript final version and agreed to be accountable for all aspects of the work and for ensuring that questions related to the accuracy or integrity of any part of the work were appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Supplementary Material
Acknowledgments
We thank Jeffery M. Smith for virus production and Valerie P. Tan for isolation of neonatal rat ventricular myocytes.
This work was supported by grants from NHLBI, National Institutes of Health Grants HL114949 (to N. H. P.) and HL091799 (to W. J. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3.
- GPCR
- G-protein–coupled receptor
- PHLPP
- PH domain leucine-rich repeat protein phosphatase
- AGC kinase
- PKA, PKG, and PKC
- GRK
- G-protein coupled receptor kinase
- PE
- phenylephrine
- NRVM
- neonatal rat ventricular myocytes
- ANF
- atrial natriuretic factor
- BNP
- brain natriuretic peptide
- NFAT
- calcineurin-nuclear factor of activated T cells
- PP2C
- protein phosphatase 2C
- HDAC
- histone deacetylase
- AngII
- angiotensin II
- siControl
- control siRNA
- CaM
- calmodulin
- DMEM
- Dulbecco's modified Eagle's medium
- MOI
- multiplicity of infection
- DAPI
- 4′,6′-diamino-2-phenylindole
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- IP
- immunoprecipitation.
References
- 1. Frey N., and Olson E. N. (2003) Cardiac hypertrophy: the good, the bad and the ugly. Annu. Rev. Physiol. 65, 45–79 10.1146/annurev.physiol.65.092101.142243 [DOI] [PubMed] [Google Scholar]
- 2. Grzechnik A. T., and Newton A. C. (2016) PHLPPing through history: a decade in the life of PHLPP phosphatases. Biochem. Soc. Trans. 44, 1675–1682 10.1042/BST20160170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kehat I., and Molkentin J. D. (2010) Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation 122, 2727–2735 10.1161/CIRCULATIONAHA.110.942268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Molkentin J. D., Lu J. R., Antos C. L., Markham B., Richardson J., Robbins J., Grant S. R., and Olson E. N. (1998) A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93, 215–228 10.1016/S0092-8674(00)81573-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bossuyt J., Helmstadter K., Wu X., Clements-Jewery H., Haworth R. S., Avkiran M., Martin J. L., Pogwizd S. M., and Bers D. M. (2008) Ca2+/calmodulin-dependent protein kinase IIdelta and protein kinase D overexpression reinforce the histone deacetylase 5 redistribution in heart failure. Circ. Res. 102, 695–702 10.1161/CIRCRESAHA.107.169755 [DOI] [PubMed] [Google Scholar]
- 6. Zhang T., Kohlhaas M., Backs J., Mishra S., Phillips W., Dybkova N., Chang S., Ling H., Bers D. M., Maier L. S., Olson E. N., and Brown J. H. (2007) CaMKII isoforms differentially affect calcium handling but similarly regulate HDAC/MEF2 transcriptional responses. J. Biol. Chem. 282 35078–35087 [DOI] [PubMed] [Google Scholar]
- 7. Kehat I., Davis J., Tiburcy M., Accornero F., Saba-El-Leil M. K., Maillet M., York A. J., Lorenz J. N., Zimmermann W. H., Meloche S., and Molkentin J. D. (2011) Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ. Res. 108 176–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Purcell N. H., Wilkins B. J., York A., Saba-El-Leil M. K., Meloche S., Robbins J., and Molkentin J. D. (2007) Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc. Natl. Acad. Sci. U.S.A. 104, 14074–14079 10.1073/pnas.0610906104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim Y. K., Seo D. W., Kang D. W., Lee H. Y., Han J. W., and Kim S. N. (2006) Involvement of HDAC1 and the PI3K/PKC signaling pathways in NF-κB activation by the HDAC inhibitor apicidin. Biochem. Biophys. Res. Commun. 347, 1088–1093 10.1016/j.bbrc.2006.06.196 [DOI] [PubMed] [Google Scholar]
- 10. Traynham C. J., Hullmann J., and Koch W. J. (2016) Canonical and non-canonical actions of GRK5 in the heart. J. Mol. Cell. Cardiol. 92, 196–202 10.1016/j.yjmcc.2016.01.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shioi T., McMullen J. R., Kang P. M., Douglas P. S., Obata T., Franke T. F., Cantley L. C., and Izumo S. (2002) Akt/protein kinase B promotes organ growth in transgenic mice. Mol. Cell. Biol. 22, 2799–2809 10.1128/MCB.22.8.2799-2809.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sussman M. A., Völkers M., Fischer K., Bailey B., Cottage C. T., Din S., Gude N., Avitabile D., Alvarez R., Sundararaman B., Quijada P., Mason M., Konstandin M. H., Malhowski A., Cheng Z., et al. (2011) Myocardial AKT: the omnipresent nexus. Physiol. Rev. 91, 1023–1070 10.1152/physrev.00024.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Newton A. C., and Trotman L. C. (2014) Turning off AKT: PHLPP as a drug target. Annu. Rev. Pharmacol. Toxicol. 54, 537–558 10.1146/annurev-pharmtox-011112-140338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brognard J., Sierecki E., Gao T., and Newton A. C. (2007) PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol. Cell 25, 917–931 10.1016/j.molcel.2007.02.017 [DOI] [PubMed] [Google Scholar]
- 15. Gao T., Brognard J., and Newton A. C. (2008) The phosphatase PHLPP controls the cellular levels of protein kinase C. J. Biol. Chem. 283, 6300–6311 10.1074/jbc.M707319200 [DOI] [PubMed] [Google Scholar]
- 16. Gao T., Furnari F., and Newton A. C. (2005) PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell 18, 13–24 10.1016/j.molcel.2005.03.008 [DOI] [PubMed] [Google Scholar]
- 17. Liu J., Stevens P. D., Li X., Schmidt M. D., and Gao T. (2011) PHLPP-mediated dephosphorylation of S6K1 inhibits protein translation and cell growth. Mol. Cell. Biol. 31, 4917–4927 10.1128/MCB.05799-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miyamoto S., Purcell N. H., Smith J. M., Gao T., Whittaker R., Huang K., Castillo R., Glembotski C. C., Sussman M. A., Newton A. C., and Brown J. H. (2010) PHLPP-1 negatively regulates Akt activity and survival in the heart. Circ. Res. 107, 476–484 10.1161/CIRCRESAHA.109.215020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu J., Weiss H. L., Rychahou P., Jackson L. N., Evers B. M., and Gao T. (2009) Loss of PHLPP expression in colon cancer: role in proliferation and tumorigenesis. Oncogene 28, 994–1004 10.1038/onc.2008.450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lv D., Yang H., Wang W., Xie Y., Hu W., Ye M., and Chen X. (2015) High PHLPP expression is associated with better prognosis in patients with resected lung adenocarcinoma. BMC Cancer 15, 687 10.1186/s12885-015-1711-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mathur A., Pandey V. K., and Kakkar P. (2017) PHLPP: a putative cellular target during insulin resistance and type 2 diabetes. J. Endocrinol. 233, R185–R198 10.1530/JOE-17-0081 [DOI] [PubMed] [Google Scholar]
- 22. Li X., Liu J., and Gao T. (2009) β-TrCP-mediated ubiquitination and degradation of PHLPP1 are negatively regulated by Akt. Mol. Cell. Biol. 29, 6192–6205 10.1128/MCB.00681-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Warfel N. A., and Newton A. C. (2012) Pleckstrin homology domain leucine-rich repeat protein phosphatase (PHLPP): a new player in cell signaling. J. Biol. Chem. 287, 3610–3616 10.1074/jbc.R111.318675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matsui T., Li L., Wu J. C., Cook S. A., Nagoshi T., Picard M. H., Liao R., and Rosenzweig A. (2002) Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J. Biol. Chem. 277 22896–22901 [DOI] [PubMed] [Google Scholar]
- 25. Shiojima I., Sato K., Izumiya Y., Schiekofer S., Ito M., Liao R., Colucci W. S., and Walsh K. (2005) Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J. Clin. Invest. 115, 2108–2118 10.1172/JCI24682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moc C., Taylor A. E., Chesini G. P., Zambrano C. M., Barlow M. S., Zhang X., Gustafsson Å. B., and Purcell N. H. (2015) Physiological activation of Akt by PHLPP1 deletion protects against pathological hypertrophy. Cardiovasc. Res. 105, 160–170 10.1093/cvr/cvu243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brognard J., and Newton A. C. (2008) PHLiPPing the switch on Akt and protein kinase C signaling. Trends Endocrinol. Metab. 19, 223–230 10.1016/j.tem.2008.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen B., Van Winkle J. A., Lyden P. D., Brown J. H., and Purcell N. H. (2013) PHLPP1 gene deletion protects the brain from ischemic injury. J. Cereb. Blood Flow Metab. 33, 196–204 10.1038/jcbfm.2012.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pearce L. R., Komander D., and Alessi D. R. (2010) The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 11, 9–22 10.1038/nrn2754,10.1038/nrm2822 [DOI] [PubMed] [Google Scholar]
- 30. Martini J. S., Raake P., Vinge L. E., DeGeorge B. Jr., Chuprun J. K., Harris D. M., Gao E., Eckhart A. D., Pitcher J. A., and Koch W. J. (2008) Uncovering G protein-coupled receptor kinase-5 as a histone deacetylase kinase in the nucleus of cardiomyocytes. Proc. Natl. Acad. Sci. U.S.A. 105, 12457–12462 10.1073/pnas.0803153105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang Y., Matkovich S. J., Duan X., Gold J. I., Koch W. J., and Dorn G. W. 2nd. (2011) Nuclear effects of G-protein receptor kinase 5 on histone deacetylase 5-regulated gene transcription in heart failure. Circ. Heart Fail. 4, 659–668 10.1161/CIRCHEARTFAILURE.111.962563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gold J. I., Martini J. S., Hullmann J., Gao E., Chuprun J. K., Lee L., Tilley D. G., Rabinowitz J. E., Bossuyt J., Bers D. M., and Koch W. J. (2013) Nuclear translocation of cardiac G protein-coupled receptor kinase 5 downstream of select Gq-activating hypertrophic ligands is a calmodulin-dependent process. PLoS One 8, e57324 10.1371/journal.pone.0057324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hullmann J., Traynham C. J., Coleman R. C., and Koch W. J. (2016) The expanding GRK interactome: implications in cardiovascular disease and potential for therapeutic development. Pharmacol. Res. 110, 52–64 10.1016/j.phrs.2016.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Islam K. N., Bae J. W., Gao E., and Koch W. J. (2013) Regulation of nuclear factor κB (NF-κB) in the nucleus of cardiomyocytes by G protein-coupled receptor kinase 5 (GRK5). J. Biol. Chem. 288, 35683–35689 10.1074/jbc.M113.529347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Islam K. N., and Koch W. J. (2012) Involvement of nuclear factor κB (NF-κB) signaling pathway in regulation of cardiac G protein-coupled receptor kinase 5 (GRK5) expression. J. Biol. Chem. 287, 12771–12778 10.1074/jbc.M111.324566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Catalucci D., Latronico M. V., Ellingsen O., and Condorelli G. (2008) Physiological myocardial hypertrophy: how and why? Front. Biosci. 13, 312–324 10.2741/2681 [DOI] [PubMed] [Google Scholar]
- 37. Cittadini A., Monti M. G., Iaccarino G., Di Rella F., Tsichlis P. N., Di Gianni A., Stromer H., Sorriento D., Peschle C., Trimarco B., Saccà L., and Condorelli G. (2006) Adenoviral gene transfer of Akt enhances myocardial contractility and intracellular calcium handling. Gene Ther. 13, 8–19 10.1038/sj.gt.3302589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cook S. A., Matsui T., Li L., and Rosenzweig A. (2002) Transcriptional effects of chronic Akt activation in the heart. J. Biol. Chem. 277, 22528–22533 10.1074/jbc.M201462200 [DOI] [PubMed] [Google Scholar]
- 39. Gold J. I., Gao E., Shang X., Premont R. T., and Koch W. J. (2012) Determining the absolute requirement of G protein-coupled receptor kinase 5 for pathological cardiac hypertrophy: short communication. Circ. Res. 111, 1048–1053 10.1161/CIRCRESAHA.112.273367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johnson L. R., Robinson J. D., Lester K. N., and Pitcher J. A. (2013) Distinct structural features of G protein-coupled receptor kinase 5 (GRK5) regulate its nuclear localization and DNA-binding ability. PLoS One 8, e62508 10.1371/journal.pone.0062508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang Z. Z., Tschopp O., Baudry A., Dümmler B., Hynx D., and Hemmings B. A. (2004) Physiological functions of protein kinase B/Akt. Biochem. Soc. Trans. 32, 350–354 10.1042/bst0320350 [DOI] [PubMed] [Google Scholar]
- 42. Newton A. C., Antal C. E., and Steinberg S. F. (2016) Protein kinase C mechanisms that contribute to cardiac remodelling. Clin. Sci. 130, 1499–1510 10.1042/CS20160036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liao Y., Deng Y., Liu J., Ye Z., You Z., Yao S., and He S. (2016) MiR-760 overexpression promotes proliferation in ovarian cancer by downregulation of PHLPP2 expression. Gynecol. Oncol. 143, 655–663 10.1016/j.ygyno.2016.09.010 [DOI] [PubMed] [Google Scholar]
- 44. Sallese M., Iacovelli L., Cumashi A., Capobianco L., Cuomo L., and De Blasi A. (2000) Regulation of G protein-coupled receptor kinase subtypes by calcium sensor proteins. Biochim. Biophys. Acta 1498, 112–121 10.1016/S0167-4889(00)00088-4 [DOI] [PubMed] [Google Scholar]
- 45. Pronin A. N., Satpaev D. K., Slepak V. Z., and Benovic J. L. (1997) Regulation of G protein-coupled receptor kinases by calmodulin and localization of the calmodulin binding domain. J. Biol. Chem. 272, 18273–18280 10.1074/jbc.272.29.18273 [DOI] [PubMed] [Google Scholar]
- 46. Pronin A. N., and Benovic J. L. (1997) Regulation of the G protein-coupled receptor kinase GRK5 by protein kinase C. J. Biol. Chem. 272 3806–3812 [DOI] [PubMed] [Google Scholar]
- 47. Zhang Y., Matkovich S. J., Duan X., Diwan A., Kang M. Y., and Dorn G. W. 2nd (2011) Receptor-independent protein kinase Cα (PKCα) signaling by calpain-generated free catalytic domains induces HDAC5 nuclear export and regulates cardiac transcription. J. Biol. Chem. 286, 26943–26951 10.1074/jbc.M111.234757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schumacher S. M., and Koch W. J. (2017) Noncanonical roles of G protein-coupled receptor kinases in cardiovascular signaling. J. Cardiovasc. Pharmacol. 70, 129–141 10.1097/FJC.0000000000000483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hogan P. G., Chen L., Nardone J., and Rao A. (2003) Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17, 2205–2232 10.1101/gad.1102703 [DOI] [PubMed] [Google Scholar]
- 50. Macian F. (2005) NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol. 5, 472–484 10.1038/nri1632 [DOI] [PubMed] [Google Scholar]
- 51. Hullmann J. E., Grisanti L. A., Makarewich C. A., Gao E., Gold J. I., Chuprun J. K., Tilley D. G., Houser S. R., and Koch W. J. (2014) GRK5-mediated exacerbation of pathological cardiac hypertrophy involves facilitation of nuclear NFAT activity. Circ. Res. 115, 976–985 10.1161/CIRCRESAHA.116.304475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dusaban S. S., Purcell N. H., Rockenstein E., Masliah E., Cho M. K., Smrcka A. V., and Brown J. H. (2013) Phospholipase Cϵ links G protein-coupled receptor activation to inflammatory astrocytic responses. Proc. Natl. Acad. Sci. U.S.A. 110, 3609–3614 10.1073/pnas.1217355110 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.