

Abstract
During neuronal development, the microtubule-associated protein tau becomes enriched in the axon, where it remains concentrated in the healthy brain. In tauopathies such as Alzheimer's disease, tau redistributes from the axon to the somatodendritic compartment. However, the cellular mechanism that regulates tau's localization remains unclear. We report here that tau interacts with the Ca2+-regulated plasma membrane–binding protein annexin A2 (AnxA2) via tau's extreme N terminus encoded by the first exon (E1). Bioinformatics analysis identified two conserved eight-amino-acids-long motifs within E1 in mammals. Using a heterologous yeast system, we found that disease-related mutations and pseudophosphorylation of Tyr-18, located within E1 but outside of the two conserved regions, do not influence tau's interaction with AnxA2. We further observed that tau interacts with the core domain of AnxA2 in a Ca2+-induced open conformation and interacts also with AnxA6. Moreover, lack of E1 moderately increased tau's association rate to microtubules, consistent with the supposition that the presence of the tau–annexin interaction reduces the availability of tau to interact with microtubules. Of note, intracellular competition through overexpression of E1-containing constructs reduced tau's axonal enrichment in primary neurons. Our results suggest that the E1-mediated tau–annexin interaction contributes to the enrichment of tau in the axon and is involved in its redistribution in pathological conditions.
Keywords: microtubule-associated protein (MAP), annexin, tau protein, axon, neuron, polarity
Introduction
The tau proteins belong to the tau/MAP2/MAP4 family of microtubule-associated proteins (MAPs),4 which share a similar microtubule-binding region at their C-terminal end (1). Tau and MAP2 are predominantly present in neurons, whereas MAP4 is a nonneuronal MAP. Whereas MAP2 is mainly localized in the somatodendritic compartment, tau becomes enriched in axons early during the development of polarity and remains concentrated in this compartment in the healthy brain (2–4). The compartment-specific distribution of the neuronal MAPs may have a role in regulating the balance of microtubule-dependent transport in axons versus dendrites (5). Remarkably, during development of tauopathies such as Alzheimer's disease (AD), tau redistributes from the axon to the somatodendritic compartment, where it aggregates into filamentous structures (paired or straight helical filaments), which form neurofibrillary tangles (6). The enrichment of tau in the axon may at least partially be mediated by the axon initial segment (AIS), which is thought to act as a selective diffusion barrier for various proteins (7–9) In fact, the integrity of the AIS is disrupted in animal models of AD (10), which may contribute to the pathologic mislocalization of tau during disease. However, it is still a matter of debate how tau becomes enriched in the axon, how it is retained in this compartment, and what causes its redistribution during disease.
Tau belongs to the class of intrinsically disordered proteins (IDPs), which are known to interact with a large number of unrelated partners. As such, a minimal interactome of 73 binding partners has been estimated (11). In tau immunoprecipitates, >500 proteins have been identified by MS (12); however, it is unclear to what extent this number reflects proteins, which directly interact with tau. It is likely that interactions other than tau's binding to microtubules are involved in retaining tau in the axonal compartment, because microtubules are ubiquitously present in neurons and tau shows a highly dynamic interaction with microtubules (13, 14). Such a dynamic interaction would result in a rapid redistribution of tau in the cell if microtubules were the sole interaction partners of tau. Therefore, the identity of tau's interaction partner(s) in the axon and how it might contribute to tau's localization still needs to be revealed.
It has been known for some time that tau interacts with components of the neuronal plasma membrane through its nonmicrotubule-binding projection domain (15). Such an interaction could provide a specific mechanism to retain MAP tau rather than other MAPs in the axonal compartment, the latter being characterized by a high (membrane) surface/volume ratio close to a high-density microtubule array. We have previously shown that the tip of a neurite acts as an adsorber trapping tau protein and that binding was mediated by tau's N-terminal projection domain (16). We also identified the membrane-binding protein annexin A2 (AnxA2) as a potential interaction partner of tau (17). However, it is not known how tau interacts with AnxA2 and which regions of the two proteins are involved in binding.
The annexins constitute a multigene family of Ca2+-regulated membrane binding proteins, which are thought to organize the interface between the cytoplasm and the cytoplasmic face of cellular membranes (18). In vertebrates, 12 annexin subfamilies (A1–A11 and A13) have been identified. The Ca2+-dependent membrane interaction occurs through the annexin core domain as a conserved binding module. The N-terminal region precedes the core domain and is diverse in length and sequence between the different members of the annexin family (19). AnxA2 and A6 have been shown to reside in lipid rafts, and, in particular, AnxA2 appears to be involved in organizing cholesterol-rich microdomains and linking them to cytoskeletal proteins (20). In neurons, AnxA2 is present in high concentrations in growth cones and axonal branches (21). AnxA6 becomes concentrated in the AIS during neuronal development (22). Its presence in the AIS is independent of neuronal activity and resistant against detergent extraction, consistent with an interaction with cytoskeletal proteins (23). Interestingly, in pathological states, the expression of AnxA6 is altered, and its distribution is changed (24, 25). Whether tau only interacts with AnxA2 or also with AnxA6 is not known.
In this study, we mapped the interaction of tau with AnxA2 and AnxA6 using a heterologous yeast system. We identified the extreme N terminus of tau encoded by the first exon (E1) as an interaction site and demonstrated that the interaction is not affected by familial tau mutations in the first coding exon or by introducing a phospho-mimicking or -blocking mutation of tyrosine 18. By bioinformatics analysis, we identified two motifs that are conserved in mammals but are absent in fish. Using an in-cell competition assay, we provide evidence that the interaction via E1 is involved in tau's axonal retention. We believe that our results contribute to an understanding of the processes, which lead to the enrichment of tau in the axon and are involved in its redistribution during pathology.
Results
Tau's N-terminal projection domain interacts with annexin A2
Previously, we have identified the calcium-regulated plasma membrane–binding protein AnxA2 as an interaction partner of tau by tandem-affinity purification tag purification and MS (17). Notably, the interaction required the presence of Ca2+. To systematically identify the interacting domain and potential regulatory mechanisms involved in the binding, we employed pulldown assays in a heterologous yeast expression system. The choice to use a yeast system was motivated by our previous observations using mammalian cell lines that the presence of Ca2+ resulted in nonspecific precipitation of AnxA2 in neural cell lysates under control conditions, probably due to the formation of nonspecific complexes in the presence of neural membrane components (17).
We first confirmed the interaction of full-length tau (FLAGtau441WT) with GFP-tagged human AnxA2 in the heterologous yeast system (Fig. 1A, left). In a control experiment where tau was co-expressed with GFP alone, tau did not precipitate (Fig. 1A, right). To test whether we could reproduce the calcium dependence of the interaction also in the heterologous yeast expression system, we performed the same pulldown experiment in the absence of Ca2+. Indeed, tau did not co-precipitate with AnxA2 under these conditions (Fig. 1A, bottom).
Figure 1.

Tau interacts with AnxA2 through E1. A, exon structure of tau441WT with conventional nomenclature is shown at the top. Exons that are alternatively spliced in the CNS are indicated in dark gray. Tau's functional organization is shown below. The repeat regions, which constitute the basic microtubule-interacting unit, are indicated in yellow, and the N-terminal projection domain is shown in light green. Immunoblots showing the result of pulldown assays of exogenously expressed tau and AnxA2 in the presence or absence of calcium ions in the heterologous yeast system are displayed below. Tau co-precipitated after pulldown of AnxA2-GFP in the presence but not the absence of Ca2+ (bottom left). No co-precipitation was observed in control experiments (pulldown of GFP; middle right). Numbers at the sides of the gel blots indicate molecular mass standards in kilodaltons. CTR, C-terminal region; PRR, proline-rich region. B, pulldown assays of annexin (AnxA2-GFP) and controls (GFP) with 2HA-tagged tau and a panel of tau deletion constructs. Tau deletion constructs containing the sequence of E1 (aa 1–44) co-precipitated after pulldown of AnxA2-GFP, whereas the C-terminal half (aa 256–441) or the carrier (Gpm1) did not. All co-precipitation experiments were confirmed by at least two independent experiments.
For detection of tau, we used an antibody (Tau5) that recognizes an epitope in the middle of the protein (aa 218–225) (26) because the FLAG tag allowed only inefficient detection in immunoblots. To be able to detect also constructs that did not contain the Tau5 epitope, we prepared a panel of tau deletion constructs with an N-terminal tandem human influenza hemagglutinin (HA) tag for immunodetection with an anti-HA antibody. We confirmed that the presence of the short tag (18 aa) did not interfere with tau's binding to AnxA2 (Fig. 1B, top). 2HA-tagged tau constructs were therefore employed in further experiments. To map the interaction to a specific region within tau, we first split tau into two parts. A tau fragment containing the N-terminal projection region and the proline-rich region (aa 1–255) showed interaction, whereas the C-terminal half containing the microtubule-binding region (MBR) and the C-terminal region did not. A further truncation of the N-terminal half showed that the N-terminal projection region (aa 1–171) was sufficient to bind to AnxA2. To test whether the amino acid sequence encoded by the first expressed exon (E1) was sufficient for the tau–AnxA2 interaction, we prepared a construct with a C-terminal fusion to the cytosolic yeast protein Gpm1 (phosphoglycerate mutase) as a carrier, because the remaining tau alone was not stably produced in yeast. We observed that the fusion construct containing E1 co-precipitated with AnxA2, whereas the HA-tagged carrier alone (2HA-Gpm1) did not. To exclude a potential influence of the FLAG epitope on binding, we prepared an additional construct lacking the FLAG sequence (2HA-tau(1–44)-Gpm1). Also, this construct co-precipitated with AnxA2, indicating that the sequence that is encoded by tau's first exon is sufficient for tau's binding to AnxA2. This is also consistent with our previous observation that fetal as well as adult tau bind to AnxA2, because E1 (in contrast to the amino acid sequences encoded by the alternatively spliced exons 2 and 3 at the N terminus) is present in all isoforms. Constructs containing the proline-rich region but lacking the projection domain could not be tested because they tended to nonspecifically precipitate in our pulldown assays, probably due to high aggregation propensity (data not shown).
Tau's first coding exon contains evolutionarily conserved sequence motifs
Although tau, at least in its nonphosphorylated state, is a basic protein, E1 is acidic with a theoretical pI of 4.26. To determine whether tau's first coding exon contains sequence motifs, which are evolutionarily conserved and may therefore also be of functional relevance, we performed bioinformatics analyses. We performed sub-HMM analysis of the pHMM from 49 mammalian full-length sequences of MAPT (27). We identified two 8-aa-long motifs, which are highlighted in Fig. 2A (top). By comparing these motifs with the pHMMs of birds, reptiles, and ray-finned fishes (Actinopterygii), we could follow their development during evolution. Neither of the two motifs was present in ray-finned fishes, whereas motif I was clearly evident also in reptiles, and, with a much lower expectation value, in birds (Fig. 2A, right). The fact that motif II is exclusively present in mammalian sequences may indicate that it represents a functional region peculiar to mammalian evolution. Motif I showed a clear overrepresentation of negatively charged amino acids (glutamate, aspartate), suggesting an involvement in protein–protein interactions through electrostatic forces.
Figure 2.

Bioinformatics analysis and effects of familial and phospho-site mutations on tau's binding to AnxA2. A, pHMM logo of E1 of mammalian tau and pHMMs of birds (Aves), reptiles (Reptilia), and ray-finned fishes (Actinopterygii). Conserved motifs are indicated by boxes in magenta and designated as I and II. Acidic amino acids are represented in red and basic ones in blue. Positions where individual amino acids are mutated in tauopathies and where potentially disease-relevant tyrosine phosphorylation had been reported are indicated by blue boxes. Probabilities (Prob.) and expectation values (E-value) for the two motifs are indicated on the right. B, pulldown assays of annexin (GFP-AnxA2) with the HA-tagged tau projection region (aa 1–171) harboring FTDP-17 mutations R5H and R5L (left) and phospho-blocking and -mimicking mutations of tyrosine 18 (Y18F and Y18E) (right). The mutated constructs co-precipitated to a similar extent (quantitation of bound versus total signal revealed 13.6 ± 9.4 and 15.7 ± 13.8% for Y18F and Y18E, respectively, and 15.0 ± 5.1 and 15.7 ± 9.4% for R5H and R5L constructs, respectively; mean ± S.D. (n = 3)). E1 is indicated in dark green with the conserved sequence motifs shown in magenta.
Tau's binding to annexin A2 is not affected by disease-associated mutations and phospho-mimicking or -blocking mutations of tyrosine 18 within E1
The interaction between tau and annexin might be influenced by disease-associated mutations or phosphorylation in some amino acid residues from the first coding exon. Previously mutations of arginine at position 5 (R5H and R5L) had been observed in tauopathies (28, 29), and phosphorylation of tyrosine 18 had been reported in paired helical filaments from AD brains (30). To test an effect of these modifications on the tau–annexin interaction, we performed pulldown assays from yeast extracts containing tau's N-terminal projection region with R5H and R5L mutations as well as phosphorylation-mimicking and -blocking mutations at tyrosine 18 (Y18E and Y18F). We observed that all constructs co-precipitated with AnxA2 to a similar extent, indicating that the mutations do not affect the interaction of tau with AnxA2 (Fig. 2B). It is noteworthy that both residues are located outside of the evolutionarily conserved motifs in E1, and the data suggest that the tau–annexin interaction is robust against changes in these positions. So far, no changes located within the two conserved regions have been described.
Tau binds to the core domain of AnxA2 in its Ca2+-induced open conformation and interacts also with annexin A6
Annexins consist of a conserved Ca2+- and membrane-binding core domain and a preceding N-terminal region, which is diverse in sequence and length (19) (Fig. 3A, left). To test which part of AnxA2 interacts with tau, we prepared deletion constructs coding only for the N-terminal region (aa 1–34) or the core domain (aa 35–339) of AnxA2, both as C-terminal GFP fusions for co-precipitation assays. We observed that tau co-precipitated with the construct coding for the core domain but failed to do so with the N terminus of AnxA2 (Fig. 3A, right). AnxA2 is known to exist in a closed (absence of Ca2+) and an open conformation (presence of Ca2+), and the N-terminal region may mask tau's binding site to the AnxA2 core domain in the closed conformation (31). Therefore, to test whether the interaction between tau and annexin's core domain remained Ca2+-dependent also in the absence of the N-terminal region, we performed co-precipitation assays of tau's N-terminal projection region with the construct coding for annexin's core domain in the presence and absence of Ca2+. In fact, we observed that binding to the AnxA2 core domain is independent of the presence of Ca2+ (Fig. 3B).
Figure 3.

Tau binds to the core domain of AnxA2 and also interacts with AnxA6. A, pulldown assays of the core domain of AnxA2 (AnxA2(35–339)-GFP) and the N-terminal region (AnxA2(1–34)-GFP) with HA-tagged tau. A schematic representation of the structure of annexin A2 is shown at the top left. Annexin repeats (R1–R4) of the core domain are indicated. Calcium ions are indicated as pink balls. B, pulldown assays of the core domain of AnxA2 (AnxA2(35–339)-GFP) with the HA-tagged tau project region (aa 1–171) in the presence or absence of Ca2+. Tau interacts with the core domain of AnxA2 independent of the presence of Ca2+. The schematic structure of tau is color-coded as described in the legend of Fig. 2. C and D, pulldown assays of AnxA6 (AnxA6-GFP) with the HA-tagged tau projection region (aa 1–171) (C) or HA-tagged tau(1–44)-Gpm1 (D) indicate co-precipitation of tau. Binding of AnxA6 to E1 requires the presence of Ca2+ (D).
The core domain, which consists of the annexin repeats, is conserved among the different annexin subfamilies. Because we have shown that tau binds to the core domain of AnxA2, it might also interact with other members of the annexin family. In mammalian neurons, AnxA6 might be an interesting candidate due to its presence in the AIS. Indeed, tau's N-terminal projection region clearly co-precipitated with AnxA6 after expression in the heterologous yeast system, indicating physical interaction (Fig. 3C). To confirm that the interaction occurs via E1 and to test for a potential Ca2+ dependence, we performed pulldown assays also with the 2HA-tau(1–44)-Gpm1 construct with AnxA6 in the presence and absence of Ca2+. We observed that the construct co-precipitated with AnxA6 in the presence of Ca2+, whereas it failed to do so without Ca2+ (Fig. 3D).
Lack of E1 moderately increases tau's association rate in axon-like processes
As demonstrated, the E1 region of tau is involved in binding of tau to the plasma membrane components AnxA2 and AnxA6. To test whether this additional interaction affects tau's interaction with microtubules, we prepared a tau construct lacking E1 (tauΔE1) and compared it with the behavior of full-length tau (tauWT), both as PAGFP-tagged versions. The constructs were present as single polypeptides in transfected PC12 cells, indicating their integrity (Fig. 4A, left). The difference in electrophoretic mobility was much higher than calculated from the sequence (16.9 kDa versus 5.2 kDa), suggesting that E1 largely contributes to the unusual low electrophoretic mobility of tau protein and forms a stiff domain. To scrutinize the interaction of the two constructs with microtubules in axon-like processes of living cells, we used a fluorescence decay after photoactivation (FDAP) approach (Fig. 4A, right). We had previously shown that the PAGFP fusion does not interfere with tau's interaction with axonal membrane components (16, 17). After neuronal differentiation of transfected PC12 cells, PAGFP was activated within a segment in the middle of the process by a laser flash at 407-nm wavelength, and FDAP was recorded in the activated region as a function of time. Both constructs showed a much slower decay than a non-MT-binding control protein of similar size (3×PAGFP), indicating binding to microtubules (Fig. 4B). Based on the effective diffusion constants, >90% of the constructs were bound to microtubules, which is consistent with previous data on the tau–MT interaction in processes of living cells (16, 32). To directly estimate the pseudo-first-order association rate (k*on = kon[MT]eq, where [MT]eq is the equilibrium concentration of tau-binding sites on MTs) and the dissociation rate (koff), we used a previously developed refined reaction–diffusion model of the tau–MT interaction (33). Absence of E1 led to a moderate but significant increase in k*on but did not influence koff (Fig. 4C), which is consistent with the slight decrease in FDAP of tauΔE1 compared with tauWT (Fig. 4B). The data are consistent with the supposition that the presence of the tau–annexin interaction moderately reduces the availability of tau to interact with MTs (as indicated by the lower k*on value of tauWT compared with tauΔE1) but does not affect its dwell time (the inverse value of koff), once tau is bound on the MT surface.
Figure 4.

Lack of E1 moderately increases tau's association rate with microtubules in axon-like processes. A, schematic representation of WT tau (tauWT) and a tau deletion construct lacking first coding exon (tauΔE1) with N-terminal PAGFP fusion. The PAGFP tag is indicated in turquoise, the schematic structure of tau is color-coded as described in the legend of Fig. 2. An immunoblot of cellular lysates after transfection with the respective tau constructs is shown below. Molecular weights as determined from the electrophoretic separation are indicated below. A schematic representation of the photoactivation approach is shown to the right. Photoactivation was performed in neuronally differentiated PC12 cells. A segment 2R in length in the middle of a cellular process of length L (L ≫ R) was photoactivated, and the fluorescence distribution was monitored over time. B, FDAP plots of PAGFP-tagged tauWT, tauΔE1, and 3×PAGFP as a non-MT-binding control of similar size are shown. Curves represent the mean values of 30–43 experiments. C, bar plots showing k*on and koff values of tauWT and tauΔE1. The numbers are expressed relative to tauWT. The box represents 50% of the population, whiskers range from 5 to 95%, crosses correspond to the minimal and maximal values, the horizontal line shows the median, and the black squares show the mean value. Statistical analysis was performed using Student's t test. Statistically significant differences between the means are indicated. *, p < 0.05 (n = 39 and 43 for tauWT and tauΔE1, respectively).
Competition with the tau–annexin interaction compromises tau's axonal enrichment
AnxA2 is present in high concentrations in neuronal growth cones, whereas AnxA6 becomes concentrated during development in the AIS (21, 22). This localization could implicate a contribution of the tau–annexin interaction to tau's retention in the axon. To test this hypothesis, we prepared Sindbis virus constructs providing transient overexpression of the interacting domain to compete with a potential interaction of endogenous tau with annexin in living primary cortical neurons. We prepared a fusion construct of tau's E1 with triple mCherry (3×mCherry) as a fluorescence marker and a second construct, where we added E2 as a spacer between E1 and the fluorescence marker. As a control, we used 3×mCherry alone. Based on Western blot analysis, we estimated a level of overexpression of the constructs compared with endogenous tau protein of at least 3-fold.
Similar to its distribution in the brain, endogenous tau shows enrichment in one process in cultured primary neurons, which could be identified as axon by morphological criteria (3). Sindbis virus–mediated expression of the control construct (3×mCherry) did not change this distribution, and the majority of infected neurons showed endogenous tau staining, which was largely restricted to the axon (Fig. 5A, top, arrowhead). In contrast, after expression of the competing constructs coding for E1 or for E1 and the amino acid sequence of exon 2 (E2), most of the infected neurons lost the preferential staining of endogenous tau in the axon, and the tau signal was present in multiple processes (Fig. 5A, middle and bottom). Quantification confirmed that overexpression of the competing constructs abolishes the preferential distribution of endogenous tau in one process (Fig. 5A, right; F(2,9) = 179.3, p < 0.001), suggesting that tau's interaction via its first coding exon contributes to its enrichment in the axon.
Figure 5.
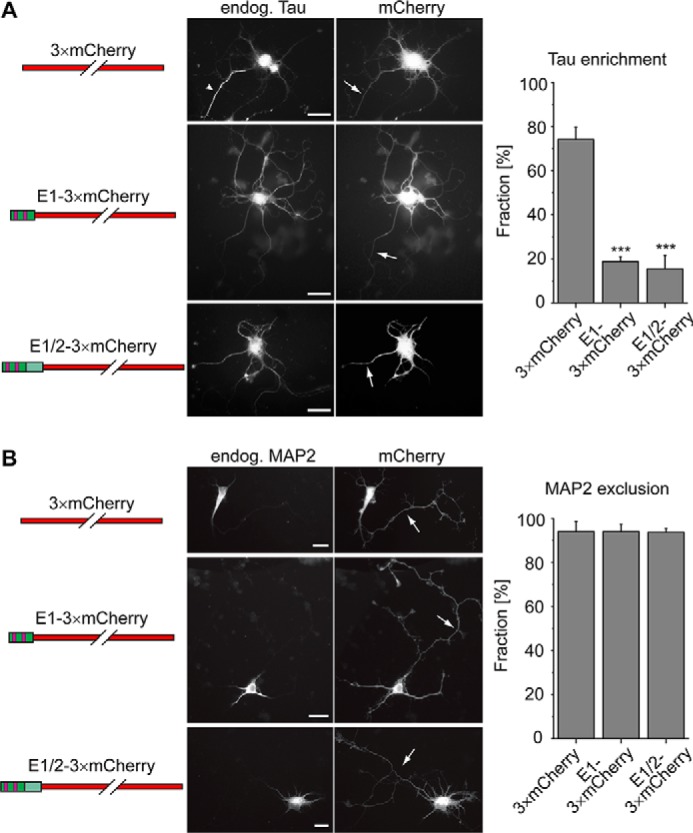
Overexpression of tau's E1 decreases axonal retention of endogenous tau but does not affect MAP2 exclusion from the axon. A, distribution of endogenous tau in primary cortical mouse neurons after viral overexpression of the indicated constructs. Representative fluorescence micrographs are shown in the middle. Arrows indicate axons in the infected neurons as they are evident by morphological criteria. The arrowhead indicates axonal enrichment of tau after expression of the control construct (3×mCherry), which is not present after expression of constructs containing tau's E1. Quantification of the fraction of infected neurons, which exhibit axonal tau enrichment, is shown on the right. B, distribution of endogenous MAP2 in neurons overexpressing the indicated constructs. Arrows, axons. Note the exclusion of MAP2 from the axon after expression of all constructs. Quantification of the fraction of infected neurons, which exhibit axonal exclusion of MAP2, is shown on the right. Tau and MAP2 distribution were classified by visual inspection of a total of 129–199 (tau) and 117–146 (MAP2) infected neurons per construct from four independent experiments. Values are shown as mean ± S.D. (error bars). Statistical analysis was performed using one-way analysis of variance with Tukey's post hoc test for multiple comparison. ***, p < 0.001, compared with the control experiment (expression of 3×mCherry) for tau enrichment; statistical analysis revealed no significant difference between the constructs for MAP2 distribution. Scale bar, 20 μm.
To determine whether the overexpression of constructs containing E1 influence axonal integrity in general, we performed similar infection experiments and stained for the distribution of the somatodendritic marker MAP2, which is known to segregate into dendrites after development of polarity (34) (Fig. 5B). We observed axonal exclusion of MAP2 in the vast majority of neurons after expression of the control construct (3×mCherry), which did not change with constructs containing E1, suggesting that overexpression of E1 specifically affected the distribution of tau.
Discussion
We have previously shown that the neuronal microtubule-associated protein tau interacts with the calcium-regulated plasma membrane-binding protein AnxA2; however, the interaction sites of both proteins and potential functional consequences remained unknown. In this study, we mapped the tau–annexin interaction using a heterologous yeast system, performed bioinformatics analysis of the interacting tau domain, and applied an in-cell competition assay to determine effects on the localization of tau. Our major findings are as follows: 1) tau interacts with AnxA2 via E1 of its N-terminal projection domain, 2) tau binds to the core domain of AnxA2 in the Ca2+-induced open conformation and interacts also with AnxA6 via E1, and 3) competition with the tau–annexin interaction compromises tau's axonal enrichment.
Tau belongs to the class of IDPs, which are known to interact with many partners (11). Whereas the C-terminal half containing the MBR is very similar among the members of the tau/MAP2/MAP4 family (27, 32), interactions of tau's N-terminal projection domain, which extends from the MT surface when tau is bound to microtubules, are likely to mediate the more specific interactions of tau. We have previously shown that tau's projection domain mediates enrichment of tau at distal neurites, probably via interaction with plasma membrane components (15, 16, 35). A recent proteomic study indicated that various membrane-bound proteins interact with N-terminal inserts of tau, providing further evidence for potentially relevant interactions of tau's projection domain with membrane components (36). In this study, we have identified tau's first coding exon as the region that interacts with AnxA2 in a Ca2+-dependent manner. E1 exhibits the maximum distance from tau's MBR with a spacing of ∼19 nm from the microtubule surface, from which the projection domains extend as armlike elements according to previous electron microscopic studies (37) (Fig. 6A). Thus, E1 is well positioned to bridge microtubules with plasma membrane components, which may be of particular importance for the axonal compartment, where the membrane surface/volume ratio is highest. Alternatively, or in addition, AnxA2-bound tau could represent an additional pool of axonal tau, thereby reducing the amount of tau that is available for the interaction with microtubules, which is known to be highly dynamic in axons (14). This is consistent with our observation that a truncated construct of tau, which lacks E1 and is therefore incapable of interacting with AnxA2, shows a moderately increased k*on rate of microtubule binding compared with WT tau (see Fig. 4C). It should, however, be noted that also other factors may affect the change in tau's microtubule interaction (e.g. binding of E1 to components other than annexins or induction of structural changes of tau).
Figure 6.

Schematic representation visualizing the major findings of the study. A, potential 3D structure of tau (tau441WT) based on the Random Coil Generator model. The MBR (yellow) and the position of exon 1 (dark green) together with the two evolutionarily conserved sequence motifs (magenta) were mapped on the structure. The end-to-end distance (distance between amino acids 1 and 441) is ∼25 nm. Tau interacts with the core domain of AnxA2 via the first exon of its N-terminal projection domain in a Ca2+-dependent manner. The interaction with annexin moderately reduces kon. Tau links microtubules to the axonal plasma membrane through its N-terminal projection domain, which protrudes ∼19 nm from the MT surface. B, Tau interacts also with AnxA6, which carries two annexin cores within a single physical entity. AnxA6 localizes to the AIS, where binding of tau may produce a bottleneck contributing to the retention of tau in the axonal compartment of higher vertebrates.
Tau is subject to a variety of post-translational modifications and can carry disease-associated mutations, some of which are located in E1 (38). Phosphorylation of tyrosine 18, which is located between two 8-aa-long conserved motifs, which we identified by bioinformatics analysis, had been observed in paired helical filaments from AD brains (30), and phosphorylation of this residue may be involved in regulating axonal transport (39). Mutations of arginine at position 5 had been identified in a late age of onset case of FTDP-17 (R5H) (28) and progressive supranuclear palsy (R5L) (29). We observed that both phospho-blocking and -mimicking tau mutants at Tyr-18 bound to AnxA2, indicating that the interaction is robust against a negative charge at this position. We also did not observe a change in the annexin interaction of the disease-associated mutations R5H and R5L. It would be informative to model the interaction between tau's E1 and AnxA2 to identify other critical residues and to deduce which modifications may affect the tau–annexin interaction. However, our attempts to predict the 3D structure of tau's E1 using popular (including one of the most efficient) tools like QuickPhyre and Tasser were unsuccessful. Both algorithms reported that the fraction of potential unordered regions is highly flexible and thus did not allow the construction of a suitable 3D model. For the time being, this impedes the presentation of an adequate tau–annexin interaction model.
We observed that the interaction of tau with AnxA2 depended on the presence of Ca2+. It had previously been suggested that annexin exists in a closed (no Ca2+, no membrane) and an open conformation (in the presence of Ca2+ and membrane binding). In the closed conformation, the N-terminal region is thought to integrate into the folded core, whereas Ca2+ binding and membrane binding can then trigger exposure of the N-terminal region in the open conformation (31). This implies that Ca2+ may be required for making annexin's core domain available for the interaction with tau (Fig. 6A). In support of such a hypothesis, we observed that tau interacts with the core domain of AnxA2 and that the tau–AnxA2 interaction becomes Ca2+-independent when the N-terminal region is removed. Interaction with the annexin core domain as a conserved binding module also implied that tau may bind to other members of the annexin family. In mammalian neurons, especially AnxA6 might be an interesting candidate because it is present in the AIS and shows an altered distribution in pathological states (23, 25). Indeed, we here observed that tau also binds to AnxA6 via E1 of its N-terminal projection domain in a Ca2+-dependent manner in the heterologous yeast system. Notably, AnxA6 is the only annexin that contains two annexin core domains within a single physical entity (18), which may imply that even two tau proteins bind to one molecule of AnxA6 (Fig. 6B).
Members of the tau/MAP2/MAP4 family share the conserved C-terminal domain containing the MBR (27) but exhibit distinct localizations in cells, pointing to a role of the N terminus in mediating proper subcellular localization. This is evident for the neuronal MAPs tau and MAP2, which exhibit an axonal and somatodendritic distribution, respectively. Our data provide evidence that the interaction of tau's N-terminal projection domain with neuronal annexins through E1 contributes to its axonal localization. We have shown that E1 contains two 8-aa-long sequence motifs (motif I and II; Fig. 2A), which are evolutionarily conserved and may therefore also be of functional relevance. Notably, both motifs were absent in fish tau, which may indicate that certain interactions of tau developed later during evolution, when nervous systems became more complex and tau and MAP2 developed compartment-specific functions. This may explain the observation that, unlike the predominantly axonal localization of tau in most mammalian species in situ, exogenously expressed tau was equally found in all compartments of lamprey anterior bulbar cells, the most studied neurons in sea lampreys (40). Supporting this view, no evidence for axon-specific localization of tau has been reported in zebrafish (41). Interestingly, among mammals, naked mole rats maintain axonal tau localization during their extraordinary long lifetime (∼32 years), indicating effective mechanisms of axonal retention (42). Motif I is highly conserved in tau from naked mole rats, whereas motif II is partially absent, suggesting that motif I has a primary role in keeping tau in the axon.
It is thought that the AIS plays a role in the selective localization of tau in the axonal compartment (7, 9). We hypothesize that the interaction of tau with annexins, in particular with AnxA6 which is enriched in the AIS, generates a bottleneck, which leads to retention of tau in the axonal compartment of higher vertebrates (Fig. 6B). It was reported previously that axonal retention requires binding of tau to microtubules and that tau redistributes when it is phosphorylated in its repeat domain and detached from microtubules (7). This indicates a requirement for both interaction with annexins and microtubule binding for tau's axonal retention.
Gene-edited endogenous tau also displays strong axonal enrichment, which is distorted when exogenous tau is overexpressed (43), consistent with a competition for annexin or MT binding. Remarkably, recent data also indicate that transgenic expression of tau causes a relocation of the AIS down the axon (44). This suggests that the AIS does not only influence tau distribution but that tau also influences the structure of the AIS. Whether such an effect is mediated via tau's interaction with annexins or whether other interaction partners are involved needs to be shown.
Our results may also contribute to an understanding of the processes that are involved in tau's redistribution during pathology, where tau leaves the axon and becomes enriched in the somatodendritic compartment. Aging alone does not appear to affect the structure of the AIS and the localization of tau, at least not in an aged rat model (45). However, during disease conditions, tau is subject to proteolytic cleavage by the Ca2+-activated cysteine protease, calpain, which is activated by Aβ and cleaves tau at lysine 44 and arginine 230, producing tau fragments with potential neurotoxicity (46–48). Calpain-mediated cleavage at Lys-44 would produce a tau fragment lacking E1, which would be defective in interacting with annexins, thereby losing axonal retention. In addition, disease-associated post-translational modifications may disturb the tau–annexin interaction. A potential candidate is tau acetylation, which has been shown to cause a missorting of tau into the somatodendritic compartment associated with a perturbation of the AIS (9). Finally, a disturbed calcium homeostasis, which is known to be associated with several neurodegenerative diseases (49), may be a common mechanism underlying pathological changes because we have shown that calcium is required for the binding of tau to the neuronal membrane via annexins.
Experimental procedures
Materials and antibodies
Chemicals were obtained from Sigma-Aldrich; cell culture media and supplements from Sigma-Aldrich and Invitrogen; and culture flasks, plates, and dishes from Thermo Fisher Scientific, unless stated otherwise. Synthetic yeast media were from BD Biosciences and applied as described previously (50). The following antibodies were used: anti-tau (Tau5 (mouse; BD Biosciences); ab75714 (chicken; Abcam)), anti-GFP (rabbit; Invitrogen), anti-annexin A2 (H-5; mouse; Santa Cruz Biotechnology, Inc.), and anti-HA (rat and mouse; kindly provided by Anja Lorberg, University of Osnabrück). As secondary antibodies, peroxidase-conjugated goat anti-mouse and goat anti-rabbit (Jackson ImmunoResearch Laboratories, Inc.), and Alexa Fluor 488–conjugated goat anti-chicken antibodies (ab150173, Abcam) were used.
Construction of expression vectors and Sindbis virus preparation
Eukaryotic expression plasmids for tau441WT (tauWT) with N-terminally fused PAGFP tag were constructed in pRc/cytomegalovirus (CMV)-based expression vectors (Life Technologies, Inc.) containing a CMV promoter and kanamycin and neomycin resistance genes. Deletion of E1 from tau (tauWT) generating tauΔE1 was achieved by using site-directed mutagenesis with the following primers: forward, 5′-TCTCCCCTGCAGACCCCC-3′; reverse, 5′-AGATCTGAGTCCGGACTTGTACAG-3′. The pCMV-3×PAGFP plasmid was described previously (16). Sequences for 3×mCherry (pJJH1295, with the fluorophore obtained by PCR from pCM79 (51)), tau(1–44) (pJJH1601), and tau(1–75) (pJJH1602) were cloned into pSinRep5 vector. The pSinRep5 vectors and helper DH(26S)DNA were then transcribed in vitro and co-electroporated into baby hamster kidney (BHK-21) cells, and pseudovirions were harvested as described previously (52).
Cell culture, transfection, and infection
PC12 cells were cultured in serum-Dulbecco's modified Eagle's medium, and transfections were performed with Lipofectamine 2000 (Invitrogen) essentially as described previously (53). For imaging, cells were plated on poly-l-lysine– and collagen–coated glass-bottom culture dishes in Dulbecco's modified Eagle's medium with 1% (v/v) serum and neuronally differentiated with 100 ng/ml 7S mouse nerve growth factor for 4 days as described previously (32). Primary cortical cultures were prepared from cerebral cortices of mouse embryos (day 14–16 of gestation) and cultured as described previously (52). The cultures were obtained by breeding C57BL/6 mice. Cells were plated at 5 × 103 cells/cm2 on polylysine- and laminin-coated coverslips. Sindbis virus was applied at 9 days in vitro, and the cell fixation was performed 24 h later as described previously (52).
Immunocytochemistry
Immunocytochemistry after fixation with 4% paraformaldehyde was performed as described previously (17), using the anti-tau antibody from chicken. Fluorescence microscopy was performed using an oil immersion ×40 (numerical aperture 1.0) objective lens on a fluorescence microscope (Eclipse TE2000-U, Nikon) equipped with a digital camera (COOL-1300, Vosskühler). Infected cells were classified by visual inspection for the presence of tau or MAP2 in neuronal processes. The axonal process was identified by morphological criteria as described previously (32).
Expression of tau and annexin in yeast
The constructs that were expressed in yeast are described in Table 1. In short, recombinant tau constructs were expressed in the yeast strain DHD5 (54) from episomal vectors based on YEp352 (2 μm, URA3 (55)) under the control of the GAL1/10 promoter (pJJH447 (56)). The coding sequences for full-length AnxA2-GFP fusion protein, the AnxA2 core domain (aa 35–339), the AnxA2 N-terminal region (aa 1–34), or AnxA6 were expressed under the control of the constitutive PFK2 promoter (57) from a vector based on YEplac181 (2 μm, LEU2 (58)). Smaller fragments of tau were produced as fusion proteins with the yeast phosphoglycerate mutase Gpm1 (59).
Table 1.
List of constructs expressed in yeast
| Expression plasmid | Insert | Backbone/Promoter | Comment |
|---|---|---|---|
| pJJH1207 | GFP | YEplac181/GAL1-10 | Control without annexin |
| pJJH1190 | AnxA2-GFP | YEplac181/PFK2 | |
| pJJH1443 | AnxA2 (1–34)-GFP | YEplac181/PFK2 | |
| pJJH1431 | AnxA2 (35–339)-GFP | YEplac181/PFK2 | |
| pJJH1597 | AnxA6-GFP | YEplac181/PFK2 | |
| pJJH900 | FLAGtau441WT | YEp352/GAL1-10 | |
| pJJH1283 | 2HA-FLAGtau441WT | YEp352/GAL1-10 | |
| pJJH1288 | 2HA- FLAGtau (1–255) | YEp352/GAL1-10 | |
| pJJH1441 | 2HA-tau (256–441) | YEp352/GAL1-10 | No FLAG tag |
| pJJH1289 | 2HA- FLAGtau (1–171) | YEp352/GAL1-10 | |
| pJJH1418 | 2HA- FLAGtau (1–44)-Gpm1 | YEp352/GAL1-10 | The short tau peptide itself is unstable in yeast, thus the fusion with a small glycolytic enzyme |
| pJJH2236 | 2HA-tau (1–44)-Gpm1 | YEp352/GAL1-10 | Same as pJJH1418 but without FLAG tag |
| pJJH1439 | 2HA-Gpm1 | YEp352/GAL1-10 | Control without tau |
| pJJH1494 | 2HA-FLAGtau (1–171;R5H) | YEp352/GAL1-10 | |
| pJJH1496 | 2HA-FLAGtau (1–171;R5L) | YEp352/GAL1-10 | |
| pJJH1541 | 2HA-FLAGtau (1–171;Y18F) | YEp352/GAL1-10 | |
| pJJH1542 | 2HA-FLAGtau (1–171;Y18E) | YEp352/GAL1-10 |
GFP pulldown assays
For preparation of yeast extracts, cells were grown overnight in 5 ml of synthetic complete medium with 2% glucose (w/v) with omissions of uracil and/or leucine as required for selection of plasmid maintenance. These cultures were used to inoculate 50 ml of fresh synthetic complete medium with 2% galactose (w/v) as a sole carbon source for high-level expression of the tau constructs and incubated for another 14–15 h at 30 °C with shaking. Cells were harvested by centrifugation and washed twice with buffer (50 mm potassium phosphate buffer, pH 7.0) before the preparation of crude extracts with glass beads, as described previously (50). Yeast extracts were prepared from yeast cells expressing recombinant GFP or GFP-annexin constructs and untagged or 2HA-tagged tau constructs in lysis buffer (10 mm Tris-HCl, 150 mm NaCl, 0.15% Nonidet P-40, pH 7.5) in the presence or absence of 1 mm CaCl2. Lysates were diluted to 500 μl with immunoprecipitation buffer (10 mm Tris-HCl, 150 mm NaCl, and 1 mm phenylmethylsulfonyl fluoride, pH 7.5) with or without 1 mm CaCl2. Pulldown assays were performed with GFP-Trap_A beads (ChromoTek) as described previously (17). In short, 50 μl of the lysates (“input”) were saved, the remaining lysate was incubated with the GFP-Trap_A beads, and GFP or GFP-fusion proteins were pulled down by centrifugation. 50 μl of the supernatant were saved. The pellet was washed and resuspended in 100 μl of 2×SDS sample buffer, and the immunocomplexes were dissociated from the beads by boiling. Beads were separated by centrifugation, and the supernatant was saved (GFP pulldown). For immunoblot analysis, 2% of the lysate (input) and 10% each of the supernatant and GFP pulldown fraction were loaded and detected as described previously (60).
Bioinformatics analysis
pHMM was generated by HMMBUILD from collected and manually cured MAPT amino acid sequences and visualized as a HMM logo by the SKYLIGN package (61, 62). The HMM of mammals (49 sequences) was compared with the HMM of other classes (Aves, 15 sequences; Reptilia, 12 sequences; Actinopterygii, 15 sequences). A potential 3D structure of tau (441-aa isoform) was generated by Random Coil Generator software (63), and the domain organization was mapped onto the 3D structure. Visualization and structure rendering was performed, using the Visual Molecular Dynamics package, as a surface representation (64). The random coil model is frequently used to generate conformational ensembles of IDPs.
Live imaging and FDAP analysis of tau–microtubule interaction
Live imaging was performed using a laser-scanning microscope (Eclipse TE2000-U inverted; Nikon, Tokyo, Japan) equipped with argon (488-nm) and violet diode (407-nm) lasers. PAGFP-tau–expressing cells were visualized with a Fluor ×60 (numerical aperture 1.4) UV-corrected objective lens. The microscope was enclosed in an incubation chamber maintained at 37 °C and 5% CO2 (Solent Scientific, Fareham, UK). Photoactivation of a neurite's segment 6 μm in length and automated image acquisition of 112 frames after photoactivation with a frame rate of 1/s were performed as described previously (33). Analysis of individual FDAP curves to estimate directly the pseudo-first-order association rate (k*on) and the dissociation rate (koff) of tau to/from microtubules was performed as described previously (32).
Other methods
Preparation of PC12 cell lysates, protein determination, SDS-PAGE, and immunoblot analysis by enhanced chemiluminescence was performed as described previously (32). Statistical analysis was performed using Student's t test for comparing two means or one-way analysis of variance with Tukey's post hoc test for multiple comparisons. The α levels were defined as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Author contributions
A. G.-K., M. S. A., F. S., B. N., L. B., and R. B. contributed to the acquisition, analysis, and interpretation of data; J. J. H., M.-P. F., and R. B. contributed to the conception and design of the work; R. B. drafted the work. All authors revised the work critically and approved the final version of the manuscript.
Acknowledgments
We thank Drs. Roman Efremov and Anton Chugunov (M. M. Shemyakin and Yu. A. Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, Moscow, Russia) for estimations concerning a potential model for the tau–annexin interaction and Vanessa Herkenhoff for technical assistance.
This work was supported by the DAAD (“Deutscher Akademischer Austauschdienst”) and performed as part of the IPID (“International Promovieren in Deutschland”) doctoral student exchange program from the University of Osnabrück and the University of Oviedo, the FICYT (“Fundación para el Fomento en Asturias de la Investigación Científica Aplicada y la Tecnología”), by computing time by the HLRN (“Norddeutscher Verbund für Hoch- und Höchstleistungsrechnen”), the UV2000/GPU cluster at the computing center of the University of Osnabrück, and the Z-project of the SFB 944, University of Osnabrück. The authors declare that they have no conflicts of interest with the contents of this article.
- MAP
- microtubule-associated protein
- AD
- Alzheimer's disease
- AIS
- axon initial segment
- IDP
- intrinsically disordered protein
- AnxA2 and AnxA6
- annexin A2 and A6, respectively
- aa
- amino acid(s)
- HA
- hemagglutinin
- MBR
- microtubule-binding region
- FDAP
- fluorescence decay after photoactivation
- MT
- microtubule
- E1 and E2
- exon 1 and 2, respectively
- CMV
- cytomegalovirus
- PAGFP
- photoactivatable GFP
- HMM
- hidden Markov model
- pHMM
- profile HMM.
References
- 1. Chapin S. J., and Bulinski J. C. (1992) Microtubule stabilization by assembly-promoting microtubule-associated proteins: a repeat performance. Cell Motil. Cytoskeleton 23, 236–243 10.1002/cm.970230403 [DOI] [PubMed] [Google Scholar]
- 2. Binder L. I., Frankfurter A., and Rebhun L. I. (1985) The distribution of tau in the mammalian central nervous system. J. Cell Biol. 101, 1371–1378 10.1083/jcb.101.4.1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kempf M., Clement A., Faissner A., Lee G., and Brandt R. (1996) Tau binds to the distal axon early in development of polarity in a microtubule- and microfilament-dependent manner. J. Neurosci. 16, 5583–5592 10.1523/JNEUROSCI.16-18-05583.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mandell J. W., and Banker G. A. (1996) A spatial gradient of tau protein phosphorylation in nascent axons. J. Neurosci. 16, 5727–5740 10.1523/JNEUROSCI.16-18-05727.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dixit R., Ross J. L., Goldman Y. E., and Holzbaur E. L. (2008) Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086–1089 10.1126/science.1152993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Spillantini M. G., and Goedert M. (2013) Tau pathology and neurodegeneration. Lancet Neurol. 12, 609–622 10.1016/S1474-4422(13)70090-5 [DOI] [PubMed] [Google Scholar]
- 7. Li X., Kumar Y., Zempel H., Mandelkow E. M., Biernat J., and Mandelkow E. (2011) Novel diffusion barrier for axonal retention of Tau in neurons and its failure in neurodegeneration. EMBO J. 30, 4825–4837 10.1038/emboj.2011.376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sun X., Wu Y., Gu M., Liu Z., Ma Y., Li J., and Zhang Y. (2014) Selective filtering defect at the axon initial segment in Alzheimer's disease mouse models. Proc. Natl. Acad. Sci. U.S.A. 111, 14271–14276 10.1073/pnas.1411837111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sohn P. D., Tracy T. E., Son H. I., Zhou Y., Leite R. E., Miller B. L., Seeley W. W., Grinberg L. T., and Gan L. (2016) Acetylated tau destabilizes the cytoskeleton in the axon initial segment and is mislocalized to the somatodendritic compartment. Mol. Neurodegener. 11, 47 10.1186/s13024-016-0109-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marin M. A., Ziburkus J., Jankowsky J., and Rasband M. N. (2016) Amyloid-β plaques disrupt axon initial segments. Exp. Neurol. 281, 93–98 10.1016/j.expneurol.2016.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Uversky V. N. (2015) Intrinsically disordered proteins and their (disordered) proteomes in neurodegenerative disorders. Front. Aging Neurosci. 7, 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gunawardana C. G., Mehrabian M., Wang X., Mueller I., Lubambo I. B., Jonkman J. E., Wang H., and Schmitt-Ulms G. (2015) The Human Tau interactome: binding to the ribonucleoproteome, and impaired binding of the proline-to-leucine mutant at position 301 (P301L) to chaperones and the proteasome. Mol. Cell. Proteomics 14, 3000–3014 10.1074/mcp.M115.050724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Konzack S., Thies E., Marx A., Mandelkow E. M., and Mandelkow E. (2007) Swimming against the tide: mobility of the microtubule-associated protein tau in neurons. J. Neurosci. 27, 9916–9927 10.1523/JNEUROSCI.0927-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Janning D., Igaev M., Sündermann F., Brühmann J., Beutel O., Heinisch J. J., Bakota L., Piehler J., Junge W., and Brandt R. (2014) Single-molecule tracking of tau reveals fast kiss-and-hop interaction with microtubules in living neurons. Mol. Biol. Cell 25, 3541–3551 10.1091/mbc.E14-06-1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brandt R., Léger J., and Lee G. (1995) Interaction of tau with the neural plasma membrane mediated by tau's amino-terminal projection domain. J. Cell Biol. 131, 1327–1340 10.1083/jcb.131.5.1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weissmann C., Reyher H. J., Gauthier A., Steinhoff H. J., Junge W., and Brandt R. (2009) Microtubule binding and trapping at the tip of neurites regulate tau motion in living neurons. Traffic 10, 1655–1668 10.1111/j.1600-0854.2009.00977.x [DOI] [PubMed] [Google Scholar]
- 17. Gauthier-Kemper A., Weissmann C., Golovyashkina N., Sebö-Lemke Z., Drewes G., Gerke V., Heinisch J. J., and Brandt R. (2011) The frontotemporal dementia mutation R406W blocks tau's interaction with the membrane in an annexin A2-dependent manner. J. Cell Biol. 192, 647–661 10.1083/jcb.201007161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gerke V., Creutz C. E., and Moss S. E. (2005) Annexins: linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 6, 449–461 10.1038/nrm1661 [DOI] [PubMed] [Google Scholar]
- 19. Rescher U., and Gerke V. (2004) Annexins: unique membrane binding proteins with diverse functions. J. Cell Sci. 117, 2631–2639 10.1242/jcs.01245 [DOI] [PubMed] [Google Scholar]
- 20. Bharadwaj A., Bydoun M., Holloway R., and Waisman D. (2013) Annexin A2 heterotetramer: structure and function. Int. J. Mol. Sci. 14, 6259–6305 10.3390/ijms14036259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao W. Q., and Lu B. (2007) Expression of annexin A2 in GABAergic interneurons in the normal rat brain. J. Neurochem. 100, 1211–1223 10.1111/j.1471-4159.2006.04311.x [DOI] [PubMed] [Google Scholar]
- 22. Yamatani H., Kawasaki T., Mita S., Inagaki N., and Hirata T. (2010) Proteomics analysis of the temporal changes in axonal proteins during maturation. Dev. Neurobiol. 70, 523–537 [DOI] [PubMed] [Google Scholar]
- 23. Sánchez-Ponce D., DeFelipe J., Garrido J. J., and Muñoz A. (2011) In vitro maturation of the cisternal organelle in the hippocampal neuron's axon initial segment. Mol. Cell Neurosci. 48, 104–116 10.1016/j.mcn.2011.06.010 [DOI] [PubMed] [Google Scholar]
- 24. Eberhard D. A., Brown M. D., and VandenBerg S. R. (1994) Alterations of annexin expression in pathological neuronal and glial reactions. Immunohistochemical localization of annexins I, II (p36 and p11 subunits), IV, and VI in the human hippocampus. Am. J. Pathol. 145, 640–649 [PMC free article] [PubMed] [Google Scholar]
- 25. Hamre K. M., Chepenik K. P., and Goldowitz D. (1995) The annexins: specific markers of midline structures and sensory neurons in the developing murine central nervous system. J. Comp. Neurol. 352, 421–435 10.1002/cne.903520308 [DOI] [PubMed] [Google Scholar]
- 26. Porzig R., Singer D., and Hoffmann R. (2007) Epitope mapping of mAbs AT8 and Tau5 directed against hyperphosphorylated regions of the human tau protein. Biochem. Biophys. Res. Commun. 358, 644–649 10.1016/j.bbrc.2007.04.187 [DOI] [PubMed] [Google Scholar]
- 27. Sündermann F., Fernandez M. P., and Morgan R. O. (2016) An evolutionary roadmap to the microtubule-associated protein MAP Tau. BMC Genomics 17, 264 10.1186/s12864-016-2590-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hayashi S., Toyoshima Y., Hasegawa M., Umeda Y., Wakabayashi K., Tokiguchi S., Iwatsubo T., and Takahashi H. (2002) Late-onset frontotemporal dementia with a novel exon 1 (Arg5His) tau gene mutation. Ann. Neurol. 51, 525–530 10.1002/ana.10163 [DOI] [PubMed] [Google Scholar]
- 29. Poorkaj P., Muma N. A., Zhukareva V., Cochran E. J., Shannon K. M., Hurtig H., Koller W. C., Bird T. D., Trojanowski J. Q., Lee V. M., and Schellenberg G. D. (2002) An R5L tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann. Neurol. 52, 511–516 10.1002/ana.10340 [DOI] [PubMed] [Google Scholar]
- 30. Lee G., Thangavel R., Sharma V. M., Litersky J. M., Bhaskar K., Fang S. M., Do L. H., Andreadis A., Van Hoesen G., and Ksiezak-Reding H. (2004) Phosphorylation of tau by fyn: implications for Alzheimer's disease. J. Neurosci. 24, 2304–2312 10.1523/JNEUROSCI.4162-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gerke V., and Moss S. E. (2002) Annexins: from structure to function. Physiol. Rev. 82, 331–371 10.1152/physrev.00030.2001 [DOI] [PubMed] [Google Scholar]
- 32. Niewidok B., Igaev M., Sündermann F., Janning D., Bakota L., and Brandt R. (2016) Presence of a carboxy-terminal pseudorepeat and disease-like pseudohyperphosphorylation critically influence tau's interaction with microtubules in axon-like processes. Mol. Biol. Cell 27, 3537–3549 10.1091/mbc.E16-06-0402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Igaev M., Janning D., Sündermann F., Niewidok B., Brandt R., and Junge W. (2014) A refined reaction-diffusion model of tau-microtubule dynamics and its application in FDAP analysis. Biophys. J. 107, 2567–2578 10.1016/j.bpj.2014.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dotti C. G., Sullivan C. A., and Banker G. A. (1988) The establishment of polarity by hippocampal neurons in culture. J. Neurosci. 8, 1454–1468 10.1523/JNEUROSCI.08-04-01454.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maas T., Eidenmüller J., and Brandt R. (2000) Interaction of tau with the neural membrane cortex is regulated by phosphorylation at sites that are modified in paired helical filaments. J. Biol. Chem. 275, 15733–15740 10.1074/jbc.M000389200 [DOI] [PubMed] [Google Scholar]
- 36. Liu C., Song X., Nisbet R., and Götz J. (2016) Co-immunoprecipitation with Tau isoform-specific antibodies reveals distinct protein interactions and highlights a putative role for 2N Tau in disease. J. Biol. Chem. 291, 8173–8188 10.1074/jbc.M115.641902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hirokawa N., Shiomura Y., and Okabe S. (1988) Tau proteins: the molecular structure and mode of binding on microtubules. J. Cell Biol. 107, 1449–1459 10.1083/jcb.107.4.1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Heinisch J. J., and Brandt R. (2016) Signaling pathways and posttranslational modifications of tau in Alzheimer's disease: the humanization of yeast cells. Microb. Cell 3, 135–146 10.15698/mic2016.04.489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stern J. L., Lessard D. V., Hoeprich G. J., Morfini G. A., and Berger C. L. (2017) Phosphoregulation of Tau modulates inhibition of kinesin-1 motility. Mol. Biol. Cell 28, 1079–1087 10.1091/mbc.E16-10-0728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hall G. F., Yao J., and Lee G. (1997) Human tau becomes phosphorylated and forms filamentous deposits when overexpressed in lamprey central neurons in situ. Proc. Natl. Acad. Sci. U.S.A. 94, 4733–4738 10.1073/pnas.94.9.4733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Paquet D., Bhat R., Sydow A., Mandelkow E. M., Berg S., Hellberg S., Fälting J., Distel M., Köster R. W., Schmid B., and Haass C. (2009) A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J. Clin. Invest. 119, 1382–1395 10.1172/JCI37537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Orr M. E., Garbarino V. R., Salinas A., and Buffenstein R. (2015) Sustained high levels of neuroprotective, high molecular weight, phosphorylated tau in the longest-lived rodent. Neurobiol. Aging 36, 1496–1504 10.1016/j.neurobiolaging.2014.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xia D., Gutmann J. M., and Götz J. (2016) Mobility and subcellular localization of endogenous, gene-edited Tau differs from that of over-expressed human wild-type and P301L mutant Tau. Sci. Rep. 6, 29074 10.1038/srep29074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hatch R. J., Wei Y., Xia D., and Götz J. (2017) Hyperphosphorylated tau causes reduced hippocampal CA1 excitability by relocating the axon initial segment. Acta Neuropathol. 133, 717–730 10.1007/s00401-017-1674-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kneynsberg A., and Kanaan N. M. (2017) Aging does not affect axon initial segment structure and somatic localization of tau protein in hippocampal neurons of Fischer 344 rats. eNeuro 4 10.1523/ENEURO.0043-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mercken M., Grynspan F., and Nixon R. A. (1995) Differential sensitivity to proteolysis by brain calpain of adult human tau, fetal human tau and PHF-tau. FEBS Lett. 368, 10–14 10.1016/0014-5793(95)00590-6 [DOI] [PubMed] [Google Scholar]
- 47. Park S. Y., and Ferreira A. (2005) The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates β-amyloid-induced neurodegeneration. J. Neurosci. 25, 5365–5375 10.1523/JNEUROSCI.1125-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lang A. E., Riherd Methner D. N., and Ferreira A. (2014) Neuronal degeneration, synaptic defects, and behavioral abnormalities in tau45–230 transgenic mice. Neuroscience 275, 322–339 10.1016/j.neuroscience.2014.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pchitskaya E., Popugaeva E., and Bezprozvanny I. (2018) Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 70, 87–94 10.1016/j.ceca.2017.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rodicio R., Koch S., Schmitz H. P., and Heinisch J. J. (2006) KlRHO1 and KlPKC1 are essential for cell integrity signalling in Kluyveromyces lactis. Microbiology 152, 2635–2649 10.1099/mic.0.29105-0 [DOI] [PubMed] [Google Scholar]
- 51. Maeder C. I., Hink M. A., Kinkhabwala A., Mayr R., Bastiaens P. I., and Knop M. (2007) Spatial regulation of Fus3 MAP kinase activity through a reaction-diffusion mechanism in yeast pheromone signalling. Nat. Cell Biol. 9, 1319–1326 10.1038/ncb1652 [DOI] [PubMed] [Google Scholar]
- 52. Bakota L., Brandt R., and Heinisch J. J. (2012) Triple mammalian/yeast/bacterial shuttle vectors for single and combined Lentivirus- and Sindbis virus-mediated infections of neurons. Mol. Genet. Genomics 287, 313–324 10.1007/s00438-012-0680-1 [DOI] [PubMed] [Google Scholar]
- 53. Fath T., Eidenmüller J., and Brandt R. (2002) Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer's disease. J. Neurosci. 22, 9733–9741 10.1523/JNEUROSCI.22-22-09733.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kirchrath L., Lorberg A., Schmitz H. P., Gengenbacher U., and Heinisch J. J. (2000) Comparative genetic and physiological studies of the MAP kinase Mpk1p from Kluyveromyces lactis and Saccharomyces cerevisiae. J. Mol. Biol. 300, 743–758 10.1006/jmbi.2000.3916 [DOI] [PubMed] [Google Scholar]
- 55. Hill J. E., Myers A. M., Koerner T. J., and Tzagoloff A. (1986) Yeast/E. coli shuttle vectors with multiple unique restriction sites. Yeast 2, 163–167 10.1002/yea.320020304 [DOI] [PubMed] [Google Scholar]
- 56. Lorberg A., Schmitz H. P., Jacoby J. J., and Heinisch J. J. (2001) Lrg1p functions as a putative GTPase-activating protein in the Pkc1p-mediated cell integrity pathway in Saccharomyces cerevisiae. Mol. Genet. Genomics 266, 514–526 10.1007/s004380100580 [DOI] [PubMed] [Google Scholar]
- 57. Raben N., Exelbert R., Spiegel R., Sherman J. B., Nakajima H., Plotz P., and Heinisch J. (1995) Functional expression of human mutant phosphofructokinase in yeast: genetic defects in French Canadian and Swiss patients with phosphofructokinase deficiency. Am. J. Hum. Genet. 56, 131–141 [PMC free article] [PubMed] [Google Scholar]
- 58. Gietz R. D., and Sugino A. (1988) New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74, 527–534 10.1016/0378-1119(88)90185-0 [DOI] [PubMed] [Google Scholar]
- 59. Heinisch J., von Borstel R. C., and Rodicio R. (1991) Sequence and localization of the gene encoding yeast phosphoglycerate mutase. Curr. Genet. 20, 167–171 10.1007/BF00312781 [DOI] [PubMed] [Google Scholar]
- 60. Leschik J., Welzel A., Weissmann C., Eckert A., and Brandt R. (2007) Inverse and distinct modulation of tau-dependent neurodegeneration by presenilin 1 and amyloid-β in cultured cortical neurons: evidence that tau phosphorylation is the limiting factor in amyloid-β-induced cell death. J. Neurochem. 101, 1303–1315 10.1111/j.1471-4159.2006.04435.x [DOI] [PubMed] [Google Scholar]
- 61. Eddy S. R. (1998) Profile hidden Markov models. Bioinformatics 14, 755–763 10.1093/bioinformatics/14.9.755 [DOI] [PubMed] [Google Scholar]
- 62. Wheeler T. J., Clements J., and Finn R. D. (2014) Skylign: a tool for creating informative, interactive logos representing sequence alignments and profile hidden Markov models. BMC Bioinformatics 15, 7 10.1186/1471-2105-15-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jha A. K., Colubri A., Freed K. F., and Sosnick T. R. (2005) Statistical coil model of the unfolded state: resolving the reconciliation problem. Proc. Natl. Acad. Sci. U.S.A. 102, 13099–13104 10.1073/pnas.0506078102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]