

Abstract
Benzodiazepines are positive allosteric modulators of the GABAA receptor (GABAAR), acting at the α–γ subunit interface to enhance GABAAR function. GABA or benzodiazepine binding induces distinct conformational changes in the GABAAR. The molecular rearrangements in the GABAAR following benzodiazepine binding remain to be fully elucidated. Using two molecular models of the GABAAR, we identified electrostatic interactions between specific amino acids at the α–γ subunit interface that were broken by, or formed after, benzodiazepine binding. Using two-electrode voltage clamp electrophysiology in Xenopus laevis oocytes, we investigated these interactions by substituting one or both amino acids of each potential pair. We found that Lys104 in the α1 subunit forms an electrostatic bond with Asp75 of the γ2 subunit after benzodiazepine binding and that this bond stabilizes the positively modified state of the receptor. Substitution of these two residues to cysteine and subsequent covalent linkage between them increased the receptor's sensitivity to low GABA concentrations and decreased its response to benzodiazepines, producing a GABAAR that resembles a benzodiazepine-bound WT GABAAR. Breaking this bond restored sensitivity to GABA to WT levels and increased the receptor's response to benzodiazepines. The α1 Lys104 and γ2 Asp75 interaction did not play a role in ethanol or neurosteroid modulation of GABAAR, suggesting that different modulators induce different conformational changes in the receptor. These findings may help explain the additive or synergistic effects of modulators acting at the GABAAR.
Keywords: allosteric regulation, GABA receptor, electrostatics, electrophysiology, Xenopus, cysteine-mediated cross-linking, Cys-loop receptor, benzodiazepines, ionotropic receptor, sedative
Introduction
The ionotropic GABAA receptor (GABAAR)2 is a pentameric protein belonging to the Cys-loop superfamily family of ligand-gated ion channels. Various subunits (α1–6, β1–3, γ1–3, δ, ϵ, θ, and π) combine in multiple combinations to form GABAARs. GABA is the predominant inhibitory neurotransmitter in the central nervous system, and its activation of the GABAAR results in anion movement through the integral ion channel pore. Benzodiazepines are used clinically for their sedative, anxiolytic, and anticonvulsant effects. These drugs act at an allosteric site of the GABAAR to positively modulate the channel when activated by an agonist acting at the orthosteric site. Several hypotheses have been suggested to explain the molecular mechanisms of this benzodiazepine enhancement of function, including an increase in the GABA binding affinity of the receptor (1–3), an increase in GABA efficacy (4, 5), and a shift of the receptor toward a “preactivated” state (6).
Different α subunit–containing GABAA receptors account for the various therapeutic indications of benzodiazepines. GABAARs containing α1 subunits are thought to be primarily responsible for the sedative and anticonvulsive effects of benzodiazepines, whereas α2-containing GABAARs are responsible for their anxiolytic effects (7–10). The inability of classic benzodiazepines to distinguish between receptors comprising different α subtypes suggests a conserved molecular mechanism of action. Histidine 101 in the α1,2,3 subunits (103 in α5) plays an important role in benzodiazepine binding with substitution of this residue with arginine rendering receptors less sensitive to benzodiazepines (11). However, the conformational changes in the GABAAR that occur subsequent to benzodiazepine binding are less well understood.
Inter- and intrasubunit electrostatic interactions play important roles in Cys-loop receptor function. For example, electrostatic interactions between residues of adjacent α subunits in the glycine receptor play an important role in its activation (12). Specifically, the aspartate 97 residue is thought to interact with arginine 119 to stabilize the closed state of the glycine receptor, and once this bond is broken after agonist binding, the channel opens. Additionally, electrostatic interactions between aspartic acid 149 and lysine 279 within the same α subunit as well as between aspartic acid 146 and lysine 215 within the same β subunit are implicated in the coupling of GABA binding to the opening of the GABAAR (13, 14). Furthermore, glutamic acid 153 and lysine 196 within the same β subunit of the GABAAR may be involved in stabilizing the open state of the receptor (15). Disulfide trapping experiments have led to insights into the conformational changes that benzodiazepines produce in the GABAAR after binding (16); however, thus far an electrostatic interaction has not been identified in the GABAAR that occurs because of this conformational change.
In the current study, we used homology modeling with published structures to produce models of α1β2γ2 GABAAR. We used these models to identify potential electrostatic interactions occurring before or after the conformational changes produced by benzodiazepine binding, identifying a pair of residues that appear to be interacting in a manner specific for benzodiazepine modulation of the GABAAR.
Results
Molecular modeling identifies possible electrostatic interactions present before and after benzodiazepine binding at the α1–γ2 subunit interface of the GABAAR
As a starting point for our studies, we used two different models to identify potential electrostatic interactions at the α1–γ2 subunit interface. The first was based on molecular dynamic modeling performed by Yoluk et al. (17) on the GluCl ligand-gated Cys-loop receptor in the absence of ivermectin (Fig. 1, left). This first model corresponds to the closed, GABA- and benzodiazepine-unbound state of the GABAAR in our studies. The second model (Fig. 1, right) is based on the GABA- and diazepam-bound GABAA receptor model described by Bergmann et al. (18). Choosing to investigate only charged residues predicted to be 6 Å or less apart, we identified seven interactions that could occur before benzodiazepine binding as well as four interactions that could occur after benzodiazepine binding. Two of these pairs, aspartic acid 56 of the α1 subunit (α1 Asp56) with arginine 197 of the γ2 subunit (γ2 Arg197) and glutamic acid 58 of the α1 subunit (α1 Glu58) with γ2 Arg197 were predicted to form electrostatic pairs both before and after diazepam binding. In the present study, we focused on the electrostatic interactions that were predicted to interact closest to the benzodiazepine binding site (Fig. 1, interactions B–F and H–K).
Figure 1.
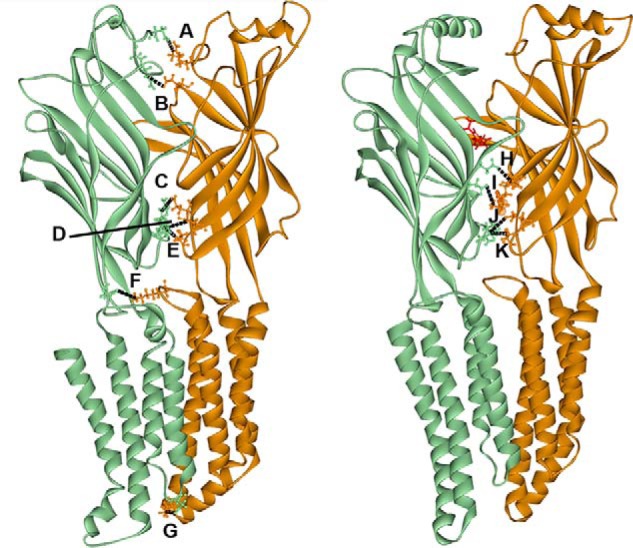
Two different homology models of the α1 (orange)–γ2 (green) interface of the GABAAR. The model on the left is based on a modified ivermectin-unbound GluCl crystal structure (17) and represents the GABA-unbound closed state of the channel. The model on the right is based on the glutamate-bound GluCl crystal structure with a contribution from the ELIC crystal structure (18) and represents the diazepam (in red)-bound receptor. Both models depict the inside of the interface. Labeled interactions represent putative electrostatic interactions of residues 6 Å or less apart that are predicted to occur between residues in the α1 and γ2 subunits before (A–F) or after (H–K) diazepam binding. A, α1 Arg28–γ2 Asp26; 5 Å. B, α1 Glu165–γ2 Arg97; 4 Å. C, α1 Glu137–γ2 Arg194; 5 Å. D, α1 Glu58–γ2 Arg197; 5 Å. E, α1 Asp56–γ2 Arg197; 5 Å. F, α1 Lys278–γ2 Asp161; 5 Å. G, α1 Lys311–γ2 Asp260; 3 Å. H, α1 Lys105–γ2 Asp120; 5 Å. I, α1 Lys104–γ2 Asp75; 5 Å. J, α1 Glu58–γ2 Arg197; 5 Å. K, α1 Asp56–γ2 Arg197; 6 Å. Black dashed lines represent intersubunit bonds.
Effects of cysteine substitution on diazepam potentiation of GABAAR function
Diazepam (1 μm) enhancement of the effects of a GABA concentration required to produce 5–10% of the maximal response (EC5–10), was tested on a series of cysteine mutants. Cysteine substitution of residues of the α1 or γ2 subunit predicted to be involved in electrostatic interactions before and/or after diazepam binding resulted in a significant effect of mutation on diazepam potentiation (see Fig. 2 legend for statistics). Replacing α1 Glu58 with cysteine (α1(E58C)) resulted in a significant increase in diazepam potentiation, whereas the α1(K104C), α1(E137C), γ2(D75C), and γ2(R197C) substitutions all resulted in significant decreases in diazepam enhancement (Fig. 2). Six of the other residues substituted with cysteine, α1(D56C), α1(K105C), α1(E165C), γ2(R97C), γ2(D120C), and γ2(R194C), resulted in no significant changes in receptor enhancement by diazepam compared with WT GABAAR. Of the pairs probed, the only hypothesized pair that produced similar changes in diazepam effects upon mutation to cysteine were α1(K104C) and γ2(D75C) (Fig. 1, interaction I). If an electrostatic interaction was occurring between two residues, one would expect similar changes in receptor function if that bond was broken by mutating either residue. For this reason, we focused on the α1 Lys104–γ2 Asp75 pair. Before diazepam binding, α1 Lys104 was predicted to be ∼9 Å from γ2 Asp75 (Fig. 3, A and B), but after diazepam binding these residues were predicted to move much closer together, to ∼5 Å apart (Fig. 3C).
Figure 2.
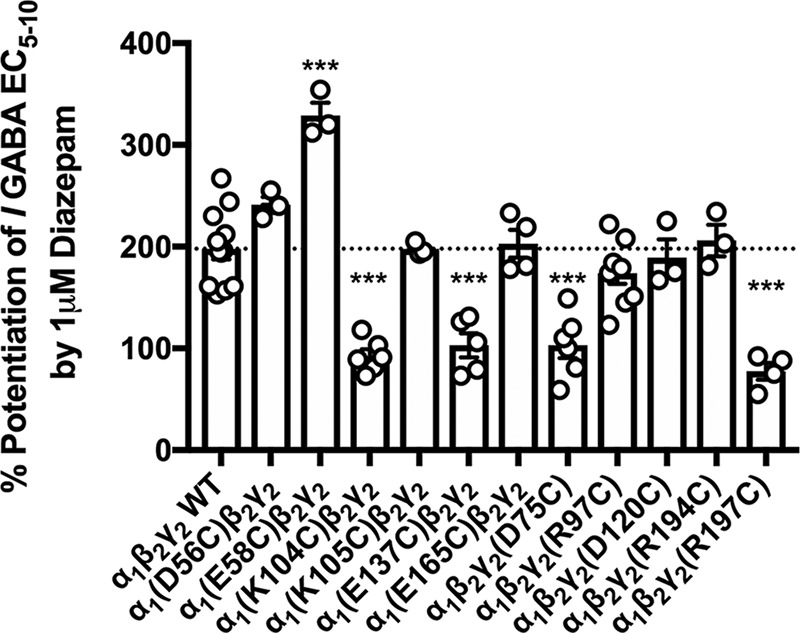
Diazepam enhancement of GABAAR function is altered in some cysteine mutations of residues predicted to form electrostatic interactions at the α1–γ2 subunit interface. EC5–10 GABA was applied alone as well as in the presence of 1 μm diazepam to WT and multiple cysteine-substituted receptors. The horizontal dashed line indicates the level of potentiation produced by diazepam in WT receptors. A one-way ANOVA showed a significant effect of cysteine substitution on receptor enhancement by 1 μm diazepam (F(11,60) = 26.310, p < 0.001). A post hoc Tukey's test showed a significant change (p < 0.001) in potentiation by 1 μm diazepam in α1(E58C, α1(K104C)-, α1(E137C)-, γ2(D75C)-, and γ2(R197C)-containing GABAAR. Each symbol represents the percent potentiation of the GABA EC5–10 seen in one oocyte, and each bar represents the mean percent potentiation. Error bars represent the S.E.
Figure 3.
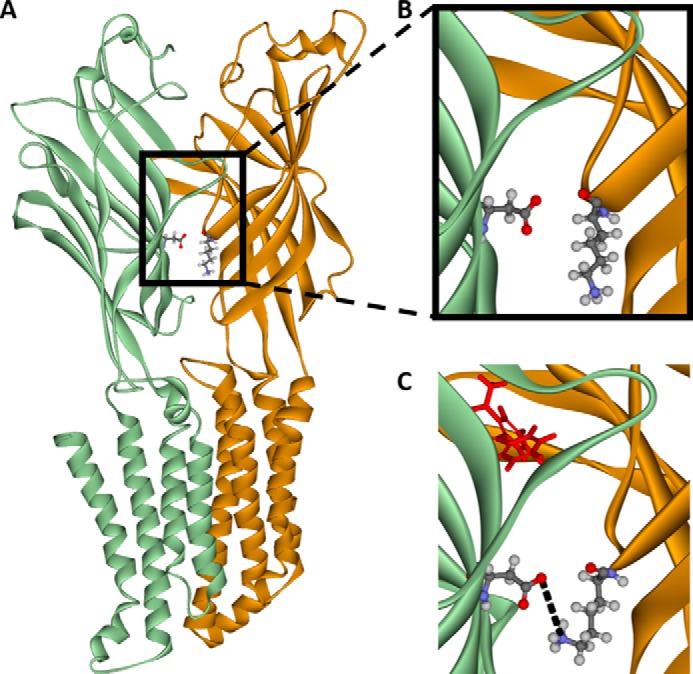
Homology models of the α1 (orange)–γ2 (green) interface inside the GABAA receptor in both the GABA-unbound closed state of the channel and the diazepam-bound open state of the channel. These models predict that the nitrogen atom of α1 lysine 104 (orange residue) and oxygen atom of γ2 aspartic acid 75 (green residue) are within 9 Å of each other before GABA and diazepam bind (A and enlarged in B) but move to within 5 Å of each other after GABA and diazepam (in red) bind to the receptor (C).
Effects of cysteine substitution on GABA sensitivity at α1 Lys104 and γ2 Asp75 residues
GABA concentration-response curves for α1(K104C)β2γ2, α1β2γ2(D75C), and α1(K104C)β2γ2(D75C) receptors did not significantly differ from those of WT receptors (Fig. 4A). However, one-way ANOVAs revealed that lower GABA concentrations (3 and 10 μm) produced greater responses in α1(K104C)β2γ2(D75C) receptors compared with the single mutants and WT receptors (see Fig. 4 legend for statistics). Despite the model-based hypothesis that the electrostatic interaction between α1 Lys104 and γ2 Asp75 is predicted to occur after diazepam binding, substituting these residues with cysteines could allow a disulfide bond to form spontaneously, which would be able to form between residues at greater distances apart than an electrostatic bond. Therefore, we tested whether the disulfide bond between α1(K104C) and γ2(D75C) had spontaneously occurred. The reducing agent dithiothreitol (DTT) is able to break accessible disulfide bonds. Application of 2 mm DTT to the α1(K104C)β2γ2(D75C) receptor resulted in an increase in the GABA EC5 from 3.6 ± 0.4 μm before DTT application to 10.5 ± 0.35 μm after DTT (Fig. 4B). This is due to the breakage of a single intersubunit disulfide bond as shown in Fig. 5A.
Figure 4.

Formation and breakage of the disulfide bond between α1(K104C) and γ2(D75C) affects responses to GABA. A, GABA concentration-response curves were generated in WT α1β2γ2, single mutants α1(K104C)β2γ2 and α1β2γ2(D75C), and double mutant α1(K104C)β2γ2(D75C) GABAA receptors. A repeated-measures ANOVA revealed no difference in the concentration-response curve between WT and mutant receptors. However, one-way ANOVAs showed significant effects of mutation at 3 μm (F(3,26) = 15.504, p < 0.001) and 10 μm GABA (F(3,26) = 18.163, p < 0.001) with a Tukey's post hoc test at both concentrations showing a significant increase in response in α1(K104C)β2γ2(D75C) receptors compared with the other three receptors (***, p < 0.001). Some symbols are hidden behind other symbols. B, DTT (2 mm; dark symbols and bars) increased the absolute concentration of GABA required to produce an EC5 response in α1(K104C)β2γ2(D75C) but not WT receptors. A two-way ANOVA followed by a Tukey's post hoc test revealed a significant effect of DTT treatment on α1(K104C)β2γ2(D75C) receptors (***, p < 0.001). Each symbol represents the GABA EC5 of one oocyte, and each bar represents the mean GABA EC5. Error bars represent the S.E.
Figure 5.

α1(K104C)β2γ2(D75C) receptors spontaneously cross-link and reform this cross-link after DTT application. A, illustration depicting the disulfide bond that spontaneously cross-links the α1 and γ2 subunits of the α1(K104C)β2γ2(D75C) GABAAR, is broken after DTT application, and slowly reforms over time. B, effect of PMTS application on currents elicited by the GABA EC5–10 of WT, α1(K104C)β2γ2, α1β2γ2(D75C), and α1(K104C)β2γ2(D75C) GABAA receptors. The change in GABA EC5–10 currents by PMTS was decreased in single mutant receptors both before (F(3,18) = 14.56, p < 0.05) and after (F(3,18) = 45.45, p < 0.001) DTT application compared with those seen in WT receptors. The change in EC5–10 currents produced by PMTS was not significantly altered after DTT application to WT or single mutant receptors but did significantly change in double mutant receptors (F(3,37) = 41.698, p < 0.001)). A one-way ANOVA showed a significant effect of time after DTT treatment (pre-DTT treatment, 5 min after, and 60 min after) on α1(K104C)β2γ2(D75C) GABAAR (F(2,13) = 108.363, p < 0.001), and a Tukey's post hoc test showed a significant difference between pre-DTT and 5 min after DTT application and a significant difference between 5 min after DTT and 60 min after DTT (***, p < 0.001; each symbol represents an oocyte, and each bar represents the mean response. Error bars represent the S.E.). C, sample tracing showing spontaneous reformation of the α1–γ2 intersubunit disulfide bond in the α1(K104C)β2γ2(D75C) GABAAR. The GABA EC5 measured in the oocyte before DTT application was 3 μm GABA, but after DTT application 3 μm GABA elicited a much smaller response. After ∼60 min, the response to 3 μm GABA had returned to pre-DTT levels. D, time courses of EC5 values plotted for five oocytes expressing α1(K104C)β2γ2(D75C) receptors returning to their pre-DTT values. This can also be interpreted as the time required to reform the disulfide bond after DTT application. The average time to return to half of the pre-DTT EC5 was 26.9 ± 2.5 min.
To further probe whether α1(K104C) and γ2(D75C) spontaneously form a disulfide bond in the α1(K104C)β2γ2(D75C) receptor, we tested propyl methanethiosulfonate (PMTS) for its effects. PMTS is able to covalently bind to free cysteine residues to which it has access. PMTS caused a significant decrease in GABA EC5 current in the single and double mutant receptors (Fig. 5B, hollow bars with open circles). In the WT and both single cysteine mutant receptors, the effect of PMTS remained unchanged after a prior DTT application (Fig. 5B, hollow bars with triangles). This indicates that in single mutant receptors the cysteine-substituted residues do not form disulfide bonds with endogenous cysteines in GABAAR. Because these single mutant and WT receptors exhibited similar changes in response to PMTS before and after DTT application, we did not test these receptors again 60 min after DTT treatment. For the α1(K104C)β2γ2(D75C) receptor, a one-way ANOVA revealed a significant effect of PMTS treatment before, 5 min after, and 60 min after DTT treatment (F(2,13) = 108.363, p < 0.001). Without prior exposure to DTT, application of PMTS resulted in a decrease in current (Fig. 5B, white bar, open circles). However, DTT application before PMTS resulted in an increase in current (solid bar). Waiting 60 min after DTT washout and then applying PMTS resulted in a decrease in current similar to that seen with PMTS application before DTT application. For the double cysteine mutant receptor, the white bar with open circles represents PMTS binding to the single available cysteine residue situated between the α and β subunit interfaces as shown in the illustration on the left in Fig. 5A. When DTT breaks the sole disulfide bond between α and γ subunits, PMTS can now bind to up to three free cysteines. Because there was no significant difference between PMTS application before DTT application and 60 min after DTT application, we hypothesize that the disulfide bond breakage produced by DTT is only temporary and that the receptor spontaneously returns to its pre-DTT form within an hour. The reformation of the disulfide bond in the double mutant receptor was also seen experimentally by repeatedly applying the GABA EC5 to the DTT-treated receptor and observing a gradual increase in current (Fig. 5, C and D). The current produced by a maximally effective concentration of GABA was not changed by applying DTT (data not shown).
Effect of cysteine substitution at α1 Lys104 and γ2 Asp75 on benzodiazepine-site responses
There were no effects of DTT application on the modulation produced by 1 μm diazepam, flunitrazepam, Ro 15-4513, or zolpidem on WT receptors as expected. This was also the case for α1(K104C)β2γ2 and α1β2γ2(D75C) receptors. However, application of DTT to the α1(K104C)β2γ2(D75C) receptor produced an increase in diazepam potentiation (from 76.6 ± 6.3 to 136.7 ± 9%) and flunitrazepam potentiation (from 121.2 ± 9.1 to 201 ± 22.3%) and a decrease in potentiation by Ro 15-4513 (from 1 ± 3.2 to −17.8 ± 1.9%) (Fig. 6, A–C). DTT treatment rescued responses of α1(K104C)β2γ2(D75C) receptors to WT levels by flunitrazepam and Ro 15-4513 but not by diazepam. Application of the nonclassical benzodiazepine zolpidem did not produce a significant interaction between receptor mutant and DTT treatment, but a Tukey's post hoc test revealed a small significant difference between pre- and post-DTT treatment in α1(K104C)β2γ2(D75C) receptors (p < 0.05) (Fig. 6D). Interestingly, Ro 15-4513 produced a greater inhibitory response in the α1β2γ2(D75C) mutant compared with WT receptors both before and after DTT treatment (Fig. 6C). Treatment with 0.3% hydrogen peroxide (H2O2) for 90 s, which would favor cysteine bond reformation, before the benzodiazepine application reversed the effects of DTT in α1(K104C)β2γ2(D75C) receptors but produced no changes in responses by WT receptors (Fig. 7, A and B).
Figure 6.

Benzodiazepine responses of WT and mutant GABAA receptors before (white symbols and bars) and after (dark symbols and bars) DTT application. Bar graphs show the percent potentiation of GABA EC5–10 in WT, α1(K104C)β2γ2, α1β2γ2(D75C), and α1(K104C)β2γ2(D75C) GABAA receptors produced by 1 μm diazepam (A), flunitrazepam (B), Ro 15-4513 (C), and zolpidem (D). A two-way ANOVA followed by a Tukey's multiple comparison post hoc test showed a significant increase in all benzodiazepine-site responses after DTT application to α1(K104C)β2γ2(D75C) GABAA receptors but not WT or single mutant receptors (*, p < 0.05; ***, p < 0.001 with each symbol representing the percent potentiation of the GABA EC5–10 seen in one oocyte, and each bar representing the mean percent potentiation. Error bars represent the S.E.).
Figure 7.

Sample tracings showing the effects of DTT and H2O2 treatment on potentiation by 1 μm diazepam and 1 μm flunitrazepam. The top panels show tracings obtained from oocytes expressing WT receptors, and the bottom panels show tracings of oocytes expressing α1(K104C)β2γ2(D75C) GABAA receptors. DTT application to α1(K104C)β2γ2(D75C) receptors increased both diazepam (A) and flunitrazepam (B) potentiation, and hydrogen peroxide application reversed this increase back to pre-DTT levels.
Effects of cysteine substitution on nonbenzodiazepine modulators of the GABAAR
Allosteric modulators of the GABAAR acting at sites other than the benzodiazepine-binding site were next tested to determine the specificity of the electrostatic interactions between α1(K104C) and γ2(D75C). Ethanol and the neurosteroid allopregnanolone produced similar potentiation of the effects of GABA on WT, α1(K104C)β2γ2, α1β2γ2(D75C), and α1(K104C)β2γ2(D75C) GABAARs (Fig. 8). There was no significant effect of DTT treatment on the enhancement of WT or mutant receptors by 200 mm ethanol, 100 nm allopregnanolone (Fig. 8), or 1 μm allopregnanolone (data not shown).
Figure 8.
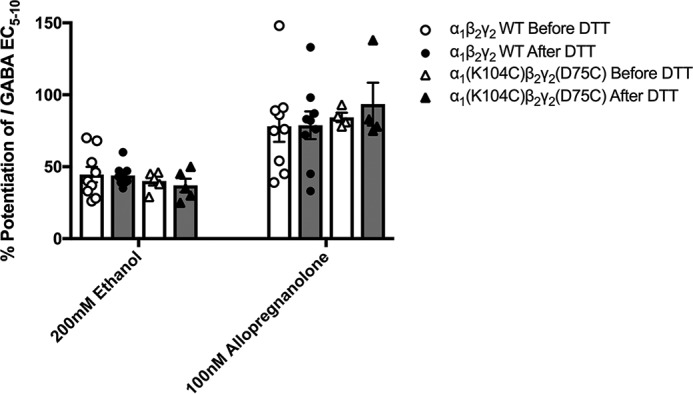
Modulators acting at sites other than the benzodiazepine site at WT and cysteine-substituted GABAA receptors are unaffected by DTT treatment. Before modulator effects were tested, the EC5–10 concentration of GABA was determined in each oocyte. Effects of 200 mm ethanol and 100 nm allopregnanolone were measured in WT and α1(K104C)β2γ2(D75C) GABAA receptors before (white symbols and bars) and after (dark symbols and bars) application of DTT with no significant changes being observed (two-way ANOVAs; 200 mm ethanol (F(3,43) = 0.031); 100 nm allopregnanolone (F(3,45) = 0.176). Each symbol represents the percent potentiation of the GABA EC5–10 seen in one oocyte, and each bar represents the mean percent potentiation. Error bars represent the S.E.
Effects of alanine substitution at α1 Lys104 and γ2 Asp75 on GABA and benzodiazepine responses
To examine the effects of alanine substitutions at the α1 Lys104 and/or γ2 Asp75 residues, we compared receptors containing these alanine residues to WT receptors in their responses to GABA, 1 μm diazepam, and 1 μm flunitrazepam. The GABA concentration-response curve for α1β2γ2(D75A) was slightly right-shifted (EC50, 133.6 ± 19.4 μm), whereas that of the α1(K104A)β2γ2 receptor was slightly left-shifted (EC50, 61.9 ± 12.2 μm) compared with the WT receptor curve (EC50, 77 ± 8.8 μm) (Fig. 9A). A repeated-measures ANOVA revealed a significant difference among the four concentration-response curves (see Fig. 9A legend for statistics). Interestingly, the α1(K104A)β2γ2(D75A) GABA concentration-response curve (EC50, 87.3 ± 12.4 μm) was not left-shifted at lower concentrations (3 and 10 μm), unlike the α1(K104C)β2γ2(D75C) receptor (Fig. 9A compared with Fig. 4A). One-way ANOVAs revealed that there was a significant effect of alanine substitution on the enhancement of GABA EC5–10 by 1 μm diazepam or flunitrazepam. Although the single substitution α1(K104A)β2γ2 and α1β2γ2(D75A) receptors exhibited a decreased response to diazepam and flunitrazepam compared with WT receptors, the α1(K104A)β2γ2(D75A) receptors displayed a level of potentiation not significantly different from that of WT receptors (Fig. 9B).
Figure 9.

Effect of alanine substitution at α1 Lys104 and γ2 Asp75 on GABA sensitivity and benzodiazepine responses. A, GABA concentration-response curves of WT, α1(K104A)β2γ2, α1β2γ2(D75A), and α1(K104A)β2γ2(D75A) receptors. The concentration-response curves were significantly different (F(21,132) = 1.937, p < 0.05). Each symbol represents the data from three to six oocytes, and error bars represent the S.E. In some cases, error bars fall within symbols. B, bar graph comparing levels of diazepam and flunitrazepam potentiation between WT and alanine-substituted receptors. Potentiation of GABA EC5–10 by 1 μm diazepam and 1 μm flunitrazepam was decreased for single but not double alanine substitution mutants compared with WT receptors. A one-way ANOVA revealed a significant effect of mutant on receptor potentiation by diazepam (F(3,23) = 51.407, p < 0.001) and flunitrazepam (F(3,19) = 26.926, p < 0.001). Each symbol represents the percent potentiation observed in one oocyte, and each bar represents the mean percent potentiation. Error bars represent the S.E.
Effects of charge reversal of α1 Lys104 and γ2 Asp75 residues on GABA and GABA receptor modulator responses
To test whether reversing the charges of α1 Lys104 and γ2 Asp75 would restore GABA sensitivity, GABA concentration-response curves of α1(K104D)β2γ2(D75K) were compared with those of WT receptors (Fig. 10A). A repeated-measures ANOVA found a significant difference between WT and α1(K104D)β2γ2(D75K) concentration-response curves (see Fig. 10A legend for statistics). The average EC50 value for WT receptors was 86.8 ± 16.5 μm, whereas the EC50 for α1(K104D)β2γ2(D75K) was increased to 146.3 ± 23.1 μm. The charge reversal did not restore to WT levels receptor potentiation by 1 μm diazepam or Ro 15-4513 but did restore potentiation by 1 μm flunitrazepam and zolpidem (Fig. 10B). Other GABAA receptor modulators (200 mm ethanol and 100 nm allopregnanolone) displayed no changes in potentiation of GABA EC5–10 after charge reversal compared with WT receptors (data not shown).
Figure 10.

Charge reversal at α1 Lys104 and γ2 Asp75 does not rescue GABA sensitivity or benzodiazepine responses to WT responses. A, the α1(K104D)β2γ2(D75K) receptor GABA concentration-response curve is significantly right-shifted compared with WT receptors (F(8,105) = 2.8, p < 0.01). The EC50 for WT receptors was 86.8 ± 16.5 μm, increasing to 146.3 ± 23.1 μm for the α1(K104D)β2γ2(D75K) GABAAR. Each symbol represents the mean from five to six oocytes, and error bars represent the S.E. B, bar graph comparing levels of benzodiazepine enhancement between α1(K104D)β2γ2(D75K) and WT receptors. The α1(K104D)β2γ2(D75K) GABAAR was unable to fully rescue responses to WT levels of potentiation by 1 μm diazepam and Ro 15-4513 but was able to rescue the responses to 1 μm flunitrazepam and zolpidem. Each symbol represents the percent potentiation of the GABA EC5–10 seen in one oocyte, and each bar represents the mean potentiation observed. Error bars represent the S.E.
Discussion
Signal transduction of ligand-gated ion channels after neurotransmitter binding to its orthosteric site is believed to involve a wave of structural rearrangements (19) in the receptor, and this rearrangement is thought to be separate from the signal transduction pathway produced by allosteric modulators (20). Using molecular modeling to identify potential electrostatic interactions between the α1 and γ2 subunits, we identified an interaction between α1 Lys104 and γ2 Asp75 that occurs after diazepam binding (Fig. 3). It is likely that these residues interact to stabilize the positively modified state of the receptor and that this interaction is specific for the benzodiazepine signal transduction pathway.
Low concentrations of GABA produced greater responses in α1(K104C)β2γ2(D75C) than in WT receptors, but this was not seen at higher GABA concentrations (Fig. 4A). This increased response at low GABA concentrations is similar to what one would expect to see in response to coapplication of GABA with a benzodiazepine in WT receptors. Benzodiazepine site agonists increase the effects of low but not higher concentrations of GABA because the ion channel approaches its maximal open probability at saturating GABA concentrations (21).
One would predict that a receptor that is behaving as though a benzodiazepine molecule has already bound would exhibit a decreased response to a coapplication of benzodiazepine with GABA. In the α1(K104C)β2γ2(D75C) receptor, the two cysteine residues spontaneously formed a disulfide bond. Accordingly, we saw the expected decrease in diazepam, flunitrazepam, and zolpidem potentiation in the double cysteine–substituted receptors (Fig. 6A, B, and D). After the disulfide bond is broken with DTT, responses to these benzodiazepines increase, suggesting that an electrostatic bond between these residues in WT receptors formed in response to benzodiazepine binding. After DTT application, the response of α1(K104C)β2γ2(D75C) receptors to flunitrazepam potentiation was rescued to WT levels (Fig. 6B). One reason why potentiation by flunitrazepam, but not diazepam or zolpidem, may be completely rescued following DTT application is that the latter two show weaker modulatory responses after α1(K104C)–γ2(D75C) disulfide bond formation than flunitrazepam, i.e. lower potentiation of GABA responses before DTT application. The conformational rearrangement within the GABAA receptor after benzodiazepine binding most likely depends on the formation of multiple bonds not just α1 Lys104–γ2 Asp75. Thus, the α1 Lys104–γ2 Asp75 bond may be less important for flunitrazepam potentiation than for diazepam or zolpidem.
Zolpidem is a nonclassical benzodiazepine and at low concentrations is selective for the α1 subunit–containing GABAAR over those containing other α subunits (22). Disulfide trapping within the γ2 subunit has shown that the conformational change produced by classical benzodiazepines may not be the same as that produced by zolpidem (16). Similarly, there are mutations in the α1 and γ2 subunits that affect classical but not nonclassical benzodiazepines or vice versa (23–26). The magnitude of the increase in α1(K104C)β2γ2(D75C) receptor potentiation by zolpidem after DTT application was far smaller than the increase seen with the classical benzodiazepines diazepam and flunitrazepam (Fig. 6). We speculate that this may be due to classical and nonclassical benzodiazepines producing overlapping but distinct conformational changes in the GABAAR after binding.
One might hypothesize that the conformational changes produced by benzodiazepines would be different from those produced by inverse agonists such as Ro 15-4513. Indeed, previous studies have shown that this might be the case where disulfide trapping at the α–γ interface of the GABAAR, which affected benzodiazepine potentiation, had no effect on inverse benzodiazepine inhibition (16). Our work supports this hypothesis as the α1(K104C)β2γ2(D75C) mutant GABAAR, which traps the receptor in a “positively modified” state, was not inhibited by Ro 15-4513 as much as WT receptors (Fig. 6C). Once DTT is applied, the α1(K104C)β2γ2(D75C) receptor is relieved of this positively modified state, and Ro 15-4513 is able to produce inhibition to levels similar to that of WT receptors (Fig. 6C). Ro 15-4513 produced more inhibition in the α1β2γ2(D75C) receptor than in the WT, α1(K104C)β2γ2, and α1(K104C)β2γ2(D75C) receptors. Substituting the γ2 Asp75 residue with a lysine or alanine residue increased the inhibition by Ro 15-4513 even more so than the cysteine replacement at that residue (data not shown). This decrease suggests that the γ2 Asp75 residue may be involved in the conformational change produced by Ro 15-4513 as well as a distinct conformational change produced by potentiating benzodiazepines.
After DTT breaks the disulfide bond in α1(K104C)β2γ2(D75C) receptors, one might expect the receptor to behave similarly to the α1(K104A)β2γ2(D75A) receptor. Although the α1(K104A)β2γ2(D75A) receptor exhibited levels of diazepam and flunitrazepam potentiation similar to WT receptors (Fig. 9B), DTT treatment to α1(K104C)β2γ2(D75C) receptors did not fully restore levels of diazepam potentiation (Fig. 6A). One possible explanation for this is that DTT treatment, which breaks the disulfide bond by reducing each mutant cysteine, results in two hydrogen-bound cysteine residues that would occupy more volume than alanine residues at those positions, preventing the conformational change produced by diazepam from occurring. Another possibility is that, in the α1(K104C)β2γ2(D75C) receptor, the spontaneous reformation of a disulfide bond after DTT treatment (Fig. 5, C and D) prevents one from experimentally capturing the maximal amount of enhancement produced by diazepam.
The α1(K104D)β2γ2(D75K) receptor, bearing two charge-reversing substitutions, displayed a right-shifted GABA concentration-response curve compared with WT receptors (Fig. 10A). Additionally, the α1(K104D)β2γ2(D75K) receptor did not restore GABA, diazepam, or Ro 15-4513 sensitivity to WT levels (Fig. 10B). This is likely because the α1 Lys104 and γ2 Asp75 residues lie within a pocket of charges and that modifying these residues is preventing other interactions from occurring; i.e. although K104D and D75K substitutions may restore the electrostatic interaction between these residues, there are other charged residues near these sites that may now interact differently with the reversed charge residues compared with the original WT amino acids. Evidently, the α1 Lys104–γ2 Asp75 interaction is not the only interaction that is important for producing the positively modified state of the receptor. If it were, one would see no benzodiazepine potentiation of the receptor after mutating the α1 Lys104 and γ2 Asp75 residues.
One might argue that the data obtained from the alanine substitution experiments at α1 Lys104 and γ2 Asp75 do not fit our overall hypothesis that formation of a bond between these two residues facilitates benzodiazepine effects at the GABAA receptor. Perhaps what is happening is that during the conformational changes produced by benzodiazepine site agonists at WT receptors these two charged residues come close enough together to at least partially neutralize each other's charges. This hypothesis is supported by results obtained using the single alanine substitutions, which retain single charged residues in each pair and display weaker effects of benzodiazepines than those seen in the double alanine mutant (Fig. 9B). A possible explanation may be that the retained charged residue in the single mutants may still be interacting with other nearby charged residues (e.g. α1 Lys105, γ2 Asp148, and γ2 Arg197), thus retarding the ability of the receptor to adopt the benzodiazepine-activated conformational state. This would also apply to the single cysteine substitutions, which also display decreased responses to diazepam and flunitrazepam. In scenarios in which the α1 104 and γ2 75 residues are in close proximity (e.g. cysteines cross-linked), the receptor has already adopted a benzodiazepine positively modified state, and thus adding exogenous benzodiazepine does not have much effect. In cases where these two residues are not initially close together but are capable of moving closer together (e.g. the double alanine substitutions or the double uncross-linked cysteines), a greater effect of applied benzodiazepine will be seen. Lastly, in scenarios in which one or the other of these residues is constrained in its movement (e.g. single substitutions), benzodiazepine effects would be smaller due to the remaining charged residue.
An initial concern was that the receptor mutants were not being incorporated correctly on cell surfaces and that oocytes were expressing primarily α1β2 receptors, not α1β2γ2 receptors. Previous studies used ZnCl2 to test for γ2 subunit incorporation as zinc inhibits α1β2 receptors to a greater extent than α1β2γ2 GABAARs (27). However, using this test may not be the most accurate way to test for αβ contamination as even a small fraction of α1β2 receptors present may produce a significant inhibitory effect by zinc (28). Interestingly, the α1β2 receptors display an increase in their GABA-evoked currents after DTT treatment, but α1β2γ2 receptor currents are unchanged (29). In our study, we saw no change in GABA-evoked currents after DTT treatment in WT and mutant receptors except for α1(K104C)β2γ2(D75C) receptors for which we actually saw a decrease in GABA-evoked currents (Fig. 5D). This, together with the fact that we injected receptor cDNAs in a 1:1:10 α1:β2:γ2 cDNA ratio and still saw a benzodiazepine effect, engenders confidence that the receptors are incorporating WT and mutated γ2 subunits.
One interesting and clinically relevant aspect of this study revolves around the additive and synergistic properties of GABAAR modulators. Benzodiazepines are often coabused with ethanol (30), and the two classes of compounds are thought to act additively or synergistically as central nervous system depressants. Although ethanol is thought to act at the α+–β− interface in αβδ GABAARs, it is not clear whether this is necessarily the case in αβγ receptors (31). We tested whether mutations that affect both GABA and benzodiazepine responses also produced changes in ethanol responses. In the α1(K104C)β2γ2(D75C) positively modified receptor, no changes in ethanol potentiation were observed (Fig. 8). Similarly, the charge reversal α1(K104D)β2γ2(D75K) receptor exhibits ethanol potentiation similar to that of WT receptors (data not shown). These data suggest that the conformational changes in the GABAAR produced by ethanol are experimentally separable from the conformational changes produced by benzodiazepines and that both can occur simultaneously to further enhance receptor function. This provides a possible molecular mechanism for the synergistic/additive effects of benzodiazepines and alcohol.
The neurosteroid allopregnanolone acts as a potent modulator of the GABAAR as well as a direct activator at high concentrations. The binding site for this enhancing action is thought to be within a cavity formed by transmembrane domains 1 and 4 within a single α subunit (32, 33). The α1(K104C)β2γ2(D75C) positively modified receptor and α1(K104D)β2γ2(D75K) charge reversal receptor displayed no differences in their sensitivities to the potentiating effects of allopregnanolone compared with WT receptors (Fig. 8). As well as having distinct binding sites, our data suggest that allopregnanolone and benzodiazepines produce distinct conformational changes in the GABAAR.
In summary, our study suggests that an intersubunit electrostatic interaction between α1 Lys104 and γ2 Asp75 occurs after benzodiazepine site agonist binding to help stabilize the GABAAR in a positively modified state. This interaction seems to be more important for classical (nonselective between GABAAR α subunits) benzodiazepines than nonclassical (α1-selective) compounds. Additionally, this interaction does not seem to be important for modulators of the GABAAR acting at nonbenzodiazepine sites, suggesting that the α1 Lys104–γ2 Asp75 interaction is specific for benzodiazepine site agents.
Experimental procedures
Reagents
All chemicals were purchased from Sigma-Aldrich unless otherwise stated below.
Structural modeling
Two homology models of the GABAAR were generated using the Modeler module of Discovery Studio 2016 (Biovia, San Diego, CA) as described previously (34). The first model was of the GABAAR in the benzodiazepine-unbound state. This was built using the GluCl X-ray structure in the absence of ivermectin (17) as a template. This template was produced by starting with the structure of GABAAR with five ivermectin molecules bound (Protein Data Bank code 3RHW), removing the five ivermectin molecules, and then running extensive constrained molecular dynamics simulations using GROMACS 4.5. The resulting model was judged to be in the closed/resting state because the subunits moved closer by 2.0 Å and the pore diameter decreased by 1.2 Å (17). The second homology model illustrated GABAAR after diazepam was bound. This model was based on a GluCl/ELIC X-ray structure that modeled diazepam binding (18). It should be noted that other investigators have proposed a different orientation of diazepam docking at this α–γ interface (35). Because the latter template was built using a novel method of combining coordinates from two X-ray structures, it deserves some comment. The model is based primarily on the glutamate-bound GluCl crystal structure (Protein Data Bank code 3RIF) with a contribution of the ELIC crystal structure (Protein Data Bank code 2VLO) that the authors identified as leading to the best alignment and the best composite structure. Of interest for the present results, in GABAAR α1, lysine 104 is in β strand 4, and all coordinates are from GluCl. However, in GABAAR γ2, aspartic acid 75 is in β strand 2; this residue is conserved in ELIC but not in GluCl. As a result, Bergman et al. (18) used the ELIC structure as a template for residues 75–77.
Because both templates are homopentamers and our goal was to measure intersubunit interactions, we prepared a composite sequence by linking GABAAR α1/β2/α1/β2/γ2 and aligning the composite with the sequence of the two templates (36). Then the GABAAR sequences were trimmed to match the length of the template sequences as needed. The two pairs of aligned sequences were submitted to the Modeler module of Discovery Studio 2016.
Both of the resulting homology models were assigned the CHARMm force field in Discovery Studio 2016, minimized, and then subjected to molecular dynamics simulations at 300 K as described previously (34). These two models were analyzed for possible electrostatic interactions using Discovery Studio 2016.
Site-directed mutagenesis
Human cDNAs encoding α1, β2, and γ2 GABAAR subunits, subcloned into a pBK-CMV vector, were used in this study. Point mutations were introduced in the α1 and γ2 subunits using a QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). These mutations were confirmed with dsDNA sequencing.
Harvesting, isolation, and injection of Xenopus laevis oocytes
X. laevis (Nasco, Fort Atkinson, WI) were housed in an Association for Assessment and Accreditation of Laboratory Animal Care International–accredited facility in a room kept at 17 °C and under a 12-h light/dark cycle in tanks monitored for water pH and conductivity. Oocytes were surgically removed in accordance with the National Institutes of Health guidelines under a protocol approved by the Institutional Animal Care and Use Committee of the University of Texas at Austin and placed in a hypertonic solution (108 mm NaCl, 1 mm EDTA, 2 mm KCl, and 10 mm HEPES). The thecal and epithelial layers of Stage V and VI oocytes were manually removed using forceps. Isolated oocytes were transferred to a solution (83 mm NaCl, 2 mm KCl, 1 mm MgCl2, and 5 mm HEPES) containing 0.5 mg/ml collagenase from Clostridium histolyticum for 10 min to enzymatically remove the follicular layer of the oocytes. The animal poles of oocytes were then injected using a Nanoject II (Drummond Scientific Co., Broomall, PA) with 1.5 ng/30 nl human α1, β2, and γ2 GABAAR subunit cDNAs in a 1:1:10 ratio. Oocytes were stored singly in 96-well plates containing incubation medium (88 mm NaCl, 1 mm KCl, 2.4 mm NaHCO3, 10 mm HEPES, 0.82 mm MgSO4·7H2O, 0.33 mm Ca(NO3)2, 0.91 mm CaCl2, 2 mm sodium pyruvate, 0.5 mm theophylline, 10 units/liter penicillin, and 10 mg/liter streptomycin). The oocytes were kept at room temperature (20 °C) and away from light.
Two-electrode voltage clamp electrophysiology
Oocytes expressed GABAARs 1–3 days postinjection with cDNA, and all electrophysiological recordings were completed within this time. An oocyte was placed in a 100-μl bath containing ND-96 buffer (96 mm NaCl, 2 mm KCl, 1 mm MgCl2, 1.8 mm CaCl2, and 5 mm HEPES, pH 7.5). The bath was continuously perfused with ND-96 buffer at a rate of 2 ml/min through 18-gauge polyethylene tubing connected to a Masterflex peristaltic pump (Cole Parmer Instruments, Vernon Hills, IL). The tips of two KCl-filled borosilicate glass electrodes, with a resistance of 0.5–10 megaohms, were placed into the animal pole of the oocyte, and it was voltage-clamped at −80 mV using an OC-725C oocyte clamp (Warner Instruments, Hamden, CT). Electrophysiological data were collected at a rate of 1 kHz using a digitizer (PowerLab ML866) and LabChart (version 7.4.7) software (both from ADInstruments, Australia).
Concentration-response curve generation and analysis
Concentration-response data were collected for WT α1β2γ2 GABAAR or the α1(K104C)β2γ2, α1(K104A)β2γ2, α1β2γ2(D75C), α1β2γ2(D75A), α1(K104C)β2γ2(D75C), α1(K104A)β2γ2(D75A), or α1(K104D)β2γ2(D75K) mutants. Once voltage-clamped, the oocyte was exposed to a maximally effective concentration of GABA (100 mm) for 10 s. Following a 10-min washout with ND-96 buffer to allow resensitization of the receptors, increasing concentrations of GABA (3 μm–10 mm) were applied for 20–30 s, allowing 5–10 min of washout between applications. Another maximally effective concentration of GABA (100 mm) was applied at the end of the experiment so that any drift (up or down) of current throughout the experiment could be corrected. The responses to increasing concentrations of GABA were fit to the Hill equation using SigmaPlot 11.0 (Systat Software, San Jose, CA).
GABAAR modulator responses
Responses to modulators (1 μm diazepam, 1 μm flunitrazepam, 1 μm flumazenil, 1 μm Ro 15-4513, 1 μm zolpidem, 100 nm and 1 μm allopregnanolone, and 200 mm ethanol) were recorded in oocytes expressing WT or mutant receptors. 10 mm stock solutions of all modulators (made with 0.1% DMSO in ND-96 buffer) except ethanol were stored at −20 °C and diluted in ND-96 buffer before use.
The GABA EC5–10, the concentration of GABA that produces 5–10% of the maximal response, was first determined and then repeatedly applied for 30 s followed by 3-min ND-96 buffer washouts until responses were stable. Once stable, oocytes were preincubated for 30 s with a modulator followed immediately by a coapplication of modulator plus GABA EC5–10. The allosteric modulation was calculated as ((IGABA + Modulator/IGABA) − 1) × 100.
DTT and H2O2 treatment
DTT and H2O2 were made fresh in ND-96 buffer before each experiment. The GABA EC5–10 was determined and applied at 3-min intervals until stable responses were obtained. This was repeated after a 2-min DTT (2 mm) application during which the oocyte was unclamped from −80 mV during the 5-min washout and after a 90-s application of 0.3% H2O2 (oocyte unclamped during the 7-min washout). To measure the effects of DTT and H2O2 on allosteric modulation, the GABA EC5–10 was determined and ensured to be stable. GABA was then applied in the presence of allosteric modulator as described previously.
PMTS treatment
A 300 mm PMTS (Toronto Research Chemicals, Canada) stock solution in DMSO was stored at −20 °C and diluted to 0.5 mm in ND-96 buffer before each experiment. The GABA EC5–10 was determined and applied at 3-min intervals until stable responses were observed. Oocytes were then unclamped from −80 mV and treated with 0.5 mm PMTS for 60 s. After a 2-min wash, oocytes were reclamped to −80 mV, and the same GABA EC5–10 was reapplied. Percent changes in current were calculated as ((IGABA after PMTS/IGABA before PMTS) − 1) ×100. This was repeated after a 2-min treatment with 2 mm DTT, waiting 5 or 60 min after application before applying PMTS.
Author contributions
N. C. P., G. L. C., and S. J. M. conceptualization; N. C. P., A. W. D., and J. R. T. data curation; N. C. P., G. L. C., and S. J. M. formal analysis; N. C. P. and S. J. M. funding acquisition; N. C. P. and J. R. T. visualization; N. C. P. methodology; N. C. P. writing-original draft; N. C. P., A. W. D., G. L. C., J. R. T., and S. J. M. writing-review and editing; S. J. M. resources; S. J. M. supervision; S. J. M. project administration.
This work was supported by National Institutes of Health Ruth L. Kirschstein National Research Service Award F31DA042564 (to N. C. P.) from the National Institute on Drug Abuse. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- GABAAR
- γ-aminobutyric acid receptor type A
- EC
- effective concentration
- PMTS
- propyl methanethiosulfonate
- ANOVA
- analysis of variance
- GluCl
- glutamate-gated chloride channel
- ELIC
- Erwinia chrysanthemi ligand-gated ion channel.
References
- 1. Rogers C. J., Twyman R. E., and Macdonald R. L. (1994) Benzodiazepine and β-carboline regulation of single GABAA receptor channels of mouse spinal neurones in culture. J. Physiol. 475, 69–82 10.1113/jphysiol.1994.sp020050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Twyman R. E., Rogers C. J., and Macdonald R. L. (1989) Differential regulation of γ-aminobutyric acid receptor channels by diazepam and phenobarbital. Ann. Neurol. 25, 213–220 10.1002/ana.410250302 [DOI] [PubMed] [Google Scholar]
- 3. Study R. E., and Barker J. L. (1981) Diazepam and pentobarbital: fluctuation analysis reveals different mechanisms for potentiation of γ-aminobutyric acid responses in cultured central neurons. Proc. Natl. Acad. Sci. U.S.A. 78, 7180–7184 10.1073/pnas.78.11.7180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Downing S. S., Lee Y. T., Farb D. H., and Gibbs T. T. (2005) Benzodiazepine modulation of partial agonist efficacy and spontaneously active GABAA receptors supports an allosteric model of modulation. Br. J. Pharmacol. 145, 894–906 10.1038/sj.bjp.0706251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Campo-Soria C., Chang Y., and Weiss D. S. (2006) Mechanism of action of benzodiazepines on GABAA receptors. Br. J. Pharmacol. 148, 984–990 10.1038/sj.bjp.0706796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gielen M. C., Lumb M. J., and Smart T. G. (2012) Benzodiazepines modulate GABAA receptors by regulating the preactivation step after GABA binding. J. Neurosci. 32, 5707–7515 10.1523/JNEUROSCI.5663-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Löw K., Crestani F., Keist R., Benke D., Brünig I., Benson J. A., Fritschy J. M., Rülicke T., Bluethmann H., Möhler H., and Rudolph U. (2000) Molecular and neuronal substrate for the selective attenuation of anxiety. Science 290, 131–134 10.1126/science.290.5489.131 [DOI] [PubMed] [Google Scholar]
- 8. Mckernan R. M., Rosahl T. W., Reynolds D. S., Sur C., Wafford K. A., Atack J. R., Farrar S., Myers J., Cook G., Ferris P., Garrett L., Bristow L., Marshall G., Macaulay A., Brown N., et al. (2000) Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABAA receptor α1 subtype. Nat. Neurosci. 3, 587–592 10.1038/75761 [DOI] [PubMed] [Google Scholar]
- 9. Morris H. V., Dawson G. R., Reynolds D. S., Atack J. R., and Stephens D. N. (2006) Both α2 and α3 GABAA receptor subtypes mediate the anxiolytic properties of benzodiazepine site ligands in the conditioned emotional response paradigm. Eur. J. Neurosci. 23, 2495–2504 10.1111/j.1460-9568.2006.04775.x [DOI] [PubMed] [Google Scholar]
- 10. Rudolph U., Crestani F., Benke D., Brünig I., Benson J. A., Fritschy J. M., Martin J. R., Bluethmann H., and Möhler H. (1999) Benzodiazepine actions mediated by specific γ-aminobutyric acidA receptor subtypes. Nature 401, 796–800 10.1038/44579 [DOI] [PubMed] [Google Scholar]
- 11. Wieland H. A., Lüddens H., and Seeburg P. H. (1992) A single histidine in GABAA receptors is essential for benzodiazepine agonist binding. J. Biol. Chem. 267, 1426–1429 [PubMed] [Google Scholar]
- 12. Todorovic J., Welsh B. T., Bertaccini E. J., Trudell J. R., and Mihic S. J. (2010) Disruption of an intersubunit electrostatic bond is a critical step in glycine receptor activation. Proc. Natl. Acad. Sci. U.S.A. 107, 7987–7992 10.1073/pnas.1001845107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kash T. L., Jenkins A., Kelley J. C., Trudell J. R., and Harrison N. L. (2003) Coupling of agonist binding to channel gating in the GABAA receptor. Nature 421, 272–275 10.1038/nature01280 [DOI] [PubMed] [Google Scholar]
- 14. Kash T. L., Dizon M. J., Trudell J. R., and Harrison N. L. (2004) Charged residues in the β2 subunit involved in GABAA receptor activation. J. Biol. Chem. 279, 4887–4893 10.1074/jbc.M311441200 [DOI] [PubMed] [Google Scholar]
- 15. Venkatachalan S. P., and Czajkowski C. (2008) A conserved salt bridge critical for GABAA receptor function and loop C dynamics. Proc. Natl. Acad. Sci. U.S.A. 105, 13604–13609 10.1073/pnas.0801854105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hanson S. M., and Czajkowski C. (2011) Disulphide trapping of the GABAA receptor reveals the importance of the coupling interface in the action of benzodiazepines. Br. J. Pharmacol. 162, 673–687 10.1111/j.1476-5381.2010.01073.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yoluk O., Brömstrup T., Bertaccini E. J., Trudell J. R., and Lindahl E. (2013) Stabilization of the GluCl ligand-gated ion channel in the presence and absence of ivermectin. Biophys. J. 105, 640–647 10.1016/j.bpj.2013.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bergmann R., Kongsbak K., Sørensen P. L., Sander T., and Balle T. (2013) A unified model of the GABAA receptor comprising agonist and benzodiazepine binding sites. PLoS One 8, e52323 10.1371/journal.pone.0052323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grosman C., Zhou M., and Auerbach A. (2000) Mapping the conformational wave of acetylcholine receptor channel gating. Nature 403, 773–776 10.1038/35001586 [DOI] [PubMed] [Google Scholar]
- 20. Venkatachalan S. P., and Czajkowski C. (2012) Structural link between γ-aminobutyric acid type A (GABAA) receptor agonist binding site and inner β-sheet governs channel activation and allosteric drug modulation. J. Biol. Chem. 287, 6714–6724 10.1074/jbc.M111.316836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Newland C. F., Colquhoun D., and Cull-Candy S. G. (1991) Single channels activated by high concentrations of GABA in superior cervical ganglion neurones of the rat. J. Physiol. 432, 203–233 10.1113/jphysiol.1991.sp018382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Langer S. Z., Faure-Halley C., Seeburg P. H., Graham D., and Arbilla S. (1992) The selectivity of zolpidem and alpidem for the α1-subunit of the GABAA receptor. Eur. Neuropsychopharmacol. 2, 232–234 10.1016/0924-977X(92)90081-I [DOI] [Google Scholar]
- 23. Mihic S. J., Whiting P. J., Klein R. L., Wafford K. A., and Harris R. A. (1994) A single amino acid of the human γ-aminobutyric acid type A receptor γ2 subunit determines benzodiazepine efficacy. J. Biol. Chem. 269, 32768–32773 [PubMed] [Google Scholar]
- 24. Buhr A., and Sigel E. (1997) A point mutation in the γ2 subunit of γ-aminobutyric acid type A receptors results in altered benzodiazepine binding site specificity. Proc. Natl. Acad. Sci. U.S.A. 94, 8824–8829 10.1073/pnas.94.16.8824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bowser D. N., Wagner D. A., Czajkowski C., Cromer B. A., Parker M. W., Wallace R. H., Harkin L. A., Mulley J. C., Marini C., Berkovic S. F., Williams D. A., Jones M. V., and Petrou S. (2002) Altered kinetics and benzodiazepine sensitivity of a GABAA receptor subunit mutation [γ2(R43Q)] found in human epilepsy. Proc. Natl. Acad. Sci. U.S.A. 99, 15170–15175 10.1073/pnas.212320199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hanson S. M., and Czajkowski C. (2008) Structural mechanisms underlying benzodiazepine modulation of the GABAA receptor. J. Neurosci. 28, 3490–3849 10.1523/JNEUROSCI.5727-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Draguhn A., Verdorn T. A., Ewert M., Seeburg P. H., and Sakmann B. (1990) Functional and molecular distinction between recombinant rat GABAA receptor subtypes by Zn2+. Neuron 5, 781–788 10.1016/0896-6273(90)90337-F [DOI] [PubMed] [Google Scholar]
- 28. Boileau A. J., Baur R., Sharkey L. M., Sigel E., and Czajkowski C. (2002) The relative amount of cRNA coding for γ2 subunits affects stimulation by benzodiazepines in GABAA receptors expressed in Xenopus oocytes. Neuropharmacology 43, 695–700 10.1016/S0028-3908(02)00036-9 [DOI] [PubMed] [Google Scholar]
- 29. Amato A., Connolly C. N., Moss S. J., and Smart T. G. (1999) Modulation of neuronal and recombinant GABAA receptors by redox reagents. J. Physiol. 517, 35–50 10.1111/j.1469-7793.1999.0035z.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Substance Abuse and Mental Health Services Administration, Center for Behavioral Health Statistics and Quality (2014) The DAWN Report: Benzodiazepines in Combination with Opioid Pain Relievers or Alcohol: Greater Risk of More Serious ED Visit Outcomes, Rockville, MD: [PubMed] [Google Scholar]
- 31. Wallner M., Hanchar H. J., and Olsen R. W. (2014) Alcohol selectivity of β3-containing GABAA receptors: evidence for a unique extracellular alcohol/imidazobenzodiazepine Ro 15-4513 binding site at the α+β− subunit interface in αβ3δ GABAA receptors. Neurochem. Res. 39, 1118–1126 10.1007/s11064-014-1243-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Akk G., Covey D. F., Evers A. S., Steinbach J. H., Zorumski C. F., and Mennerick S. (2007) Mechanisms of neurosteroid interactions with GABAA receptors. Pharmacol. Ther. 116, 35–57 10.1016/j.pharmthera.2007.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hosie A. M., Wilkins M. E., da Silva H. M., and Smart T. G. (2006) Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature 444, 486–489 10.1038/nature05324 [DOI] [PubMed] [Google Scholar]
- 34. McCracken M. L., Gorini G., McCracken L. M., Mayfield R. D., Harris R. A., and Trudell J. R. (2016) Inter- and intra-subunit butanol/isoflurane sites of action in the human glycine receptor. Front. Mol. Neurosci. 9, 45 10.3389/fnmol.2016.00045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ci S., Ren T., and Su Z. (2008) Investigating the putative binding-mode of GABA and diazepam within GABAA receptor using molecular modeling. Protein J. 27, 71–78 10.1007/s10930-007-9109-9 [DOI] [PubMed] [Google Scholar]
- 36. Trudell J. (2002) Unique assignment of inter-subunit association in GABAA α1β3γ2 receptors determined by molecular modeling. Biochim. Biophys. Acta 1565, 91–96 10.1016/S0005-2736(02)00512-6 [DOI] [PubMed] [Google Scholar]