

Abstract
Opine dehydrogenases (ODHs) from the bacterial pathogens Staphylococcus aureus, Pseudomonas aeruginosa, and Yersinia pestis perform the final enzymatic step in the biosynthesis of a new class of opine metallophores, which includes staphylopine, pseudopaline, and yersinopine, respectively. Growing evidence indicates an important role for this pathway in metal acquisition and virulence, including in lung and burn-wound infections (P. aeruginosa) and in blood and heart infections (S. aureus). Here, we present kinetic and structural characterizations of these three opine dehydrogenases. A steady-state kinetic analysis revealed that the three enzymes differ in α-keto acid and NAD(P)H substrate specificity and nicotianamine-like substrate stereoselectivity. The structural basis for these differences was determined from five ODH X-ray crystal structures, ranging in resolution from 1.9 to 2.5 Å, with or without NADP+ bound. Variation in hydrogen bonding with NADPH suggested an explanation for the differential recognition of this substrate by these three enzymes. Our analysis further revealed candidate residues in the active sites required for binding of the α-keto acid and nicotianamine-like substrates and for catalysis. This work reports the first structural kinetic analyses of enzymes involved in opine metallophore biosynthesis in three important bacterial pathogens of humans.
Keywords: siderophore, dehydrogenase, metal homeostasis, bacterial pathogenesis, protein structure, metallophore, opine, pseudopaline, staphylopine, yersinopine
Introduction
Opine dehydrogenases (ODHs)2 are a family of NAD(P)H-dependent oxidoreductases (EC 1.5.1.-) that catalyze the condensation of an α or ω amino group from an amino acid with an α-keto acid followed by reduction with NAD(P)H, producing a family of products known as N-(carboxyalkyl) amino acids or opines (Fig. 1) (1). Opines are composed of a variety α-keto acid and amino acid substrates and have diverse functional roles. Octopine, isolated from octopus muscle by Morizawa in 1927, was the first described opine, composed of pyruvate and arginine (2). ODHs are widespread in cephalopods and mollusks, such as Pecten maximus, the king scallop, where they allow the continuation of glycolysis under anaerobic conditions by shunting pyruvate into opine products and regenerating NAD+ (3). Opines are also associated with crown gall tumor pathogenesis caused by Agrobacterium tumifaciens. In these plant infections, ODHs are encoded by the Ti plasmid. When expressed in the infected plant cell, they convert plant metabolites into opines providing nutrients to the pathogen. Variant strains of A. tumifaciens have been shown to encode ODHs utilizing α-keto acids such as glyoxylate, pyruvate, and α-ketoglutarate and amino acids such as l-histidine, l-arginine, l-lysine, and l-methionine (4). ODHs have also been characterized in soil bacteria such as Pseudomonas putida (5), the yeast Saccharomyces cerevisiae (6), and Arthrobacter sp. strain 1C, which incorporates glyoxylate or pyruvate and several different amino acids into a variety of opines (7, 8). These examples highlight the existence of diverse products by this enzyme family.
Figure 1.
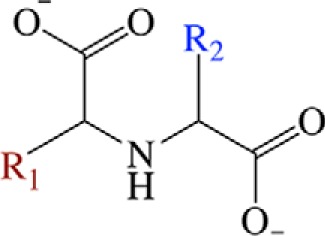
Generalized α-NH2 opine. R1 (red) corresponds to the amino acid side chain. R2 (blue) corresponds to the variable position for the α-keto acid substrate.
Recently, novel opine compounds acting as metallophores have been identified (9–11), with variants found in Staphylococcus aureus (staphylopine) and Pseudomonas aeruginosa (pseudopaline). The occurrence of an additional variant in Yersinia pestis (yersinopine) has also been predicted and is explored further in this work. These opine metallophores play an important role in metal acquisition, with staphylopine involved in zinc (12), cobalt, nickel, and iron uptake (9) and pseudopaline involved in zinc, cobalt (13), and iron (14) uptake. These metallophores are also associated with the pathogenesis of several disease states. In S. aureus, murine bacteremia and urinary tract infections are attenuated when the staphylopine receptor CntA is deleted (15). Additionally, staphylopine production is up-regulated in an endocarditis rat model (16). P. aeruginosa burn wound and respiratory infections are attenuated for pseudopaline export transport mutants (14), and the pseudopaline pathway is up-regulated in a murine acute pneumonia model (17) and in clinical isolates from burn and respiratory infections in humans (18, 19). These findings suggest an important role for opine metallophores in infectious diseases.
Opine metallophore biosynthesis requires two enzymes, a nicotianamine synthase (NAS) and an ODH. The nicotianamine synthase is an S-adenosyl-l-methionine (SAM)-dependent aminoalkyl transferase that forms a secondary amine between an amino acid and the aminobutyrate moiety of SAM (Fig. 2). These enzymes are commonly associated with the production of the plant metallophore nicotianamine, composed of three aminobutyrate moieties derived from SAM (20). P. aeruginosa NAS uses available l-histidine along with SAM in the production of l-His-nicotianamine (l-HisNA), the substrate for PaODH. S. aureus, however, encodes a third enzyme, a histidine racemase, that generates d-histidine for use by the SaNAS in the production of d-His-nicotianamine (d-HisNA), the substrate for SaODH. Each ODH binds HisNA substrate performing a reductive condensation with an α-keto acid, followed by reduction using NAD(P)H, and generating the opine metallophore product.
Figure 2.

Opine metallophore biosynthesis. Generalized reaction scheme for opine metallophore production by NAS and ODH. SaODH recognizes d-HisNA (nicotianamine incorporating d-histidine), whereas YpODH and PaODH recognize l-HisNA (nicotianamine incorporating l-histidine). R represents the α-keto acid side chain incorporated by the ODH enzymes. MTA, methylthioadenosine.
We observe differential substrate specificity by yersinopine dehydrogenase (YpODH, Y. pestis), pseudopaline dehydrogenase (PaODH, P. aeruginosa), and staphylopine dehydrogenase (SaODH, S. aureus). The substrate specificity for each enzyme was demonstrated using a steady-state kinetic approach, and the structural determinants of this specificity were investigated using X-ray crystallography.
Results and discussion
Substrate specificity
We previously reported the enzymatic production of pseudopaline by the P. aeruginosa NAS and ODH (10), and confirmed the production of staphylopine by the homologous S. aureus enzymes as first demonstrated by Ghssein et al. (9). In that work, we examined the utilization of different α-keto acids by the ODH enzymes observing differential specificity. To better define this specificity, we determined steady-state kinetic parameters for the ODHs in the presence of pyruvate, oxaloacetate, and glyoxylate (YpODH and SaODH) or α-ketoglutarate (PaODH). Each ODH requires d- or l-HisNA substrate made by NAS. To produce this substrate in excess, NAS was first incubated with SAM and d-His (SaNAS) or l-His (PaNAS) before adding ODH and NAD(P)H to the parent mix. Reactions were then initiated by combining the parent mix with α-keto acid and measuring a loss of absorbance at 340 nm as NAD(P)H is oxidized to NAD(P)+.
SaODH selects the α-keto acid substrate pyruvate (Table 1 and Fig. S1) but also exhibits limited turnover with oxaloacetate and glyoxylate. The SaODH kcat for oxaloacetate is equivalent to pyruvate, but kcat/Km is 49-fold lower. For glyoxylate, kcat is 3.5-fold lower, and kcat/Km is 370-fold lower. Catalysis in the presence of α-ketoglutarate was within an error of 0 as previously reported (10). In contrast, PaODH uses α-ketoglutarate (Table 1 and Fig. S1). It is able to incorporate α-ketoglutarate with either NADH or NADPH as a substrate with a 2-fold higher catalytic efficiency using NADPH. This suggests that either NADPH or NADH may be used in vivo, dependent upon availability. We observe very limited catalysis for PaODH in the presence of glyoxylate (apparent kcat = 0.015 s−1; 3-fold higher than the detection limit of 0.005 s−1). Catalysis is within error of 0 for pyruvate and oxaloacetate as previously reported (10). YpODH selects pyruvate as its primary substrate (Table 1 and Fig. S1). The YpODH kcat for oxaloacetate is equivalent to pyruvate, but kcat/Km is 28-fold lower. For glyoxylate, kcat is 1.4-fold lower, and kcat/Km is 35-fold lower. Catalysis with α-ketoglutarate was within error of the detection limit. These data indicate that glyoxylate and oxaloacetate are not effective substrates for these enzymes. YpODH and SaODH are specific for pyruvate, whereas PaODH is specific for α-ketoglutarate.
Table 1.
Kinetic parameters
Reaction buffer was 50 mm potassium phosphate, pH 8.0 (Yp and Pa) or Tris, pH 8.0 (Sa), and 5% glycerol. HisNA was produced by reacting histidine with S-adenosyl-l-methionine and PaNAS (l-HisNA) or SaNAS (d-HisNA). For reactions initiated with α-keto acid, ODH at 2 μm and 400 μm (Pa and Sa) or 600 μm (Yp) NAD(P)H were combined with the NAS reaction mixture in one syringe and mixed 1:1 in a stopped-flow spectrometer with α-keto acid in reaction buffer in a second syringe. For reactions initiated with NAD(P)H, ODH at 2 μm and 4 mm pyruvate (Sa and Yp) or α-ketoglutarate (Pa) were combined with the NAS reaction mixture in one syringe and mixed 1:1 with 400 μm (Pa and Sa) or 600 μm NAD(P)H (Yp). Secondary plots are found in the supporting information.
| Protein | Initiating substrate | NAD(P)H | α-Keto acid | HisNA substrate | kcat (s−1) | Km (μm) | kcat/Km (m−1s−1) |
|---|---|---|---|---|---|---|---|
| SaODH | Pyruvate | NADPH | d-HisNA | 0.26 ± 0.01 | 29 ± 2 | 8900 ± 700 | |
| SaODH | Pyruvate | NADPH | l-HisNA | 0.85 ± 0.01 | 280 ± 2 | 3000 ± 40 | |
| SaODH | Oxaloacetate | NADPH | d-HisNA | 0.26 ± 0.01 | 1400 ± 100 | 180 ± 10 | |
| SaODH | Glyoxylate | NADPH | d-HisNA | 0.074 ± 0.003 | 3000 ± 200 | 24 ± 2 | |
| PaODH | α-Ketoglutarate | NADPH | l-HisNA | 0.42 ± 0.01 | 14 ± 1 | 31000 ± 2000 | |
| PaODH | α-Ketoglutarate | NADH | l-HisNA | 0.92 ± 0.01 | 61 ± 3 | 15000 ± 800 | |
| YpODH | Pyruvate | NADPH | l-HisNA | 0.30 ± 0.01 | 73 ± 1 | 4200 ± 200 | |
| YpODH | Oxaloacetate | NADPH | l-HisNA | 0.29 ± 0.03 | 1900 ± 400 | 150 ± 40 | |
| YpODH | Glyoxylate | NADPH | l-HisNA | 0.22 ± 0.01 | 1900 ± 100 | 120 ± 8 | |
| SaODH | NADPH | Pyruvate | d-HisNA | 0.26 ± 0.01 | |||
| SaODH | NADH | Pyruvate | d-HisNA | 0.009 ± 0.002 | |||
| PaODH | NADPH | α-Ketoglutarate | l-HisNA | 0.33 ± 0.02 | |||
| PaODH | NADH | α-Ketoglutarate | l-HisNA | 0.71 ± 0.02 | |||
| YpODH | NADPH | Pyruvate | l-HisNA | 0.38 ± 0.02 | |||
| YpODH | NADH | Pyruvate | l-HisNA | 0.013 ± 0.004 |
SaNAS produces d-HisNA, whereas PaNAS produces l-HisNA. Amino acid specificity for the final opine metallophore could be a result of the NAS alone, or the ODH may be stereospecific for one HisNA. Therefore, each ODH was tested with HisNA produced by SaNAS and by PaNAS, with excess NADPH, and initiated by α-keto acid (Sa and Yp, pyruvate; Pa, α-ketoglutarate) (Table 1). Remarkably, although SaODH produces staphylopine in vivo using d-HisNA (9), it exhibits a kcat 3.3-fold higher in the presence of l-HisNA with a kcat/Km 3-fold lower. Both PaODH and YpODH select l-HisNA as their substrate. In the presence of d-HisNA, PaODH exhibits a kcat of 0.016 s−1 (3-fold above the detection limit), and YpODH kcat is within error of the detection limit. Therefore, the ODHs have differing tolerances for the amino acid stereochemistry incorporated by the NAS into the HisNA substrate. Although YpNAS has not yet been purified preventing analysis, catalysis by YpODH in the presence of pyruvate and l-HisNA suggests the production of a yersinopine metallophore in Y. pestis (Fig. 2). Future kinetic analysis with purified YpNAS will determine whether l-histidine or an alternate l-amino acid is incorporated into yersinopine.
Although all three ODHs use NADPH, significant catalysis is observed for PaODH with NADH (Table 1). A comparison of relative kcat values for each ODH was performed in the presence of saturating amounts of α-keto acid and d- or l-HisNA product with either NADH or NADPH. Although both YpODH and SaODH show little turnover with NADH, 38- and 26-fold lower than the kcat for NADPH, respectively, the PaODH kcat is 2.2-fold higher than the kcat for NADPH. This is consistent with the kcat differences observed when α-ketoglutarate is the initiating substrate (Table 1). In summary, SaODH and YpODH show specificity for NADPH, whereas PaODH is nonspecific for the NAD(P)H substrate.
ODH structure determination
X-ray diffraction data were collected to determine crystal structures for YpODH, PaODH, and SaODH (Table 2). Initially, opine-family dehydrogenases from P. maximus (PmODH, PDB code 3C7A) and Arthrobacter sp. strain 1C (ArODH, PDB code 1BG6) were used as models for molecular replacement trials, but a satisfactory phasing solution was not found. This was unsurprising given low sequence identity (15–18%). Selenomethionine-labeled YpODH was produced, and single-wavelength anomalous dispersion data were collected to 2.15 Å with anomalous signal to 2.85 Å. Anomalous phasing and density modification were attempted with Phenix Autosol (27) and resulted in an Rfree value of 0.64, although helices and strands were clearly visible in the electron density maps. Phasing and density modification were then performed using CRANK2 (24) in CCP4 (25). CRANK2 generated a model placing 93% of YpODH residues with an initial Rfree of 0.30. After refinement, YpODH–SeMet was used as a molecular replacement model for a 2.0 Å YpODH–Apo data set (Fig. 3A). YpODH–Apo was used as a molecular replacement model for the initial phasing of PaODH and SaODH (Fig. 3, B and C). In the subsequent discussion, amino acid numbering is for YpODH unless otherwise noted.
Table 2.
Data collection and refinement statistics
All data were collected on Beamline 12-2 or 9-2 at the Stanford Synchrotron Radiation Lightsource. The values in parentheses are for the highest resolution shells.
| YpODH–SeMet | YpODH–Apo | YpODH–NADP+ | PaODH–NADP+ | SaODH–Apo | SaODH–NADP+ | |
|---|---|---|---|---|---|---|
| Data collection | ||||||
| Space group | C2221 | C2221 | C2221 | C2 | P21212 | P21212 |
| Unit cell (Å) | a = 98.0, b = 123.9, c = 88.5 | a = 97.9, b = 124.1, c = 88.0 | a = 98.4, b = 125.7, c = 88.0 | a = 180.0, b = 53.6, c = 96.8, β = 99.0° | a = 187.7, b = 49.1, c = 58.6 | a = 186.1, b = 49.3, c = 59.1 |
| Resolution range (Å) | 38.4–2.15 | 38.4–2.00 | 38.7–1.94 | 38.1–1.95 | 38.6–2.29 | 38.6–2.49 |
| Completeness (%) | 99.2 (98.1) | 99.6 (95.7) | 99.2 (95.8) | 96.7 (94.5) | 99.8 (96.7) | 99.5 (96.2) |
| Total reflections | 118,529 | 245,736 | 181,826 | 117,971 | 165,517 | 126,596 |
| Unique reflections | 29,208 | 36,673 | 40,471 | 64,907 | 25,087 | 19,677 |
| I/σ | 10.2 (2.2) | 12.2 (2.4) | 14.2 (2.0) | 10.6 (2.6) | 13.0 (2.0) | 12.2 (2.0) |
| Rmergea | 7.3 (50.9) | 8.2 (104) | 5.3 (68.8) | 7.0 (77.7) | 8.8 (86.0) | 9.0 (88.9) |
| Rpimb | 6.0 (43.5) | 5.1 (64.7) | 4.1 (55.3) | 6.5 (72.3) | 5.4 (55.3) | 5.6 (56.8) |
| Multiplicity | 4.1 (4.0) | 6.7 (6.5) | 4.5 (4.3) | 3.5 (3.4) | 6.6 (6.2) | 6.4 (6.3) |
| No. of selenium sites | 5 | |||||
| Initial FOMc | 22.7 | |||||
| FOM after density modc | 50.0 | |||||
| Final mean FOMc | 85.3 | |||||
| Refinement | ||||||
| Resolution range (Å) | 35.89–2.00 | 38.7–1.94 | 38.1–1.95 | 37.63–2.29 | 37.8–2.49 | |
| No. of reflections | 36,620 | 40,415 | 64,880 | 25,032 | 19,596 | |
| Rwork/Rfreed | 20.0/24.6 | 19.0/22.9 | 16.7/20.8 | 20.7/26.8 | 22.0/26.6 | |
| No. of non-hydrogen atoms | 3372 | 3485 | 7099 | 3557 | 3531 | |
| Protein | 3325 | 3296 | 6602 | 3468 | 3476 | |
| Ligand/ion | 48 | 100 | 32 | 45 | ||
| Water | 47 | 141 | 397 | 57 | 10 | |
| Ramachandran allowed (%) | 100 | 100 | 100 | 100 | 100 | |
| Wilson B | 32.3 | 31.5 | 24.3 | 39.6 | 51.1 | |
| Average B (Å2) | 45.5 | 38.2 | 26.6 | 47.1 | 56.3 | |
| Protein | 45.6 | 38.1 | 26.5 | 47.0 | 56.1 | |
| Ligand/ion | 41.6 | 26.4 | 74.1 | 76.6 | ||
| RMSDs | ||||||
| Bond lengths (Å) | 0.009 | 0.010 | 0.010 | 0.009 | 0.015 | |
| Bond angles (°) | 1.01 | 1.01 | 1.09 | 0.97 | 1.26 | |
a Rmerge = Σhkl|Ihkl − <I>hkl|/ΣhklIhkl, where Ihkl is the intensity of reflection hkl, and <I> is the mean intensity of related reflections.
b Rpim = Σhkl |Ihkl − <I>hkl|/ΣhklIhkl, where n is the multiplicity of related reflections.
c Figure of merit (FOM) = <αΣPαeiα/ΣPα>, where the phase = α and the phase probability distribution = Pα.
d R = Σ|Fo − |Fc||/Σ|Fo|, where Fo is the observed structure factors, and Fc is the structure factors calculated from the model. 5% of the reflections were initially reserved to create an Rfree test set used during each subsequent round of refinement.
Figure 3.

ODH structural comparisons. ODH ribbon diagrams are shown. A, YpODH–Apo, 2.0 Å (green). B, PaODH–NADP+, 1.95 Å (blue). C, SaODH–Apo, 2.3 Å (yellow). All three proteins have the same secondary structural elements with TM-align scores between 0.91 and 0.93.
ODH overall structure
YpODH is composed of three domains (Fig. 4A). NADPH binds along a canonical GXGXXA loop within the N-terminal NAD(P)H-binding domain. This domain forms one half of the active site. The other half, and the proposed location for substrate binding, is formed by the catalytic domain. These domains are separated by a central cleft. Embedded within the catalytic domain is a third domain that forms a dimerization interface.
Figure 4.

Global structure, assembly, and homolog comparisons. A, YpODH structure. Helices are labeled with letters, and strands are labeled with numbers beginning with the N terminus. B, PaODH dimeric assembly. The PISA-calculated interface (1484 Å2) between the PaODH homodimer is depicted with blue spheres. C, overlay of ODH dimers. SaODH (gray) and YpODH (same color scheme as in A) also form dimers with a symmetry mate using the same interface as PaODH (gray), each with a coiled coil forming the core of a largely hydrophobic interface. D, in ArODH (coloring as for YpODH) the dimerization domain is truncated, as is αO and β13–14. E, overlay of the ArODH (colored as in D) and SaODH (yellow/gray) dimeric assemblies demonstrates the differing interface used by these homologs.
The NAD(P)H-binding domain has a Rossmann-like fold. This domain contains twelve β-strands (1–12), five α-helices (A, B, and D–F), and one 310 helix (C). Helix G acts as a linker connecting the NAD(P)H-binding domain with the C-terminal, predominantly α-helical, domains. αH–J and N–P, as well as β13–14, form the catalytic domain, whereas αK–M form the dimerization domain. All YpODH structures solved to date have no density for two loops comprised of residues 158–181 and 349–360. The former contains three TQDIXAX sequence repeats of unknown function. The latter connects helix M to helix N between the catalytic and dimerization domains. Mass spectrometry confirmed that the missing amino acid sequences were present in both purified YpODH and YpODH crystals, indicating that these loops are present but disordered in this crystal condition (Fig. S2).
PaODH and SaODH share the same domain and secondary structural elements as YpODH (Fig. 3). RMSDs reflect this, ranging from 1.47 to 2.09 Å (PDBeFold) (29) with Tm scores (30) of 0.91–0.93 over 418–428 Cα residues. Whereas both YpODH and SaODH have one monomer in the asymmetric unit, PaODH has two. The two PaODH monomers have an RMSD of 1.47 Å and a Tm score of 0.96 over 423 Cα. The YpODH TQDIXAX repeat loop (158–181) is not found in PaODH or SaODH. Instead, a short, five-residue loop connects β9–10. The disordered YpODH loop (349–360) connecting the dimerization and catalytic domains is ordered in PaODH and SaODH and composes part of the dimerization interface. The absence of these loops in the YpODH model accounts for the slightly higher RMSDs for comparisons with YpODH, whereas the Tm-align scores suggest equivalent structural similarity between the three enzymes.
ODH dimeric assembly
All three ODHs are dimers by size-exclusion chromatography (Fig. S3) and within their crystal structures (Fig. 4, B and C). Although the dimeric assembly is evident in the asymmetric unit of PaODH, the dimers for YpODH and SaODH are built using a crystallographic 2-fold rotation. The dimeric interface is formed by αL, αM, loop M–N, and the C terminus of αJ. PDBePISA calculates a buried surface area of 870 Å2 for YpODH, 1484 Å2 for PaODH, and 1457 Å2 for SaODH. The reduced surface area in YpODH is due to the disordered 349–360 loop. The interfaces are composed of 50–61% buried hydrophobic residues with minimal (0, 9, and 1, respectively) hydrogen bonds.
Homologous ODHs
Arthrobacter sp. strain 1C opine dehydrogenase (ArODH) and P. maximus opine dehydrogenase (PmODH) represent the nearest structurally characterized homologs with RMSDs of 2.43 and 2.85 Å over 307 Cα and 302 Cα residues. The core strands and helices of the NAD(P)H binding and catalytic domains show good alignment; however, an extended 310C, αP, αO, and β13–14, found in the three ODHs described here, are absent or abridged in ArODH (Fig. 4D) and PmODH. ArODH is reported to be a dimer (33), yet its dimeric interface is formed by αJ and αO rather than αL–M as seen for Yp, Pa, and SaODH (Fig. 4E). Notably, the key helices in the YpODH dimer are missing (αL) or truncated (αM) in ArODH. Glycerol-3-phosphate dehydrogenase is also a structural homolog of opine dehydrogenases. Human glycerol-3-phosphate dehydrogenase (PDB code 1XOV) has an RMSD of 3.12 Å over 240 Cα residues (compared with YpODH), demonstrating structural conservation of core elements from both the NAD(P)H-binding and catalytic domains (Fig. S4). Any effort made in future to halt opine metallophore biosynthesis through ODH inhibition must take care to exclude compounds active against human glycerol-3-phosphate dehydrogenase.
NADP+ binding and selectivity
Structures with NADP+ bound were solved for all three ODHs. The NAD(P)H binding motif, with the conserved sequence GXGXXA, is formed by the β1–αA loop. Residues within this loop (Fig. 5A) form hydrogen bonds with the adenosine ribose sugar and the phosphates spanning the nicotinamide and adenosine ribose sugars. The nicotinamide ring has good electron density in YpODH and PaODH but was not resolved in SaODH and was omitted from the SaODH model (Fig. 5B).
Figure 5.

NAD(P)H binding. YpODH electron density is displayed as 2mFo − DFc simulated annealing omit map contoured at 1.5 σ, and PaODH is displayed as mFo − DFc simulated annealing omit map contoured at 4.0 σ. A, YpODH bound to NADP+. β1 and αA are connected by a loop in all three ODHs with the sequence GXGXXA. B, overlay of NADP+ from each species. Green, Yp; blue, Pa; yellow, Sa. SaODH has partial density for the nicotinamide ring which was left unmodeled. C, YpODH–NADP+ stereo view. The YpODH–NADP+ 2′ phosphate forms five hydrogen bonds with four amino acid side chains. D, PaODH–NADP+ stereo view. The PaODH–NADP+ 2′ phosphate forms two hydrogen bonds with two amino acid side chains.
Hydrogen bonding patterns for the 2′ phosphate of NADPH suggests an explanation for the differential specificity observed in our kinetic analysis. Stabilization of the phosphate differs between YpODH, SaODH, and PaODH (Fig. 5, C and D). In YpODH and SaODH, Lys40 hydrogen bonds with the 2′ phosphate, although in PaODH the structurally homologous Arg44 is hydrogen-bonded to the backbone oxygens of Pro169 and Leu16 preventing coordination of the phosphate. Additionally, the His40 side chain in PaODH stacks with the adenosine and hydrogen bonds with the 2′ phosphate turning it away from Arg44. In YpODH and SaODH, Arg36 stacks next to the adenosine and hydrogen bonds with the phosphate, but its longer side chain rotates the 2′ phosphate toward Lys40, positioning the phosphate to form an additional hydrogen bond. Finally, Ser38, whose side chain forms a hydrogen bond with the 2′ phosphate, is substituted with Gly42 in PaODH. Collectively, these features result in a much more extensive hydrogen-bonding network around the 2′ phosphate in YpODH and SaODH than in PaODH. This is consistent with YpODH and SaODH specificity for NADPH, whereas PaODH shows catalytic efficiency within 2-fold with either NADPH or NADH.
Domain closure
The NAD(P)H and catalytic domains of all three ODHs are separated by a large central cleft with nominal dimensions of 30 Å × 16 Å × 16 Å. This places the hydrides of the nicotinamide ring 8.5 Å distant from the catalytic domain. Britton et al. (33) reported the ArODH structure as representing an open conformation and postulated that closure between the domains would be necessary for hydride transfer. When describing the PmODH structures, Smits et al. (34) reported that both the open and closed forms had been captured. They presented an open-form, NADH-bound structure (PDB code 3C7A) and a closed-form, NADH/arginine-bound structure (PDB code 3C7C). A review of the PmODH models and electron density maps raises several concerns. First, each structure was crystallized with the C-terminal pentahistidine tag from a symmetry mate inserted into the cleft between the domains causing steric clash and preventing potential closed conformations. Second, these structures align with an RMSD of 1.1 Å over 403 Cα residues, and DynDom (35) analysis suggests no dynamic motion between the domains, which is evident by visual inspection (Fig. 6A).
Figure 6.

Domain closure. A, surface view of P. maximus ODH (PmODH) “closed form” (PDB code 3C7C) showing the active site cleft with NADH in white carbons and the pentahistidine tag in green carbons. The expanded view shows a ribbon diagram of PmODH “open form” active site (blue) overlaid with the “closed form” (gray). Note the lack of conformational change indicating that both structures have open active sites. B, surface view of E. coli KPR (PDB code 2OFP) in a closed conformation with pantoate and NADPH bound. The expanded view shows a ribbon diagram overlay of KPR using the NADPH-binding domain for alignment. The holo form is in gray, and the apo form is in red (PDB code 1KS9). Ligands are NADPH (white) and pantoate (yellow). The dashed line is 3.3 Å. The holo form shows 24° closure of the catalytic domain as calculated by DynDom.
Ketopantoate reductase (KPR) from Escherichia coli represents a useful model for domain closure and shows good structural homology with an RMSD of 3.19 Å over 253 Cα residues when compared with YpODH. KPR reduces pantoate converting the ketone to an alcohol. When both NADPH and pantoate are bound, significant closure of the α-helical domain relative to the NAD(P)H-binding domain is evident (calculated as 24° by DynDom) (Fig. 6B). We would expect a similar closure (∼5 Å) to bring the nicotinamide ring in proximity to the catalytic domain. Unfortunately, we have not yet been successful in capturing a closed ODH structure, and no significant domain motions are noted between the apo- and NADP+-bound structures for YpODH (RMSD of 0.54 Å over 416 Cα residues) or SaODH (RMSD of 0.28 Å over 424 Cα residues).
Active site
NADP+ binds in a C-shaped cleft between the NAD(P)H-binding and catalytic domains (Fig. 7A). The amide of the nicotinamide ring hydrogen bonds with Thr154 on β9 holding the ring in a syn conformation. A cavity adjacent to the nicotinamide ring is lined with amino acid residues largely conserved by YpODH, PaODH, and SaODH producing a substrate-binding site. The syn conformation of the nicotinamide projects the pro-S hydride into this space and would result in pro-S hydride transfer to the opine metallophore Schiff base intermediate consistent with octopine dehydrogenase (36, 37). Asp153 is positioned above the hydrides of the nicotinamide ring and is conserved in SaODH. The carboxylate of Asp153 may electrostatically repel the carboxylate of α-ketoglutarate in YpODH and SaODH, whereas the Ala153 substitution found in PaODH allows the longer, more negatively charged α-ketoglutarate to bind. Arg383 is conserved in all five structurally characterized ODHs. Smits et al. (34) have suggested that the residue analogous to Arg383 in ArODH acts as a sensor for the NAD(P)H phosphates during domain closure, but we note that Arg383 has little or no density past the β carbon in any of the solved ODH structures (presented here or previously). We have modeled the side chain in a favorable rotameric conformation, and we hypothesize that Arg383 acts to position and stabilize the α-keto acid substrate by coordinating its carboxylate.
Figure 7.

Active site. A, stereo view of YpODH–NADP+ active site cavity with a CastP (computed atlas of surface topography of proteins) calculated surface. White carbons, NADP+. Purple carbons, side chains conserved by YpODH, PaODH, and SaODH. White carbons, side chains not conserved by YpODH, PaODH, and SaODH. B, hypothetical mechanism for the condensation of pyruvate and l-HisNA by YpODH with His242 acting as the general acid, followed by reduction of the Schiff base by NADPH, leading to product formation.
Across from the nicotinamide ring is His242, which is hydrogen-bonded to Asp388, both with well defined electron density in all structures. His–Asp proton relays are common among dehydrogenases including lactate dehydrogenase (38) and glyceraldehyde-3-phosphate dehydrogenase (39). Therefore, we propose that the histidine is positioned to act as a general acid/general base deprotonating the nucleophile and then donating the proton back to the 2-carbon hydroxyl leading to water release and Schiff base formation (Fig. 7B). HisNA should be oriented with the imidazole moiety deep in the active site to confer stereoselectivity. This would place the primary amine of the amino butyrate proximal to the plane between the hydride and His242, positioning the substrate for nucleophilic attack. In our current structures, the nicotinamide ring hydride is 8.5 Å distant from the histidine proton, too far for catalysis, further supporting the necessity of domain closure.
Opine dehydrogenases can be categorized by the stereochemistry of their products (1). ArODH, PmODH, and their homologs produce opines with (R)-carboxyalkyl chiral centers (8, 40). Saccharopine dehydrogenase and carboxyethyl ornithine dehydrogenase generate (S)-carboxyalkyl chiral centers (41). YpODH, PaODH, and SaODH have sequence and, indeed, structural homology with (R)-carboxylalkyl opine dehydrogenases. By contrast, saccharopine dehydrogenase (PDB code 3UH1) has an RMSD of only 5.3 Å over 96 Cα residues when compared with YpODH. Although staphylopine has been synthesized with an (S)-carboxyalkyl chiral center by Toronto Research Chemicals (9) and as reported by Zhang et al. (42), we propose that YpODH, PaODH, and SaODH form (R)-carboxyalkyl opine metallophore products, given their homology with (R)-carboxylalkyl opine dehydrogenases. It will be valuable to experimentally determine the full stereochemistry of the opine metallophores in future.
Conclusion
Yersinopine, pseudopaline, and staphylopine dehydrogenases perform the final step in the production of three distinct opine metallophores. These enzymes share substantial structural homology yet are able to distinguish differing substrate stereochemistry (l- versus d-HisNA), differing α-keto acid length (pyruvate versus α-ketoglutarate) and differing NAD(P)H substrates. Structural analysis reveals a dimeric assembly using secondary structure not conserved in previously documented opine dehydrogenases. Within the active site, Arg383 is expected to align substrate for Schiff base formation and hydride transfer, whereas Asp153 is predicted to function in α-keto acid substrate selection. His242 is positioned to act as a general acid/base promoting the formation of the Schiff base intermediate. The ODH structures determined were in an open conformation, but domain closure would be necessary to promote catalysis. The full stereochemistry of the opine metallophores is unknown, but structural homology with characterized opine dehydrogenases suggests that a (R)-carboxyalkyl opine is formed.
Experimental procedures
Preparation of overexpression plasmids
S. aureus and P. aeruginosa ODH and NAS were prepared for overexpression as previously described (10). The gene sequence for the Y. pestis CO92 strain ODH (YPO1347) was acquired from NCBI with accession number YP_002346370.1. This sequence was codon optimized for expression in E. coli by GenScript and ligated into pET-15TV with an N-terminal hexahistidine affinity tag. The plasmids were transformed into New England Biolabs BL21 (DE3) cells for expression.
Protein expression and purification
S. aureus and P. aeruginosa ODH and NAS were expressed and purified as previously described (10). For YpODH, baffled flasks with 1 liter of LB Miller medium containing 200 μg/ml ampicillin were inoculated with 10 ml of overnight culture (50 ml grown at 37 °C, 225 rpm) and grown to an A600 of 0.6 at 37 °C (2–3 h.). The cultures were induced with 200 μl of 1 m isopropyl β-d-1-thiogalactopyranoside and grown for 21 h at 15 °C. The cells were centrifuged at 4225 × g for 10 min. The pellet was resuspended in 50 mm potassium phosphate, pH 8, 300 mm NaCl, 50 mm imidazole, and 10% (v/v) glycerol. The cells were French pressed three times at 950 p.s.i., and the lysate was centrifuged for 1 h at 23,426 × g before loading onto a GE Healthcare nickel-chelating fast flow Sepharose column equilibrated with the same buffer. The protein was eluted using a linear gradient of increasing imidazole up to 500 mm. YpODH elutes at 185 mm imidazole. Protein was loaded onto a GE Superdex 200 size-exclusion chromatography column equilibrated with 50 mm potassium phosphate, pH 8, 150 mm sodium citrate, and 20% (v/v) glycerol. YpODH eluted at a molecular mass of 93.1 kDa, roughly equivalent to a dimer of the 50.9-kDa YpODH protein. YpODH was concentrated using Millipore Amicon centrifugation concentrators with 30,000 molecular weight cutoff to a final concentration of 8 mg/ml as measured by Bradford assay prior to flash freezing aliquots in liquid nitrogen. This preparation yields 25 mg/liter of culture.
Selenomethionine (SeMet) substituted YpODH expression and purification
YpODH–SeMet was produced by inhibiting methionine biosynthesis following the method of Van Duyne et al. (21), with the exception that pET15TV–YpODH cultures were grown in M9 minimal medium at 37 °C to an A600 of 0.7 prior to the addition of the amino acid mix containing 60 mg of selenomethionine. Subsequent expression and purification steps were carried out as described above for native protein except that 1.5 μm Tris(2-carboxyethyl)phosphine was added to YpODH–SeMet prior to injection onto the GE Superdex 200 size-exclusion chromatography column equilibrated with 50 mm potassium phosphate pH 8, 150 mm sodium citrate, 20% glycerol, and 1 mm Tris(2-carboxyethyl)phosphine. Collected protein was concentrated to 8.4 mg/ml and frozen in liquid nitrogen with a yield of 13 mg/liter of culture.
Initial rate reactions to determine steady-state kinetic parameters
Initial rates were measured by observing NADPH oxidation as a loss of absorbance at 340 nm using a TgK stopped flow spectrometer with a xenon lamp at 22 °C. The reaction buffer was 50 mm Tris, pH 8.0 (Sa), or 50 mm potassium phosphate, pH 8.0 (Yp and Pa). NAS (46 μm SaNAS or 56 μm PaNAS) was incubated with 4 mm histidine (d-His for SaNAS or l-His for PaNAS) and 4 mm SAM for 75 min at 22 °C to produce d- or l-HisNA in nonrate-limiting excess. 2 μm ODH and 400 μm NAD(P)H (SaODH and PaODH) or 600 μm NAD(P)H (YpODH) were then added to complete the parent mix. Sodium pyruvate, glyoxylic acid, oxaloacetic acid or α-ketoglutaric acid (Sigma) were reconstituted in reaction buffer prior to serial dilution in the same buffer. The pH of glyoxylic acid and oxaloacetic acid was adjusted to 8.0 prior to serial dilution to compensate for the pH shift observed for these α-keto acids at higher concentrations. The ODH reaction was initiated by mixing an α-keto acid with the parent mix in a 1:1 ratio in the stopped flow spectrometer. Secondary plots of initial rates (supporting information) were fit to the Michaelis–Menten equation to determine kinetic parameters. For NAD(P)H initial rates, 2 mm α-keto acid (pyruvate, SaODH and YpODH; or α-ketoglutarate, PaODH) replaced NAD(P)H in the parent mix, and each reaction was initiated with NAD(P)H at the concentrations above. The non–rate-limiting concentration of all substrates was determined by comparing full progress curves for varied concentrations of substrates and ODH enzyme. Each experiment was repeated at least three times, and the error was calculated as the standard deviation of these trials.
Protein crystallization
All crystals were grown in hanging drops composed of 1.5 μl of protein and 1.5 μl of well solution at 24 °C. The crystals were transferred into well solution supplemented with 25% glycerol (YpODH and SaODH) or 20% ethylene glycol (PaODH) as a cryoprotectant and flash-cooled in liquid nitrogen prior to data collection.
YpODH–Apo crystals were grown using 8.0 mg/ml N-terminal hexahistidine-tagged purified protein in a well solution of 3.5 m sodium formate, pH 6.0. Subsequent YpODH crystals were aided by microseeding from this condition. For YpODH–NADP+, protein was preincubated with 1 mm NADP+, and crystals were grown in 2.7 m sodium formate, pH 6.0, 1.8% ethyl acetate aided by microseeding. YpODH–SeMet crystals were grown in 3.1 m sodium formate, pH 7.5, aided by microseeding. After drops were mixed, 2-mercaptoethanol was added to the well solution to a final concentration of 1%. All crystals grew as thin plates within 2 days and reached full size within 10 days. Samples of purified YpODH and YpODH from dissolved protein crystals were submitted to the Mass Spectrometry Research Center Proteomics Laboratory at Vanderbilt University for MALDI–TOF analysis to verify the amino acid sequence and confirm that no proteolytic cleavage had occurred (Fig. S2).
PaODH at 7.5 mg/ml, with its N-terminal hexahistidine tag, was supplemented with 2 mm NADP+ and grown in a well solution of 100 mm BisTris, pH 5.45, 200 mm ammonium acetate, 26% (w/v) PEG 3350. Small, rod-shaped crystals formed within 3 days.
SaODH at 12 mg/ml, with its N-terminal hexahistidine tag, was supplemented with 2 mm NADP+ prior to crystallization for both the SaODH–Apo and SaODH–NADP+ structures. SaODH–Apo crystallized in a well solution of 100 mm sodium citrate:HCl, pH 5.6, 200 mm ammonium sulfate, and 25% (w/v) PEG 4000. SaODH–NADP+ crystallized in a well solution of 100 mm Tris, pH 8.0, 100 mm lithium sulfate, 23% (w/v) PEG 3350. In both conditions, square plates began forming within 2 days and reached full size in 2 weeks. Cryoprotectant was supplemented with 2 mm NADP+ for the holo, but not the apo, structure.
Data collection and structure determination
Diffraction data were collected remotely using Blu-Ice (22) at the Stanford Synchrotron Radiation Lightsource (Menlo Park, CA). For all structures, 180° of data with 0.15° oscillation were collected at a temperature of 100 K. Statistics for data collection and refinement are listed in Table 2. The data were processed in XDS (23).
YpODH–SeMet data were collected on Beamline 12-2 at a wavelength of 0.9795 Å and 0.2-s exposure with a detector distance of 350 mm. Phasing and initial model building were performed using CRANK2 (24) in the CCP4 package (25). Substructure determination found all five selenomethionine sites. Subsequent density modification and model building placed 426 of 456 amino acids resulting in an initial Rwork of 25.3 and an Rfree of 29.6.
YpODH–Apo data were collected on Beamline 9-2 at a wavelength of 0.9794 Å and 0.2-s exposure with a detector distance of 400 mm. The YpODH–Apo structure was solved with Phenix Phaser using YpODH–SeMet as a search model with a log likelihood gain (LLG) of 9992 and a TFZ of 85.4.
YpODH–NADP+ data were collected on Beamline 9-2 at a wavelength of 0.9794 Å and 0.32-s exposure with the detector distance at 350 mm. The YpODH–NADP+ structure was solved using Phenix Phaser with YpODH–Apo as a search model with a LLG of 6081 and a TFZ of 66.2.
PaODH–NADP+ data were collected on Beamline 9-2 at a wavelength of 0.97891 Å and 0.83-s exposure with a detector distance of 410 mm. These data were processed to 2.53 Å. The YpODH–Apo structure was used as a search model for molecular replacement in Phenix Phaser resulting in a solution with a LLG of 73 and a TFZ of 8.8. Phenix Morph Model was used to improve model fit. Phenix Autobuild placed 610 of 868 amino acid residues of the two monomers in the asymmetric unit with an initial Rwork = 31.2 and Rfree = 36.4. This model was then fully refined to an Rwork = 19.3 and Rfree = 26.4. Higher resolution PaODH–NADP+ data were subsequently collected on Beamline 12-2 at a wavelength of 0.9795 Å and 0.2-s exposure with a detector distance of 350 mm. The 2.53 Å PaODH model was used to determine initial phases for the higher resolution data set using Phenix Refine resulting in an initial Rwork of 24.4 and an Rfree of 29.6.
SaODH–NADP+ data were collected on Beamline 12-2 at a wavelength of 0.97937 Å and 0.2-s exposure with a detector distance of 490 mm. The YpODH–Apo structure was used as a search model for molecular replacement in Phenix Phaser resulting in a solution with a LLG of 70.8 and a TFZ of 9.8. Phenix Autobuild placed 342 of 432 amino acid residues with an initial Rwork of 37.4 and an Rfree of 45.0. Active site density corresponding to NADP+ was incomplete for the nicotinamide ring, so the nicotinamide was omitted from the final model.
SaODH–Apo data were collected on Beamline 9-2 at a wavelength of 0.9795 Å and 0.28-s exposure with a detector distance of 404 mm. The SaODH–NADP+ structure was used as a search model for molecular replacement in Phenix Phaser giving a solution with a LLG of 6,672 and a TFZ of 57.3. Initial refinement in Phenix Refine had an Rwork of 23.0 and an Rfree of 29.5.
For each structure, rounds of model building and refinement were completed in Coot (26) and Phenix Refine (27). Waters were placed by Phenix Refine, corrected manually, and verified following a round of refinement. For each NADP+-bound structure, density was visible for NADP+ in the initial electron density map. NADP+ was added to the model by Phenix Ligandfit after polypeptide refinement was complete, followed by a round of refinement and verified by generating a simulated annealing composite omit map in Phenix with NADP+ omitted from the model.
Crystallographic models
Summary data for the models are provided in Table 3. Ramachandran analysis was performed by MolProbity (28), showing good geometry with no outliers for YpODH, PaODH, or SaODH.
Table 3.
Model components
| Protein | PDB code | ASUa | Ordered residues | Waters | Ligandsb |
|---|---|---|---|---|---|
| YpODH–Apo | 6C4L | 1 | 1–157, 182–348, 361–456 | 47 | None |
| YpODH–NADP+ | 6C4M | 1 | 1–156, 181–348, 362–455 | 141 | 1 NADP+ |
| PaODH–NADP+ | 6C4N | 2 | Chain A: 7–431 | 397 | 2 NADP+ |
| Chain B: 7–62, 70–431 | 1 ethylene glycol | ||||
| SaODH–Apo | 6C4R | 1 | 1–428 | 117 | 2 glycerol |
| 4 sulfate | |||||
| SaODH–NADP+ | 6C4T | 1 | 2–428 | 10 | 1 NADP+ |
| 1 glycerol |
a Number of monomers in the asymmetric unit.
b Glycerol and ethylene glycol were used as cryoprotectants, and sulfate was in the SaODH-Apo well solution.
Structural analysis
A comparison of structures and calculation of RMSD values were performed using PDBeFold (29). TM scores were determined by TM-align (30). Interface surface areas and interacting residues for the assembled dimers were analyzed using PDBePISA (31). The active site surface was calculated by CASTp (computed atlas of surface topography of proteins) (32). Structure figures were generated in PyMOL (PyMOL Molecular Graphics System, version 2.0, Schrödinger, LLC). The atomic coordinates and structure factors have been deposited in the Protein Data Bank (accession codes in Table 3).
Author contributions
J. S. M. and A. L. L. conceptualization; J. S. M., C. L. D., and A. L. L. data curation; J. S. M., C. L. D., and A. L. L. formal analysis; J. S. M. and A. L. L. validation; J. S. M., C. L. D., and A. L. L. investigation; J. S. M. and A. L. L. visualization; J. S. M., C. L. D., and A. L. L. methodology; J. S. M. writing-original draft; A. L. L. resources; A. L. L. supervision; A. L. L. funding acquisition; A. L. L. project administration; A. L. L. writing-review and editing.
Supplementary Material
Acknowledgments
We thank the staff at the Stanford Synchrotron Radiation Lightsource for generous assistance, Hayes McDonald at the Mass Spectrometry Proteomics Laboratory at Vanderbilt University for MS analysis, Annemarie Chilton for technical assistance during crystallization trials, and Dr. Catherine Shelton and Trey Ronnebaum for critically reading the text.
This work was supported by National Science Foundation Grant CHE-1403293, by National Institutes of Health Graduate Training Program in the Dynamic Aspects of Chemical Biology Grant T32 GM008545 (to J. S. M.), and by an Institutional Development Award (IdeA) from the NIGMS via Grant P20 GM103418 (to C. L. D.). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract DE-AC02-76SF00515. The Stanford Synchrotron Radiation Lightsource Structural Molecular Biology Program is supported by the U.S. Department of Energy Office of Biological and Environmental Research and by NIGMS, National Institutes of Health Grant P41GM103393. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 6C4L, 6C4M, 6C4N, 6C4R, and 6C4T) have been deposited in the Protein Data Bank (http://wwpdb.org/).
This article contains Figs. S1–S4.
- ODH
- opine dehydrogenase
- Ar
- Arthrobacter
- l-HisNA
- l-histidine nicotianamine
- d-HisNA
- d-histidine nicotianamine
- KPR
- ketopantoate reductase
- NAS
- nicotianamine synthase
- Pa
- P. aeruginosa
- Sa
- S. aureus
- SAM
- S-adenosyl-l-methionine
- Yp
- Y. pestis
- PDB
- Protein Data Bank
- RMSD
- root mean square deviation
- SeMet
- selenomethionine
- LLG
- log likelihood gain
- TFZ
- translation function Z-score.
References
- 1. Thompson J., and Donkersloot J. A. (1992) N-(Carboxyalkyl) amino acids: occurence, synthesis, and functions. Annu. Rev. Biochem. 61, 517–557 10.1146/annurev.bi.61.070192.002505 [DOI] [PubMed] [Google Scholar]
- 2. Van Thoai N., and Robin Y. (1959) Biosynthése de l'octopine et répartition de l'enzyme l'opérant chez les invertebrés. Biochim. Biophys. Acta 35, 446–453 10.1016/0006-3002(59)90394-4 [DOI] [PubMed] [Google Scholar]
- 3. Grieshaber M. K., Hardewig I., Kreutzer U., and Pörtner H. O. (1994) Physiological and metabolic responses to hypoxia in invertebrates. Rev. Physiol. Biochem. Pharmacol. 125, 43–147 10.1007/BFb0030909 [DOI] [PubMed] [Google Scholar]
- 4. Moore L. W., Chilton W. S., and Canfield M. L. (1997) Diversity of opines and opine-catabolizing bacteria isolated from naturally occurring crown gall tumors. Appl. Environ. Microbiol. 63, 201–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Watanabe S., Sueda R., Fukumori F., and Watanabe Y. (2015) Characterization of flavin-containing opine dehydrogenase from bacteria. PLoS One 10, e0138434 10.1371/journal.pone.0138434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Andi B., Xu H., Cook P. F., and West A. H. (2007) Crystal structures of ligand-bound saccharopine dehydrogenase from Saccharomyces cerevisiae. Biochemistry 46, 12512–12521 10.1021/bi701428m [DOI] [PubMed] [Google Scholar]
- 7. Asano Y., Yamaguchi K., and Kondo K. (1989) A New NAD+-dependent opine dehydrogenase from Arthrobacter sp. strain 1C. J. Bacteriol. 171, 4466–4471 10.1128/jb.171.8.4466-4471.1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kato Y., Yamada H., and Asano Y. (1996) Stereoselective synthesis of opine-type secondary amine carboxylic acids by a new enzyme opine dehydrogenase use of recombinant enzymes. J. Mol. Catal. B Enzym. 1, 151–160 10.1016/1381-1177(95)00011-9 [DOI] [Google Scholar]
- 9. Ghssein G., Brutesco C., Ouerdane L., Fojcik C., Izaute A., Wang S., Hajjar C., Lobinski R., Lemaire D., Richaud P., Voulhoux R., Espaillat A., Cava F., Pignol D., Borezée-Durant E., et al. (2016) Biosynthesis of a broad-spectrum nicotianamine-like metallophore in Staphylococcus aureus. Science 352, 1105–1109 10.1126/science.aaf1018 [DOI] [PubMed] [Google Scholar]
- 10. McFarlane J. S., and Lamb A. L. (2017) Biosynthesis of an opine metallophore by Pseudomonas aeruginosa. Biochemistry 56, 5967–5971 10.1021/acs.biochem.7b00804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lhospice S., Gomez N. O., Ouerdane L., Brutesco C., Ghssein G., Hajjar C., Liratni A., Wang S., Richaud P., Bleves S., Ball G., Borezée-Durant E., Lobinski R., Pignol D., Arnoux P., et al. (2017) Pseudomonas aeruginosa zinc uptake in chelating environment is primarily mediated by the metallophore pseudopaline. Sci. Rep. 7, 17132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grim K. P., San Francisco B., Radin J. N., Brazel E. B., Kelliher J. L., Párraga Solorzano P. K., Kim P. C., McDevitt C. A., and Kehl-Fie T. E. (2017) The metallophore staphylopine enables Staphylococcus aureus to compete with the host for zinc and overcome nutritional immunity. MBio 8, e01281–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mastropasqua M. C., D'Orazio M., Cerasi M., Pacello F., Gismondi A., Canini A., Canuti L., Consalvo A., Ciavardelli D., Chirullo B., Pasquali P., and Battistoni A. (2017) Growth of Pseudomonas aeruginosa in zinc poor environments is promoted by a nicotianamine-related metallophore. Mol. Microbiol. 106, 543–561 10.1111/mmi.13834 [DOI] [PubMed] [Google Scholar]
- 14. Gi M., Lee K. M., Kim S. C., Yoon J. H., Yoon S. S., and Choi J. Y. (2015) A novel siderophore system is essential for the growth of Pseudomonas aeruginosa in airway mucus. Sci. Rep. 5, 14644 10.1038/srep14644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Remy L., Carrière M., Derré-Bobillot A., Martini C., Sanguinetti M., and Borezée-Durant E. (2013) The Staphylococcus aureus Opp1 ABC transporter imports nickel and cobalt in zinc-depleted conditions and contributes to virulence. Mol. Microbiol. 87, 730–743 10.1111/mmi.12126 [DOI] [PubMed] [Google Scholar]
- 16. Hanses F., Roux C., Dunman P. M., Salzberger B., and Lee J. C. (2014) Staphylococcus aureus gene expression in a rate model of infective endocarditis. Genome Med. 6, 93 10.1186/s13073-014-0093-3,10.1186/PREACCEPT-4819325051343079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Damron F. H., Oglesby-Sherrouse A. G., Wilks A., and Barbier M. (2016) Dual-seq transcriptomics reveals the battle for iron during Pseudomonas aeruginosa acute murine pneumonia. Sci. Rep. 6, 39172 10.1038/srep39172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bielecki P., Puchalka J., Wos-Oxley M. L., Loessner H., Glik J., Kawecki M., Nowak M., Tümmler B., Weiss S., and dos Santos V. A. (2011) In-vivo expression profiling of Pseudomonas aeruginosa infections reveals niche-specific and strain-independent transcriptional programs. PLoS One 6, e24235 10.1371/journal.pone.0024235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bielecki P., Komor U., Bielecka A., Müsken M., Puchalka J., Pletz M. W., Ballmann M., Martins dos Santos V. A., Weiss S., and Häussler S. (2013) Ex vivo transcriptional profiling reveals a common set of genes important for the adaptation of Pseudomonas aeruginosa to chronically infected host sites. Environ. Microbiol. 15, 570–587 10.1111/1462-2920.12024 [DOI] [PubMed] [Google Scholar]
- 20. von Wiren N., Klair S., Bansal S., Briat J. F., Khodr H., Shioiri T., Leigh R. A., and Hider R. C. (1999) Nicotianamine chelates both FeIII and FeII implications for metal transport in plants. Plant Physiol. 119, 1107–1114 10.1104/pp.119.3.1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Van Duyne G. D., Standaert R. F., Karplus P. A., Schreiber S. L., and Clardy J. (1993) Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 229, 105–124 10.1006/jmbi.1993.1012 [DOI] [PubMed] [Google Scholar]
- 22. McPhillips T. M., McPhillips S. E., Chiu H.-J., Cohen A. E., Deacon A. M., Ellis P. J., Garman E., Gonzalez A., Sauter N. K., Phizackerley R. P., Soltis S. M., and Kuhn P. (2002) Blu-Ice and the distributed control system: software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 9, 401–406 10.1107/S0909049502015170 [DOI] [PubMed] [Google Scholar]
- 23. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Skubák P., and Pannu N. S. (2013) Automatic protein structure solution from weak X-ray data. Nat. Commun. 4, 2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Krissinel E., and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 10.1107/S0907444904026460 [DOI] [PubMed] [Google Scholar]
- 30. Zhang Y., and Skolnick J. (2005) TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309 10.1093/nar/gki524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 32. Dundas J., Ouyang Z., Tseng J., Binkowski A., Turpaz Y., and Liang J. (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 34, W116–W118 10.1093/nar/gkl282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Britton K. L., Asano Y., and Rice D. W. (1998) Crystal structure and active site location of N-(1-d-carboxylethyl)-l- norvaline dehydrogenase. Nat. Struct. Biol. 5, 593–601 10.1038/854 [DOI] [PubMed] [Google Scholar]
- 34. Smits S. H., Mueller A., Schmitt L., and Grieshaber M. K. (2008) A structural basis for substrate selectivity and stereoselectivity in octopine dehydrogenase from Pecten maximus. J. Mol. Biol. 381, 200–211 10.1016/j.jmb.2008.06.003 [DOI] [PubMed] [Google Scholar]
- 35. Girdlestone C., and Hayward S. (2016) The DynDom3D webserver for the analysis of domain movements in multimeric proteins. J. Comput. Biol. 23, 21–26 10.1089/cmb.2015.0143 [DOI] [PubMed] [Google Scholar]
- 36. Nambiar K. P., Sauffer D. M., Kolodziej P. A., and Benner S. A. (1983) A mechanistic basis for the stereoselectivity of enzymatic transfer of hydrogen from nicotinamide cofactors. J. Am. Chem. Soc. 105, 5886–5890 10.1021/ja00356a028 [DOI] [Google Scholar]
- 37. Biellmann J. F., Branlant G., and Olomucki A. (1973) Sterochemistry of the hydrogen transfer to the coenzyme by octopine dehydrogenase. FEBS Lett. 32, 254–256 10.1016/0014-5793(73)80845-2 [DOI] [PubMed] [Google Scholar]
- 38. Holbrook J. J., Liljas A., Steindel S. J., and Rossmann M. G. (1975) Lactate dehydrogenase. In The Enzymes (Boyer P. D., ed) pp. 191–292, Academic Press, Orlando, FL [Google Scholar]
- 39. Harris J. I., and Waters M. (1976) Glyceraldehyde-3-phosphate dehydrogenase. In The Enzymes (Boyer P. D., ed) pp. 1–49, Academic Press, Orlando, FL [Google Scholar]
- 40. Biellmann J., Branlant G., and Wallen L. (1977) Stereochemistry of octopine and of its isomers and their enzymatic properties. Bioorg. Chem. 6, 89–93 10.1016/0045-2068(77)90011-6 [DOI] [Google Scholar]
- 41. Thompson J., and Miller S. P. (1988) N6-(1-Carboxyethly)lysine formation by Streptococcus lactis. J. Biol. Chem. 263, 2064–2069 [PubMed] [Google Scholar]
- 42. Zhang J., Wang S., Bai Y., Guo Q., Zhou J., and Lei X. (2017) Total syntheses of natural metallophores staphylopine and aspergillomarasmine. J. Org. Chem. 82, 13643–13648 10.1021/acs.joc.7b02342 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.