

Abstract
ADAM9 is an active member of the family of transmembrane ADAMs (a disintegrin and metalloproteases). It plays a role in processes such as bone formation and retinal neovascularization, and importantly, its expression in human cancers correlates with disease stage and poor prognosis. Functionally, ADAM9 can cleave several transmembrane proteins, thereby shedding their ectodomains from the cell surface. Moreover, ADAM9 regulates cell behavior by binding cell-surface receptors such as integrin and membrane-type matrix metalloproteases. Because these functions are mainly restricted to the cell surface, understanding the mechanisms regulating ADAM9 localization and activity at this site is highly important. To this end, we here investigated how intracellular trafficking regulates ADAM9 availability at the cell surface. We found that ADAM9 undergoes constitutive clathrin-dependent internalization and subsequent degradation or recycling to the plasma membrane. We confirmed previous findings of an interaction between ADAM9 and the intracellular sorting protein, sorting nexin 9 (SNX9), as well as its close homolog SNX18. Knockdown of either SNX9 or SNX18 had no apparent effects on ADAM9 internalization or recycling. However, double knockdown of SNX9 and SNX18 decreased ADAM9 internalization significantly, demonstrating a redundant role in this process. Moreover, SNX9 knockdown revealed a nonredundant effect on overall ADAM9 protein levels, resulting in increased ADAM9 levels at the cell surface, and a corresponding increase in the shedding of Ephrin receptor B4, a well-known ADAM9 substrate. Together, our findings demonstrate that intracellular SNX9-mediated trafficking constitutes an important ADAM9 regulatory pathway.
Keywords: ADAM, sorting nexin (SNX), endocytosis, trafficking, shedding
Introduction
ADAMs5 (a disintegrin and metalloproteases) are important cell-surface proteases involved in ectodomain shedding of numerous transmembrane proteins, such as growth factors, cytokines, and cell-surface receptors. In addition, ADAMs possess nonproteolytic activities regulating cell-cell and cell-extracellular matrix interactions. Through these processes, ADAMs are central players during development, in normal tissue homeostasis, and in many pathological conditions (1–3). ADAM9 is one of the proteolytically active ADAM family members (4), implicated in cell-surface shedding of transmembrane proteins such as epidermal growth factor (EGF) (5), fibroblast growth factor receptor 2 (FGFR2) (6), and the ephrin receptor B4 (EphB4) (7). ADAM9 has a broad tissue expression profile (8, 9), and is involved in processes like osteogenesis and retinal neovascularization (6, 7). Importantly, its expression is up-regulated in a variety of human cancers (10–12), correlating with disease stage and patient prognosis (13–15). In line with these findings, ADAM9 promotes tumor progression in mouse models (5) and several in vitro studies have implicated ADAM9 in the regulation of tumor cell proliferation and invasion, thereby contributing to the aggressive phenotype of cancer cells (16–21).
As ADAM9 functions are primarily executed at the plasma membrane (22), its cell-surface availability is key for its biological actions. It is well-described that transport of ADAM proteases through the secretory pathway and cleavage of their pro-domain by pro-protein convertases in the trans-Golgi ensure delivery of active mature proteases to the plasma membrane (4, 23, 24). However, the removal of ADAMs from the cell surface is much less studied. Recently, it has been shown that ADAMs, similarly to other membrane proteins undergo endocytosis (3, 25). This serves as an additional regulatory level, controlling surface availability and thereby ADAM actions at the plasma membrane. Several endocytic pathways have been identified, with clathrin-mediated endocytosis (CME) receiving the main attention (26). We previously reported ADAM12 to be constitutively endocytosed through a clathrin-dependent pathway (3). Similarly, ADAM10 is removed from the plasma membrane via CME (25). However, both the route and mechanism of endocytosis for ADAM9 remain unresolved.
Given the important functions of ADAM9 at the cell surface, we set out to characterize how ADAM9 cell-surface availability is regulated. We demonstrate that ADAM9 undergoes constitutive CME, followed by partial recycling to the plasma membrane. We verified previous findings demonstrating a protein-protein interaction between ADAM9 and the intracellular sorting protein and known endocytic regulator sorting nexin (SNX) 9 (27), as well as its close homolog SNX18. Importantly, siRNA-mediated SNX9 and SNX18 double knockdown decreased ADAM9 internalization. Moreover, SNX9 knockdown up-regulated ADAM9 protein levels including at the cell surface, resulting in increased ectodomain shedding of the ADAM9 substrate EphB4. Together, these findings highlight intracellular trafficking and SNX9 as important ADAM9 regulatory components.
Results
ADAM9 is constitutively internalized in a clathrin-dependent manner
ADAM9 exerts important functions at the cell surface. Yet, how cell-surface levels of ADAM9 are regulated by endocytic mechanisms is presently unknown. To investigate ADAM9 endocytosis, we used MDA-MB-231 human breast carcinoma cells, which express ample amounts of endogenous ADAM9. Cell-surface proteins were labeled with cleavable biotin and the internalization, recycling, and degradation of biotinylated proteins were investigated by streptavidin pulldown and subsequent Western blotting. A substantial amount of mature ADAM9 was observed at the plasma membrane, whereas both pro (∼100 kDa) and mature forms (∼84 kDa) of ADAM9 were detected in total cell lysates. Time-dependent constitutive internalization of ADAM9 was observed, with internalized ADAM9 detected as early as after 15 min and reaching a plateau after ∼30–60 min (Fig. 1A).
Figure 1.

ADAM9 is constitutively internalized in a clathrin-dependent manner. A, MDA-MB-231 cells were labeled with cleavable biotin at 4 °C, followed by incubation at 37 °C for the indicated times. After incubation, cells were treated with reduction reagent (GSH) to remove noninternalized biotin from the cell surface, except one sample was left to show the total amount on the cell surface at time 0 (total). The time 0 sample was kept at 4 °C following biotin removal and served as a control for complete removal of cell-surface biotin. Cells were lysed, biotinylated proteins were precipitated with streptavidin-conjugated agarose beads (pulldown), and analyzed by immunoblotting (IB). Total cell lysates were blotted for ADAM9, and actin on the same membrane served as a loading control. Blots are representative of n = 3 independent experiments. B, MDA-MB-231 cells were surface labeled with biotin as described above, treated with Pitstop2TM to block CME, followed by incubation at 37 °C for the indicated time. Samples were analyzed as in A. Membranes were re-blotted for the TfR. C, quantification of Western blots in B, showing percentage of internalized ADAM9 relative to the total surface amount from cells treated with Pitstop2TM or vehicle. ADAM9 surface levels were normalized to TCL ADAM9, n = 5. D, quantification of internalized TfR as in C. E, MDA-MB-231 cells were transiently transfected with siRNA against CHC or control siRNA. Cells were surface biotinylated, incubated at 37 °C for 30 or 60 min, and analyzed as described in A. F, quantification of Western blots in E, showing the percentage of internalized ADAM9 relative to total surface amount from CHC or control siRNA-treated cells, calculated as in C, n = 4. Plots show individual data, and average values ± S.D., *, p < 0.05; **, p < 0.01 (ANOVA).
To examine the pathway of ADAM9 internalization, we first used the small molecule inhibitor Pitstop2, inhibiting clathrin assembly (28). Treating MDA-MB-231 cells with Pitstop2 caused a statistically significant inhibition of ADAM9 internalization (Fig. 1, B and C), indicating that ADAM9 is internalized, at least in part, through CME. To validate the inhibitory effect of Pitstop2 on CME, membranes were re-blotted for the transferrin receptor (TfR) (Fig. 1, B and D), which is known to be internalized through this pathway (29). Because Pitstop2 does not selectively inhibit CME (30), we next knocked down clathrin heavy chain (CHC). Confirming that ADAM9 is internalized through CME, CHC depletion inhibited ADAM9 internalization (Fig. 1, E and F).
Because ADAM9 was found to be constitutively internalized, we next investigated whether ADAM9 can be recycled to the cell surface. Using a biotinylation-based recycling assay (Fig. 2A), it was evident that ADAM9 is recycled back to the cell surface following internalization (Fig. 2B). Quantification of ADAM9 recycling showed that ∼40% of the amount internalized after 15 min is recycled back to the plasma membrane and ∼60% after 30 min (Fig. 2C). To examine degradation of ADAM9, cells were surface labeled and treated with the proteasomal inhibitor MG132, the lysosomal inhibitor chloroquine, or vehicle control (DMSO). Clearly, both inhibitors reduced degradation of ADAM9 as compared with control, with lysosomal inhibition having the most prominent effect after 5 h treatment, whereas inhibiting the proteasome had a larger effect on ADAM9 levels at 15 h of treatment (Fig. 2D).
Figure 2.

ADAM9 is rapidly recycled or degraded. A, schematic of the recycling assay used in B, showing two rounds of stripping with reduced GSH. B, MDA-MB-231 cells were surface labeled with biotin, then incubated for 30 min at 37 °C. After treatment with reduction reagent, cells were kept at 4 °C (cont.) or incubated again at 37 °C to let protein recycle to the cell membrane and then stripped again. Cells were lysed, biotinylated proteins were precipitated with streptavidin-conjugated agarose beads (pulldown) and analyzed by immunoblotting (IB). Total cell lysates (TCL) were blotted for ADAM9, and actin on the same membrane served as a loading control. C, quantification of the recycling (loss of band intensity as compared with internalized amounts) from B, n = 3. D, MDA-MB-231 cells were surface labeled with noncleavable biotin, then incubated at 37 °C with MG132 or chloroquine (CQ) for the indicated time points and examined as in B. E, MDA-MB-231 cells were transfected with CHC or control siRNA. Cells were treated as in D and incubated for 5 h. F, quantification of ADAM9 levels from E, n = 3. Western blots are representative of n = 3 independent experiments. Plots show individual data, and average values ± S.D., *, p < 0.05 (ANOVA). IB, immunoblot.
Because the route of internalization may determine the fate of cargo proteins (31, 32), it was investigated whether ADAM9 undergoing CME is destined toward a specific degradation route. To study this, we knocked down CHC in combination with MG132, chloroquine, or vehicle treatment and incubated cells for 5 h. Silencing of CHC prevented degradation of ADAM9 to the same degree as the two inhibitors (Fig. 2E). Moreover, blocking CME diminished the effect of inhibiting both types of protein degradation (Fig. 2, E and F). Together, these findings indicate that ADAM9 internalized via CME is sent for degradation if not recycled back to the cell surface.
The interaction of ADAM9 with sorting nexin 9 and 18 regulates its endocytosis
ADAM9 was previously reported to interact with the intracellular sorting protein SNX9 (27), which plays an important role during clathrin-dependent internalization (33). To examine a potential role of SNX9 and its close homolog SNX18 in ADAM9 endocytosis, we first confirmed the protein interaction by co-immunoprecipitation (Fig. 3, A–C). To stabilize the interaction and trap endogenous protein-protein interactions in their correct cellular compartment, cells were treated with a cell-permeable cross-linker prior to lysis. Shown in Fig. 3A, both SNX9 and SNX18 were immunoprecipitated in a complex with ADAM9. Performing the reverse experiments, we found that SNX9 and SNX18 were both able to pulldown pro- and mature ADAM9, yet both apparently more efficiently the pro-form (Fig. 3, B and C).
Figure 3.
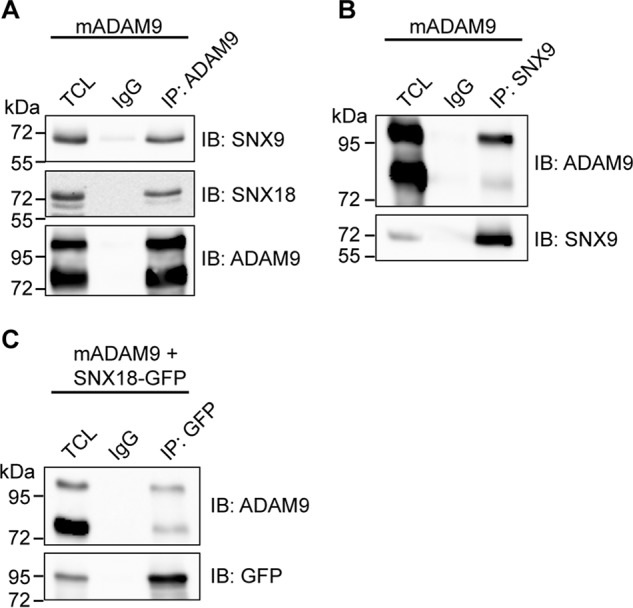
ADAM9 interacts with sorting nexin 9 and 18. A and B, HEK293-VnR cells were transiently transfected with mouse ADAM9, treated with the dithiobis(succinimidyl propionate) cross-linker to stabilize and trap protein-protein interactions in their correct cellular compartments, and lysed. A, lysate was subjected to IP with anti-ADAM9 or IgG control antibodies, examined by SNX9 and SNX18 Western blotting, and re-blotted with anti-ADAM9 antibody. Blots are representative of n = 3 independent experiments. B, lysate was subjected to IP with IgG control or anti-SNX9 antibody and examined as in A. Blots are representative of n = 3 independent experiments. C, HEK293-VnR cells were transiently transfected with mADAM9 and SNX18-tGFP. Lysate was subjected to IP with IgG control or anti-GFP antibody and examined as in A. Blots are representative of n = 3 independent experiments. IB, immunoblot.
As mentioned, SNX9 and -18 both play a role in CME (34, 35). We therefore wanted to examine how knockdown of SNX9 and SNX18 affected the internalization and recycling of ADAM9 (Fig. 4, A and D). Neither SNX9 nor SNX18 silencing alone had any effect on internalization (Fig. 4, B and E) or recycling (Fig. 4, C and F) of ADAM9. However, demonstrating a redundant role of SNX9 and SNX18 in CME, knockdown of both proteins caused a significant decrease in ADAM9 internalization without affecting recycling to the cell surface (Fig. 4, G–I).
Figure 4.

Redundant effects of sorting nexin 9 and 18 on ADAM9 endocytosis. A, D, and G, MDA-MB-231 cells were transiently transfected with SNX9, SNX18, SNX9 + -18, or control siRNA. Cells were surface labeled with cleavable biotin at 4 °C, left alone to internalize or recycle, and examined by Western blotting as described in the legends to Figs. 1 and 2. B and C, quantification of internalized and recycled ADAM9 in SNX9 knockdown cells from A. Internalized ADAM9 was normalized to total surface ADAM9, whereas recycled ADAM9 was normalized to internalized ADAM9 for either SNX9 or control siRNA. E and F, quantification of internalized and recycled ADAM9 in SNX18 knockdown cells from D was done as in B and C. H and I, quantification of internalized and recycled ADAM9 in SNX9 + -18 knockdown cells from G was described in B and C. Western blots are representative of n = 4 independent experiments. Plots show individual data, and average values ± S.D., *, p < 0.05 (Student's t test).
Loss of sorting nexin 9 increases ADAM9 cell-surface levels
In addition to its role in ADAM9 internalization, loss of SNX9 expression appeared to increase overall ADAM9 levels (Fig. 4A). To further examine this effect, we first quantified the amount of mature cell-surface localized ADAM9 after knockdown of SNX9 or SNX18. Silencing of SNX9 or -18 both resulted in a statistically significant increase in ADAM9 at the cell surface, with SNX9 showing the largest effect of ∼3-fold increase (Fig. 5, A and B). Not only cell-surface localized ADAM9, but also total amounts of both pro- and mature cellular ADAM9 levels were up-regulated in SNX9 knockdown cells as compared with control-treated cells (Fig. 5, C and D). Quantitative PCR analysis showed that siRNA-mediated silencing reduced the SNX9 mRNA expression ∼90%, whereas not significantly affecting the ADAM9 mRNA level (Fig. 5E). Together, these findings confirm that SNX9 regulates ADAM9 at the overall protein level.
Figure 5.
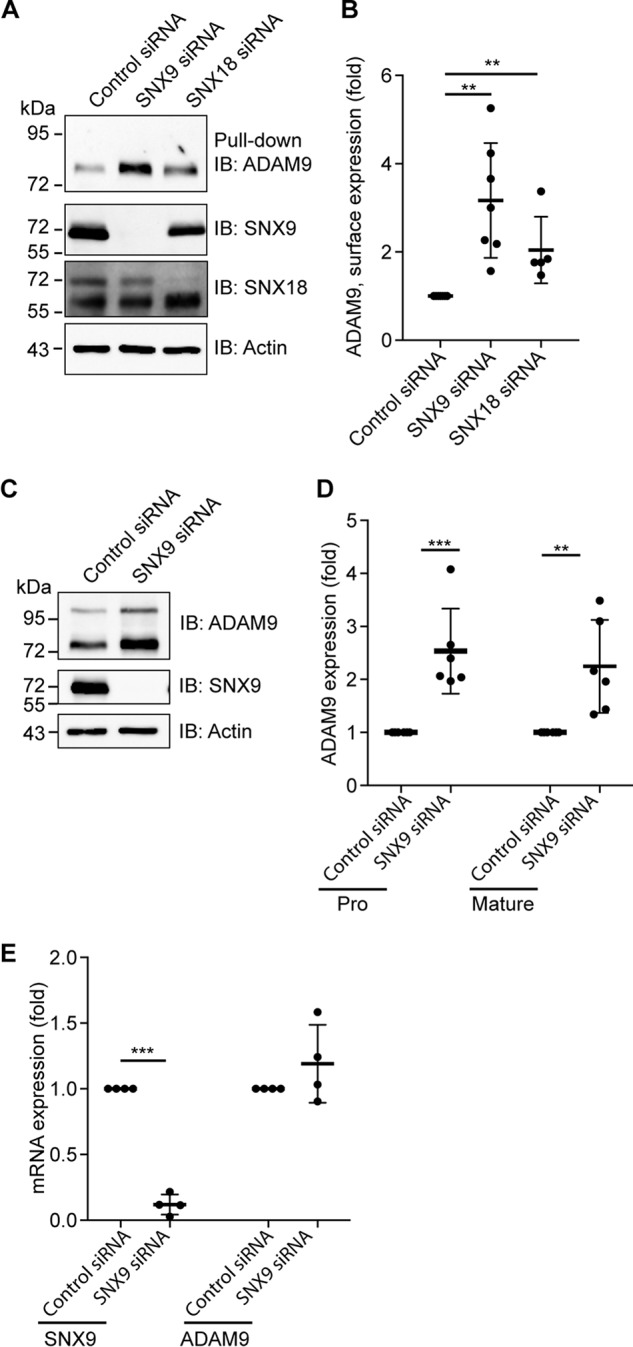
Loss of sorting nexin 9 increases ADAM9 cell-surface levels. A, MDA-MB-231 cells were transiently transfected with SNX9, SNX18, or control siRNA. Cells were surface labeled with noncleavable biotin at 4 °C, lysed, and biotinylated proteins were precipitated with streptavidin-conjugated agarose beads (pulldown) and analyzed by immunoblotting (IB). Actin blotted on the same membrane was used as a loading control. B, quantification of cell-surface ADAM9 normalized to actin, n > 4. C–E, MDA-MB-231 cells were transfected with SNX9 or control siRNA. C, total cell lysate was blotted for the expression of pro- and mature ADAM9, n = 6. D, quantification of total cell lysate of ADAM9 (pro and mature) relative to actin blotted on the same membrane, serving as a loading control. E, mRNA expression of ADAM9 and SNX9 were analyzed by qRT-PCR, n = 4. GAPDH was used as a control. Plots show individual data, and average values ± S.D., **, p < 0.01; ***, p < 0.005 (ANOVA).
Given the observed redundancy of SNX9 and SNX18 regulating ADAM9 internalization (Fig. 4, G–I), the increase in mature ADAM9 levels likely reflects decreased protein turnover (internalization and subsequent degradation). However, this does not explain the observed increase in pro-ADAM9 levels. Because SNX9 can bind the pro-form of ADAM9 (Fig. 3B), we speculated whether silencing of SNX9 affected ADAM9 proprotein processing. Indeed, previous findings suggested a functional role of SNX9 in the Golgi compartment (33) and immunofluorescent staining and confocal microscopy indicated a minor overlap between SNX9 and the Golgi marker Golgin-97 (Fig. 6A). Although hampered by the lack of suitable ADAM9 antibodies, expression of HA-tagged ADAM9 revealed a certain perinuclear co-localization with both SNX9 and Golgin-97 (Fig. 6A).
Figure 6.

Sorting nexin 9 modulates ADAM9 maturation. A, MDA-MB-231 cells were transfected with ADAM9-HA and plated on glass cover slides. Cells were fixed, permeabilized, and stained for SNX9 and HA (top row), HA and Golgin-97 (middle row), and SNX9 and Golgin-97 (bottom row), using fluorescently labeled secondary antibodies as indicated and examined by confocal microscopy. Representative pictures from 3 independent experiments are shown and right-hand pictures show merged images, including DAPI nuclear stain. Fluorescence intensity were quantified along the line on the enlarged images and depicted on the graphs. Bar = 10 μm. B, MDA-MB-231 cells were transiently transfected with SNX9 or control siRNA and incubated with 50 μg/ml of cycloheximide (CHX) for the indicated time. Cells were lysed and analyzed by immunoblotting (IB) as indicated. Western blots are representative of n = 3 independent experiments. C, quantification of loss of the pro-form relative to actin blotted on the same membrane and serving as a loading control. Individual data, and average values ± S.D. are shown; *, p < 0.05; **, p < 0.01 (ANOVA).
We then treated control and SNX9 knockdown cells with cycloheximide to block protein translation and subsequently tracked the conversion of pro- to mature ADAM9 by Western blotting (loss of Western band at ∼100 kDa) (Fig. 6B). Within less than 8 h, the substantially higher amounts of pro-ADAM9 in SXN9-depleted cells were processed to the same extend as in control cells, demonstrating an increased rate of ADAM9 maturation (Fig. 6C). Although increased ADAM9 maturation contributes to the higher ADAM9 cell-surface levels, the simultaneous increase in pro-ADAM9 is not intuitively explained.
Loss of SNX9 results in increased shedding of the ADAM9 substrate ephrin B4 receptor
ADAM9 exerts it main function at the cell surface. Thus, to examine whether SNX9, by regulating ADAM9 cell-surface levels, controls ADAM9-mediated shedding activities, we used the ephrin B4 receptor with alkaline phosphatase (AP) fused at the N terminus (EphB4-AP) as a known ADAM9 substrate (7, 36). In line with previous findings, knockdown of ADAM9 expression in MDA-MB-231 cells had no significant effect on EphB4-AP shedding (Fig. 7, A and B), yet overexpression of WT ADAM9 in HEK293-VnR cells led to a substantial loss of ephB4-AP protein from cell lysates (Fig. 7C) and a correspondingly increased AP activity in conditioned cell medium (Fig. 7D), as compared with mock transfection or cells expressing a catalytically inactive ADAM9 mutant.
Figure 7.
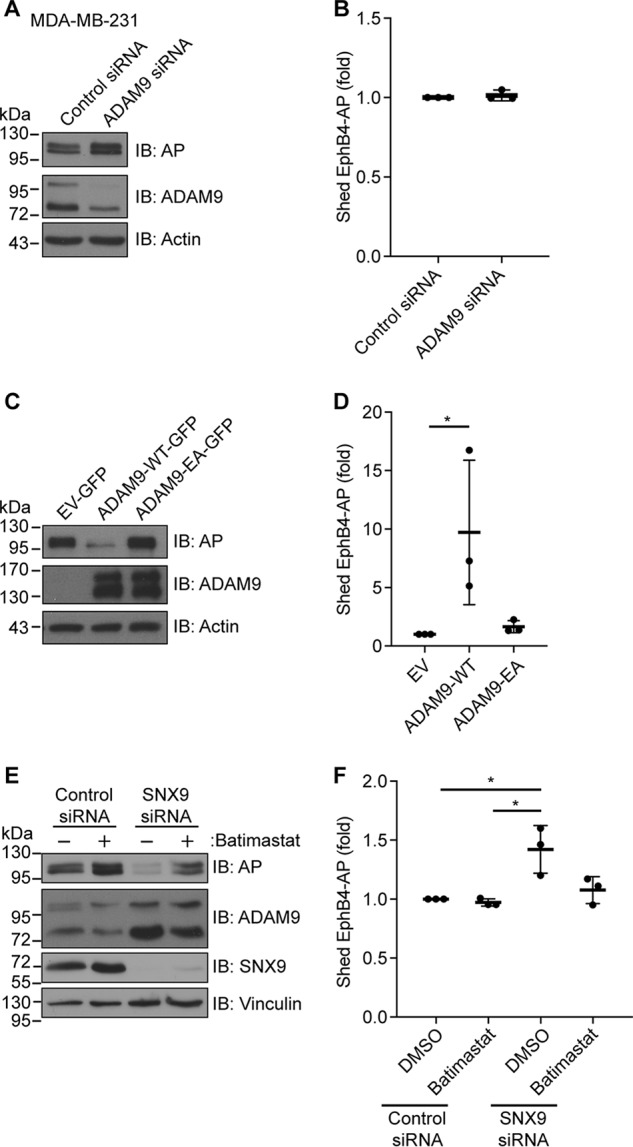
Loss of sorting nexin 9 results in increased shedding of an ADAM9 substrate. A and B, MDA-MB-231 cells were transfected with ADAM9 or control siRNAs, and subsequently with the ADAM9 substrate, ephrin B4 receptor N-terminally fused to alkaline phosphatase (EphB4-AP). Cells were incubated for 18 h, and in A, cell lysates were analyzed by immunoblotting (IB) as indicated, or B, the amount of EphB4-AP shed into the conditioned cell media was measured, n = 3. C and D, HEK293-VnR cells were co-transfected with EphB4-AP and WT GFP-tagged ADAM9 (ADAM9-WT-GFP), catalytic inactive GFP-tagged ADAM9 (ADAM9-EA-GFP), or control vector (EV-GFP). Cells were incubated for 18 h, and cell lysates were analyzed as in A and B, n = 3. E and F, HEK293-VnR cells were transfected with SNX9 or control siRNA and subsequently co-transfected with EphB4-AP. Cells were incubated for 18 h with or without Batimastat, and cell lysates were analyzed as in A and B, n = 3. Western blots are representative of n = 3 independent experiments. Plots show individual data, and average values ± S.D., *, p < 0.05 (ANOVA).
Using the same cell-based assay, cells were treated with SNX9 or control siRNAs with or without the metalloprotease inhibitor Batimastat. Interestingly, silencing SNX9 expression reduced the amount of EphB4-AP in the cell lysate (Fig. 7E), whereas causing a corresponding increase in AP activity in conditioned cell medium (Fig. 7F). The observed effects of SNX9 loss were completely blocked by treating the cells with Batimastat (Fig. 7 E and F), strongly implicating SNX9-dependent ADAM9 activity in this proteolytic event.
Discussion
ADAM9 is an important proteolytic enzyme, whose main functions are exerted at the cell surface. Here, ADAM9 sheds several transmembrane substrates and interacts with other cell-surface receptors (7, 17, 37). Under normal circumstances, ADAM9 acts to maintain tissue homeostasis (6, 38). However, ADAM9 is found overexpressed in a variety of different cancers, e.g. breast and prostate cancer (10, 13, 15, 39), likely promoting disease progression. Given these important functions, it is of great interest to understand how ADAM9 activity at the cell surface is regulated.
We here identified endocytosis and SNX9-mediated intracellular trafficking as important ADAM9 regulatory mechanisms. Using biotin-labeling to monitor ADAM9 fate, we showed that similar to the situation for ADAM10, ADAM12, and ADAM17 (3, 25, 40), ADAM9 undergoes a relatively slow constitutive internalization followed by either recycling to the plasma membrane or lysosomal degradation. Using two different ways to block clathrin-mediated internalization, i.e. the inhibitor Pitstop2 and siRNA-mediated knockdown of CHC, we showed that ADAM9 is primarily internalized through CME. Interestingly, both the observed route and relatively slow rate of ADAM9 endocytosis are very similar to the few other reports on ADAM endocytosis (3, 25, 39, 40), suggesting a common endocytic regulation of ADAM proteins.
It was recently discovered that following internalization, ADAM10 and -12 localize within distinct Rab4 and Rab11 GTPase-positive recycling endosomes (3, 41). We found that ADAM9 is recycled back to the cell surface by 15 min post-internalization, suggesting that ADAM9 traffics via a fast recycling pathway (42). However, no knowledge on what directs ADAM9 for recycling versus degradation is available and lack of appropriate antibodies against the extracellular part of ADAM9 prevents us from tracking internalized ADAM9 protein by immunofluorescence to determine the exact recycling route. If not recycled, internalized proteins traffic from late endosomes to lysosomes, as is the case for EGFR (43) or they are delivered to the proteasome as seen for the c-MET receptor (44). Inhibiting lysosomal or proteasomal degradation both resulted in increased ADAM9 protein levels. Yet, knockdown of CHC prevented the effect of blocking both lysosomal and proteasomal degradation, indicating that internalized ADAM9 is degraded via both pathways. Lysosomal degradation of endocytosed ADAM17 was recently reported (39). For unknown reasons, chloroquine treatment caused a small increase in the amount of pro-ADAM9. The effect of inhibiting the proteasome could reflect changes in de novo synthesized ADAM9 protein. Alternatively, proteasomal inhibition could affect ADAM9 indirectly by preventing the degradation of an ADAM9 regulator.
ADAM9 contains two proline-rich regions in the cytoplasmic tail, known to mediate the interaction with several Src homology 3 domain-containing proteins, such as SNX9 (27). SNX9 and SNX18 constitute the ubiquitously expressed Src homology 3 domain-containing SNX9 family and have been previously found to regulate protein internalization and intracellular protein trafficking (34, 45, 46). Thus, we asked whether SNX9 or SNX18 knockdown would disrupt ADAM9 endocytosis, as has been reported for membrane type 1 matrix metalloproteinase (MT1-MMP) (47, 48). When knocking down either SNX9 or -18, no statistically significant changes in ADAM9 internalization or recycling were detected. This could in part be due to redundancy between SNX9 and SNX18, so that when one is knocked down, the other takes over (35). Indeed, double knockdown of SNX9 and SNX18 caused a significant reduction in ADAM9 internalization without affecting recycling, thereby supporting a redundant role of these proteins in CME. Although reduced, a substantial amount of ADAM9 is still endocytosed, suggesting that additional factors are involved. Similar redundancy between sorting nexins is seen for β1 integrin, where SNX17 and -31 are both important for recycling (49).
Intriguingly, our internalization studies revealed an increase in the total amount of ADAM9, when SNX9 was silenced. Exploring this further, we found that SNX9 and to some extend also SNX18 knockdown increased the cell-surface levels of ADAM9. Such cell-surface accumulation could be an effect of blocked ADAM9 internalization. Thus, although both SNX9 and SNX18 had to be depleted to detect a reduction in ADAM9 internalization, it seems likely that SNX9 loss alone interferes enough with the process to cause some ADAM9 accumulation over time. Although this could account for the observed increase in mature ADAM9 levels, it does not intuitively explain the increase in pro-ADAM9 upon SNX9 knockdown.
The pro-form of ADAM9 is retained in intracellular compartments until removal of the pro-domain (9). In line with earlier reports (27), we found that both SNX9 and SNX18 interact more efficiently with pro-ADAM9. Immunofluorescent staining and confocal microscopy supported previous indications of a role of SNX9 in the trans-Golgi (33), and showed a partial perinuclear co-localization of SNX9 and HA-tagged ADAM9. Based on these findings, we investigated how ADAM9 maturation (proprotein processing) was affected by SNX9 knockdown. Silencing SNX9 increased the rate of ADAM9 proprotein processing, indicating that SNX9 restricts the maturation of ADAM9 in the secretory pathway. Alternatively, the increased rate of maturation may simply reflect the fact that there is more pro-ADAM9 present when SNX9 is depleted. Although this could contribute to the increase in mature cell-surface localized ADAM9 levels, it does not explain the concurrent increase in pro-ADAM9. Thus, further studies are needed to address a potential effect of SNX9 on ADAM9 biosynthesis and fully uncover the complexity of SNX9-mediated ADAM9 intracellular trafficking.
In summary, we showed that endocytosis constitutes an important ADAM9 regulatory mechanism (Fig. 8). Importantly, the well-known endocytic regulators SNX9 and SNX18 exert a somewhat redundant regulatory effect on ADAM9 endocytosis. Additionally, SNX9 seems to control ADAM9 cell-surface levels by limiting ADAM9 proprotein processing in the secretory pathway. The identification of SNX9 family proteins as regulators of ADAM9 intracellular trafficking could reflect the need to restrict the shedding activity of ADAM9, which presumably takes place at the cell surface. In contrast to most other ADAMs, ADAM9 is not inhibited by tissue inhibitors of metalloproteases (50), thus likely other ways of controlling its activity exist. Also, our findings are well in line with several recent reports highlighting the importance of intracellular trafficking in the control of ADAM functions (51). Most strikingly, iRhoms mediate ER to Golgi transit of ADAM17 (52, 53), whereas selective tetraspanins control the transport of ADAM10 (54). Moreover, the intracellular sorting protein PACS-2 regulates the endocytic fate of ADAM17 (55).
Figure 8.
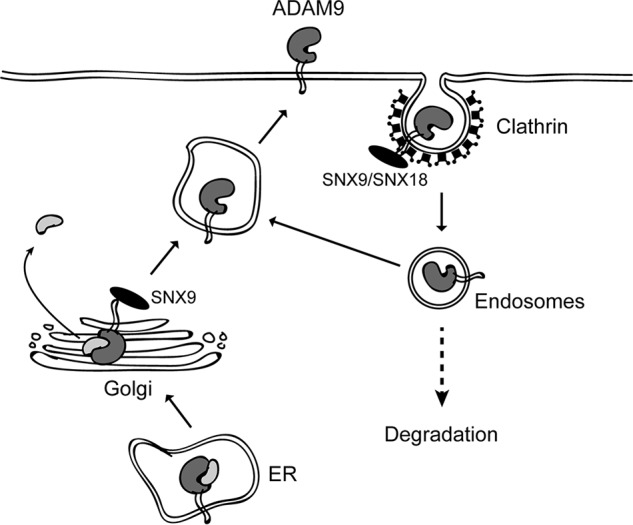
Model illustrating the regulation of ADAM9 by intracellular trafficking. Newly synthesized pro-ADAM9 is matured during secretion to the cell surface by prodomain cleavage in the Golgi compartment. Mature cell-surface localized ADAM9 is then constitutively internalized via a clathrin-dependent mechanism, followed by either recycling or intracellular degradation. Our findings indicate that an interaction between ADAM9 and the intracellular sorting proteins SNX9 or SNX18 is required for optimal internalization. Moreover, SNX9 may have an additional effect, binding pro-ADAM9 in an intracellular compartment (possibly Golgi) to regulate its processing. Combined, SNX9 serves to restrict the amount of ADAM9 and hence ADAM9-mediated shedding activities at the cell surface.
An increase in ADAM9 cell-surface levels would presumably increase the shedding of ADAM9 substrates. Indeed, our data demonstrate that loss of SNX9 function results in increased shedding of EphB4, a previously described ADAM9 substrate. Because ADAM9 is up-regulated in many human cancers (10, 12, 56), whereas SNX9 has been proposed as a tumor suppressor, down-regulated or deleted in a number of tumors (57–60), we speculate that oncogenic loss of SNX9 expression could amplify pro-tumorigenic ADAM9 functions.
Experimental procedures
Reagents and antibodies
The metalloprotease inhibitor Batimastat (BB-94) was from Calbiochem. Chloroquine (C6628), MG132 (M7449), Pitstop2 (SML1169), and cycloheximide (C7698) were purchased from Sigma. HALT phosphatase inhibitor mixture was from Thermo Fisher Scientific, and complete EDTA-free inhibitor mixture was from Roche Applied Science. Primary antibodies used for Western blotting were ADAM9 (AF949, R&D Systems), Actin (Millipore, MAB1501), transferrin receptor (Invitrogen, 13-6800), GFP (Clonetech, JL-8), SNX9 (Abcam, ab181856), SNX18 (Abcam, Ab111702), CHC (Abcam, ab21679), AP (Santa Cruz, SC15065), ubiquitin (Santa Cruz, SC8017), Golgin-97 (Cell Signaling Technology, 97537), and HA (Sigma, H6908; Covance, 16B12). Goat control IgG was from Fitzgerald. Secondary rabbit anti-goat HRP-conjugated antibody (P0449) was from Dako, donkey anti-rabbit (NA934) and sheep anti-mouse (NXA931) HRP-conjugated antibodies were purchased from GE Healthcare, whereas mouse negative IgG control and HRP-labeled secondary antibodies were from DAKO.
Cell culture
The human breast adenocarcinoma cell line MDA-MB-231 was obtained from American Type Cell Culture (ATCC). Human epithelial HEK293-VnR cell line stably expressing the vitronectin receptor (αVβ3 integrin) was a kind gift from Professor Archana Sanjay (61). All cells were grown in Dulbecco's modified Eagle's medium (DMEM) (Gibco), supplemented with 10% fetal bovine serum (HyClone). Cells were maintained at 37 °C and 5% CO2 humidified atmosphere.
Expression constructs and siRNAs
Mammalian expression constructs for full-length mouse ADAM9 (mADAM9) in pcDNA3 and EphB4-AP were kindly provided by Professor Carl Blobel (Hospital for Special Surgery, New York) and previously described (7). Mouse ADAM9-HA and ADAM9-GFP were kind gifts from William R. English and Gillian Murphy (Cambridge University, UK). The ADAM9-GFP catalytically inactive (EA) mutant was generated by mutating glutamic acid at position 348 to alanine, using the Phusion High-Fidelity kit (Thermo F530S) and the following primers: forward, 5′-cattgttgctcatgcattggggcataac-3′; reverse, 5′-gatgcaaatgtctccacagtgatttgcc-3′, and confirmed by sequencing. Sorting Nexin 9 with His6 tag (His6-SNX9) was purchased from Addgene (number 34690) and kindly shared by Sandra Schmid (UT Southwestern Medical Center, Dallas, TX). Sorting nexin 18 with a Turbo GFP tag (SNX8-tGFP) was purchased from OriGene (RG219205). pEGFP-N1 (Clontech) and empty pcDNA3 (RZPD) were used as empty vector controls. siGENOME SMARTpool siRNA's from Dharmacon against CHC, SNX9 and -18 were used to silence gene expression, whereas MISSION siRNA Universal Negative Control (Control siRNA) (Sigma) was used as control siRNA.
Transfections
HEK293-VnR cells were grown to 70% confluence and transfected with plasmids using X-tremeGENE 9 DNA Transfection Reagent (Roche Applied Science) and Opti-MEM (Gibco) according to the manufacturer's instructions. Experiments were carried out at either 24 or 48 h after transfection. For siRNA-mediated knockdown, cells were seeded in 6-cm dishes at 5 × 105 cells per dish the day before transfection. For transfection with CHC, SNX9 and -18 siRNAs (50 pm), INTERFERin® (Polyplus) and Opti-MEM Medium were used according to the manufacturer's instructions. Seventy-two hours after transfection, cells were trypsinized and re-seeded at 6 × 105 cells per 6-cm dish, and the following day re-transfected with siRNA again. Experiments were carried out 48–72 h after the second round of transfection.
Cell-surface biotinylation assays
MDA-MB-231 cells were grown in normal growth medium in 6-cm dishes to 80–100% confluence. The cells were transferred to 4 °C and washed twice with cold phosphate-buffered saline (PBS). Cell-surface proteins were labeled with 2 ml of 0.2 mg/ml of noncleavable EZ-LinkTM Sulfo-NHS-LC-Biotin (Pierce 22335) in cold PBS for 30 min at 4 °C. Unbound biotin was quenched of by washing three times in 100 mm glycine in cold PBS. Finally, cells were lysed in RIPA buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1% Triton X-100, 1 mm EDTA, 1× complete EDTA-free inhibitor mixture). Cell lysates were cleared by centrifugation for 10 min at 16,000 × g and examined by Western blotting.
Internalization and recycling assays
MDA-MB-231 cells were grown in normal growth medium in 6-cm dishes to 80–100% confluence. The cells were transferred to 4 °C and washed twice with cold PBS. Cell-surface proteins were labeled with 2 ml of 0.05 mg/ml of cleavable EZ-LinkTM Sulfo-NHS-SS-Biotin (Pierce 21331) in cold PBS for 30 min at 4 °C. Unbound biotin was quenched off by washing three times with 100 mm glycine in cold PBS. Pre-warmed serum-free DMEM was added to the cells (3 ml/dish) and biotin-labeled surface proteins were allowed to internalize at 37 °C for varying time points. Internalization was stopped by transferring cells to 4 °C. Surface biotin was removed by washing three times for 10 min in cold stripping buffer (50 mm l-GSH reduced, 75 mm NaCl, 75 mm NaOH, 1% bovine serum albumin (BSA), and 10 mm EDTA, pH 8.0). The total amount of surface biotinylation was detected by keeping a dish on ice after biotin labeling and omitting treatment with stripping buffer. To verify proper removal of surface-labeled biotin, a dish kept on ice after biotin labeling was washed three times with stripping buffer. Following treatment, cells were washed three times in cold PBS and lysed in RIPA buffer.
For inhibition of clathrin, biotin-labeled cells were preincubated at 4 °C for 15 min with 30 μm Pitstop2 in serum-free medium. After incubation, cells were transferred to 37 °C and new pre-warmed serum-free medium with 30 μm Pitstop2 was added to the cells. Cells were incubated for the indicated time points. For recycling assays, biotin-labeled surface proteins were allowed to internalize at 37 °C for 30 min. Surface biotin was removed as described above. Following treatment with stripping buffer, cells were washed once in cold medium. Pre-warmed serum-free growth medium was added and the cells incubated at 37 °C for the indicated time points. After incubation cells were washed three times at 10 min with stripping buffer to remove recycled biotin-labeled proteins, washed three times in cold PBS, and lysed in RIPA buffer. For some recycling assays, a dish was kept at 4 °C following preinternalization and subjected to stripping buffer to ensure no recycling at 4 °C.
For all experiments, cell lysates were cleared by centrifugation for 10 min at 16,000 × g. Lysates were subjected to BCA assay (Pierce) according to the manufacturer's protocol and equal protein amounts were incubated with streptavidin-conjugated agarose beads (Sigma S1638) for 2 h at 4 °C. Beads were washed three times in RIPA buffer and bound proteins eluted in 5× Laemmli sample buffer (0.3 m Tris, pH 6.8, 20% glycerol, 2% SDS, 0.01% bromphenol blue, 5% 2-mercaptoethanol). Samples were analyzed by Western blotting.
Co-immunoprecipitation
For all co-immunoprecipitation experiments, cells were treated with a cell-permeable cross-linker prior to lysis, to stabilize and trap protein-protein interactions in their correct cellular compartments. In brief, cells were washed twice in PBS and incubated in 10 μm Lomant's reagent (dithiobis(succinimidyl propionate), Pierce number 22585) in PBS at room temperature for 30 min, washed three times with cold glycine-PBS, and lysed in modified RIPA (mRIPA) (50 mm Tris, pH 7.4, 150 mm NaCl, 0.5% Igepal (Nonidet P-40), 1× cOmplete EDTA-free inhibitor mixture) supplemented with 1× Halt phosphatase inhibitor. Cell lysates were cleared by centrifugation for 10 min at 16,000 × g. Supernatants were incubated with antibodies for 2 h at 4 °C with gentle agitation. Protein G-SepharoseTM 4 Fast Flow beads (GE Healthcare) were added followed by incubation for an additional hour at 4 °C. Beads were washed three times in mRIPA buffer. Bound proteins were eluted in 5× Laemmli sample buffer, followed by Western blot analysis.
SDS-PAGE and Western blotting
Proteins were separated by 7.5 or 10% reducing SDS-PAGE and transferred to Hybond-ECL nitrocellulose (Amersham Biosciences, 0.45 μm). The membranes were blocked in 5% skim milk or BSA in Tris-buffered saline containing 0.1% Tween 20 (TBS-T). Membranes were incubated overnight at 4 °C with primary antibodies, followed by incubation with the appropriate HRP-conjugated secondary antibodies (DAKO). Images were acquired using either a LAS4000 (GE Healthcare) or Hyperfilm ECL high performance chemiluminescence film (GE Healthcare) and a Valsø X-Ray (Ferrania, Valsoe X-ray). Band intensities were determined by densitometric analysis using Total Lab. Blots shown are representative of at least three independent experiments.
Immunofluorescence staining
Glass coverslips were coated with fetal bovine serum for 1 h at 37 °C, and subsequently washed with PBS. Cells were cultured on coated glass coverslips overnight, washed twice in PBS, fixed in 4% paraformaldehyde for 10 min at room temperature, and permeabilized in 0.2% Triton X-100 for 10 min. Free aldehyde groups were quenched by incubation with 0.1 m NH4Cl in PBS for 10 min and blocked in 2% BSA in PBS-T for 30 min, followed by incubation with primary antibody overnight at 4 °C. Secondary antibody diluted in blocking buffer containing DAPI were incubated for 1 h at room temperature in the dark. Images were collected by confocal microscopy (Leica SP8) with ×63/1.4 oil objective, using Leica imaging software (Leica Application Suite X). Fluorescence intensity (line scan) were quantified along the line on the enlarged images and depicted on the graphs, using ImageJ.
Quantitative PCR
Cells were cultured in 6-well plates, washed twice in cold PBS, and RNA was isolated from cells using RNeasy mini kit (Qiagen, number 74104). cDNA was synthesized using a iScript cDNA Synthesis kit (Bio-Rad, number 170-8890), according to the manufacturer's protocol. Quantitative PCR was performed using SYBR Green (ThermoFisher, number K0221) and the following primers: SNX9, forward (5′-caacaatcaccaggttggca-3′), SNX9, reverse (5-ttcccagatttggaggcact-3′), ADAM9, forward (5′-tcccccaaattgtgagactaa-3′); ADAM9, reverse (5′-tccgtccctcaatgcagtat-3′); GAPDH, forward (5′-agggctgcttttaactctggt-3′); GAPDH, reverse (5′-ccccacttgattttggaggga-3′). The relative gene expression was calculated using the comparative ΔΔCT method. GAPDH was used as reference gene.
Shedding assay
Cells were co-transfected with EphB4-AP and either wildtype (WT) or catalytically inactive (EA) ADAM9-GFP. After 6 h, medium was exchanged to serum-free DMEM with or without 10 nm Batimastat and cells were incubated for 18 h. Supernatant was collected and cleared by spinning down at 1,000 × g for 5 min and cells were lysed in RIPA lysis buffer. AP activity was measured, using SIGMAFASTTM p-nitrophenyl phosphate tablets (Sigma, N1891) in 5 ml of ddH2O. Cell lysates (20 μl + 30 μl of PBS) and supernatant (100 μl) were added in triplicates into a flat-bottom 96-well plate, serum-free medium was used for blank values. A 1:1 substrate solution (50 μl for lysates, 100 μl for supernatant) was added and incubated in the dark at 37 °C. Absorbance was measured at 405 nm. Shedding activity was supernatant relative to lysate +supernatant.
Statistical analysis
All plots show individual data and average values ± S.D. Data were analyzed by unpaired Student's t test or analysis of variance ANOVA) with the Tukey's post-test as appropriate, using GraphPad Prism version 7.00 for Macintosh (GraphPad Software). In all cases, p < 0.05 was considered statistically significant.
Author contributions
K. J. M. and M. K. data curation; K. J. M. and M. K. formal analysis; K. J. M., T. S., M. L. F., J. S.-P., and J. S. investigation; K. J. M. and J. S.-P. visualization; K. J. M. methodology; K. J. M. and M. K. writing-original draft; O. M. A. and M. K. conceptualization; O. M. A. and M. K. writing-review and editing; M. K. supervision; M. K. funding acquisition; M. K. project administration.
The work was supported in part by The Danish Cancer Society, Lundbeck Foundation, and Københavns Universitets Fond for Kræftforskning. The authors declare that they have no conflicts of interest with the contents of this article. K. J. M. was supported by a Ph.D. fellowship from the Faculty of Health and Medical Sciences, University of Copenhagen.
- ADAM
- A disintegrin and metalloproteases
- CME
- clathrin-mediated endocytosis
- EphB4
- ephrin receptor B4
- SNX
- sorting nexin
- TfR
- transferrin receptor
- CHC
- clathrin heavy chain
- HA
- hemagglutinin
- AP
- alkaline phosphatase
- HRP
- horseradish peroxidase
- DMEM
- Dulbecco's modified Eagle's medium
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- ANOVA
- analysis of variance.
References
- 1. Wong G. E., Zhu X., Prater C. E., Oh E., and Evans J. P. (2001) Analysis of fertilin α (ADAM1)-mediated sperm-egg cell adhesion during fertilization and identification of an adhesion-mediating sequence in the disintegrin-like domain. J. Biol. Chem. 276, 24937–24945 10.1074/jbc.M101637200 [DOI] [PubMed] [Google Scholar]
- 2. Edwards D. R., Handsley M. M., and Pennington C. J. (2008) The ADAM metalloproteinases. Mol. Aspects Med. 29, 258–289 10.1016/j.mam.2008.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stautz D., Leyme A., Grandal M. V., Albrechtsen R., van Deurs B., Wewer U., and Kveiborg M. (2012) Cell-surface metalloprotease ADAM12 is internalized by a clathrin- and Grb2-dependent mechanism. Traffic 13, 1532–1546 10.1111/j.1600-0854.2012.01405.x [DOI] [PubMed] [Google Scholar]
- 4. Roghani M., Becherer J. D., Moss M. L., Atherton R. E., Erdjument-Bromage H., Arribas J., Blackburn R. K., Weskamp G., Tempst P., and Blobel C. P. (1999) Metalloprotease-disintegrin MDC9: intracellular maturation and catalytic activity. J. Biol. Chem. 274, 3531–3540 10.1074/jbc.274.6.3531 [DOI] [PubMed] [Google Scholar]
- 5. Peduto L., Reuter V. E., Shaffer D. R., Scher H. I., and Blobel C. P. (2005) Critical function for ADAM9 in mouse prostate cancer. Cancer Res. 65, 9312–9319 10.1158/0008-5472.CAN-05-1063 [DOI] [PubMed] [Google Scholar]
- 6. Chan K. M., Wong H. L., Jin G., Liu B., Cao R., Cao Y., Lehti K., Tryggvason K., and Zhou Z. (2012) MT1-MMP inactivates ADAM9 to regulate FGFR2 signaling and calvarial osteogenesis. Dev. Cell 22, 1176–1190 10.1016/j.devcel.2012.04.014 [DOI] [PubMed] [Google Scholar]
- 7. Guaiquil V., Swendeman S., Yoshida T., Chavala S., Campochiaro P. A., and Blobel C. P. (2009) ADAM9 is involved in pathological retinal neovascularization. Mol. Cell Biol. 29, 2694–2703 10.1128/MCB.01460-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hotoda N., Koike H., Sasagawa N., and Ishiura S. (2002) A secreted form of human ADAM9 has an α-secretase activity for APP. Biochem. Biophys. Res. Commun. 293, 800–805 10.1016/S0006-291X(02)00302-9 [DOI] [PubMed] [Google Scholar]
- 9. Weskamp G., Krätzschmar J., Reid M. S., and Blobel C. P. (1996) MDC9, a widely expressed cellular disintegrin containing cytoplasmic SH3 ligand domains. J. Cell Biol. 132, 717–726 10.1083/jcb.132.4.717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. O'Shea C., McKie N., Buggy Y., Duggan C., Hill A. D., McDermott E., O'Higgins N., and Duffy M. J. (2003) Expression of ADAM-9 mRNA and protein in human breast cancer. Int. J. Cancer 105, 754–761 10.1002/ijc.11161 [DOI] [PubMed] [Google Scholar]
- 11. Carl-McGrath S., Lendeckel U., Ebert M., Roessner A., and Röcken C. (2005) The disintegrin-metalloproteinases ADAM9, ADAM12, and ADAM15 are upregulated in gastric cancer. Int. J. Oncol. 26, 17–24 [PubMed] [Google Scholar]
- 12. Tao K., Qian N., Tang Y., Ti Z., Song W., Cao D., and Dou K. (2010) Increased expression of a disintegrin and metalloprotease-9 in hepatocellular carcinoma: implications for tumor progression and prognosis. Jpn. J. Clin. Oncol. 40, 645–651 10.1093/jjco/hyq030 [DOI] [PubMed] [Google Scholar]
- 13. Grutzmann R., Lüttges J., Sipos B., Ammerpohl O., Dobrowolski F., Alldinger I., Kersting S., Ockert D., Koch R., Kalthoff H., Schackert H. K., Saeger H. D., Kloppel G., and Pilarsky C. (2004) ADAM9 expression in pancreatic cancer is associated with tumour type and is a prognostic factor in ductal adenocarcinoma. Br. J. Cancer 90, 1053–1058 10.1038/sj.bjc.6601645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fritzsche F. R., Wassermann K., Jung M., Tölle A., Kristiansen I., Lein M., Johannsen M., Dietel M., Jung K., and Kristiansen G. (2008) ADAM9 is highly expressed in renal cell cancer and is associated with tumour progression. BMC Cancer 8, 179 10.1186/1471-2407-8-179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zubel A., Flechtenmacher C., Edler L., and Alonso A. (2009) Expression of ADAM9 in CIN3 lesions and squamous cell carcinomas of the cervix. Gynecol. Oncol. 114, 332–336 10.1016/j.ygyno.2009.05.005 [DOI] [PubMed] [Google Scholar]
- 16. Nath D., Slocombe P. M., Webster A., Stephens P. E., Docherty A. J., and Murphy G. (2000) Meltrin γ(ADAM-9) mediates cellular adhesion through α(6)β(1)integrin, leading to a marked induction of fibroblast cell motility. J. Cell Sci. 113, 2319–2328 [DOI] [PubMed] [Google Scholar]
- 17. Zhou M., Graham R., Russell G., and Croucher P. I. (2001) MDC-9 (ADAM-9/Meltrinγ) functions as an adhesion molecule by binding the α(v)β(5) integrin. Biochem. Biophys. Res. Commun. 280, 574–580 10.1006/bbrc.2000.4155 [DOI] [PubMed] [Google Scholar]
- 18. Mazzocca A., Coppari R., De Franco R., Cho J. Y., Libermann T. A., Pinzani M., and Toker A. (2005) A secreted form of ADAM9 promotes carcinoma invasion through tumor-stromal interactions. Cancer Res. 65, 4728–4738 10.1158/0008-5472.CAN-04-4449 [DOI] [PubMed] [Google Scholar]
- 19. Zigrino P., Steiger J., Fox J. W., Löffek S., Schild A., Nischt R., and Mauch C. (2007) Role of ADAM-9 disintegrin-cysteine-rich domains in human keratinocyte migration. J. Biol. Chem. 282, 30785–30793 10.1074/jbc.M701658200 [DOI] [PubMed] [Google Scholar]
- 20. Fry J. L., and Toker A. (2010) Secreted and membrane-bound isoforms of protease ADAM9 have opposing effects on breast cancer cell migration. Cancer Res. 70, 8187–8198 10.1158/0008-5472.CAN-09-4231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zigrino P., Nischt R., and Mauch C. (2011) The disintegrin-like and cysteine-rich domains of ADAM-9 mediate interactions between melanoma cells and fibroblasts. J. Biol. Chem. 286, 6801–6807 10.1074/jbc.M110.168617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Izumi Y., Hirata M., Hasuwa H., Iwamoto R., Umata T., Miyado K., Tamai Y., Kurisaki T., Sehara-Fujisawa A., Ohno S., and Mekada E. (1998) A metalloprotease-disintegrin, MDC9/meltrin-γ/ADAM9 and PKCδ are involved in TPA-induced ectodomain shedding of membrane-anchored heparin-binding EGF-like growth factor. EMBO J. 17, 7260–7272 10.1093/emboj/17.24.7260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Endres K., Anders A., Kojro E., Gilbert S., Fahrenholz F., and Postina R. (2003) Tumor necrosis factor-α converting enzyme is processed by proprotein-convertases to its mature form which is degraded upon phorbol ester stimulation. Eur. J. Biochem. 270, 2386–2393 10.1046/j.1432-1033.2003.03606.x [DOI] [PubMed] [Google Scholar]
- 24. Seals D. F., and Courtneidge S. A. (2003) The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 17, 7–30 10.1101/gad.1039703 [DOI] [PubMed] [Google Scholar]
- 25. Marcello E., Saraceno C., Musardo S., Vara H., de la Fuente A. G., Pelucchi S., Di Marino D., Borroni B., Tramontano A., Pérez-Otano I., Padovani A., Giustetto M., Gardoni F., and Di Luca M. (2013) Endocytosis of synaptic ADAM10 in neuronal plasticity and Alzheimer's disease. J. Clin. Invest. 123, 2523–2538 10.1172/JCI65401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Doherty G. J., and McMahon H. T. (2009) Mechanisms of endocytosis. Annu. Rev. Biochem. 78, 857–902 10.1146/annurev.biochem.78.081307.110540 [DOI] [PubMed] [Google Scholar]
- 27. Howard L., Nelson K. K., Maciewicz R. A., and Blobel C. P. (1999) Interaction of the metalloprotease disintegrins MDC9 and MDC15 with two SH3 domain-containing proteins, endophilin I and SH3PX1. J. Biol. Chem. 274, 31693–31699 10.1074/jbc.274.44.31693 [DOI] [PubMed] [Google Scholar]
- 28. von Kleist L., Stahlschmidt W., Bulut H., Gromova K., Puchkov D., Robertson M. J., MacGregor K. A., Tomilin N., Tomlin N., Pechstein A., Chau N., Chircop M., Sakoff J., von Kries J. P., Saenger W., Kräusslich H. G., Shupliakov O., Robinson P. J., McCluskey A., and Haucke V. (2011) Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell 146, 471–484 10.1016/j.cell.2011.06.025 [DOI] [PubMed] [Google Scholar]
- 29. Hopkins C. R., and Trowbridge I. S. (1983) Internalization and processing of transferrin and the transferrin receptor in human carcinoma A431 cells. J. Cell Biol. 97, 508–521 10.1083/jcb.97.2.508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guo S., Zhang X., Zheng M., Zhang X., Min C., Wang Z., Cheon S. H., Oak M. H., Nah S. Y., and Kim K. M. (2015) Selectivity of commonly used inhibitors of clathrin-mediated and caveolae-dependent endocytosis of G protein-coupled receptors. Biochim. Biophys. Acta 1848, 2101–2110 10.1016/j.bbamem.2015.05.024 [DOI] [PubMed] [Google Scholar]
- 31. Di Guglielmo G. M., Le Roy C., Goodfellow A. F., and Wrana J. L. (2003) Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nat. Cell Biol. 5, 410–421 10.1038/ncb975 [DOI] [PubMed] [Google Scholar]
- 32. Sigismund S., Argenzio E., Tosoni D., Cavallaro E., Polo S., and Di Fiore P. P. (2008) Clathrin-mediated internalization is essential for sustained EGFR signaling but dispensable for degradation. Dev. Cell 15, 209–219 10.1016/j.devcel.2008.06.012 [DOI] [PubMed] [Google Scholar]
- 33. Soulet F., Yarar D., Leonard M., and Schmid S. L. (2005) SNX9 regulates dynamin assembly and is required for efficient clathrin-mediated endocytosis. Mol. Biol. Cell 16, 2058–2067 10.1091/mbc.E04-11-1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shin N., Lee S., Ahn N., Kim S. A., Ahn S. G., YongPark Z., and Chang S. (2007) Sorting nexin 9 interacts with dynamin 1 and N-WASP and coordinates synaptic vesicle endocytosis. J. Biol. Chem. 282, 28939–28950 10.1074/jbc.M700283200 [DOI] [PubMed] [Google Scholar]
- 35. Park J., Kim Y., Lee S., Park J. J., Park Z. Y., Sun W., Kim H., and Chang S. (2010) SNX18 shares a redundant role with SNX9 and modulates endocytic trafficking at the plasma membrane. J. Cell Sci. 123, 1742–1750 10.1242/jcs.064170 [DOI] [PubMed] [Google Scholar]
- 36. Maretzky T., Swendeman S., Mogollon E., Weskamp G., Sahin U., Reiss K., and Blobel C. P. (2017) Characterization of the catalytic properties of the membrane-anchored metalloproteinase ADAM9 in cell-based assays. Biochem. J. 474, 1467–1479 10.1042/BCJ20170075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Karadag A., Zhou M., and Croucher P. I. (2006) ADAM-9 (MDC-9/meltrin-γ), a member of the a disintegrin and metalloproteinase family, regulates myeloma-cell-induced interleukin-6 production in osteoblasts by direct interaction with the α(v)β5 integrin. Blood 107, 3271–3278 10.1182/blood-2005-09-3830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. English W. R., Corvol P., and Murphy G. (2012) LPS activates ADAM9 dependent shedding of ACE from endothelial cells. Biochem. Biophys. Res. Commun. 421, 70–75 10.1016/j.bbrc.2012.03.113 [DOI] [PubMed] [Google Scholar]
- 39. Lorenzen I., Lokau J., Korpys Y., Oldefest M., Flynn C. M., Künzel U., Garbers C., Freeman M., Grötzinger J., and Düsterhoft S. (2016) Control of ADAM17 activity by regulation of its cellular localisation. Sci. Rep. 6, 35067 10.1038/srep35067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Doedens J. R., and Black R. A. (2000) Stimulation-induced down-regulation of tumor necrosis factor-α converting enzyme. J. Biol. Chem. 275, 14598–14607 10.1074/jbc.275.19.14598 [DOI] [PubMed] [Google Scholar]
- 41. Linford A., Yoshimura S., Nunes Bastos R., Langemeyer L., Gerondopoulos A., Rigden D. J., and Barr F. A. (2012) Rab14 and its exchange factor FAM116 link endocytic recycling and adherens junction stability in migrating cells. Dev. Cell 22, 952–966 10.1016/j.devcel.2012.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Grant B. D., and Donaldson J. G. (2009) Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 10, 597–608 10.1038/nrm2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Soubeyran P., Kowanetz K., Szymkiewicz I., Langdon W. Y., and Dikic I. (2002) Cbl-CIN85-endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature 416, 183–187 10.1038/416183a [DOI] [PubMed] [Google Scholar]
- 44. Jeffers M., Taylor G. A., Weidner K. M., Omura S., and Vande Woude G. F. (1997) Degradation of the Met tyrosine kinase receptor by the ubiquitin-proteasome pathway. Mol. Cell Biol. 17, 799–808 10.1128/MCB.17.2.799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Horazdovsky B. F., Davies B. A., Seaman M. N., McLaughlin S. A., Yoon S., and Emr S. D. (1997) A sorting nexin-1 homologue, Vps5p, forms a complex with Vps17p and is required for recycling the vacuolar protein-sorting receptor. Mol. Biol. Cell 8, 1529–1541 10.1091/mbc.8.8.1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Böttcher R. T., Stremmel C., Meves A., Meyer H., Widmaier M., Tseng H. Y., and Fässler R. (2012) Sorting nexin 17 prevents lysosomal degradation of β1-integrins by binding to the β1-integrin tail. Nat. Cell. Biol. 14, 584–592 10.1038/ncb2501 [DOI] [PubMed] [Google Scholar]
- 47. Lundmark R., and Carlsson S. R. (2009) SNX9: a prelude to vesicle release. J. Cell Sci. 122, 5–11 10.1242/jcs.037135 [DOI] [PubMed] [Google Scholar]
- 48. Bendris N., Williams K. C., Reis C. R., Welf E. S., Chen P. H., Lemmers B., Hahne M., Leong H. S., and Schmid S. L. (2016) SNX9 promotes metastasis by enhancing cancer cell invasion via differential regulation of RhoGTPases. Mol. Biol. Cell 10.1019/mbc.E16-02-0101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tseng H. Y., Thorausch N., Ziegler T., Meves A., Fässler R., and Böttcher R. T. (2014) Sorting nexin 31 binds multiple β integrin cytoplasmic domains and regulates beta1 integrin surface levels and stability. J. Mol. Biol. 426, 3180–3194 10.1016/j.jmb.2014.07.003 [DOI] [PubMed] [Google Scholar]
- 50. Amour A., Knight C. G., English W. R., Webster A., Slocombe P. M., Knäuper V., Docherty A. J., Becherer J. D., Blobel C. P., and Murphy G. (2002) The enzymatic activity of ADAM8 and ADAM9 is not regulated by TIMPs. FEBS Lett. 524, 154–158 10.1016/S0014-5793(02)03047-8 [DOI] [PubMed] [Google Scholar]
- 51. Matthews A. L., Noy P. J., Reyat J. S., and Tomlinson M. G. (2017) Regulation of A disintegrin and metalloproteinase (ADAM) family sheddases ADAM10 and ADAM17: the emerging role of tetraspanins and rhomboids. Platelets 28, 333–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Adrain C., Zettl M., Christova Y., Taylor N., and Freeman M. (2012) Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 335, 225–228 10.1126/science.1214400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McIlwain D. R., Lang P. A., Maretzky T., Hamada K., Ohishi K., Maney S. K., Berger T., Murthy A., Duncan G., Xu H. C., Lang K. S., Häussinger D., Wakeham A., Itie-Youten A., Khokha R., Ohashi P. S., Blobel C. P., and Mak T. W. (2012) iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science 335, 229–232 10.1126/science.1214448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Haining E. J., Yang J., Bailey R. L., Khan K., Collier R., Tsai S., Watson S. P., Frampton J., Garcia P., and Tomlinson M. G. (2012) The TspanC8 subgroup of tetraspanins interacts with A disintegrin and metalloprotease 10 (ADAM10) and regulates its maturation and cell surface expression. J. Biol. Chem. 287, 39753–39765 10.1074/jbc.M112.416503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dombernowsky S. L., Samsøe-Petersen J., Petersen C. H., Instrell R., Hedegaard A. M., Thomas L., Atkins K. M., Auclair S., Albrechtsen R., Mygind K. J., Fröhlich C., Howell M., Parker P., Thomas G., and Kveiborg M. (2015) The sorting protein PACS-2 promotes ErbB signalling by regulating recycling of the metalloproteinase ADAM17. Nat. Commun. 6, 7518 10.1038/ncomms8518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shintani Y., Higashiyama S., Ohta M., Hirabayashi H., Yamamoto S., Yoshimasu T., Matsuda H., and Matsuura N. (2004) Overexpression of ADAM9 in non-small cell lung cancer correlates with brain metastasis. Cancer Res. 64, 4190–4196 10.1158/0008-5472.CAN-03-3235 [DOI] [PubMed] [Google Scholar]
- 57. Sun H. S., Su I. J., Lin Y. C., Chen J. S., and Fang S. Y. (2003) A 2.6 Mb interval on chromosome 6q25.2-q25.3 is commonly deleted in human nasal natural killer/T-cell lymphoma. Br. J. Haematol. 122, 590–599 10.1046/j.1365-2141.2003.04419.x [DOI] [PubMed] [Google Scholar]
- 58. Monoranu C. M., Huang B., Zangen I. L., Rutkowski S., Vince G. H., Gerber N. U., Puppe B., and Roggendorf W. (2008) Correlation between 6q25.3 deletion status and survival in pediatric intracranial ependymomas. Cancer Genet. Cytogenet. 182, 18–26 10.1016/j.cancergencyto.2007.12.008 [DOI] [PubMed] [Google Scholar]
- 59. Mao X., Boyd L. K., Yáñez-Muñoz R. J., Chaplin T., Xue L., Lin D., Shan L., Berney D. M., Young B. D., and Lu Y. J. (2011) Chromosome rearrangement associated inactivation of tumour suppressor genes in prostate cancer. Am. J. Cancer Res. 1, 604–617 [PMC free article] [PubMed] [Google Scholar]
- 60. Wu G., Barnhill R. L., Lee S., Li Y., Shao Y., Easton J., Dalton J., Zhang J., Pappo A., and Bahrami A. (2016) The landscape of fusion transcripts in spitzoid melanoma and biologically indeterminate spitzoid tumors by RNA sequencing. Mod. Pathol. 29, 359–369 10.1038/modpathol.2016.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sanjay A., Houghton A., Neff L., DiDomenico E., Bardelay C., Antoine E., Levy J., Gailit J., Bowtell D., Horne W. C., and Baron R. (2001) Cbl associates with Pyk2 and Src to regulate Src kinase activity, α(v)β(3) integrin-mediated signaling, cell adhesion, and osteoclast motility. J. Cell Biol. 152, 181–195 10.1083/jcb.152.1.181 [DOI] [PMC free article] [PubMed] [Google Scholar]