

Abstract
Notch signaling plays critical roles in cancer progression, motivating efforts to identify inhibitors of this pathway. Perron et al. report a small-molecule screen intended to discover compounds that could interfere with the downstream transcription factor HES1. Target validation of their compounds unexpectedly identified a novel HES1-interacting protein, prohibitin 2. This highlights a new mechanism to block Notch signaling and prompting further exploration of HES1 biology.
Notch proteins are a family of four transmembrane receptors involved in regulatory and cellular developmental processes. Aberrant Notch signaling has been associated with cancer stemness, pro-tumorigenic tumor microenvironment, and immune evasion for lung, breast, and pancreas carcinogenesis. Accordingly, Notch signaling has been a focus for cancer therapy. The complexity of the Notch pathway provides multiple targets to interfere with signaling. For example, neutralizing antibodies have been developed against both Notch ligands (Jagged 1 and 2 and DLL-1, -3, and -4) and the Notch receptors, such as the Notch2- and Notch3-neutralizing antibody, tarextumab (OncoMed Pharmaceuticals) (Fig. 1). Unfortunately, these have not been successful in recent clinical trials despite preclinical successes (1). After ligand binding, the receptor undergoes serial proteolytic cleavage by ADAM family metalloproteinases and the transmembrane γ-secretase complex. γ-Secretase inhibitors have been widely tested as anticancer therapy, and results from clinical studies show that these inhibitors in combination with chemotherapeutic agents can sensitize cells to chemotherapy (2). Nevertheless, their continuous administration causes severe diarrhea in patients, due to the prevalence of Notch1 receptors in the gut, and becomes intolerable (3). Finally, the Notch intracellular domain (NICD),2 once released by γ-secretase, translocates to the nucleus and interacts in complexes with transcription factors, including RBP-Jκ, CSL, and MAML (4). These complexes are now the targets of the next generation of Notch signaling antagonists. A new study by Perron et al. (5) joins in this effort, seeking to identify a small molecule that could stabilize a repressive transcription factor complex. Surprisingly, characterization of their small molecules led to the serendipitous discovery of a new heterodimer and a new mechanism of inhibition for the Notch pathway.
Figure 1.
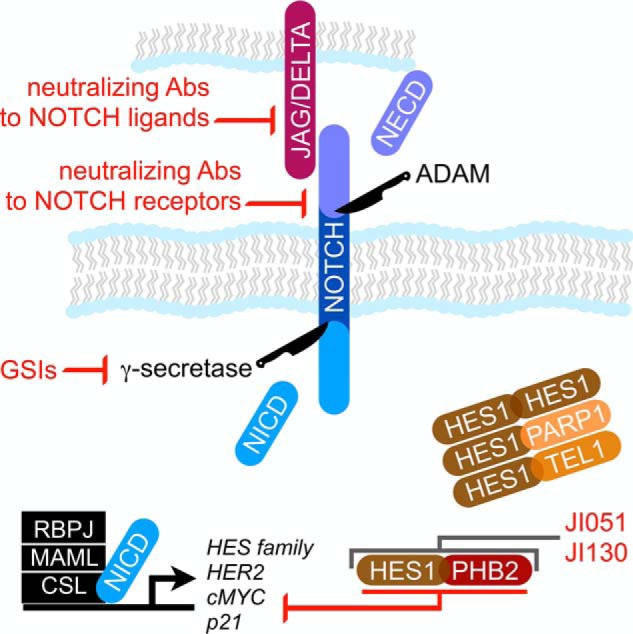
A novel NOTCH signaling antagonist targets the newly discovered HES1–PHB2 repressive complex. A Notch receptor–ligand complex initiates the signaling cascade involving sequential cleavage of the extracellular and intercellular domains of the Notch receptor (NECD and NICD, respectively). In the nucleus, NICD complexes promote expression of several genes, including those of the HES family. The HES proteins can, in turn, repress NICD activity as homo- and heterodimers. Inhibitors against γ-secretase, the Notch receptor and its ligands (Jagged and Delta proteins), have had limited clinical benefit. However, small molecules that stabilize the HES1–PHB2 heterodimer (JI051 and JI130) reveal a novel mechanism of Notch inhibition that could provide a clinical advantage.
The transcription factor hairy and enhancer of split 1 (HES1, homolog of the Drosophila gene Hairy) is a Notch target gene and a transcriptional repressor that promotes cell proliferation and differentiation when complexed with certain cofactors. For example, when HES1 couples with transducin-like enhancer of split 1 (TLE1, homolog of the Drosophila gene Groucho), it promotes cell cycle progression by repressing p27Kip1, a cyclin-dependent kinase inhibitor (6). Due to the role of HES1 in tissue development, researchers have studied inhibition of specific HES1 dimers in models of neural differentiation and cancer, using both siRNA and small molecule inhibitors. A combined approach modulating HES1 using both shRNA and a HES1 antagonist was able to reduce T-cell acute lymphoblastic leukemia proliferation (7), providing support for this direction.
In their study, Perron et al. (5) sought to develop small molecules to target the HES1–TLE1 interaction. They focused their chemical library on compounds containing indole moieties, which they anticipated would complement the WRPW (Trp–Arg–Pro–Trp) consensus interaction domain of HES1. They screened a library of 1800 compounds, finding 3 that caused elevated Notch reporter activity and demonstrated antiproliferative activity. The authors then synthesized and tested 130 derivatives of their initial hits, an indolylacrylamide molecule. They provided clear structure–activity relationships and identified several compounds with improved activity. One of these, JI051, was demonstrated to cause G2/M cell cycle arrest in a HES1-dependent manner.
In an interesting development, when characterizing JI051's interaction with HES1/TLE1, Perron et al. (5) found that JI051 did not interact with TLE1, but rather identified prohibitin 2 (PHB2) as its target through nanoLC-MS/MS. PHB2 is reported to repress estrogen receptor-α–dependent transcriptional activity, mediate antiapoptotic signals, and most recently recognized to facilitate mitophagy (8, 9). To confirm this target in vitro, the authors used siRNA to study the effect of PHB2 silencing on JI051 activity, observing that JI051's ability to induce condensed chromatin— a hallmark of mitosis—was lost in the absence of PHB2. However, the G2/M cell cycle inhibition observed with JI051 treatment was not mimicked by PHB2 knockdown alone, suggesting a new role for PHB2 that is apparent only when coupled to HES1. Furthermore, co-immunoprecipitation assays demonstrated that JI051 stabilized the HES1–PHB2 interaction while confocal microscopy indicated that the complex is primarily in the cytoplasm, suggesting JI051 may interfere with normal HES1 trafficking. Finally, JI051 and its derivative JI130 were tested on pancreatic cancer cell lines and in xenograft experiments, showing suppression of cell growth and cell cycle arrest in vitro and a significant decrease in tumor growth in vivo (Fig. 1).
Inhibiting Notch signaling in cancer has been studied extensively. However, the efficacy of these drugs has not been realized in the clinic due their adverse side effects. Addressing downstream targets, such as HES1, could interrupt differentiation and maintenance of cancer stem cells, a critical aspect of chemoresistance (10). HES1 functions in homo- or heterodimers to target and repress specific tumor-suppressive pathways (7). The work by Perron et al. (5) identified a new heterodimer with an important role in HES1 biology and demonstrated that stabilizing the HES1–PHB2 can antagonize tumor expansion. Given that the targeted WRPW moiety is a common motif for HES1 dimerization, it will be important to test whether these small molecules have effects on other complexes. The surprising fact that JI051 did not affect the HES1–TLE1 dimer could suggest that signaling specificity for the many Notch effectors is possible and limit undesirable side effects. It is also possible that some of the other small molecule derivatives made by Perron et al. (5) could interrupt or even stabilize other HES1 dimers; these derivatives could serve as important tools in interrogating new signaling mechanisms. Specific targeting of the HES1-repressive complex can also be applied to many other diseases. For example, being able to block the HES1–HES1 homodimer could be instrumental in inhibiting neuronal stem cells, thus providing a new direction for regenerative drug candidates for spinal cord injury or neurodegenerative disorders. The discovery of these chemicals, biomolecular partners, and mechanisms of action will enable many new investigations as the research for Notch inhibition continues.
This work was supported by NCI, National Institutes of Health Grant CA098912 and U. S. Department of Veterans Affairs Grant I01BX001040. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- NICD
- Notch intracellular domain
- NECD
- Notch extracellular domain.
References
- 1. Yen W. C., Fischer M. M., Axelrod F., Bond C., Cain J., Cancilla B., Henner W. R., Meisner R., Sato A., Shah J., Tang T., Wallace B., Wang M., Zhang C., Kapoun A. M., Lewicki J., Gurney A., and Hoey T. (2015) Targeting Notch signaling with a Notch2/Notch3 antagonist (tarextumab) inhibits tumor growth and decreases tumor-initiating cell frequency. Clinical Cancer Res. 21, 2084–2095 10.1158/1078-0432.CCR-14-2808 [DOI] [PubMed] [Google Scholar]
- 2. Yuan X., Wu H., Xu H., Xiong H., Chu Q., Yu S., Wu G. S., and Wu K. (2015) Notch signaling: An emerging therapeutic target for cancer treatment. Cancer Lett. 369, 20–27 10.1016/j.canlet.2015.07.048 [DOI] [PubMed] [Google Scholar]
- 3. Locatelli M. A., Aftimos P., Dees E. C., LoRusso P. M., Pegram M. D., Awada A., Huang B., Cesari R., Jiang Y., Shaik M. N., Kern K. A., and Curigliano G. (2017) Phase I study of the gamma secretase inhibitor PF-03084014 in combination with docetaxel in patients with advanced triple-negative breast cancer. Oncotarget 8, 2320–2328 10.18632/oncotarget.13727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kumar R., Juillerat-Jeanneret L., and Golshayan D. (2016) Notch antagonists: Potential modulators of cancer and inflammatory diseases. J. Med. Chem. 59, 7719–7737 10.1021/acs.jmedchem.5b01516 [DOI] [PubMed] [Google Scholar]
- 5. Perron A., Nishikawa Y., Iwata J., Shimojo H., Takaya J., Kobayashi K., Imayoshi I., Mbenza N. M., Takenoya M., Kageyama R., Kodama Y., and Uesugi M. (2018) Small-molecule screening yields a compound that inhibits the cancer-associated transcription factor Hes1 via the PHB2 chaperone. J. Biol. Chem. 293, 8285–8294 10.1074/jbc.RA118.002316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Murata K., Hattori M., Hirai N., Shinozuka Y., Hirata H., Kageyama R., Sakai T., and Minato N. (2005) Hes1 directly controls cell proliferation through the transcriptional repression of p27Kip1. Mol. Cell. Biol. 25, 4262–4271 10.1128/MCB.25.10.4262-4271.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schnell S. A., Ambesi-Impiombato A., Sanchez-Martin M., Belver L., Xu L., Qin Y., Kageyama R., and Ferrando A. A. (2015) Therapeutic targeting of HES1 transcriptional programs in T-ALL. Blood 125, 2806–2814 10.1182/blood-2014-10-608448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bavelloni A., Piazzi M., Raffini M., Faenza I., and Blalock W. L. (2015) Prohibitin 2: At a communications crossroads. IUBMB Life 67, 239–254 10.1002/iub.1366 [DOI] [PubMed] [Google Scholar]
- 9. Wei Y., Chiang W. C., Sumpter R. Jr., Mishra P., and Levine B. (2017) Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 168, 224–238.e210 10.1016/j.cell.2016.11.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu Z. H., Dai X. M., and Du B. (2015) Hes1: A key role in stemness, metastasis and multidrug resistance. Cancer Biol. Ther. 16, 353–359 10.1080/15384047.2015.1016662 [DOI] [PMC free article] [PubMed] [Google Scholar]