

Abstract
The type I cGMP-dependent protein kinases (PKG I) serve essential physiological functions, including smooth muscle relaxation, cardiac remodeling, and platelet aggregation. These enzymes form homodimers through their N-terminal dimerization domains, a feature implicated in regulating their cooperative activation. Previous investigations into the activation mechanisms of PKG I isoforms have been largely influenced by structures of the cAMP-dependent protein kinase (PKA). Here, we examined PKG Iα activation by cGMP and cAMP by engineering a monomeric form that lacks N-terminal residues 1–53 (Δ53). We found that the construct exists as a monomer as assessed by whole-protein MS, size-exclusion chromatography, and small-angle X-ray scattering (SAXS). Reconstruction of the SAXS 3D envelope indicates that Δ53 has a similar shape to the heterodimeric RIα–C complex of PKA. Moreover, we found that the Δ53 construct is autoinhibited in its cGMP-free state and can bind to and be activated by cGMP in a manner similar to full-length PKG Iα as assessed by surface plasmon resonance (SPR) spectroscopy. However, we found that the Δ53 variant does not exhibit cooperative activation, and its cyclic nucleotide selectivity is diminished. These findings support a model in which, despite structural similarities, PKG Iα activation is distinct from that of PKA, and its cooperativity is driven by in trans interactions between protomers.
Keywords: cyclic GMP (cGMP), protein kinase, allosteric regulation, signal transduction, small-angle X-ray scattering (SAXS), surface plasmon resonance (SPR), AGC kinases, cGMP-dependent protein kinase
Introduction
cGMP-dependent protein kinases (PKGs)5 are members of the AGC kinase family (PKA/PKG/PKC). They are conserved in eukaryotes and ubiquitously expressed throughout the human body (1, 2). These kinases regulate critical processes such as vascular tone, cardiac remodeling, and platelet aggregation (1, 3–5). The primary activator of PKG isoforms is the second messenger cyclic 3′,5′-guanosine monophosphate (cGMP) (1). In vascular smooth muscle, activated PKG I phosphorylates intracellular targets, including myosin phosphatase target subunit 1 (MYPT1), regulator of G-protein signaling 2 (RGS2), inositol trisphosphate receptor-associated cGMP-kinase (IRAG), and the large-conductance calcium-activated potassium channel (KCa 1.1 or BK), resulting in vasodilation (6–11).
PKG I is expressed as two splice variants that form homodimers (α/β) (2, 12, 13). In both isoforms, each protomer is composed of a dimerization domain formed by a leucine zipper (LZ) followed by an autoinhibitory domain segment, a regulatory domain containing two cGMP-binding sites (A and B), and a catalytic domain (see Fig. 1A) (14). PKG Iα and Iβ have low sequence conservation in their dimerization and autoinhibitory domains, and this difference is thought to both control the activity and response of the kinase to cGMP and mediate its targeting to specific substrates (6, 15–20). How the regulatory and catalytic domains interact in the inactive state and communicate to control cooperative activation have remained unsettled questions for the field (21, 22). In the inactive state, it has been hypothesized that the autoinhibitory domain of PKG I occupies the catalytic cleft formed between the N- and C-terminal lobes and acts as a pseudosubstrate in a manner similar to the cAMP-dependent protein kinase (PKA) (23).
Figure 1.

Design of the Δ53 construct based on sequence homology with PKA R-domains. A, the domain architecture of PKG Iα (charcoal) depicting the truncation of the dimerization domain to generate Δ53 (red) alongside PKA RIα (cyan) and PKA catalytic (gray) domains. The domain architecture is described as follows: D/D, dimerization domain; AI, autoinhibitory domain; cGMP-A/B, cGMP-binding sites; SW, switch helix (25); N/C-lobe, catalytic domain. B, a multiple sequence alignment shaded by BLOSUM62 score is shown for PKG Iα relative to PKA regulatory domain isoforms. The autoinhibitory sequences of PKG I (red) and PKA–RIα (cyan) are outlined. C, the crystal structure of the RIα–C heterodimer (Protein Data Bank code 2QCS) detailing the contacts between the autoinhibitory domain segment beginning at Lys92 (cyan) and the catalytic domain (gray).
PKG isoforms share sequence homology with PKA (28 and 41% identity in its regulatory domain and catalytic domain, respectively). Consequently, current models of the relationship between PKG I structure and function have been overwhelmingly influenced by studies of PKA. However, PKA is expressed as separate catalytic and regulatory domains and PKG exists as a single polypeptide chain (24–26). Structural and biochemical studies of PKA have demonstrated an in cis mechanism for PKA activation (27–29). The sequential binding of cAMP to two coupled binding sites (A and B) within the regulatory subunit constitutes the physical basis for cooperativity in both cAMP binding and kinase activation. Although it is known that full-length PKG I dimers also exhibit cooperative activation, it is unknown whether cooperativity is driven by the regulatory domain (in a fashion similar to PKA) or whether the dimeric state influences this effect (15, 25, 30).
It has been suggested that the N-terminal domains in the hinge region between the leucine zipper and autoinhibitory domains in PKG I are highly flexible, and structural models of PKG I have depicted the N terminus protruding from the holoenzyme (regulatory and catalytic domain complex) (14, 23, 31). Although it has been widely suggested that dimerization is essential for catalytic function, it has never been experimentally tested in the Iα variant. Informed by previous models, we hypothesized that PKG Iα could be truncated to generate a functional monomer by removing the flexible N terminus (zipper and hinge). Moreover, we surmised that this construct would retain cGMP-dependent activation due to the inclusion of the autoinhibitory domain. In this study, we provide the biochemical and biophysical characterization of PKG Iα Δ53 (subsequently referred to as Δ53) by demonstrating its monomeric architecture, phosphorylation state, cGMP binding properties, and the kinetic characteristics of its cGMP-dependent activation. Using the Δ53 construct, we address the following: 1) whether PKG Iα is inhibited in cis or in trans, 2) how cyclic nucleotide binding and selectivity of the A-site is linked to activation, and 3) a putative mechanism by which cGMP-mediated cooperativity is derived in PKG. Our data indicate that PKG Iα can form an in cis autoinhibited complex. Furthermore, cooperative activation of PKG Iα by cGMP, in contrast to PKA, relies on interactions between the homodimeric regulatory domains. We conclude that cooperativity is driven in part by the N-terminal dimerization domain by localizing PKG monomers within close proximity during cGMP binding to form in trans interprotomer interactions.
Results
Design, expression, and purification of Δ53
The design of a monomeric form of PKG Iα was accomplished using a bioinformatics approach by comparing with a previously solved structure of the homolog, PKA, in a heterodimeric complex with its regulatory domain (Fig. 1, A and B). The complex RIα–C (Protein Data Bank code 2QCS) was chosen because of the high sequence conservation of its autoinhibitory segment with PKG Iα and the availability of its three-dimensional coordinates (Fig. 1C). A multiple sequence alignment of the PKG Iα autoinhibitory fragment with that of known isoforms of PKA was performed. The distance from the first residue (Lys92) to the start of the autoinhibitory sequence in the RIα–C structure was used to determine that the residue equidistant from the autoinhibitory sequence in PKG Iα was Pro56 (Fig. 1B). We hypothesized that, in a similar fashion to RIα–C, the autoinhibitory segment preceding the canonical (A/G)ISAE pseudosubstrate segment would confine the P + 1 loop of the catalytic domain and make further contacts with the C-terminal tail, glycine-rich loop, and the F-helix from the large lobe (32, 33). As a consequence and so as not to exclude residues that may be important for autoinhibition, Δ53 was engineered to begin at Ile54.
Following expression and purification of Δ53 (Fig. 2A), we examined the kinase by size-exclusion chromatography (Fig. 2B). Both the apo and cGMP-bound forms exhibited reproducible elution profiles centered at 14.8 and 14.7 ml, respectively, indicative of a 70-kDa species according to molecular mass standards (Fig. S1). The addition of cGMP appeared to have a negligible effect on the elution volume of the protein, suggesting that dimer formation was not present in the apo or cGMP-saturated samples. Δ53 was further examined by time-of-flight (TOF) MS to determine its mass and phosphorylation state (Fig. 2C). A mass of 70,511 Da, corresponding to a monophosphorylated, monomeric species, was observed. We also discovered the presence of a doubly phosphorylated species comprising 20% of the total ion content (70,591 Da). Tandem MS analyses of trypsin-digested samples were used to confirm the primary phosphorylation site at Thr517 (full-length enzyme numbering scheme) located in the activation loop of the catalytic domain (Table 1). This phosphorylation site has been previously identified in full-length PKG Iα and is required for catalytic activity (34). To confirm this, we also examined preparations of full-length PKG Iα and found a single phosphorylation site corresponding to Thr517 (Table S1). Based on previous studies, the minor phosphorylation site observed for Δ53 was suspected to reside in the autoinhibitory domain (35, 36). However, the peaks corresponding to the peptide containing the autophosphorylated pseudosubstrate sequence, R↓AQGISAEPQTYR↓S (residues 61–72), were of high intensity and suggested no phosphorylations were present.
Figure 2.
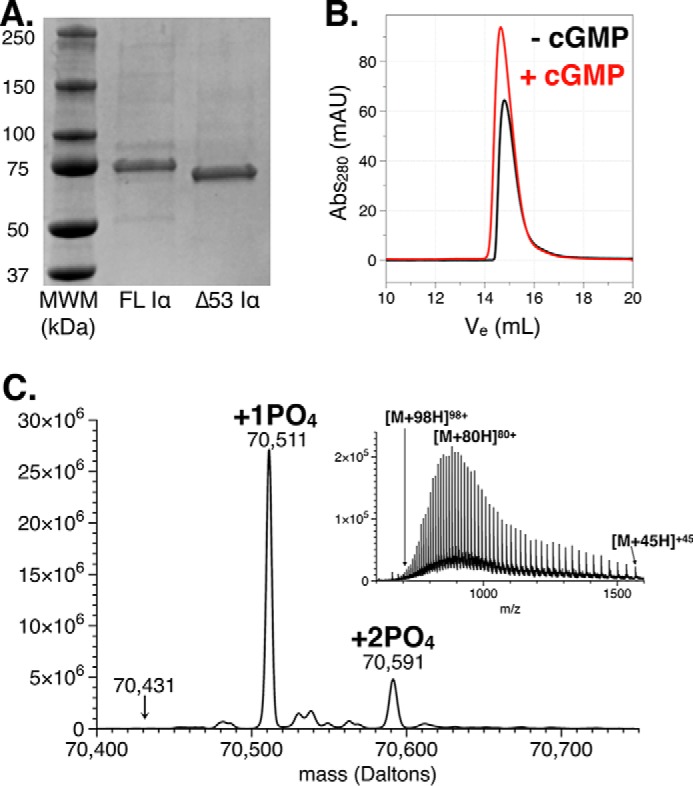
Biophysical characterization of Δ53. A, Coomassie-stained 12% SDS-PAGE of PKG Iα full length and Δ53 under denaturing and reducing conditions. B, size-exclusion chromatography traces of Δ53 in its apo (black) and cGMP-saturated (red) forms. C, a raw m/z trace (inset) and the deconvoluted TOF-MS displaying the two predominant masses observed for Δ53 corresponding to singly and doubly phosphorylated species. The mass correlated to the location of an unphosphorylated monomer is also denoted. Abs, absorbance; mAU, milli-absorbance units.
Table 1.
Phosphopeptides of Δ53 PKG I
| Precursor MH+ | z | Start | End | Sequence | Modifications | Retention time | Intensity (×103) |
|---|---|---|---|---|---|---|---|
| Da | min | ||||||
| 1320.665 | 2.0 | 61 | 72 | R↓AQGISAEPQTYR↓S | 19.71 | 9,523 | |
| 2262.055 | 2.7 | 515 | 533 | K↓TWTFCGTPEYVAPEIILNK↓G | PO4-Thr517 | 44.89 | 1,591 |
| 2182.082 | 2.7 | 515 | 533 | K↓TWTFCGTPEYVAPEIILNK↓G | 42.80 | 123 | |
| 2390.150 | 3.0 | 514 | 533 | K↓KTWTFCGTPEYVAPEIILNK↓G | PO4-Thr517 | 41.03 | 260 |
| 2310.173 | 3.0 | 514 | 533 | K↓KTWTFCGTPEYVAPEIILNK↓G | 39.02 | 10 |
Small angle X-ray scattering (SAXS) analysis
The results presented thus far indicate that Δ53 exists as a monomer in solution. To characterize the low-resolution solution structure of the autoinhibited complex, we utilized size-exclusion chromatography (SEC) coupled to SAXS wherein during analysis a single species associated with Δ53 was observed (Table 2). Concurrent Guinier calculation during data collection indicated a constant Rg across the sample peak (Fig. 3A). The averaged scattering intensity curve (Iq versus q) for Δ53, which was obtained by averaging frames 410–459 of the SEC-SAXS peak, exhibited a linear Guinier plot (Fig. 3, B and inset, and Table 2). The Guinier plot indicated the presence of a monodisperse system with no evidence of aggregation. A Kratky plot of the data suggested the presence of a well-folded species (Fig. 3C). The P(r) curve calculated from the scattering intensity contained a single peak at ∼30 Å that smoothly decayed to a maximum linear dimension (Dmax) of 97 Å (Fig. S2). When compared with the P(r) curve of the homologous PKA heterodimer upon which we based our construct, RIα–C (green), both showed a single peak at 34 Å, but the RIα–C heterodimer had an extended Dmax of ∼130 Å. Comparison of the Δ53 P(r) with that observed for a different PKA heterodimer, RIIβ–C (red), demonstrated that they have nearly identical dimensions (Table S2) (37).
Table 2.
Small angle X-ray scattering data collection and analysis
C, concentration; SAS, small-angle scattering; N/A, not applicable; SSRL, Stanford Synchrotron Radiation Lightsource; PDB, Protein Data Bank; Vol, volume; SASBDB, Small Angle Scattering Biological Data Bank; Vol, optimal excluded volume; Ra, optimal atomic group radius; Dro, optimal hydration shell contrast.
| Δ53 PKG Iα | |||
|---|---|---|---|
| (a) Sample details | |||
| Organism | Bos taurus | ||
| Source | Sf9 | ||
| UniProt sequence ID (residues in construct) | NP_776861 (54–671) | ||
| Extinction coefficient ϵ (A280, 0.1% (w/v)) | 1.069 | ||
| Molecular mass M from chemical composition (Da) | 70,434 | ||
| SEC-SAS | |||
| Loading concentration (mg/ml−1) | 6 | ||
| Injection volume (μl) | 35 | ||
| Flow rate (ml/min−1) | 0.05 | ||
| Average C in combined data frames (mg/ml−1) | 0.6 | ||
| Solvent (solvent blanks taken from SEC flow-through prior to elution of protein) | 50 mm MES, 300 mm NaCl, 1 mm TCEP, 5 mm DTT, pH 6.9 | ||
| (b) SAS data collection parameters | |||
| Source, instrument, and description or reference | SSRL BL4-2 with Rayonix MX225-HE detector | ||
| Wavelength (Å) | 1.127 | ||
| Beam geometry: size (sample-to-detector distance) | 0.3 × 0.3 mm (1.7 m) | ||
| q measurement range (Å−1) | 0.0087–0.5126 | ||
| Absolute scaling method | N/A | ||
| Basis for normalization to constant counts | Transmission intensity measured by pin diode beamstop | ||
| Method for monitoring radiation damage, X-ray dose where relevant | fplcplots (available at BL4-2) | ||
| Exposure time, number of exposures | 1 s, 600 images | ||
| Sample configuration, including path length and flow rate where relevant | 1.5-mm quartz capillary, 0.05 ml/min | ||
| Sample temperature (°C) | 22 | ||
| (c) Software used for SAS data reduction, analysis, and interpretation | |||
| SAS data reduction to sample-solvent scattering and extrapolation, merging, desmearing, etc. as relevant | Sastool | ||
| Calculation of ϵ from sequence | Protparam (69) | ||
| Basic analyses: Guinier, P(r), scattering particle volume | PRIMUS from ATSAS v2.8.3 | ||
| Shape/bead modeling | DAMMIF (65) and DAMMIN (70) via ATSAS online (https://www.embl-hamburg.de/biosaxs/atsas-online/)a | ||
| Atomic structure modeling (homology, rigid body, ensemble) | FoXS (38) via web server (https://modbase.compbio.ucsf.edu/foxs/)a and CRYSOL from PRIMUSqt in ATSAS 2.8.1 (71) | ||
| Molecular graphics | PyMOL v1.7 Mac | ||
| (d) Structural parameters | |||
| Guinier analysis | |||
| I(0) (cm−1) | 321.37 ± 1.23 | ||
| Rg (Å) | 29.71 ± 0.58 | ||
| q range (Å−1) | 0.0160–0.1904 | ||
| qRg maximum | 1.32 | ||
| Coefficient of correlation, R2 | 0.98 | ||
| M from I(0) (ratio to expected value) | 75,814 (1.08) | ||
| P(r) analysis | |||
| I(0) (cm−1) | 323.9 ± 1.8 | ||
| Rg (Å) | 30.23 ± 0.25 | ||
| dmax (Å) | 96.77 | ||
| q range (Å−1) | 0.0160–0.1904 | ||
| χ2 (total estimate from GNOM) | 0.979 (0.766) | ||
| M from I(0) (ratio to expected value) | 72,865 (1.03) | ||
| Porod volume (Å−3) (ratio VP/Mcalc) | 105,000 (1.49) | ||
| (e) Shape modeling results (a complete panel for each method) | |||
| DAMMIF (default parameters, 20 calculations) | |||
| q range for fitting (Å−1) | 0.00–0.190 | ||
| Symmetry, anisotropy assumptions | P1, none | ||
| NSD (S.D.), no. of clusters | 0.904 (0.151), 6 | ||
| χ2 value range | 0.601–0.602 | ||
| Resolution (Å) (from SASRES) | 35 ± 3 | ||
| M estimate as 0.5 × volume of models (Da) (ratio to expected) | 73,473 (1.04) | ||
| DAMMIN (default parameters) | |||
| q range for fitting (Å−1) | 0.0160–0.190 | ||
| Symmetry, anisotropy assumptions | P1, none | ||
| χ2, CORMAP p value | 0.602, 0.999 | ||
| (f) Atomistic modeling | |||
| Crystal structures | PDB code 2QCS | PDB code 4WBB | PDB code 5DYK |
| PKA RIα–C heterodimer | PKA RIIβ–C heterodimer | PfPKG | |
| CRYSOL (with default parameters) | |||
| q range for fitting | 0–0.1904 | 0–0.1904 | 0–0.1904 |
| No constant subtraction | |||
| χ2 value, p value | 1.29, 0.004 | 1.83, 0.000 | 10.35, 0.000 |
| Predicted Rg (Å) | 29.47 | 28.75 | 32.74 |
| Vol (Å), Ra (Å), Dro (e Å−3) | 98,423, 1.8, 0.025 | 94,847, 1.8, 0.030 | 105,437, 1.8, 0.00 |
| Constant subtraction allowed | |||
| χ2 value, p value | 0.82, 0.963 | 1.25, 0.010 | 9.21, 0.000 |
| Predicted Rg (Å) | 29.84 | 29.11 | 32.74 |
| Vol (Å), Ra (Å), Dro (e Å−3) | 98,423, 1.4, 0.033 | 94,847, 1.5, 0.037 | 105,437, 1.8, 0.00 |
| FoXS (with default parameters) | |||
| q range for fitting | 0.0160–0.1904 | 0.0160–0.1904 | 0.0160–0.1904 |
| χ2 value | 0.89 | 1.19 | 1.29 |
| Predicted Rg (Å) | 27.61 | 26.88 | 32.35 |
| c1, c2 | 1.05, 1.19 | 1.05, 1.79 | 1.05, −2.00 |
| (g) Data and model deposition ID | |||
| SASBDB | SASDDS4 | ||
a Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party-hosted site.
Figure 3.

SEC-SAXS analysis of PKG Iα Δ53 in the autoinhibited state. A, SEC-SAXS trace of Δ53 depicting scattering intensity (red circles; right axis) and Rg values (black circles; left axis) estimated from Guinier plots of each detector exposure frame. B, the intensity profile (Iq versus q) and Guinier plot (inset) for Δ53 was derived from the averaging of frames 410–459 from A. C, Kratky plot of the scattering profile of Δ53 determined using the averaged scattering data from B. D, the resulting fit and residual plot of the intensity profile (Iq versus q) for Δ53 by CRYSOL (allowing constant subtraction) for RIα–C (Protein Data Bank code 2QCS; green) and RIIβ–C (Protein Data Bank code 4WBB; red). The 3D envelope of the scattering curve derived from Δ53 (gray balls) was fit to the crystal structures of RIα–C (NSD = 1.06) (E) and RIIβ–C (NSD = 1.09) (F) heterodimers.
Predicted X-ray scattering from the crystal structures of the PKA RIα–C, PKA RIIβ–C, and PfPKG from Plasmodium falciparum (a monomeric species containing four cGMP-binding sites and a catalytic domain) was conducted using both CRYSOL and FoXS (Table 2f) (38, 39). The best fit of the experimental SAXS data to the predicted scattering was obtained from RIα–C using CRYSOL with constant subtraction (χ2 = 0.82; Fig. 3D and Table 2). Using the RIIβ–C heterodimer in the same analysis resulted in a poorer fit to the experimental data (χ2 = 1.25). The PfPKG fits were poor using CRYSOL (χ2 = 9.21) but acceptable with the FoXS server (χ2 = 1.29; Table 2f and Fig. S3). We also found that the Rg values calculated using the predicted scattering curves for both the PKA RIα–C and RIIβ–C heterodimer crystal structures were in closer agreement with the measured and calculated values for Δ53 (Table 2f). Next, DAMMIF and DAMMIN modeling was implemented. Using SUPCOMB, the resulting averaged and filtered DAMMIF/DAMMIN ab initio three-dimensional envelope accommodated both crystal structure models of the RIα–C and RIIβ–C heterodimers with little protrusion outside of the envelope (Table 2e and Fig. 3, E and F). Among the 3D models examined, these data cumulatively suggest that Δ53 adopts a shape most similar to PKA RIα–C.
Binding and activation of full-length PKG Iα and Δ53 with cNMP
Finally, we studied the activation kinetics of Δ53. No significant differences in basal activity were observed between PKG Iα full length and Δ53, indicating that the monomeric construct was autoinhibited at levels comparable with previous reports (15, 30). Moreover, the cGMP-dependent activation of Δ53 and full-length PKG Iα were analogous with respect to their maximum velocities of 4.0 and 3.9 μmol/min × mg, respectively (Fig. 4B, panel a). These results indicate that the cGMP-dependent -fold stimulation of Δ53 is indistinguishable from full length. However, differences between the two constructs were observed with respect to their activation constants and degree of cooperativity. We observed Ka values and Hill coefficients corresponding to 182 nm (nH = 1.6) for PKG Iα full length and 250 nm (nH = 1.0) for Δ53 (p < 0.0001 for both measurements). When we tested the activation profiles of full-length and Δ53 PKG Iα with cAMP, we observed that the full-length enzyme exhibited cooperative activation, whereas again Δ53 was noncooperative. Comparisons of the -fold difference in activation by cGMP and cAMP for both enzymes demonstrated that the removal of the N terminus of PKG Iα reduced the selectivity for cyclic nucleotide from 58- (full length) to 3.5-fold (Δ53), representing an overall 16-fold decrease in selectivity for cGMP over cAMP.
Figure 4.

Biochemical characterization of Δ53. A, domain diagram depicting the constructs used for the activation and binding studies. The domain architecture is described as follows: D/D, dimerization domain; AI, autoinhibitory domain; cGMP-A/B, cGMP-binding sites; SW, switch helix (25); N/C-lobe, catalytic domain. B, activation of PKG constructs. Data are represented as the mean ± S.D. (error bars). B, panel a, activation of PKG Iα full length (black) and Δ53 (red) with cGMP. Significant differences in activation by individual cGMP concentrations were confirmed by two-way analysis of variance (p < 0.005) and are denoted by an asterisk. B, panel b, normalized activation of PKG Iα (black) and Δ53 (red) with cGMP (solid lines) and cAMP (dotted lines). Shaded areas depict the mean KD ± S.D. of the high-affinity cyclic nucleotide–binding site for cGMP (solid) and cAMP (dashed). C, cNMP binding curves associated with PKG constructs in (A) as measured by SPR spectroscopy. Data are represented as the mean ± S.D. (error bars) and fit with a two-site binding model. C, panel a, binding curves showing cGMP (solid lines) and cAMP (dotted lines) association with full-length PKG Iα (black) and Δ53 (red). C, panel b, binding curves denoting PKG Iα(1–326) (blue) and PKG I(78–326) (green) with cGMP (solid lines) and cAMP (dotted lines).
To probe how binding of cyclic nucleotides correlates with the activities of the enzymes, we measured cGMP and cAMP binding to PKG Iα constructs by surface plasmon resonance (SPR) spectroscopy. Binding of cyclic nucleotides was fit using a two-site binding model, because the regulatory domain of PKG Iα contains two cGMP sites per monomer. To determine cooperativity of the binding, we also fit the data using a one-site model with Hill coefficient. PKG Iα full length exhibited an almost 3-fold weaker affinity for cGMP than Δ53 (KD(FL) = 7.9 μm and KD(Δ53) = 2.9 μm) when fit with a one-site model (Table 3). However, when fit with a two-site binding model, the ratio dropped to 1.6-fold. Moreover, both constructs displayed negative cooperativity in binding cGMP (nH(FL) = 0.60 and nH(Δ53) = 0.74). When cAMP binding was measured for the two constructs, the KD values for their high-affinity sites increased by over 30-fold. However, cAMP binding remained negatively cooperative in both enzymes. In binding to either cyclic nucleotide, we found that the low-affinity site demonstrated much weaker binding than the high-affinity site.
Table 3.
Binding and activation of PKG Iα constructs
The data are represented as the mean ± S.D. —, not measured.
| PKG Iα construct | cNMP | Binding (SPR) |
Activation (32PO4 assay) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| One-site model |
Two-site model |
||||||||
| KD | nH | KDHi | KDLo | N | Ka | nH | N | ||
| μm | μm | μm | μm | ||||||
| Full length | cGMP | 7.9 ± 1.1 | 0.60 ± 0.04 | 1.6 ± 0.3 | 90.2 ± 33.9 | 6 | 0.182 ± 0.004 | 1.58 ± 0.06 | 12 |
| Full length | cAMP | 387 ± 37 | 0.76 ± 0.03 | 51.7 ± 25.4 | 685 ± 174 | 5 | 10.80 ± 0.67 | 1.75 ± 0.09 | 6 |
| Δ53 | cGMP | 2.9 ± 0.4 | 0.74 ± 0.06 | 1.0 ± 0.4 | 19.1 ± 16.7 | 6 | 0.250 ± 0.010 | 0.97 ± 0.03 | 5 |
| Δ53 | cAMP | 297 ± 21 | 0.76 ± 0.03 | 36.6 ± 9.2 | 509 ± 62 | 4 | 0.860 ± 0.157 | 0.81 ± 0.06 | 6 |
| 1–326 | cGMP | 21.3 ± 1.3 | 0.81 ± 0.03 | 8.4 ± 2.5 | 74.4 ± 38.6 | 6 | — | — | |
| 1–326 | cAMP | 64.3 ± 5.3 | 0.72 ± 0.03 | 16.9 ± 3.9 | 312 ± 112 | 4 | — | — | |
| 78–326 | cGMP | 30.0 ± 1.1 | 0.90 ± 0.02 | 3.2 ± 3.1 | 35.5 ± 4.4 | 6 | — | — | |
| 78–326 | cAMP | 340 ± 10 | 0.92 ± 0.02 | 4.8 ± 3.1 | 350 ± 10 | 4 | — | — | |
To distinguish the cyclic nucleotide sites responsible for activation in the full-length PKG Iα kinase, we mutated the glutamic acid residues in the Phe-Gly-Glu (FGE) motif of the phosphate-binding cassettes within the A-site (E168G) and the B-site (E292A). Because the glutamate residue is involved in binding to the 2′-hydroxyl from the ribose, mutation of this site in PKA has been shown to disrupt cyclic nucleotide binding (28, 40–43). Mutation of the A-site (PKG Iα(E168G)) completely abolished cGMP-dependent activation of the full-length kinase (Fig. S4). These results demonstrate that a functional A-site is necessary for kinase activity. Moreover, when we examined the cGMP-dependent activation of PKG Iα(E292A), we observed cooperative (nH = 1.45) stimulation of the kinase with a Ka of 95 nm.
In an effort to further examine the contribution of the N terminus and the catalytic domain on cyclic nucleotide binding selectivity, we purified two constructs of the regulatory domain of PKG Iα. The first construct, PKG Iα(1–326), contains the N terminus and cGMP-binding regulatory domain, whereas the second construct, PKG I(78–326), contains only the regulatory domain. When we examined binding of cGMP to PKG Iα(1–326) and PKG I(78–326), we found that the one-site model KD shifted significantly compared with values observed for full-length PKG Iα and Δ53 (Fig. S5). For PKG Iα(1–326), the two-site binding model showed that the KD value for cGMP binding to the A-site increased by 8-fold compared with the full-length enzyme (Table 3). Furthermore, binding of cAMP showed a drastic decrease in selectivity for cGMP over cAMP. The B-site showed a clear selectivity for cGMP over cAMP (4-fold). However, when the N terminus was excluded in PKG I(78–326), we observed no difference in the affinities for cGMP and cAMP in the high-affinity A-site. The B-site retained a clear selectivity for cGMP over cAMP (10-fold).
Discussion
PKG activation has been extensively studied by traditional biochemical methods and, more recently, using modern biophysical approaches (18, 24, 25, 44, 45). These investigations sought to determine the architecture of the homodimeric kinase in its basal and activated states, the order and selectivity of cGMP binding, and, by extension, the origin of its cooperativity. The generation of a functional, monomeric form of PKG Iα is a useful tool for investigating the molecular basis of these long-observed biochemical phenotypes.
PKG Iα Δ53 purified from Sf9 cell extracts exists as a mixture of mono- and diphosphorylated forms; no unphosphorylated form was detected. The primary phosphorylation site, Thr517, was found in high abundance by TOF-MS (Fig. 2C and Table 1). Phosphorylation of Thr517, which is located in the activation loop of the catalytic domain, is essential for catalytic activity (34). In the PKA catalytic domain, the analogous phosphorylation at Thr197 forms contacts with other activation loop residues, the catalytic loop, and the α C-helix to integrate the active site components (46, 47). It has been hypothesized that phosphorylated Thr517 in PKG serves a similar function because the lack of phosphorylation or mutation of this residue renders PKG inactive (34). The presence of this phosphorylation site in Δ53 corroborates the cGMP-dependent activation observed by our phosphotransferase assays. In regard to the second, less abundant phosphorylation site, we had hypothesized that this residue would be located in the autoinhibitory domain based on previous published data (35, 36, 48). Analysis of autoinhibitory domain residues suggested that Ser64 and Thr70 were not phosphorylated (parent peptide Ala61–Arg72; Table 1). Another potential site, Thr58, has previously been identified as the major site that is autophosphorylated most rapidly in the presence of cGMP in vitro or cAMP in native PKG I preparations isolated from bovine lung (35, 48). Because Sf9 cells endogenously express adenylyl cyclases, we hypothesized that Thr58 could be the minor phosphorylation site (49, 50). However, our LC-MS/MS data could not confirm the precise location. These results are in agreement with previously published results that observed the phosphorylation at Thr517, but were also unsuccessful in identifying the second site (23). Furthermore, these results suggest that neither its dimerization through the N-terminal dimerization domain nor exposure to cGMP are required for activation loop phosphorylation.
Despite the presence of these two phosphorylation states, we consistently observed a compact, monodisperse, monomeric species in solution by SEC (Fig. 2B). In addition, the elution profile of Δ53 did not change with the addition of cGMP, suggesting that it maintains its monomeric state even during long-lived exposure. To reconstruct the three-dimensional shape of inactive Δ53 and confirm its monomeric character, we collected data by SEC-SAXS and compared the reconstructed 3D envelope with the crystal structures and SAXS data from two PKA heterodimers (Protein Data Bank codes 2QCS and 4WBB) and PfPKG (Protein Data Bank code 5DYK). We found that Δ53 adopts a similar shape to that of the PKA RIα–C heterodimer (Protein Data Bank code 2QCS) based on the best fit of the atomistic modeling in CRYSOL to the scattering data (χ2 0.82) (Fig. 3D and Table 2e). In addition, the crystal structure is readily accommodated by the 3D envelope calculated from the Δ53 SAXS data (NSD = 1.06; Fig. 4F). These data concurred with previously published results for the apo state of monomeric PKG Iβ Δ1–52 (Table S2) (45). PKG Iβ Δ1–52 contains 18 additional residues upstream of its autoinhibitory domain compared with PKG Iα Δ53. Considering the high overlap between these two truncated constructs in the SAXS analyses, this suggests that the linker in Iβ Δ1–52 adopts a compact conformation relative to the regulatory–catalytic domain complex. Based upon these data, we propose a model wherein each monomer within the dimeric PKG Iα complex can be autoinhibited by its own regulatory domain and autoinhibitory sequence (Fig. 5, top).
Figure 5.
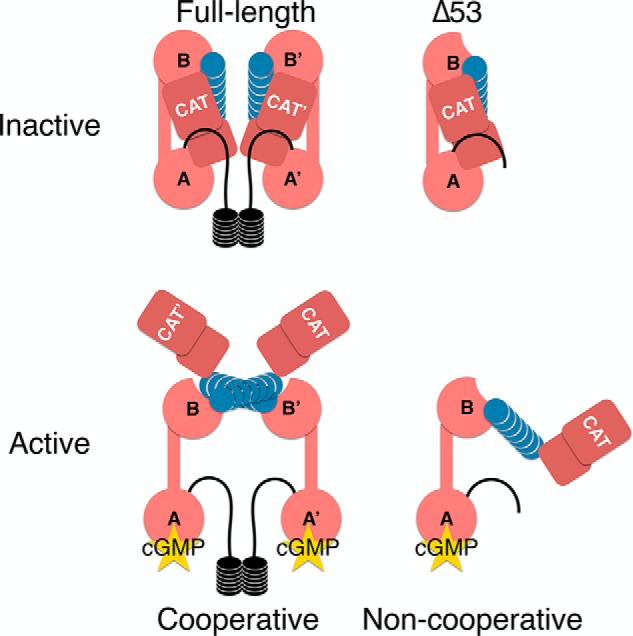
Putative model of activation of FL and Δ53 PKG Iα. The model depicted hypothesizes that one cyclic nucleotide bound to the high-affinity A-site is sufficient to activate both the full-length and Δ53 PKG Iα constructs. Cooperativity arises from the in trans interaction of protomers at the knob–nest site and not from cooperative cyclic nucleotide binding. In the Δ53 construct, cooperativity is not observed and cannot be facilitated due to the lack of dimerization. The organization of the full-length enzyme must also lend itself to generate a cGMP selectivity filter for the A-site based upon our full-length and truncated PKG Iα data. CAT, catalytic domain.
Finally, we report that Δ53 is activated by cGMP at concentrations similar to the full-length kinase (Fig. 4B, panel a). A previous study by Richie-Jannetta et al. (51) and a more recent study by Kim et al. (52) expressed truncated forms of PKG Iβ (Δ1–53 and Δ1–55) to investigate the influence of the dimeric interface on cGMP-dependent activation. Both studies described significant shifts in the activation constants of the truncated Iβ constructs. These results contrast with our observations of PKG Iα. Δ53 does not exhibit an appreciable shift in the activation constant compared with the full-length enzyme. These results support the longstanding conclusion that PKG Iα and -β are enzymatically distinct due to differences in their N termini (1, 15, 20, 30, 50).
Interestingly, Δ53 does not exhibit cooperative cGMP-dependent activation (Fig. 4B, panel a). The loss of cooperativity has been previously reported for PKG Iβ wherein full-length PKG Iβ (nH = 2.1) becomes noncooperative when the N terminus is removed (Δ1–52 PKG Iβ) (30, 52). In addition, we observed that cAMP also cooperatively activates the full-length kinase (nH = 1.75) but not Δ53 (nH = 0.81). These results suggest that the cooperative activation is mediated by the N terminus through the facilitation of homodimer formation. The N-terminal dimerization domains of PKG Iα and Iβ are left-handed, coiled-coil, LZ motifs. The LZ is characterized by a heptad repeat of amino acids (abcdefg) where residues a and d are typically hydrophobic and residues e and g are typically hydrophilic. The leucine zipper of PKG Iα spans residues 1–47. In addition to its leucine and isoleucine residues, there are also four non-leucine/isoleucine residues at the d positions that provide additional nonhydrophobic interhelical contacts (Phe7, Lys14, Lys28, and Cys42) (53). One of these residues, Cys42, is unique to the Iα isoform and forms an interprotomer disulfide bond in the presence of oxidizing agents such as hydrogen peroxide (54, 55). The leucine zipper of PKG Iβ spans residues 4–53 and also contains four non-leucine/isoleucine residues at the d positions (Lys13, Arg20, Lys34, and Tyr48) (18). Although the majority of the noncanonical residues are basic, their differences in position within the helices and the overall discrepancy in length of the helices are reasons why PKG I protomers have not been shown to form heterodimers either in vitro or in vivo.
It is well-characterized that the cGMP A-site in the regulatory domain is the high-affinity binding site and thus is the first to bind cyclic nucleotide (24, 26, 44). Our data of PKG Iα full length and Δ53 agree with these previously published results and indicate a clear selectivity for cGMP over cAMP in the A-site (Table 3). To further test the contributions of the A- and B-sites in PKG Iα activity, we measured the activities of PKG Iα(E167G) and PKG Iα(E292A). We found that disruption of cyclic nucleotide binding to the B-site did not have an appreciable effect on activity (Fig. S4). In contrast, disruption of cyclic nucleotide binding to the A-site completely abolished activity. These data collectively support the importance of the A-site in regulating activation of PKG Iα. However, previous studies using isolated regulatory domain constructs have suggested that the A-site is nonselective for cyclic nucleotide (44, 25). These results suggest that selectivity for cGMP binding to the A-site must be controlled through regions outside of the regulatory domain.
To investigate the influence of regions outside of the regulatory domain on the cyclic nucleotide–dependent characteristics of PKG Iα, we expressed and purified two additional constructs, PKG Iα(1–326) and PKG Iα(78–326). PKG Iα without its catalytic domain (PKG Iα(1–326)) showed a significant decrease in binding affinity for cGMP and loss of selectivity for cGMP over cAMP in the A-site compared with the full-length construct (Table 3). Selectivity was only slightly reduced in the B-site, and the values agree with those measured previously for the isolated B-domain (44). These results differ from the binding and nucleotide selectivity results observed for PKG Iα full length and Δ53. Thus, these characteristics seem to be integrated into the architecture of the inactive holoenzyme.
The initial comparison of the activation curves for full-length and Δ53 with the one-site binding model indicates a large disparity between the concentration of cGMP required for full saturation of the A- and B-sites and the minimum concentration required for full activation. In early measurements of cyclic nucleotide binding with [3H]cGMP, a significant difference in the KD values for cGMP binding at 4 and 30 °C was observed (56). Our measurements for cGMP are consistent with these early experiments. When a two-site model is applied to the binding data, we can directly correlate the KD for the A-site with the Vmax for activation. This model is reasonable because there are two cGMP-binding sites with distinct binding affinities. Furthermore, this effect is observed for both cGMP and cAMP equally (Fig. 4). This implies that full-length PKG Iα only requires that half of the A-sites are occupied to stimulate full activation. These data, which also suggest that the B-site is nonessential for kinase activity, are further supported by the fact that the KD for the B-site (∼90 μm) is higher than estimated intracellular concentrations of cGMP (∼10 nm–10 μm) (57, 58, 59). Moreover, we observed that mutation of the nucleotide-binding cassette in the B-site does not appreciably affect activation of the full-length kinase (Fig. S4). Thus, we can also conclude that binding studies that are either directed toward the isolated PKG regulatory domains or use equilibrium exchange of radiolabeled cyclic nucleotides are limited, and future studies should examine these effects in the context of the intact holoenzymes.
These SPR measurements also indicate that binding of cGMP to both full length and Δ53 is negatively cooperative (nH < 1), which indicates that binding of cGMP to subsequent sites becomes more difficult with increasing concentrations. These results suggest a lack of avidity between cyclic nucleotide–binding domains A and B, and this has been corroborated by recent structural evidence (52). These observations stand in contrast to PKA where the origins of cooperativity have been directly linked to the regulatory domain wherein binding of cAMP into the B-site enhances binding for the A-site through intrasubunit contacts (27, 28, 32). It has been suggested that the cooperativity observed for PKG activation arises from a similar mechanism (23, 24, 44). However, the loss of cooperative activation in PKG Iα through disruption of the dimer via two mechanisms, 1) by removing residues that allow for N-terminal dimerization (this study) and 2) via mutation of residues forming the knob–nest interface, suggests that this is not the case (25).
Finally, we propose a model based on our findings wherein autoinhibition of PKG Iα is driven in cis. The high-affinity A-site provides cyclic nucleotide selectivity through contacts outside of the regulatory domain, and cooperative activation of PKG Iα is driven in trans, facilitated by the N terminus, which ensures monomers are close enough to form necessary interprotomer contacts (Fig. 5). These data reinforce our earlier hypothesis that cooperative activation of the kinase is not driven through cooperative binding of cyclic nucleotides to the regulatory domain within the same protomer. Rather, it is driven through an inducible dimeric interface mediated by the switch helix (30, 60). Therefore, we conclude that the mechanism driving cooperativity in PKG Iα lies in the interface between protomers and not between cGMP-binding sites within the same protomer.
Experimental procedures
Sequence alignments
Sequences for PKG Iα and PKA regulatory domain isoforms were downloaded from the NCBI and UniProt repositories and uploaded to Jalview Desktop (61). The following accession numbers were used: NP_001091982 (PKG Iα, Homo sapiens), NP_776861 (PKG Iα, Bos taurus), NP_001013855 (PKG Iα, Mus musculus), NP_001099201 (PKG Iα, Rattus norvegicus), NP_037313 (PKA RIα, R. norvegicus), NP_997637 (PKA RIα, H. sapiens), NP_001068669 (PKA RIβ, B. taurus), NP_001240819 (PKA RIβ, M. musculus) NP_062137 (PKA RIIα, R. norvegicus), P12367 (PKA RIIα, M. musculus), NP_001178296 (PKA RIIα, B. taurus), NP_004148 (PKA RIIα, H. sapiens), P31324 (PKA RIIβ, M. musculus), NP_001025191 (PKA RIIβ, R. norvegicus), NP_002727 (PKA RIIβ, H. sapiens), and P31322 (PKA RIIβ, B. taurus). A multiple sequence alignment using ClustalO with the default parameters was accomplished using the JalView Desktop interface (62).
Maintenance of Sf9 cells
Sf9 cells were purchased from Life Technologies. Frozen cell stocks were prepared according to the Growth and Maintenance of Insect Cell Lines Manual (Life Technologies). Typically, one vial containing Sf9 cells (1 × 106 cells/vial in freezing medium (80% supplemented Grace's insect medium (Gibco), 10% fetal bovine serum (FBS; Sigma), and 10% DMSO) was suspended in 15 ml of Grace's medium (Life Technologies) supplemented with 5% fetal bovine serum (Sigma) and 10 μg/ml gentamicin (Sigma). Cells were grown in a 75-cm2 flask until cells reached 80% confluence. Cells were detached from the flask and suspended in Sf900 III medium (Life Technologies) supplemented with 1× lipids (Sigma), 1% poloxamer 81 (Sigma), and 10 μg/ml gentamicin (Sigma). Sf9 cells were maintained in suspension at 27 °C at 80 rpm and passaged every 3.5 days to 1.2 × 106 cells/ml.
Expression of PKG Iα constructs in Sf9 cells
Full-length PKG Iα (B. taurus) was amplified and cloned into pFAST Bac-HTA (Invitrogen) as described previously (63). PKG Iα(54–671) (Δ53) was amplified by PCR from bovine PKG Iα(1–671) using forward and reverse primers containing NcoI- and XhoI-cut sites, respectively: 5′-GGC GCC ATG GAT ATC GGC CCC CGG ACC ACC-3′ (sense) and 5′-ATG CCT CGA GAC CTA TTA GAA GTC TAT GTC CCA TCC TGA-3′ (antisense) (Integrated DNA Technologies). The resulting product was cloned into pFastBac-HTA (Invitrogen) using NcoI, XhoI, and DNA ligase (New England Biolabs). Site-directed mutagenesis was performed on the full-length PKG Iα construct in pFAST Bac-HTA to produce E167G and E292A using the following primers: 5′-AAG GTG TTT GGA GGG TTG GCT A-3′ (E167G-sense), 5′-ATA GCC AAC CCT CCA AAC ACT T-3′ (E167G-antisense), 5′-GGA AAA GGA GAT TGG TTT GGA GCG AAA GCC TTG-3′ (E292A-sense), and 5′-TTC CCC CTG CAA GGC TTT CGC TCC AAA CC-3′ (E292A-antisense). Transposition of the full-length PKG Iα, mutant, and Δ53 genes into bacmid using DH10 Bac Escherichia coli, confirmation of its insertion by PCR, and transfection into Sf9 cells to produce baculovirus were performed following the Bac-to-Bac Baculovirus Expression System Manual (Life Technologies). Third amplification viruses were used at dilution ratios of ∼1:500 to express both constructs (virus:Sf9 cells). At 72 h postinfection, cultures were harvested by centrifugation at 500 × g for 10 min. Pellets were resuspended in lysis buffer (10 mm TES, 300 mm NaCl, 5 mm imidazole, pH 7) at a volume of 20 ml/liter Sf9 cells, flash frozen in liquid nitrogen, and stored at −80 °C.
Expression of PKG I regulatory domain constructs in E. coli
The PKG I regulatory domain constructs (PKG I(78–326) and PKG Iα(1–326)) were amplified by PCR from the full-length PKG Iα gene using forward and reverse primers containing BamHI- and EcoRI-cut sites, respectively: PKG I-78-f, 5′-CGG GAT CCA TGC AGG CAT TCC GGA AGT TC-3′ (sense); PKG I-1-f, 5′-TGG GGA TCC AGC GAG CTG GAG-3′ (sense); PKG I-326-r, 5′-TCG AAT TCC ATG CTA TTA TAA TCC TCC AAT CAA ATG-3′ (antisense). The respective fragments were ligated into BamHI/EcoRI–digested pRSET-A using T4 ligase (New England Biolabs). Both constructs were expressed in Rosetta2 E. coli (Novagen). Overnight cultures were used to inoculate 1 liter of terrific broth supplemented with 50 μg/ml ampicillin and 25 μg/ml chloramphenicol and subsequently grown at 37 °C at 220 rpm to an A600 of 0.8. Expression was induced by the addition of isopropyl 1-thio-β-d-galactopyranoside (1 mm final), and the induced cultures were incubated overnight at 25 °C at 220 rpm. E. coli were harvested by centrifugation at 1500 × g for 30 min, and the resulting bacterial pellets were flash frozen in liquid nitrogen and stored at −80 °C.
Purification of PKG Iα constructs
Protease inhibitors (Roche Applied Science) were dissolved in 50 ml of lysis buffer and added to 1 liter of pelleted Sf9 cells containing either PKG Iα full length, E167G, E292A, or Δ53 (B. taurus). Sf9 cell pellets were thawed on ice for 40 min followed by gentle mixing. Resuspended cell pellets were lysed using a French pressure cell (two passes at >1,500 bars; SLM-Aminco) or by passing cells through a 22.5-gauge needle (three passes). Bacterial pellets containing PKG I(78–326) were resuspended by vortexing in 50 ml of lysis buffer supplemented with protease inhibitors (Roche Applied Science) and lysed using a French pressure cell (five passes at >1,500 bars; SLM-Aminco).
All cell lysates were clarified by centrifugation at 30,000 × g for 30 min at 4 °C and passed through a 0.22-μm polyethersulfone syringe filter (Millipore). The resulting clarified, filtered lysates were loaded onto a 5-ml prepacked nickel immobilized metal affinity chromatography column (Bio-Rad) using a P-1 peristaltic pump (GE Healthcare) and washed with 5 column volumes of lysis buffer and 6 column volumes of mid-wash buffer (lysis buffer supplemented with 30 mm imidazole). Proteins were eluted from the column with elution buffer (lysis buffer supplemented with 250 mm imidazole) using a Profinia FPLC (Bio-Rad). Peak fractions containing PKG Iα were pooled and analyzed by SDS-PAGE. PKG Iα was dialyzed against 4 liters of 50 mm MES, 300 mm NaCl, 1 mm TCEP, pH 6.9, using 12–14-kDa molecular mass cutoff dialysis tubing and against 1 liter of 50 mm MES, 300 mm NaCl, 1 mm TCEP, 10% glycerol, pH 6.9. Aliquots of PKG Iα were flash frozen in liquid nitrogen and stored at −80 °C.
Analytical SEC
Samples containing 0.5 mg of PKG Iα Δ53 (apo or preincubated with 5 μm cGMP) were loaded sequentially onto a Superdex 200 10/300 column (GE Healthcare) connected to an Äkta PURE FPLC (GE Healthcare) and eluted isocratically in 50 mm MES, 300 mm NaCl, 1 mm TCEP, pH 6.9 (apo), or buffer supplemented with 5 μm cGMP (bound). Peak fractions were examined for the presence of PKG Iα Δ53 by SDS-PAGE. Peaks were analyzed with Unicorn 6.4 (GE Healthcare) and plotted using DataGraph (Visual Data Tools).
Phosphotransferase assays
Activation of PKG Iα constructs by cGMP (BioLog) was assessed by measuring phosphorylation of a synthetic peptide substrate (W15; TQAKRKKSLAMA) with [γ-32P]ATP similar to the method described previously (63). Each 100-μl reaction contained 20 μl of 5× MES mixture (250 mm MES, 5 mm MgOAc, 50 mm NaCl, pH 6.9), 10 μl of 100 mm DTT, 10 μl of 10 mg/ml BSA (Cohn's fraction V; Sigma), 10 μl of 100 μm W15, 10 μl of cGMP (0–5 μm), 20 μl of 5 nm PKG Iα (in 50 mm MES, 1 mm TCEP, pH 6.9), 10 μl of ATP mixture ([γ-32P]ATP in 1 mm ATP; approximate specific activity, 200 cpm/min), and 10 μl of H2O. Reactions were performed at 30 °C and initiated with the addition of either PKG Iα or ATP mixture. Blank reactions were performed by substituting the PKG I and cGMP with H2O. Reactions were quenched after 1.5 or 3 min by spotting onto P81 phosphocellulose (Whatman and Reaction Biology Corp.) and washing with 0.8% phosphoric acid. Determination of 32P incorporation into W15 substrate was measured by liquid scintillation. Data were analyzed using Excel (Microsoft) and Prism v7 (GraphPad Software) and then plotted using DataGraph.
Measurement of cGMP binding by SPR spectroscopy
SPR measurements were conducted on an SR7500 dual-channel surface plasmon resonance spectrometer connected to an SR7100 autosampler utilizing a standard, dual-channel flow cell (Reichert Technologies). All steps were performed at 25 °C. Gold sensorchips coated with a 1500-nm linear polycarboxylate hydrogel (HC1500M, Xantec) were installed on the instrument and pre-equilibrated with SPR buffer (50 mm MES, 150 mm NaCl, 1 mm EDTA, 0.05% Tween 20, 1 mm TCEP, pH 6.9) at 20 μl/min. Sensorchips were washed sequentially with a solution of 2 m NaCl and 10 mm NaOH for 5 min each followed by 2 min with SPR buffer. The sensorchip was activated for 5 min with a solution of freshly prepared, degassed 100 μm N-hydroxysuccinimide (NHS) and 200 μm 1-ethyl-3-(-3-dimethylaminopropyl)carbodiimide hydrochloride dissolved in 1 ml of 500 mm MES, pH 5.5. PKG constructs were diluted to 1 μm in 20 mm NaOAc, pH 5.0 (500 μl), and injected into the left channel for 10 min until 10,000–15,000 micro-refractive index units was observed in the difference channel. Excess protein was washed from the surface for 5 min. Any remaining reactive groups on the sensorchips were deactivated by injecting 1 m ethanolamine, pH 8.5, into both channels for 5 min and then washing with SPR buffer for 1 min. Solutions of cyclic nucleotides were prepared using the same SPR buffer used during the PKG I coupling steps. During cNMP binding experiments, solutions were injected at 50 μl/min for 1 min (association) and then washed with SPR buffer for 3 min (dissociation). All injected solutions were prepared in clear or amber borosilicate screw-top glass vials with robotic screw-top vial closures (Fisher Scientific). Data were reduced using Scrubber (BioLogic). Data were fit to one-site (with Hill coefficient) and two-site (specific binding) models using GraphPad Prism and plotted using DataGraph.
Mass spectrometry
All LC-MS measurements were performed using a Xevo G2-XS quadrupole TOF mass spectrometer connected to an Acquity ultraperformance LC (UPLC) system (Waters Corp.) in positive electrospray ionization mode. Mass spectrometry of intact protein was achieved using a Jupiter 5-μm 300-Å, 1 × 150-mm C4 column (Phenomenex). Conditions were as follows: LC flow, 80 μl/min; A, water with 0.1% formic acid; B, acetonitrile; isocratic flow, 0–1 min with 90% A and 10% B, 1–4 min ramp to 10% A and 90% B; scan range, 500–2000 m/z. Approximately 2 pmol of PKG Δ53 was injected per run. The resulting spectra were processed using MassLynx v4.1 and the deconvolution software MaxEnt (Waters Corp.).
LC-MS/MS was performed to determine specific phosphorylation sites as follows. Two 150-pmol aliquots of dried PKG Δ53 or PKG Iα full-length enzyme were resuspended in 25 μl of 50 mm ammonium bicarbonate buffer, pH 8.5, and 30 μl of 0.02% ProteaseMAX (Promega). A 5-μl aliquot of sequencing grade trypsin (Promega) was added at a 1:20 ratio (enzyme to substrate). Samples were incubated overnight at 37 °C and acidified with formic acid to obtain a final concentration of 0.5%. The samples were evaporated and resuspended in 10 μl of 98% water, 2% acetonitrile, 0.1% formic acid prior to LC-MS analyses. UPLC separation of the tryptic peptides was performed on an Acquity high-strength silica T3 1.8-μm, 1.0 × 150-mm column (Waters Corp.). UPLC conditions were as follows: LC flow, 50 μl/min; column temperature, 45 °C; A, water and 0.1% formic acid; B, acetonitrile and 0.1% formic acid; linear gradient from 98% A and 2% B to 65% A and 35% B over 50 min. Approximately 30 pmol of digested PKG Δ53 was injected. All samples were analyzed by data-independent acquisition between 100 and 2000 m/z using the MSE mode with alternating low- (4-V) and high-energy (15–40-V) acquisitions. The MSE data were processed using ProteinLynx Global Server v3.0.1 (Waters Corp.) that searched against a consolidated database that included human cGMP-dependent protein kinase 1 (Q13976) and possible modifications, including phosphorylations.
SEC-SAXS
SAXS data pertaining to Δ53 was collected at the Stanford Synchrotron Radiation Lightsource BL4-2 using an MX225-HE detector (Rayonix). A 35-μl sample of 6 mg/ml PKG Iα Δ53 was injected onto a Superdex 200 3.2/30 column (GE Healthcare) and eluted isocratically using an Ettan FPLC (GE Healthcare) at 0.05 ml/min in 50 mm MES, 300 mm NaCl, 1 mm TCEP, pH 6.9, supplemented with 5 mm DTT as a radical scavenger. The eluant stream was connected in line to a 1.5-mm quartz capillary positioned 1.7 m from the detector. Data were collected using a wavelength of 1.127 Å with a 1 s/frame exposure rate at 22 °C. A total of 600 frames were collected with a measurement range of 0.0087–0.5126 Å−1. Frames 410–459 containing the peak corresponding to Δ53 PKG were averaged and subtracted from the background (45 averaged frames of buffer alone). The data were truncated to exclude the range of 0.0087–0.016 Å−1 due to radiation-induced aggregation and above 0.19 Å−1 for all subsequent analyses. The scattering curve, Guinier and P(r) functions, and Porod volume were calculated using the PRIMUS program suite (64). DAMMIF/DAMMIN/DAMMAVER/DAMFILT was used to generate a total of 20 models of which 19 were used to calculate the final 3D envelope. Models were excluded based upon their NSD to the mean using DAMSEL. The excluded model was determined to have an NSD that was 2 times the standard deviation from the mean of all NSD scores using a cross-correlation matrix. SUPCOMB was further used to fit the averaged 3D envelope to existing heterodimeric structures of PKA RIα–C (Protein Data Bank code 2QCS), RIIβ–C (Protein Data Bank code 4WBB), and PfPKG (Protein Data Bank code 5DYK) (65–67). Radii of gyration were calculated from the predicted scattering curves of the RIα–C and RIIβ–C heterodimers (using Protein Data Bank codes 2QCS and 4WBB, respectively) using the FoXS and ATSAS servers (https://modbase.compbio.ucsf.edu/foxs/ and https://www.embl-hamburg.de/biosaxs/atsas-online)6 (38, 39). Data were deposited in the Small Angle Scattering Biological Data Bank (SASBDB) using the identifier SASDDS4 (68).
Author contributions
T. M. M. conceptualization; T. M. M., J. L. S., D. E. M., and D. K. B. data curation; T. M. M., J. L. S., P. N., L. W. N., D. E. M., and W. R. D. formal analysis; T. M. M., D. E. M., D. K. B., and W. R. D. supervision; T. M. M., J. L. S., D. E. M., and D. K. B. validation; T. M. M., J. L. S., P. N., L. W. N., J. W., and D. K. B. investigation; T. M. M. visualization; T. M. M., D. E. M., D. K. B., and W. R. D. methodology; T. M. M., J. L. S., and W. R. D. writing-original draft; T. M. M. and W. R. D. project administration; T. M. M., J. L. S., D. E. M., D. K. B., and W. R. D. writing-review and editing; W. R. D. resources; D. E. M., T. M. M., J. L. S., and W. R. D. funding acquisition.
Supplementary Material
Acknowledgments
We thank the beamline scientists, Tsutomu Mitsui and Thomas Weiss, at Stanford Synchrotron Radiation Lightsource (SSRL) BL4-2 for assistance with SEC-SAXS data collection. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the United States Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences under Contract DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institute of General Medical Sciences at the National Institutes of Health (NIH) (including Grant P41GM103393). Mass spectrometry measurements were supported by Centers of Biomedical Research Excellence (COBRE) Grant P30-GM118228 from the NIGMS, National Institutes of Health. Mass spectrometry measurements were assisted by Bruce O'Rourke. We thank Werner Tegge at the Helmholtz Centre for Infection Research (HZI) in Braunschweig, Germany for synthesizing the W15 peptide substrate used in the phosphotransferase assays. We also thank Wolfgang Peti at the University of Arizona for the generous use of a Superdex 200 10/300 analytical column and Äkta PURE FPLC system and Ross Buchan for helpful discussions regarding this manuscript.
This work was supported by National Institutes of Health Grants 5T32 HL007647 (to T. M. M.), 5T32 HL007594-30 (to J. L. S.), and S10-OD018126 (to D. E. M.) and by the Totman Trust for Biomedical Research. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5 and Tables S1 and S2.
Data were deposited in the Small Angle Scattering Biological Data Bank (SASBDB) using the identifier SASDDS4.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party-hosted site.
- PKG
- cGMP-dependent protein kinase
- NSD
- normalized spatial discrepancy
- TCEP
- tris(2-carboxyethyl)phosphine
- Rg
- radius of gyration
- SEC
- size-exclusion chromatography
- SAXS
- small-angle X-ray scattering
- SPR
- surface plasmon resonance
- LZ
- leucine zipper
- R
- regulatory subunit
- Dmax
- maximum linear dimension
- Pf
- P. falciparum
- TES
- 2-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}ethanesulfonic acid
- UPLC
- ultraperformance LC
- FL
- full length.
References
- 1. Hofmann F., Bernhard D., Lukowski R., and Weinmeister P. (2009) cGMP regulated protein kinases (cGK). Handb. Exp. Pharmacol. 137–162 10.1007/978-3-540-68964-5_8 [DOI] [PubMed] [Google Scholar]
- 2. Keilbach A., Ruth P., and Hofmann F. (1992) Detection of cGMP dependent protein kinase isozymes by specific antibodies. Eur. J. Biochem. 208, 467–473 10.1111/j.1432-1033.1992.tb17209.x [DOI] [PubMed] [Google Scholar]
- 3. Bian K., and Murad F. (2007) Nitric oxide signaling in vascular biology. J. Am. Soc. Hypertens. 1, 17–29 10.1016/j.jash.2006.11.007 [DOI] [PubMed] [Google Scholar]
- 4. Kemp-Harper B., and Schmidt H. H. (2009) cGMP in the vasculature. Handb. Exp. Pharmacol. 447–467 10.1007/978-3-540-68964-5_19 [DOI] [PubMed] [Google Scholar]
- 5. Potter L. R., Yoder A. R., Flora D. R., Antos L. K., and Dickey D. M. (2009) Natriuretic peptides: their structures, receptors, physiologic functions and therapeutic applications. Handb. Exp. Pharmacol. 341–366 10.1007/978-3-540-68964-5_15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ammendola A., Geiselhöringer A., Hofmann F., and Schlossmann J. (2001) Molecular determinants of the interaction between the inositol 1,4,5-trisphosphate receptor-associated cGMP kinase substrate (IRAG) and cGMP kinase Iβ. J. Biol. Chem. 276, 24153–24159 10.1074/jbc.M101530200 [DOI] [PubMed] [Google Scholar]
- 7. Antl M., von Brühl M. L., Eiglsperger C., Werner M., Konrad I., Kocher T., Wilm M., Hofmann F., Massberg S., and Schlossmann J. (2007) IRAG mediates no/cGMP-dependent inhibition of platelet aggregation and thrombus formation. Blood 109, 552–559 10.1182/blood-2005-10-026294 [DOI] [PubMed] [Google Scholar]
- 8. Robertson B. E., Schubert R., Hescheler J., and Nelson M. T. (1993) cGMP-dependent protein kinase activates Ca-activated K channels in cerebral artery smooth muscle cells. Am. J. Physiol. Cell Physiol. 265, C299–C303 10.1152/ajpcell.1993.265.1.C299 [DOI] [PubMed] [Google Scholar]
- 9. Sausbier M., Schubert R., Voigt V., Hirneiss C., Pfeifer A., Korth M., Kleppisch T., Ruth P., and Hofmann F. (2000) Mechanisms of NO/cGMP-dependent vasorelaxation. Circ. Res. 87, 825–830 10.1161/01.RES.87.9.825 [DOI] [PubMed] [Google Scholar]
- 10. Schmidtko A., Gao W., Sausbier M., Rauhmeier I., Sausbier U., Niederberger E., Scholich K., Huber A., Neuhuber W., Allescher H. D., Hofmann F., Tegeder I., Ruth P., and Geisslinger G. (2008) Cysteine-rich protein 2, a novel downstream effector of cGMP/cGMP-dependent protein kinase I-mediated persistent inflammatory pain. J. Neurosci. 28, 1320–1330 10.1523/JNEUROSCI.5037-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schubert R., and Nelson M. T. (2001) Protein kinases: tuners of the BKCa channel in smooth muscle. Trends Pharmacol. Sci. 22, 505–512 10.1016/S0165-6147(00)01775-2 [DOI] [PubMed] [Google Scholar]
- 12. Orstavik S., Natarajan V., Taskén K., Jahnsen T., and Sandberg M. (1997) Characterization of the human gene encoding the type I α and type I β cGMP-dependent protein kinase (PRKG1). Genomics 42, 311–318 10.1006/geno.1997.4743 [DOI] [PubMed] [Google Scholar]
- 13. Wernet W., Flockerzi V., and Hofmann F. (1989) The cDNA of the two isoforms of bovine cGMP-dependent protein kinase. FEBS Lett. 251, 191–196 10.1016/0014-5793(89)81453-X [DOI] [PubMed] [Google Scholar]
- 14. Pfeifer A., Ruth P., Dostmann W., Sausbier M., Klatt P., and Hofmann F. (1999) Structure and function of cGMP-dependent protein kinases. Rev. Physiol. Biochem. Pharmacol. 135, 105–149 10.1007/BFb0033671 [DOI] [PubMed] [Google Scholar]
- 15. Ruth P., Pfeifer A., Kamm S., Klatt P., Dostmann W. R., and Hofmann F. (1997) Identification of the amino acid sequences responsible for high affinity activation of cGMP kinase Iα. J. Biol. Chem. 272, 10522–10528 10.1074/jbc.272.16.10522 [DOI] [PubMed] [Google Scholar]
- 16. Blanton R. M., Takimoto E., Aronovitz M., Thoonen R., Kass D. A., Karas R. H., and Mendelsohn M. E. (2013) Mutation of the protein kinase Iα leucine zipper domain produces hypertension and progressive left ventricular hypertrophy: a novel mouse model of age-dependent hypertensive heart disease. J. Gerontol. A Biol. Sci. Med. Sci. 68, 1351–1355 10.1093/gerona/glt042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Casteel D. E., Boss G. R., and Pilz R. B. (2005) Identification of the interface between cGMP-dependent protein kinase Iβ and its interaction partners TFII-I and IRAG reveals a common interaction motif. J. Biol. Chem. 280, 38211–38218 10.1074/jbc.M507021200 [DOI] [PubMed] [Google Scholar]
- 18. Casteel D. E., Smith-Nguyen E. V., Sankaran B., Roh S. H., Pilz R. B., and Kim C. (2010) A crystal structure of the cyclic GMP-dependent protein kinase Iβ dimerization/docking domain reveals molecular details of isoform-specific anchoring. J. Biol. Chem. 285, 32684–32688 10.1074/jbc.C110.161430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kato M., Blanton R., Wang G. R., Judson T. J., Abe Y., Myoishi M., Karas R. H., and Mendelsohn M. E. (2012) Direct binding and regulation of RhoA protein by cyclic GMP-dependent protein kinase Iα. J. Biol. Chem. 287, 41342–41351 10.1074/jbc.M112.421040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee E., Hayes D. B., Langsetmo K., Sundberg E. J., and Tao T. C. (2007) Interactions between the leucine-zipper motif of cGMP-dependent protein kinase and the C-terminal region of the targeting subunit of myosin light chain phosphatase. J. Mol. Biol. 373, 1198–1212 10.1016/j.jmb.2007.08.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dostmann W. R., Koep N., and Endres R. (1996) The catalytic domain of the cGMP-dependent protein kinase Iα modulates the cGMP-binding characteristics of its regulatory domain. FEBS Lett. 398, 206–210 10.1016/S0014-5793(96)01242-2 [DOI] [PubMed] [Google Scholar]
- 22. Richie-Jannetta R., Busch J. L., Higgins K. A., Corbin J. D., and Francis S. H. (2006) Isolated regulatory domains of cGMP-dependent protein kinase Iα and Iβ retain dimerization and native cGMP-binding properties and undergo isoform-specific conformational changes. J. Biol. Chem. 281, 6977–6984 10.1074/jbc.M510886200 [DOI] [PubMed] [Google Scholar]
- 23. Alverdi V., Mazon H., Versluis C., Hemrika W., Esposito G., van den Heuvel R., Scholten A., and Heck A. J. (2008) cGMP-binding prepares PKG for substrate binding by disclosing the C-terminal domain. J. Mol. Biol. 375, 1380–1393 10.1016/j.jmb.2007.11.053 [DOI] [PubMed] [Google Scholar]
- 24. Huang G. Y., Kim J. J., Reger A. S., Lorenz R., Moon E. W., Zhao C., Casteel D. E., Bertinetti D., Vanschouwen B., Selvaratnam R., Pflugrath J. W., Sankaran B., Melacini G., Herberg F. W., and Kim C. (2014) Structural basis for cyclic-nucleotide selectivity and cGMP-selective activation of PKG I. Structure 22, 116–124 10.1016/j.str.2013.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Osborne B. W., Wu J., McFarland C. J., Nickl C. K., Sankaran B., Casteel D. E., Woods V. L. Jr., Kornev A. P., Taylor S. S., and Dostmann W. R. (2011) Crystal structure of cGMP-dependent protein kinase reveals novel site of interchain communication. Structure 19, 1317–1327 10.1016/j.str.2011.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhao J., Hoye E., Boylan S., Walsh D. A., and Trewhella J. (1998) Quaternary structures of a catalytic subunit-regulatory subunit dimeric complex and the holoenzyme of the cAMP-dependent protein kinase by neutron contrast variation. J. Biol. Chem. 273, 30448–30459 10.1074/jbc.273.46.30448 [DOI] [PubMed] [Google Scholar]
- 27. Byeon I. J., Dao K. K., Jung J., Keen J., Leiros I., Døskeland S. O., Martinez A., and Gronenborn A. M. (2010) Allosteric communication between cAMP binding sites in the RI subunit of protein kinase A revealed by NMR. J. Biol. Chem. 285, 14062–14070 10.1074/jbc.M110.106666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Su Y., Dostmann W. R., Herberg F. W., Durick K., Xuong N. H., Ten Eyck L., Taylor S. S., and Varughese K. I. (1995) Regulatory subunit of protein kinase A: structure of deletion mutant with cAMP binding domains. Science 269, 807–813 10.1126/science.7638597 [DOI] [PubMed] [Google Scholar]
- 29. Wu J., Brown S., Xuong N. H., and Taylor S. S. (2004) RIα subunit of PKA: a cAMP-free structure reveals a hydrophobic capping mechanism for docking cAMP into site B. Structure 12, 1057–1065 10.1016/j.str.2004.03.022 [DOI] [PubMed] [Google Scholar]
- 30. Moon T. M., Tykocki N. R., Sheehe J. L., Osborne B. W., Tegge W., Brayden J. E., and Dostmann W. R. (2015) Synthetic peptides as cGMP-independent activators of cGMP-dependent protein kinase Iα. Chem. Biol. 22, 1653–1661 10.1016/j.chembiol.2015.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Francis S. H., and Corbin J. D. (1994) Structure and function of cyclic nucleotide-dependent protein kinases. Annu. Rev. Physiol. 56, 237–272 10.1146/annurev.ph.56.030194.001321 [DOI] [PubMed] [Google Scholar]
- 32. Kim C., Cheng C. Y., Saldanha S. A., and Taylor S. S. (2007) PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell 130, 1032–1043 10.1016/j.cell.2007.07.018 [DOI] [PubMed] [Google Scholar]
- 33. Kim C., Xuong N. H., and Taylor S. S. (2005) Crystal structure of a complex between the catalytic and regulatory (RIα) subunits of PKA. Science 307, 690–696 10.1126/science.1104607 [DOI] [PubMed] [Google Scholar]
- 34. Feil R., Kellermann J., and Hofmann F. (1995) Functional cGMP-dependent protein kinase is phosphorylated in its catalytic domain at threonine-516. Biochemistry 34, 13152–13158 10.1021/bi00040a029 [DOI] [PubMed] [Google Scholar]
- 35. Aitken A., Hemmings B. A., and Hofmann F. (1984) Identification of the residues on cyclic GMP-dependent protein kinase that are autophosphorylated in the presence of cyclic AMP and cyclic GMP. Biochim. Biophys. Acta 790, 219–225 10.1016/0167-4838(84)90025-6 [DOI] [PubMed] [Google Scholar]
- 36. van de Waterbeemd M., Lössl P., Gautier V., Marino F., Yamashita M., Conti E., Scholten A., and Heck A. J. (2014) Simultaneous assessment of kinetic, site-specific, and structural aspects of enzymatic protein phosphorylation. Angew. Chem. Int. Ed. Engl. 53, 9660–9664 10.1002/anie.201404637 [DOI] [PubMed] [Google Scholar]
- 37. Vigil D., Blumenthal D. K., Taylor S. S., and Trewhella J. (2005) The conformationally dynamic C helix of the RIα subunit of protein kinase A mediates isoform-specific domain reorganization upon C subunit binding. J. Biol. Chem. 280, 35521–35527 10.1074/jbc.M506769200 [DOI] [PubMed] [Google Scholar]
- 38. Schneidman-Duhovny D., Hammel M., Tainer J., and Sali A. (2013) Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys. J. 105, 962–974 10.1016/j.bpj.2013.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schneidman-Duhovny D., Hammel M., Tainer J. A., and Sali A. (2016) FoXS, FoXSDock and MultiFoXS: single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res. 44, W424–W429 10.1093/nar/gkw389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Diller T. C., Madhusudan, Xuong N. H., and Taylor S. S. (2001) Molecular basis for regulatory subunit diversity in cAMP-dependent protein kinase: crystal structure of the type IIβ regulatory subunit. Structure 9, 73–82 10.1016/S0969-2126(00)00556-6 [DOI] [PubMed] [Google Scholar]
- 41. Ogreid D., Døskeland S. O., Gorman K. B., and Steinberg R. A. (1988) Mutations that prevent cyclic nucleotide binding to binding sites A or B of type I cyclic AMP-dependent protein kinase. J. Biol. Chem. 263, 17397–17404 [PubMed] [Google Scholar]
- 42. Steinberg R. A., Russell J. L., Murphy C. S., and Yphantis D. A. (1987) Activation of type I cyclic AMP-dependent protein kinases with defective cyclic AMP-binding sites. J. Biol. Chem. 262, 2664–2671 [PubMed] [Google Scholar]
- 43. Zawadzki K. M., and Taylor S. S. (2004) cAMP-dependent protein kinase regulatory subunit type IIβ: active site mutations define an isoform-specific network for allosteric signaling by cAMP. J. Biol. Chem. 279, 7029–7036 10.1074/jbc.M310804200 [DOI] [PubMed] [Google Scholar]
- 44. Kim J. J., Casteel D. E., Huang G., Kwon T. H., Ren R. K., Zwart P., Headd J. J., Brown N. G., Chow D. C., Palzkill T., and Kim C. (2011) Co-crystal structures of PKG Iβ (92–227) with cGMP and cAMP reveal the molecular details of cyclic-nucleotide binding. PLoS One 6, e18413 10.1371/journal.pone.0018413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wall M. E., Francis S. H., Corbin J. D., Grimes K., Richie-Jannetta R., Kotera J., Macdonald B. A., Gibson R. R., and Trewhella J. (2003) Mechanisms associated with cGMP binding and activation of cGMP-dependent protein kinase. Proc. Natl. Acad. Sci. U.S.A. 100, 2380–2385 10.1073/pnas.0534892100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Steichen J. M., Kuchinskas M., Keshwani M. M., Yang J., Adams J. A., and Taylor S. S. (2012) Structural basis for the regulation of protein kinase A by activation loop phosphorylation. J. Biol. Chem. 287, 14672–14680 10.1074/jbc.M111.335091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zheng J., Trafny E. A., Knighton D. R., Xuong N. H., Taylor S. S., Ten Eyck L. F., and Sowadski J. M. (1993) 2.2 Å refined crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MnATP and a peptide inhibitor. Acta Crystallogr. D Biol. Crystallogr. 49, 362–365 10.1107/S0907444993000423 [DOI] [PubMed] [Google Scholar]
- 48. Takio K., Smith S. B., Walsh K. A., Krebs E. G., and Titani K. (1983) Amino acid sequence around a “hinge” region and its “autophosphorylation” site in bovine lung cGMP-dependent protein kinase. J. Biol. Chem. 258, 5531–5536 [PubMed] [Google Scholar]
- 49. Kawabe J. i., Toya Y., Schwencke C., Oka N., Ebina T., and Ishikawa Y. (1996) Soluble adenylyl cyclase from Spodoptera frugiperda (Sf9) cells. Purification and biochemical characterization. J. Biol. Chem. 271, 20132–20137 [DOI] [PubMed] [Google Scholar]
- 50. Lee Y. C., Martin E., and Murad F. (2000) Human recombinant soluble guanylyl cyclase: expression, purification, and regulation. Proc. Natl. Acad. Sci. U.S.A. 97, 10763–10768 10.1073/pnas.190333697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Richie-Jannetta R., Francis S. H., and Corbin J. D. (2003) Dimerization of cGMP-dependent protein kinase Iβ is mediated by an extensive amino-terminal leucine zipper motif, and dimerization modulates enzyme function. J. Biol. Chem. 278, 50070–50079 10.1074/jbc.M306796200 [DOI] [PubMed] [Google Scholar]
- 52. Kim J. J., Lorenz R., Arold S. T., Reger A. S., Sankaran B., Casteel D. E., Herberg F. W., and Kim C. (2016) Crystal structure of PKG I:cGMP complex reveals a cGMP-mediated dimeric interface that facilitates cGMP-induced activation. Structure 24, 710–720 10.1016/j.str.2016.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Qin L., Reger A. S., Guo E., Yang M. P., Zwart P., Casteel D. E., and Kim C. (2015) Structures of cGMP-dependent protein kinase (PKG) Iα leucine zippers reveal an interchain disulfide bond important for dimer stability. Biochemistry 54, 4419–4422 10.1021/acs.biochem.5b00572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Burgoyne J. R., Madhani M., Cuello F., Charles R. L., Brennan J. P., Schröder E., Browning D. D., and Eaton P. (2007) Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science 317, 1393–1397 10.1126/science.1144318 [DOI] [PubMed] [Google Scholar]
- 55. Landgraf W., Regulla S., Meyer H. E., and Hofmann F. (1991) Oxidation of cysteines activates cGMP-dependent protein kinase. J. Biol. Chem. 266, 16305–16311 [PubMed] [Google Scholar]
- 56. Landgraf W., and Hofmann F. (1989) The amino terminus regulates binding to and activation of cGMP-dependent protein kinase. Eur. J. Biochem. 181, 643–650 10.1111/j.1432-1033.1989.tb14771.x [DOI] [PubMed] [Google Scholar]
- 57. Francis S. H., Blount M. A., Zoraghi R., and Corbin J. D. (2005) Molecular properties of mammalian proteins that interact with cGMP: protein kinases, cation channels, phosphodiesterases, and multi-drug anion transporters. Front. Biosci. 10, 2097–2117 10.2741/1684 [DOI] [PubMed] [Google Scholar]
- 58. Nausch L. W., Ledoux J., Bonev A. D., Nelson M. T., and Dostmann W. R. (2008) Differential patterning of cGMP in vascular smooth muscle cells revealed by single GFP-linked biosensors. Proc. Natl. Acad. Sci. U.S.A. 105, 365–370 10.1073/pnas.0710387105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Trivedi B., and Kramer R. H. (1998) Real-time patch-cram detection of intracellular cGMP reveals long-term suppression of responses to no and muscarinic agonists. Neuron 21, 895–906 10.1016/S0896-6273(00)80604-2 [DOI] [PubMed] [Google Scholar]
- 60. Moon T. M., Osborne B. W., and Dostmann W. R. (2013) The switch helix: a putative combinatorial relay for interprotomer communication in cGMP-dependent protein kinase. Biochim. Biophys. Acta 1834, 1346–1351 10.1016/j.bbapap.2013.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Waterhouse A. M., Procter J. B., Martin D. M., Clamp M., and Barton G. J. (2009) Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 10.1093/bioinformatics/btp033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dostmann W. R., Taylor M. S., Nickl C. K., Brayden J. E., Frank R., and Tegge W. J. (2000) Highly specific, membrane-permeant peptide blockers of cGMP-dependent protein kinase Iα inhibit no-induced cerebral dilation. Proc. Natl. Acad. Sci. U.S.A. 97, 14772–14777 10.1073/pnas.97.26.14772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., and Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 10.1107/S0021889803012779 [DOI] [Google Scholar]
- 65. Franke D., and Svergun D. I. (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 10.1107/S0021889809000338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Volkov V. V., and Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 10.1107/S0021889803000268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kozin M. B., and Svergun D. I. (2001) Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34, 33–41 10.1107/S0021889800014126 [DOI] [Google Scholar]
- 68. Valentini E., Kikhney A. G., Previtali G., Jeffries C. M., and Svergun D. I. (2015) SASBDB, a repository for biological small-angle scattering data. Nucleic Acids Res. 43, D357–D363 10.1093/nar/gku1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., and Bairoch A. (2005) Protein identification and analysis tools on the ExPASy server, in The Proteomics Protocols Handbook (Walker J. M., ed) pp. 571–607, Humana Press, Totowa, NJ [Google Scholar]
- 70. Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 10.1016/S0006-3495(99)77443-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Svergun D., Barberato C., and Koch M. H. J. (1995) CRYSOL—a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 10.1107/S0021889895007047 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.