

ABSTRACT
Two common signal transduction mechanisms used by bacteria to sense and respond to changing environments are two-component systems (TCSs) and eukaryote-like Ser/Thr kinases and phosphatases (eSTK/Ps). Enterococcus faecalis is a Gram-positive bacterium and a serious opportunistic pathogen that relies on both a TCS and an eSTK/P pathway for intrinsic resistance to cell wall-targeting antibiotics. The TCS consists of a histidine kinase (CroS) and a response regulator (CroR) that become activated upon exposure of cells to cell wall-targeting antibiotics, leading to a modulation of gene expression. The eSTK/P pathway consists of a transmembrane kinase (IreK) and its cognate phosphatase (IreP), which act antagonistically to mediate antibiotic resistance through an unknown mechanism. Because both CroS/R and IreK/P contribute to enterococcal resistance toward cell wall-targeting antibiotics, we hypothesized that these signaling systems are intertwined. To test this hypothesis, we analyzed CroR phosphorylation and CroS/R-dependent gene expression to probe the influence of IreK and IreP on CroS/R signaling. In addition, we analyzed the phosphorylation state of CroS, which revealed the IreK-dependent phosphorylation of a Thr residue important for CroS function. Our results are consistent with a model in which IreK positively influences CroR-dependent gene expression through the phosphorylation of CroS to promote antimicrobial resistance in E. faecalis.
IMPORTANCE Two-component signaling systems (TCSs) and eukaryote-like Ser/Thr kinases (eSTKs) are used by bacteria to sense and adapt to changing environments. Understanding how these pathways are regulated to promote bacterial survival is critical for a more complete understanding of bacterial stress responses and physiology. The opportunistic pathogen Enterococcus faecalis relies on both a TCS (CroS/R) and an eSTK (IreK) for intrinsic resistance to cell wall-targeting antibiotics. We probed the relationship between CroS/R and IreK, revealing the convergence of IreK and the sensor kinase CroS to enhance signaling through CroS/R and increase antimicrobial resistance in E. faecalis. This newly described example of eSTK/TCS convergence adds to our understanding of the signaling networks mediating antimicrobial resistance in E. faecalis.
KEYWORDS: Enterococcus, PASTA kinase, cell wall stress, eSTK, two-component signaling system
INTRODUCTION
Most bacteria are well equipped to sense and respond to their environment. Two types of conserved signal transduction systems used by bacteria are two-component systems (TCSs) and eukaryote-like Ser/Thr kinases and phosphatases (eSTK/Ps). TCSs are ubiquitous in bacteria and respond to a variety of environmental cues. Typically, a TCS consists of a transmembrane sensor histidine kinase (HK) and a cytoplasmic response regulator (RR). Upon sensing a signal, the HK autophosphorylates on a conserved histidine, which subsequently donates its phosphoryl group to a conserved aspartate on the cognate RR. Many RRs bind DNA and modulate gene expression upon phosphorylation in response to specific stimuli. In addition to promoting the phosphorylation of a cognate RR, many HKs dephosphorylate their cognate RR (1–3). Therefore, the balance of kinase and phosphatase activities by an HK regulates the output of a given TCS. Unlike TCSs, eSTK/P pathways use distinct enzymes to regulate the phosphorylation state of effector proteins and are thought to be more promiscuous in substrate phosphorylation (i.e., capable of phosphorylating more than one substrate) (4). This appears to be true for a particular family of eSTKs known as the PASTA kinases, which are transmembrane eSTKs containing extracellular PASTA (penicillin-binding protein- and Ser/Thr kinase-associated) domains that are widespread among the Actinobacteria and Firmicutes. Protein substrates identified for PASTA kinases are involved in a variety of cellular processes, including cell wall homeostasis, protein synthesis, and transcriptional regulation (5). However, much is still to be learned about PASTA kinase substrates and their roles in regulating bacterial physiology.
Although TCS and eSTK/P pathways are often conceptualized as distinct systems, they have coevolved in bacterial cells, and examples of convergence have emerged. Staphylococcus aureus, group A and group B streptococci, and Bacillus subtilis carry a homologous PASTA kinase (known as Stk1 or PrkC) capable of directly phosphorylating TCS RRs to modulate RR DNA binding (6–11). Although conceptually similar observations have been made for multiple bacterial species, generalization is difficult, and it remains challenging to predict new interactions between signaling systems. For example, in different species, the PASTA kinase RR substrates are part of different TCSs, Thr phosphorylation occurs at different sites within the RR, and phosphorylation by Stk1/PrkC can have positive or negative effects on the RR output.
Enterococcus faecalis is a Gram-positive bacterium and a serious opportunistic pathogen that relies on both a TCS and a PASTA kinase for intrinsic resistance to cell wall-targeting antibiotics. The TCS consists of the HK CroS and the RR CroR. The phosphorylation of CroR by CroS is stimulated by cell wall-targeting antibiotics and is required for resistance (12, 13). Although CroR modulates gene expression in response to cell wall-targeting antibiotics (12, 14), the genes regulated by CroR that promote resistance remain unknown. The PASTA kinase IreK (originally known as PrkC [15]) is the only eSTK readily identifiable in the genome of E. faecalis. IreK acts antagonistically to its cognate phosphatase IreP to mediate antibiotic resistance (15, 16). One substrate for IreK has been identified (IreB), which acts as a negative regulator of the pathway (17). Cell wall stresses lead to the enhanced phosphorylation of IreK, resulting in the activation and, subsequently, the phosphorylation of IreB (18), although it remains unclear how IreB influences antimicrobial resistance. The CroS/R and IreK/P pathways have been characterized for their ability to dynamically influence resistance to cephalosporins (a subset of beta-lactam antibiotics targeting peptidoglycan biosynthesis). For example, E. faecalis strains lacking IreK or CroR are more susceptible to cephalosporins than are wild-type strains (14, 15). Conversely, strains lacking negative regulators of IreK or CroR (i.e., the phosphatase activity of IreP or CroS) display a dramatic increase in cephalosporin resistance compared to wild-type strains (12, 16).
The parallel effects of the IreK/P and CroS/R pathways on cephalosporin resistance prompted us to hypothesize that these signaling systems converge to coordinately regulate cephalosporin resistance in enterococci. We probed the relationship between the IreK/P and CroS/R pathways, yielding a model in which IreK positively affects CroS/R signaling by promoting the phosphorylation of CroS. CroS was found to be phosphorylated in response to cell wall-targeting antimicrobial agents in an IreK-dependent manner, at a site distinct from that of the autophosphorylated His residue. Furthermore, substitutions at the newly identified site altered CroR phosphorylation and cephalosporin resistance. This newly described example of TCS/eSTK convergence at the level of HK phosphorylation extends our knowledge of the signaling networks mediating antimicrobial resistance in E. faecalis and diversifies the known mechanisms by which eSTKs influence TCS function.
RESULTS
IreK and IreP reciprocally affect CroS/R signaling.
The output of the IreK signaling pathway is unknown. Previously, we showed that IreK is constitutively active in the absence of its cognate phosphatase IreP, resulting in hyperresistance to cephalosporins (16, 18). To determine if CroR was required for this effect, we analyzed the resistance of an E. faecalis mutant lacking both IreP and CroR. The double mutant was no longer highly resistant (Table 1), demonstrating that the ΔcroR mutation is epistatic and suggesting that the IreK and CroS/R pathways might be integrated. To test this hypothesis, we examined signal transduction through CroS/R in strains exhibiting altered IreK activity. Phos-tag SDS-PAGE and immunoblot analyses of lysates from unstressed cells revealed a more slowly migrating isoform of CroR in the ΔireP mutant (Fig. 1A). Our previous work demonstrated that this more slowly migrating isoform corresponds to phosphorylated (activated) CroR (CroR-P) (12). Additionally, CroR was overexpressed in the ΔireP mutant, reflecting constitutively enhanced CroS/R production due to the CroR-P-dependent autoregulation of its own synthesis (14). Consistent with this, a lacZ reporter fusion revealed enhanced expression from the CroR-dependent PcroR promoter in the ΔireP mutant (Fig. 1B, black bars). Analysis of the ΔirePK double mutant confirmed that IreK was required for the activation of CroR in the ΔireP mutant. Collectively, these observations indicate that CroS/R signaling is upregulated in the presence of constitutively activated IreK.
TABLE 1.
Ceftriaxone resistancea of E. faecalis strains

Median MIC from ≥2 biological replicates.
b Strains analyzed were wild-type strain OG1, ΔireK strain JL206, ΔireB strain JL367, ΔireP strain JL455, ΔireP ΔcroR strain JL457, ΔcroR strain SB23, ΔireP ΔcisS strain SB95, and ΔireP ΔcisS ΔcroS strain SB97.
FIG 1.
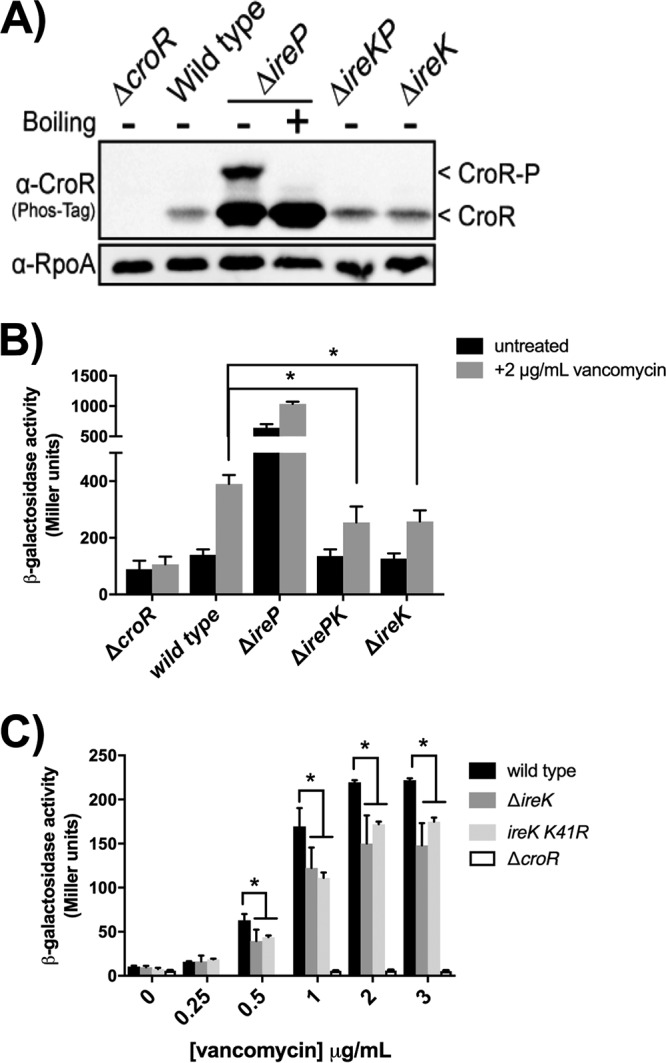
IreK enhances CroS/R signaling. (A) Phos-tag SDS-PAGE and immunoblot analyses of whole-cell lysates from unstressed, exponentially growing E. faecalis strains carrying different combinations of ireP and ireK deletions. Samples were not boiled before electrophoresis except where indicated. A subunit of RNA polymerase (RpoA) was used as a loading control. Data are representative of results from ≥2 experiments. (B) Beta-galactosidase activity from Pcro′-lacZ was determined in exponentially growing cells in the presence and absence of vancomycin (2 μg/ml). (C) Beta-galactosidase activity was determined in exponentially growing cells in response to various concentrations of vancomycin using a lacZ fusion with the promoter of OG1RF_12500. Error bars represent the standard deviations from ≥3 biological replicates; * indicates a P value of <0.05, as determined by a t test. Strains were ΔcroR strain SB21, wild-type strain OG1RF, ΔireP strain CK121, ΔirePK strain CK125, ΔireK strain CK119, and ireK K41R strain SK131. Strains carried no plasmid (A), pJLL170 (B), or pSLK235 (C).
In contrast, ΔireK mutant cells exhibited attenuated expression of a CroR-dependent reporter gene compared to the wild-type strain after treatment with a known stimulant of CroS/R signaling, vancomycin (Fig. 1B, gray bars). This attenuation of CroS/R signaling was apparent over a range of vancomycin concentrations and with a distinct CroR-dependent reporter fusion (Fig. 1C), suggesting a global defect in CroS/R signal transduction in the absence of IreK. Moreover, a strain expressing an IreK variant with a substitution at its catalytic site that severely impairs in vitro kinase activity and ablates the in vivo functions of IreK (IreK K41R) (16, 18) is also impaired in CroS/R signal transduction (Fig. 1C), indicating the kinase activity of IreK is required for the full induction of CroS/R signaling. Together, these data demonstrate that IreK and IreP reciprocally influence CroS/R signal transduction and that the kinase activity of IreK enhances, but is not essential for, signaling through CroS/R.
The IreK-dependent activation of CroR in the absence of ireP indicates that IreK can directly or indirectly influence the phosphorylation of CroR. To determine if CroR was phosphorylated at the canonical Asp residue in its receiver domain or at a Ser/Thr residue elsewhere, the ΔireP whole-cell lysate sample was subjected to heating before Phos-tag SDS-PAGE analysis. Because phosphoryl-Asp bonds are heat sensitive and phosphoryl-Ser/Thr bonds are heat resistant, we can discriminate between these types of phosphorylation by Phos-tag SDS-PAGE. The CroR-P isoform disappeared upon heating (Fig. 1A), indicating that CroR is likely phosphorylated at the canonical Asp residue. Although CroS is the cognate histidine kinase for CroR, our previous study revealed the possibility of CroR phosphorylation by another histidine kinase, CisS, under certain conditions (12). To determine which histidine kinase is responsible for the phosphorylation of CroR in the ΔireP mutant, double and triple ΔireP ΔcisS ΔcroS mutants were constructed. Phos-tag SDS-PAGE and antimicrobial susceptibility analyses revealed that CroS is specifically required for the phosphorylation of CroR and for the cephalosporin hyperresistance of the ΔireP mutant (Fig. 2 and Table 1). Together, these results suggest that IreK does not modulate CroR through direct Ser/Thr phosphorylation but rather influences CroR indirectly through the activity of CroS.
FIG 2.
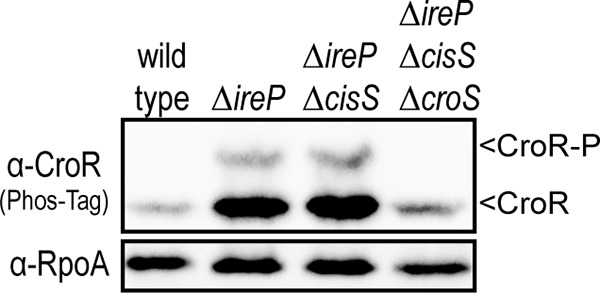
CroS phosphorylates CroR in the ΔireP mutant. Shown are data from Phos-tag SDS-PAGE and immunoblot analyses of whole-cell lysates from unstressed, exponentially growing E. faecalis strains. A subunit of RNA polymerase (RpoA) was used as a loading control. Data are representative of results from ≥2 experiments. Strains were wild-type strain OG1, ΔireP strain JL455, ΔireP ΔcisS strain SB95, and ΔireP ΔcisS ΔcroS strain SB97.
CroS is modified in an IreK-dependent manner.
One way in which IreK could modify the activity of CroS is by direct phosphorylation. To test for phosphorylation, CroS was analyzed by Phos-tag SDS-PAGE. Because we do not have an antibody specific for CroS, we used epitope-tagged variants of CroS. To rule out the possibility that an epitope tag itself could be subject to spurious phosphorylation, we independently fused 2 distinct tags (hemagglutinin [HA] [YPYDVPDYA] or Strep-tag [WSHPQFEK]) to the C terminus of CroS. Plasmids coexpressing croR and croS together (to maintain the appropriate stoichiometric ratio) were introduced into E. faecalis lacking the chromosomal croRS locus. The expression of croR croS-HA is driven by the native Pcro promoter and yields CroR levels comparable to those of wild-type strains (12). Additionally, the expression of croR croS-HA drives cephalosporin resistance in the ΔcroRS deletion mutant, indicating that CroS is functional with the HA tag extension (Table 2). Anti-HA immunoblot analyses of lysates from unstressed cells after Phos-tag SDS-PAGE revealed two isoforms of CroS, indicating the presence of a small fraction of the phosphorylated isoform (Fig. 3A). The abundance of the phosphorylated CroS isoform was increased when cells were treated with the same cell wall-targeting stressors that induced the phosphorylation of CroR (Fig. 3B) (13, 14, 19). Stressors that did not stimulate the phosphorylation of CroR also did not enhance the phosphorylation of CroS, such as the beta-lactam ampicillin and the DNA gyrase inhibitor norfloxacin. The result with ampicillin is perhaps unexpected, given that ampicillin is a cell wall-targeting antibiotic, but in previous studies of CroS/R signal transduction, we observed a similar disconnect (13). This disconnect remains unexplained, but it may result from a more robust stimulation of signaling by antibiotics that act earlier in the peptidoglycan synthesis pathway than ampicillin or may simply be due to the specific experimental conditions used (as a previous study demonstrated CroS/R-dependent changes in gene expression after ampicillin exposure [14]).
TABLE 2.
Ceftriaxone resistancea of strains carrying CroS variants
| croS alleleb | Median MIC (μg/ml) for host strain genotypeb |
|
|---|---|---|
| ΔcroRS | ΔcroRS ΔcisRS | |
| None | 8 | 8 |
| Wild type | 32 | 32 |
| T346A | 1,024 | 1,024 |
| T346A D173A | 8 | ND |
| T346V | 1,024 | ND |
| T346E | >1,024 | ND |
| S343A | 512 | 512 |
| S341A | 1,024 | 1,024 |
Median MIC from ≥2 biological replicates. ND, not determined.
Strains analyzed were SB35 strains with pJRG8 (empty vector), pSLK134, pSLK141-3, pSLK225-7, or pCEM1. All plasmids except pJRG8 express the croRS locus with an HA tag on CroS.
FIG 3.

CroS is phosphorylated in response to cell wall stressors. Exponentially growing cells treated with various stressors were analyzed for CroS-HA (A) and CroR (B) by Phos-tag and standard SDS-PAGE and immunoblot analyses. Stressors were ceftriaxone (Cx), ampicillin (Amp), vancomycin (Van), bacitracin (Bac), hydrogen peroxide (H2O2), and norfloxacin (NF). A subunit of RNA polymerase, RpoA, was used as a loading control. Data are representative of results from ≥2 experiments. The strain used is SB35(pSLK134). The empty vector strain is SB35(pJRG8).
The production of phosphorylated CroS was not an artifact of the HA tag, because CroS with Strep-tag also exhibited a phosphorylated isoform upon treatment with ceftriaxone (Fig. 4A). However, no phosphorylated CroS was observed after treatment of the ΔireK mutant with ceftriaxone, indicating that IreK is directly or indirectly required for CroS phosphorylation. To determine if the phosphorylated CroS isoform observed after Phos-tag SDS-PAGE was phosphorylated at the canonical, conserved histidine residue in the HisKA domain of CroS, we expressed a CroS H172A variant in E. faecalis. The phosphorylated CroS isoform was still observed, indicating that phosphorylation was occurring elsewhere (Fig. 4B). It remains unclear whether the His-phosphorylated isoform of CroS is lost during sample preparation or if it is not resolved under our Phos-tag SDS-PAGE conditions. Regardless, we conclude that CroS is phosphorylated in vivo in response to cell wall stress, in an IreK-dependent manner, at a site distinct from H172.
FIG 4.
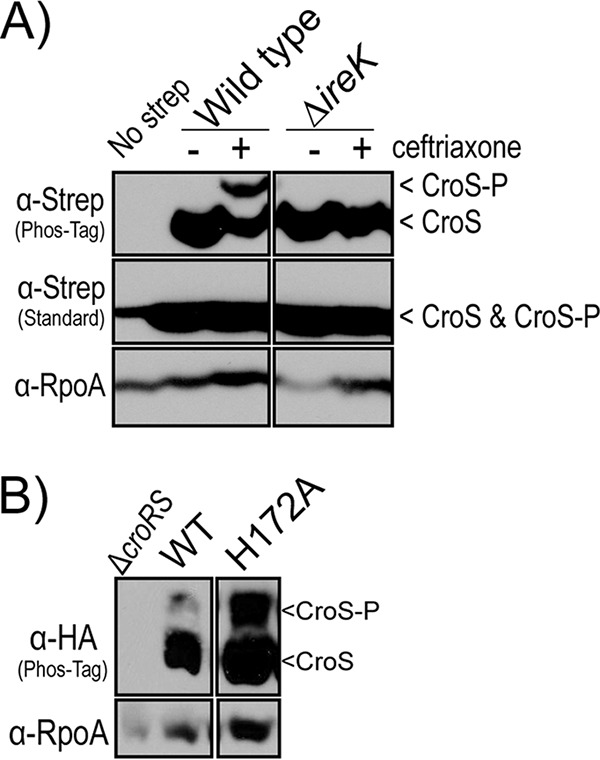
CroS modification requires IreK. (A) Phos-tag SDS-PAGE and immunoblot analyses of CroS-Strep-tag from strains containing or lacking ireK. Cells were harvested during exponential growth with or without exposure to ceftriaxone. Strains are SB6(pSLB30) (Wild type) and ΔireK strain SB17(pSLB30). A subunit of RNA polymerase, RpoA, was used as a loading control. (B) Phos-tag SDS-PAGE and immunoblot analyses of CroS-HA (wild type [WT]) and a variant substituted at the site of autophosphorylation, H172A. Cells were harvested during exponential growth without the addition of cell wall stressors. Strains were SB35(pSLK134) and SB35(pSLK135). A subunit of RNA polymerase, RpoA, was used as a loading control. Data are representative of results from ≥2 experiments.
Because the presence of CroS-P depends on IreK (Fig. 4A), we hypothesized that IreK modulates the activities of CroS in response to cell wall stress through an as-yet-uncharacterized phosphorylation event. One possibility is that CroS is a direct substrate for IreK and IreP. To test this, we sought to perform in vitro kinase and phosphatase reactions by using CroS as a substrate. However, purified cytoplasmic fragments of CroS have proven to be insoluble (12). As an alternative approach, membrane preparations from E. faecalis cells expressing epitope-tagged CroS were used as a source of CroS for reactions with recombinant IreK (cytoplasmic kinase fragment) or IreP in vitro. Membranes derived from otherwise wild-type cells initially contained a small fraction of phosphorylated CroS, and the addition of recombinant IreK did not lead to increased CroS phosphorylation (Fig. 5). In contrast, membranes derived from ΔireP mutant cells initially contained predominantly phosphorylated CroS, and the addition of recombinant IreP led to a shift in mobility consistent with the loss of CroS phosphorylation. Although it remains unclear why IreK was unable to act on CroS under our in vitro conditions (perhaps the truncated form of IreK cannot efficiently recognize CroS, or perhaps a cofactor was missing), these results suggest that CroS is indeed phosphorylated in vivo and can be dephosphorylated by IreP, the cognate phosphatase of IreK.
FIG 5.
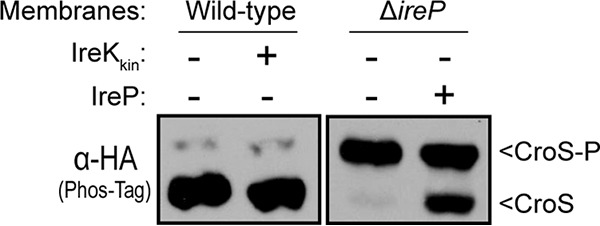
Recombinant IreP can promote dephosphorylation of CroS. Membrane fractions containing CroS-HA were used as substrates in reactions with recombinant IreK and IreP and analyzed by Phos-tag SDS-PAGE and immunoblot analyses. Membranes from a wild-type strain were incubated with recombinant IreK (the cytoplasmic kinase domain of IreK) and ATP, while membranes from an ΔireP mutant were incubated with recombinant IreP. Results from a 60-min incubation with recombinant IreK and a 10-min incubation with recombinant IreP are shown. Images are representative of results from ≥2 experiments. Strains were SB35(pSLK134) and JL457(pSLK134).
Thr-346 is required for CroS phosphorylation.
To define where CroS is phosphorylated, mass spectrometry was performed on CroS-His6 that was affinity purified from the E. faecalis ΔireP mutant. We obtained 70% sequence coverage of CroS (including coverage of the entire cytoplasmic domain) and identified four phosphopeptides with a mass shift consistent with a single Ser/Thr/Tyr phosphoryl group (Fig. 6A), although we were not able to unambiguously define the specific site of phosphorylation in all peptides. To determine which of the candidate sites were phosphorylated in vivo, we generated a series of CroS variants in which each of the potential sites of phosphorylation was individually replaced with a nonphosphorylatable residue (Ala). These mutants were individually expressed in the E. faecalis ΔireP mutant to determine if any of the substitutions would result in an altered mobility of CroS during Phos-tag SDS-PAGE. Two variants initially were of interest: CroS Y223A and CroS T346A (Fig. 6B). Of note, CroS Y223A was clearly less abundant than the other mutants, and longer exposures of the immunoblot revealed the presence of a phosphorylated CroS isoform with this mutant (see Fig. S1 in the supplemental material). In contrast, even with long immunoblot exposures, the phosphorylated CroS isoform was not detected with the CroS T346A variant, suggesting that the phosphorylated isoform of CroS resolved under our Phos-tag SDS-PAGE conditions represents CroS that has been phosphorylated at Thr-346. Thr-346 was also required for the appearance of the phosphorylated CroS isoform detected when wild-type cells were exposed to ceftriaxone (Fig. 6C). It remains unclear if the additional putative sites of phosphorylation that emerged from the mass spectrometry analysis are simply not phosphorylated under our experimental conditions, if they are phosphorylated but unresolved under our Phos-tag SDS-PAGE conditions, or if phosphorylation at other sites is dependent on Thr-346.
FIG 6.

Identification of Thr-346 as the critical site of CroS phosphorylation. (A) Mass spectrometry identified four peptides with a mass shift consistent with the addition of one phosphoryl group. Specific sites of phosphorylation could not be unambiguously resolved; phosphorylatable residues within each peptide are underlined. Numbers correspond to the peptide sequence of CroS. (B) Phos-tag and standard SDS-PAGE and immunoblot analyses of an E. faecalis ΔireP mutant carrying variants of CroS-HA. Samples were harvested during exponential growth without the addition of cell wall stressors. The strain is JL457 with pSLK134, pSLK135, or pSLK137-145. (C) Phos-tag SDS-PAGE and immunoblot analyses of otherwise wild-type cells carrying wild-type CroS-HA or a T346A variant exposed to ceftriaxone (Cx). Images are representative of results from ≥2 experiments. The strain is SB35 with pSLK134 or pSLK143.
Thr-346 and the ATP lid influence CroS activity.
Based on these results, we hypothesized that the reversible phosphorylation of Thr-346 alters the activity of CroS in times of cell wall stress. Thr-346 is located within a structure of the CroS ATPase domain known as the ATP lid. This structure is important for nucleotide positioning and exchange during HK autophosphorylation and makes contact with cognate RRs during phosphoryltransfer events (20, 21). To test if Thr-346 is important for the proper regulation of CroS activity, we analyzed the accumulation of CroR-P and ceftriaxone resistance in strains expressing CroS variants with substitutions at Thr-346. Replacement of Thr-346 with the nonphosphorylatable residue Ala resulted in an accumulation of CroR-P in unstressed cells (Fig. 7); moreover, replacement with Ala or Val resulted in a 32-fold enhancement of ceftriaxone resistance over that of the wild-type strain (Table 2), reflecting a marked dysregulation of CroS activity. Replacement of Thr-346 with Glu, which often acts as a “phosphomimetic” analogous to a permanently phosphorylated site, also led to an accumulation of CroR-P and to a dramatic enhancement of ceftriaxone resistance, suggesting that the T346E mutation does not behave as a phosphomimetic in CroS or, alternatively, that the reversible phosphorylation of CroS is critical for the proper regulation of CroS activity in vivo.
FIG 7.
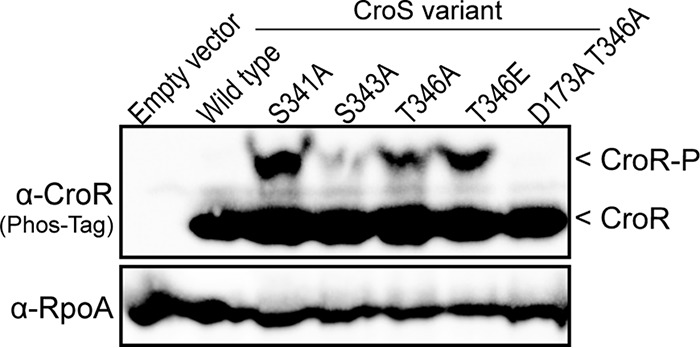
Substitutions within the ATP lid of CroS alter CroR phosphorylation. Shown are data from Phos-tag SDS-PAGE and immunoblot analyses of whole-cell lysates from unstressed, exponentially growing E. faecalis strains carrying variants of CroS. A subunit of RNA polymerase (RpoA) was used as a loading control. Data are representative of results from ≥2 experiments. Strains were empty vector strain SB35(pJRG8), wild-type strain SB35(pSLK134), S341A mutant strain SB35(pSLK141), S343A mutant strain SB35(pSLK142), T346A mutant strain SB35(pSLK143), T346E mutant strain SB35(pSLK226), and D173A T346A mutant strain SB35(pCEM1).
These results suggest that the ATP lid plays an important role in the regulation of CroS activity. Substitutions with Ala at two other sites in the ATP lid (Ser-341 and Ser-343), which do not prevent IreK-mediated CroS phosphorylation in vivo (Fig. 6B), also led to an accumulation of CroR-P and substantially increased ceftriaxone resistance (Fig. 7 and Table 2), again reflecting the dysregulation of CroS activity. Previous studies revealed that the loss of CroS, or a mutation that specifically impairs the phosphatase activity of CroS, also leads to ceftriaxone hyperresistance due to the cross-phosphorylation of CroR by a noncognate TCS kinase (CisS) (12). We confirmed that the hyperresistance of the CroS ATP lid mutants was not due to CisS (Table 2), indicating that hyperresistance is indeed due to alterations in the activity of CroS itself rather than an altered stability of the protein. Moreover, a substitution that specifically ablated the kinase activity of CroS (D173A) (12) prevented the accumulation of CroR-P in the T346A mutant and rendered it sensitive to ceftriaxone (Fig. 7 and Table 2). These observations indicate that the kinase activity of CroS drives the accumulation of CroR-P and enhanced cephalosporin resistance when Thr-346 is substituted and furthermore that the phosphatase activity of the CroS T346A mutant is intact (i.e., capable of eliminating any cross-phosphorylation of CroR by CisS). Together, our results suggest that the ATP lid plays an important role in regulating the kinase activity of CroS in vivo, and the IreK-dependent phosphorylation of the ATP lid at T346 may serve to regulate this function.
DISCUSSION
Bacteria use TCS and eSTK/P signaling systems to sense and adapt to changing environments. Understanding how these pathways are regulated to influence bacterial survival under unfavorable conditions is critical for a more complete understanding of bacterial stress responses and physiology. Although convergence between eSTKs and TCSs has been documented previously, it is challenging to predict which systems interact with each other and the molecular logic of these connections. For example, the PASTA kinases from streptococci, S. aureus, and B. subtilis each phosphorylate a TCS RR, but the identities of the target RRs, the sites of phosphorylation, and the effects on the signaling output in each case are not equivalent (6–11). Therefore, experimental studies are required to dissect interactions between TCS and eSTK systems and reveal the nature and extent of cross-communication between these distinct signaling systems in bacterial cells.
In this study, we investigated the effect of the PASTA kinase IreK and its cognate phosphatase IreP on CroS/R signal transduction. Consistent with both systems being positive regulators of cephalosporin resistance, we showed that IreK positively influences CroS/R signal transduction by promoting the phosphorylation of CroS, phosphoryltransfer to CroR, and, ultimately, CroS/R-dependent gene expression. Furthermore, we demonstrated that CroS is phosphorylated at a site distinct from the autophosphorylated His-172, likely at Thr-346 located within the ATP lid. Genetic perturbation of the ATP lid of CroS (including the substitution of Thr-346) resulted in an accumulation of CroR-P and a dramatic increase in the cephalosporin resistance of E. faecalis that was dependent on the kinase activity of CroS. Additionally, the phosphorylation of CroS at Thr-346 was enhanced in the presence of cell wall stressors, an effect that was dependent on IreK. Coupled with data from previous work demonstrating the enhanced activity of IreK upon cell wall stress in E. faecalis (18), these data suggest a model in which IreK promotes the phosphorylation of CroS at T346 in response to cell wall damage, which in turn enhances CroS/R signal transduction by modulating the function of the ATP lid of CroS.
It remains unclear if this communication with CroS/R is the only mechanism by which IreK drives cephalosporin resistance or if other factors downstream of IreK influence resistance independent of CroS/R. Of note, the ΔireK mutant is more susceptible to ceftriaxone than the ΔcroR mutant (Table 1), suggesting that IreK contributes to cephalosporin resistance at least in part independently of CroS/R. Such a mechanism could involve IreB, a known substrate for phosphorylation by IreK (17, 18). Mutants lacking IreB exhibit elevated resistance to cephalosporins (17) although not to the extent of the ΔireP mutant (16) (Table 1), which implies the existence of multiple IreK-dependent effectors to promote resistance. Thus, the IreK “pathway” may be a branched signaling network that influences cephalosporin resistance through CroS/R signal transduction and through other mechanisms that are yet to be elucidated.
Despite the identification of several CroS phosphopeptides by mass spectrometry, Thr-346 was the only residue to modulate the phosphorylation state of CroS, as detected by Phos-tag SDS-PAGE. It remains unclear if the additional sites are simply not phosphorylated under our experimental conditions, if they are phosphorylated but unresolved under our Phos-tag SDS-PAGE conditions, or if phosphorylation at other sites is dependent on Thr-346. Biochemical approaches would allow us to further explore the relationship between Thr-346 and other potential sites of phosphorylation and their effect on the activity of CroS. Unfortunately, we have thus far been unable to obtain a soluble, active form of CroS that would enable such studies (12). However, the phosphorylation of CroS at Thr-346 is well positioned to influence the activity of CroS through modulation of the ATP lid, as ATP lids are considered to be critical for nucleotide binding and positioning before HK autophosphorylation (22–24). Although our in vivo data suggest that the kinase activity of CroS is enhanced when the ATP lid is perturbed, biochemical data are required to thoroughly probe the molecular effects of Thr-346 phosphorylation.
The communication between IreK and CroS/R is distinct from previously described examples of TCS/eSTK convergence (6–11) in that the kinase CroS, rather than the response regulator CroR, is the target of eSTK-dependent phosphorylation. One previous report described the phosphorylation of an HK by an eSTK in B. subtilis. B. subtilis possesses 3 eSTKs, 2 of which (YbdM and YabT, neither of which is a PASTA kinase) were reported to phosphorylate the atypical cytosolic HK DegS in vitro (25). Although the site of phosphorylation by YabT was not identified, YbdM was reported to phosphorylate DegS on a serine residue in the sensing domain. Phosphorylation by YbdM enhanced DegS kinase activity and phosphoryl transfer to the RR DegU in vitro, and the substitution of the site of phosphorylation for a phosphomimetic residue promoted phenotypes consistent with an intermediate level of DegU phosphorylation (25). Although we predict that the phosphorylation of CroS by IreK enhances CroS kinase activity and phosphoryl transfer to CroR, there are important differences between these examples of eSTK/HK convergence. First, the HKs are phosphorylated in different domains. Serine phosphorylation of DegS in the sensing domain likely alters the activity of DegS indirectly, perhaps by mimicking or augmenting the detection of the natural stimulus for DegS. On the other hand, the phosphorylation of CroS within the ATP lid likely modulates the mechanics of the ATPase domain directly. Second, IreK and YbdM belong to different protein families of eSTKs. IreK is a PASTA kinase and is homologous to eSTKs with a known ability to phosphorylate TCS response regulators (e.g., PrkC and Stk1), while YbdM does not have PASTA domains and has not previously been implicated in convergence with TCSs. These differences highlight the need for continued investigations of cross-communication between TCSs and eSTKs in bacterial signaling networks, as the emerging examples suggest that the protein players and molecular details of each system may vary.
MATERIALS AND METHODS
Bacterial strains, growth media, and chemicals.
Bacterial strains and plasmids used in the study are listed in Table 3. E. faecalis strains were grown in half-strength (0.5×) brain heart infusion (hBHI) medium or Mueller-Hinton broth (MHB) for routine maintenance. Escherichia coli strains were grown in lysogeny broth (LB). Erythromycin (Em) was used at 10 μg/ml or 100 μg/ml for plasmid selection in E. faecalis and E. coli respectively. Chloramphenicol (Cm) was used at 10 μg/ml for plasmid selection in E. faecalis and E. coli. Kanamycin (Kn) was used at 50 μg/ml for plasmid selection in E. coli. All cultures were grown aerobically with shaking.
TABLE 3.
Strains and plasmids used in this study
| Strain or plasmid | Genotype or descriptiona | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5α | Routine cloning host | Laboratory stock |
| BL21(DE3) | Protein overexpression host | Laboratory stock |
| E. faecalis | ||
| OG1 | Wild-type laboratory strain isolated from an oral sample (MLST 1) | 29 |
| OG1RF | Spontaneous rifampin- and fusidic acid-resistant derivative of OG1 | 30 |
| CK119 | OG1RF ΔireK2 | 15 |
| CK121 | OG1RF ΔireP2 | 16 |
| CK125 | OG1RF Δ(ireP ireK)2 | 16 |
| JL206 | OG1 ΔireK2 | 18 |
| JL367 | OG1 ΔireB2 | 18 |
| JL455 | OG1 ΔireP2 | 18 |
| JL457 | OG1 ΔcroR2 ΔireP2 | This work |
| SB6 | OG1RF Δ(croR croS)3 | This work |
| SB17 | OG1RF Δ(croR croS)3 ΔireK2 | This work |
| SB21 | OG1RF ΔcroR2 | This work |
| SB23 | OG1 ΔcroR2 | 31 |
| SB35 | OG1 Δ(croR croS)3 | 12 |
| SB95 | OG1 ΔcisS1 ΔireP2 | This work |
| SB97 | OG1 ΔcisS1 ΔcroS2 ΔireP2 | This work |
| SK131 | OG1RF ireK K41R | This work |
| Plasmids | ||
| pBK200 | E. faecalis expression vector (Emr) | 31 |
| pCEM1 | Pcro-croR-croS D173A T346A-HA in pJRG8 | This work |
| pCJK111 | ireK-n (kinase/juxtamembrane domain) in pET28b | 16 |
| pCJK112 | ireP in pET28b | 16 |
| pCJK218 | E. faecalis allelic exchange vector (Cmr) | 28 |
| pCJK236 | P23-croRS-his6 in pBK200 | This work |
| pCJK242 | ΔireP2 deletion allele in pCJK218 | 18 |
| pCI3340 | E. coli-E. faecalis shuttle vector (Cmr) | 27 |
| pET28b | E. coli expression vector (Knr) | Novagen |
| pJRG8 | E. faecalis expression vector (Emr) | 16 |
| pJLL69 | ΔireK2 deletion allele in pCJK218 | This work |
| pJLL170 | Pcro′-lacZ in pCI3340 | This work |
| pSLB6 | ΔcroS2 deletion allele | 12 |
| pSLB7 | Δ(croR croS)3 deletion allele | 12 |
| pSLB30 | P23S-croRS-strep in pJRG8 | This work |
| pSLB38 | ΔcroR2 deletion allele in pCJK218 | 31 |
| pSLB73 | ΔcisS1 deletion allele in pCJK245 | 12 |
| pSLK134 | Pcro-croRS-HA in pJRG8 | This work |
| pSLK135 | Pcro-croR-croS H172A-HA in pJRG8 | This work |
| pSLK136 | Pcro-croR-croS D173A-HA in pJRG8 | This work |
| pSLK137 | Pcro-croR-croS S132A-HA in pJRG8 | This work |
| pSLK138 | Pcro-croR-croS Y223A-HA in pJRG8 | This work |
| pSLK139 | Pcro-croR-croS T224A-HA in pJRG8 | This work |
| pSLK140 | Pcro-croR-croS S327A-HA in pJRG8 | This work |
| pSLK141 | Pcro-croR-croS S341A-HA in pJRG8 | This work |
| pSLK142 | Pcro-croR-croS S343A-HA in pJRG8 | This work |
| pSLK143 | Pcro-croR-croS T346A-HA in pJRG8 | This work |
| pSLK144 | Pcro-croR-croS T349A-HA in pJRG8 | This work |
| pSLK145 | Pcro-croR-croS S358A-HA in pJRG8 | This work |
| pSLK225 | Pcro-croR-croS T346V-HA in pJRG8 | This work |
| pSLK226 | Pcro-croR-croS T346E-HA in pJRG8 | This work |
| pSLK234 | Promoterless lacZ in pCI3340 | This work |
| pSLK235 | Promoter region of OG1RF_12500 in pSLK234 (includes 350 bp upstream of the translational start site) | This work |
MLST, multilocus sequence type.
Plasmid construction.
Plasmids were constructed by using either a BsaI-based seamless cloning strategy with primer-encoded restriction sites or Gibson assembly (26). All inserts in recombinant plasmids were sequenced in their entirety to confirm the absence of mutations. Plasmid pJRG8 (16) was used to express various alleles of croRS in E. faecalis cells. For pSLB30, croRS was expressed from the P23S promoter of pJRG8 with a carboxy-terminal Strep-tag fused to CroS. The remaining croRS plasmids were constructed as previously described (12), with the P23S promoter of pJRG8 being removed and replaced with the promoter region of the croRS locus and the croRS genes. During the construction of these plasmids, a carboxy-terminal HA tag was fused to CroS.
Constructs for lacZ fusions with promoter regions of CroR-dependent genes were constructed by using pCI3340 (27). First, the transcriptional terminator, multiple-cloning site, and lacZ gene of pCJK4 (28) were amplified and introduced into pCI3340, generating pSLK234. Subsequently, the region upstream of OG1RF_12500 was amplified (350 bp upstream of the start codon) and cloned into pSLK234 to generate pSLK235. Because a lacZ fusion with the promoter region of croR already existed (pCJK106) (19), the transcriptional terminator, croR promoter region, and lacZ gene were amplified from pCJK106 and inserted into pCI3340, generating pJLL170. The lacZ-promoter fusions in the pCI3340 backbone gave a larger range of beta-galactosidase activity in E. faecalis cells compared to pCJK4 derivatives.
Construction of E. faecalis mutants.
In-frame deletion mutants in various E. faecalis strains were constructed by using markerless allelic exchange, as previously described (28). Mutant alleles were constructed and introduced into pCJK218 by using either a BsaI-based seamless cloning strategy with primer-encoded restriction sites or Gibson assembly (26). Each deletion allele retains codons at the 5′ and 3′ ends of the gene in an attempt to avoid perturbing the expression of adjacent genes (2 to 10% of the gene remains). All mutant strains were constructed independently at least twice for analysis.
Antibiotic susceptibility determinations and growth curves.
The MIC for ceftriaxone was determined by using a microtiter plate serial dilution method as described previously (12). Briefly, bacteria from stationary-phase cultures in MHB (plus 10 μg/ml Em for plasmid-carrying strains) were inoculated at a cell density of ∼105 CFU/ml into wells containing 2-fold serial dilutions of ceftriaxone. Plates were incubated in a Bioscreen C plate reader at 37°C for 24 h with brief shaking before each measurement. The optical density at 600 nm (OD600) was determined every 15 min, and the lowest concentration of antibiotic that prevented growth was recorded as the MIC.
Sample collection for Phos-tag SDS-PAGE. (i) Stimulation of phosphorylated CroS/R by various stressors.
Stationary-phase cultures of SB35(pSLK134) were diluted to an OD600 of 0.01 in fresh MHB supplemented with Em and a subinhibitory concentration of various stressors: ceftriaxone (128 μg/ml), ampicillin (4 μg/ml), vancomycin (1.25 μg/ml), bacitracin (30 μg/ml), hydrogen peroxide (1.5 mM), or norfloxacin (2 μg/ml). Cultures were grown at 37°C with aeration, and exponentially growing cells were harvested at an OD600 of 0.2 for Phos-tag SDS-PAGE and immunoblot analyses.
(ii) IreK dependency of phosphorylated CroS.
Stationary-phase cultures of SB6(pSLB30) and SB17(pSLB30) were diluted to an OD600 of 0.01 in fresh MHB supplemented with Em and grown at 37°C with aeration. Cultures were split in the exponential phase (OD600 = 0.2), left untreated or treated with 1 mg/ml ceftriaxone, and harvested after 90 min for Phos-tag SDS-PAGE and immunoblot analyses.
Phos-tag SDS-PAGE and immunoblot analyses.
Bacteria were harvested by centrifugation after mixing with an equal volume of a cold ethanol-acetone (1:1) mixture to rapidly kill the bacteria and prevent any further signaling events. Pellets were washed with water and normalized based on the OD600 before lysozyme treatment and lysis with 1× SDS Laemmli sample buffer. Samples were loaded onto 8% SDS-PAGE gels with or without 30 μM Phos-tag and 60 μM MnCl2 (for analysis of CroS) or onto 10% SDS-PAGE gels with or without 20 μM Phos-tag and 40 μM MnCl2 (for analysis of CroR). Gels were run at 4°C at 200 V in Laemmli's buffer system until the dye front reached the bottom of the gel. For better separation of CroS, the Phos-tag gel was run for an additional 45 min (200 V at 4°C). After electrophoresis, gels were soaked in a 5 mM EDTA solution before being soaked in Bjerrum Schafer-Nielsen transfer buffer with SDS (48 mM Tris, 39 mM glycine, 20% methanol, 1.3 mM SDS) and transferred onto a polyvinylidene difluoride (PVDF) membrane using a Bio-Rad Trans-Blot semidry transfer apparatus. Transfer conditions were 15 V for 120 min for the detection of CroS and 12 V for 30 min for the detection of CroR. Membranes were blocked with 5% milk in Tris-buffered saline (TBS). CroS was detected by using antibodies to epitope tags used in this study: a polyclonal rabbit anti-Strep-tag antibody (catalog number A00626; GenScript) or a polyclonal rabbit anti-HA antibody (catalog number 71-5500 [Invitrogen] or ab9110 [Abcam]). CroR was detected by using custom rabbit anti-CroR antiserum. A horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody was used for antigen detection (catalog number G21234; Invitrogen). A subunit of RNA polymerase (RpoA) was detected as a loading control by using custom rabbit anti-RpoA antiserum.
Beta-galactosidase activity assays.
CroS/R-dependent gene expression was monitored by using lacZ fusion reporter plasmids (pJLL170 or pSLK235). Stationary-phase cultures of plasmid-bearing strains were diluted to an OD600 of 0.01 in MHB supplemented with Cm for plasmid selection and cultured to the exponential phase (OD600 = 0.2). Cultures were split and left unstressed or exposed to various concentrations of vancomycin for 30 min before being harvested. Beta-galactosidase activity was measured as previously described (19). Briefly, cells were permeabilized with SDS and chloroform, and β-galactosidase activity was measured by using ortho-nitrophenyl-β-galactoside. Samples were analyzed in triplicate, and experiments were performed a minimum of three times.
Isolation of E. faecalis membranes.
Stationary-phase cultures of SB35(pSLK134) and JL457(pSLK134) were diluted 1:50, grown to exponential phase in 100 ml of hBHI broth supplemented with Em (OD600 = 0.6 to 0.7), and harvested by centrifugation. Samples were resuspended in 50 mM Tris (pH 8.0) plus 1× Halt protease inhibitor cocktail and subjected to bead beating for six rounds of 30 s each at 4°C. Samples were centrifuged (16,000 × g for 3 min at 4°C), and the supernatant was collected. Ultracentrifugation was performed on the whole-cell lysates (100,000 × g for 60 min at 4°C), and the insoluble fractions were resuspended in a solution containing 50 mM Tris (pH 7.2), 25 mM NaCl, and 1× protease inhibitors to be used for kinase/phosphatase assays with recombinant IreK and IreP.
In vitro reactions with E. faecalis membranes and recombinant IreK and IreP.
In vitro IreK and IreP kinase and phosphatase assays were performed with a reaction buffer described previously by Hall et al. (17). Half of the reaction volume was comprised of the insoluble (i.e., membrane) fraction from SB35(pSLK134) or JL457(pSLK134) whole-cell lysates. The soluble kinase domain of IreK or full-length IreP expressed and isolated as previously described (16) was added to the membranes at 7 μM. When included, ATP was added at 7 mM. Reaction mixtures were incubated at 37°C, quenched with SDS loading buffer, and analyzed by Phos-tag SDS-PAGE and immunoblotting.
Isolation of CroS from E. faecalis cells and mass spectrometry analysis.
A stationary-phase culture of JL457(pCJK236) was diluted 1:50 in 500 ml of fresh hBHI medium supplemented with Em. The culture was grown to exponential phase (OD600 = 0.6 to 0.7) and harvested by centrifugation. Bacteria were treated with 5 ml of 5 mg/ml lysozyme and 100 U/ml mutanolysin in 50 mM Tris (pH 8) for 30 min at 37°C, lysed by the addition 1% SDS, and solubilized by incubation at 37°C for 2 h. At room temperature, 2 ml of an immobilized-metal affinity chromatography (IMAC)–Ni-charged slurry (Bio-Rad) was equilibrated and mixed with the lysate for 30 min. The lysate-slurry mix was centrifuged (1,000 × g), and the unbound fraction was collected. The resin was subsequently washed three times with wash buffer (5 ml per wash; wash buffer contained 50 mM Tris [pH 8.0], 300 mM NaCl, 20 mM imidazole, and 1% SDS). Elution was performed with 3 ml elution buffer (wash buffer with 500 mM imidazole). SDS-PAGE analysis was performed on all fractions, and the elution fraction was concentrated by acetone precipitation. The concentrated CroS sample was excised from a Phos-tag SDS-PAGE gel and stored in microcentrifuge tubes before analysis at the Proteomics and Mass Spectrometry Facility at the University of Massachusetts Medical School.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jaime Little for the generous provision of plasmids and strains used in this study and members of the Kristich laboratory for critical review of the manuscript.
This study was supported in part by grants R01 AI081692, OD006447, and R21 AI128219 from the National Institutes of Health (NIH). The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00086-18.
REFERENCES
- 1.Gao R, Stock AM. 2009. Biological insights from structures of two-component proteins. Annu Rev Microbiol 63:133–154. doi: 10.1146/annurev.micro.091208.073214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huynh TN, Noriega CE, Stewart V. 2010. Conserved mechanism for sensor phosphatase control of two-component signaling revealed in the nitrate sensor NarX. Proc Natl Acad Sci U S A 107:21140–21145. doi: 10.1073/pnas.1013081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Willett JW, Kirby JR. 2012. Genetic and biochemical dissection of a HisKA domain identifies residues required exclusively for kinase and phosphatase activities. PLoS Genet 8:e1003084. doi: 10.1371/journal.pgen.1003084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wright DP, Ulijasz AT. 2014. Regulation of transcription by eukaryotic-like serine-threonine kinases and phosphatases in Gram-positive bacterial pathogens. Virulence 5:863–885. doi: 10.4161/21505594.2014.983404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dworkin J. 2015. Ser/Thr phosphorylation as a regulatory mechanism in bacteria. Curr Opin Microbiol 24:47–52. doi: 10.1016/j.mib.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fridman M, Williams GD, Muzamal U, Hunter H. 2013. Two unique phosphorylation-driven signaling pathways crosstalk in Staphylococcus aureus to modulate the cell-wall charge: Stk1/Stp1 meets GraSR. Biochemistry 52:7975–7986. doi: 10.1021/bi401177n. [DOI] [PubMed] [Google Scholar]
- 7.Canova MJ, Baronian G, Brelle S, Cohen-Gonsaud M, Bischoff M, Molle V. 2014. A novel mode of regulation of the Staphylococcus aureus vancomycin-resistance-associated response regulator VraR mediated by Stk1 protein phosphorylation. Biochem Biophys Res Commun 447:165–171. doi: 10.1016/j.bbrc.2014.03.128. [DOI] [PubMed] [Google Scholar]
- 8.Horstmann N, Saldaña M, Sahasrabhojane P, Yao H, Su X, Thompson E, Koller A, Shelburne SA III. 2014. Dual-site phosphorylation of the control of virulence regulator impacts group A streptococcal global gene expression and pathogenesis. PLoS Pathog 10:e1004088. doi: 10.1371/journal.ppat.1004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rajagopal L, Vo A, Silvestroni A, Rubens CE. 2006. Regulation of cytotoxin expression by converging eukaryotic-type and two-component signalling mechanisms in Streptococcus agalactiae. Mol Microbiol 62:941–957. doi: 10.1111/j.1365-2958.2006.05431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin WJ, Walthers D, Connelly JE, Burnside K, Jewell KA, Kenney LJ, Rajagopal L. 2009. Threonine phosphorylation prevents promoter DNA binding of the group B Streptococcus response regulator CovR. Mol Microbiol 71:1477–1495. doi: 10.1111/j.1365-2958.2009.06616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Libby EA, Goss LA, Dworkin J. 2015. The eukaryotic-like Ser/Thr kinase PrkC regulates the essential WalRK two-component system in Bacillus subtilis. PLoS Genet 11:e1005275. doi: 10.1371/journal.pgen.1005275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kellogg SL, Kristich CJ. 2016. Functional dissection of the CroRS two-component system required for resistance to cell wall stressors in Enterococcus faecalis. J Bacteriol 198:1326–1336. doi: 10.1128/JB.00995-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kellogg SL, Little JL, Hoff JS, Kristich CJ. 2017. Requirement of the CroRS two-component system for resistance to cell wall-targeting antimicrobials in Enterococcus faecium. Antimicrob Agents Chemother 61:e02461-. doi: 10.1128/AAC.02461-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Comenge Y, Quintiliani R, Li L, Dubost L, Brouard JP, Hugonnet JE, Arthur M. 2003. The CroRS two-component regulatory system is required for intrinsic beta-lactam resistance in Enterococcus faecalis. J Bacteriol 185:7184–7192. doi: 10.1128/JB.185.24.7184-7192.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kristich CJ, Wells CL, Dunny GM. 2007. A eukaryotic-type Ser/Thr kinase in Enterococcus faecalis mediates antimicrobial resistance and intestinal persistence. Proc Natl Acad Sci U S A 104:3508–3513. doi: 10.1073/pnas.0608742104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kristich CJ, Little JL, Hall CL, Hoff JS. 2011. Reciprocal regulation of cephalosporin resistance in Enterococcus faecalis. mBio 2:e00199-. doi: 10.1128/mBio.00199-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall CL, Tschannen M, Worthey EA, Kristich CJ. 2013. IreB, a Ser/Thr kinase substrate, influences antimicrobial resistance in Enterococcus faecalis. Antimicrob Agents Chemother 57:6179–6186. doi: 10.1128/AAC.01472-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Labbe BD, Kristich CJ. 2017. Growth- and stress-induced PASTA kinase phosphorylation in Enterococcus faecalis. J Bacteriol 199:e00363-. doi: 10.1128/JB.00363-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Djorić D, Kristich CJ. 2015. Oxidative stress enhances cephalosporin resistance of Enterococcus faecalis through activation of a two-component signaling system. Antimicrob Agents Chemother 59:159–169. doi: 10.1128/AAC.03984-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stewart RC. 2010. Protein histidine kinases: assembly of active sites and their regulation in signaling pathways. Curr Opin Microbiol 13:133–141. doi: 10.1016/j.mib.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Casino P, Rubio V, Marina A. 2009. Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction. Cell 139:325–336. doi: 10.1016/j.cell.2009.08.032. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J, Xu Y, Shen J, Luo X, Chen J, Chen K, Zhu W, Jiang H. 2005. Dynamic mechanism for the autophosphorylation of CheA histidine kinase: molecular dynamics simulations. J Am Chem Soc 127:11709–11719. doi: 10.1021/ja051199o. [DOI] [PubMed] [Google Scholar]
- 23.Trajtenberg F, Graña M, Ruétalo N, Botti H, Buschiazzo A. 2010. Structural and enzymatic insights into the ATP binding and autophosphorylation mechanism of a sensor histidine kinase. J Biol Chem 285:24892–24903. doi: 10.1074/jbc.M110.147843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhate MP, Molnar KS, Goulian M, DeGrado WF. 2015. Signal transduction in histidine kinases: insights from new structures. Structure 23:981–994. doi: 10.1016/j.str.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jers C, Kobir A, Søndergaard EO, Jensen PR, Mijakovic I. 2011. Bacillus subtilis two-component system sensory kinase DegS is regulated by serine phosphorylation in its input domain. PLoS One 6:e14653. doi: 10.1371/journal.pone.0014653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 27.Hayes F, Daly C, Fitzgerald GF. 1990. Identification of the minimal replicon of Lactococcus lactis subsp. lactis UC317 plasmid pCI305. Appl Environ Microbiol 56:202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vesić D, Kristich CJ. 2013. A Rex family transcriptional repressor influences H2O2 accumulation by Enterococcus faecalis. J Bacteriol 195:1815–1824. doi: 10.1128/JB.02135-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gold OG, Jordan HV, van Houte J. 1975. The prevalence of enterococci in the human mouth and their pathogenicity in animal models. Arch Oral Biol 20:473–477. doi: 10.1016/0003-9969(75)90236-8. [DOI] [PubMed] [Google Scholar]
- 30.Dunny GM, Brown BL, Clewell DB. 1978. Induced cell aggregation and mating in Streptococcus faecalis: evidence for a bacterial sex pheromone. Proc Natl Acad Sci U S A 75:3479–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Snyder H, Kellogg SL, Skarda LM, Little JL, Kristich CJ. 2014. Nutritional control of antibiotic resistance via an interface between the phosphotransferase system and a two-component signaling system. Antimicrob Agents Chemother 58:957–965. doi: 10.1128/AAC.01919-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.