

ABSTRACT
Methicillin-resistant Staphylococcus aureus (MRSA) is a leading cause of deadly hospital-acquired infections. The discovery of anti-Staphylococcus antibiotics and new classes of drugs not susceptible to the mechanisms of resistance shared among bacteria is imperative. We recently showed that tomatidine (TO), a steroidal alkaloid from solanaceous plants, possesses potent antibacterial activity against S. aureus small-colony variants (SCVs), the notoriously persistent form of this bacterium that has been associated with recurrence of infections. Here, using genomic analysis of in vitro-generated TO-resistant S. aureus strains to identify mutations in genes involved in resistance, we identified the bacterial ATP synthase as the cellular target. Sequence alignments were performed to highlight the modified sequences, and the structural consequences of the mutations were evaluated in structural models. Overexpression of the atpE gene in S. aureus SCVs or introducing the mutation found in the atpE gene of one of the high-level TO-resistant S. aureus mutants into the Bacillus subtilis atpE gene provided resistance to TO and further validated the identity of the cellular target. FC04-100, a TO derivative which also possesses activity against non-SCV strains, prevents high-level resistance development in prototypic strains and limits the level of resistance observed in SCVs. An ATP synthesis assay allowed the observation of a correlation between antibiotic potency and ATP synthase inhibition. The selectivity index (inhibition of ATP production by mitochondria versus that of bacterial ATP synthase) is estimated to be >105-fold for FC04-100.
KEYWORDS: ATP synthase, small-colony variant, Staphylococcus aureus, new target, tomatidine
INTRODUCTION
Antibiotic resistance is now an overwhelming health problem, with estimates reaching 10 million deaths per year worldwide by 2050 if nothing is done to slow down the resistance epidemic and if no new antibiotics are discovered to fight resistant pathogens (1, 2). Most of the antibiotics developed in the last decade are still based on scaffolds identified 60 years ago (3). As an adaptive and evolutionary response to this limited scaffold diversity, bacteria have acquired efficient resistance mechanisms that can be transferred among microbial species (4-6). There are multiple challenges in the discovery of novel bioactive molecules (7), and only a few antibiotics based on new scaffolds reaching new validated molecular targets are present in the current pharmaceutical pipeline (3). We report here the elucidation of the mode of action of tomatidine (TO) and its analogs, which represent a new chemical scaffold targeting the bacterial ATP synthase. Noteworthy, the bacterial ATP synthase is now recognized as a clinically relevant and validated molecular target with the recent FDA approval of bedaquiline, a structurally distinct antitubercular drug having a similar mode of action (8).
Since the bacterial ATP synthase has similarity to the human mitochondrial counterpart, selectivity of drugs based on this molecular target is imperative. Like bedaquiline, which is highly selective for the mycobacterial ATP synthase (9, 10), TO selectively targets species of the Bacillales, namely, Staphylococcus, Listeria, and Bacillus spp. (11–13). Staphylococcus aureus and methicillin-resistant S. aureus (MRSA) are often associated with recurrent and difficult-to-treat hospital- or community-acquired infections (14, 15) and are among the bacterial threats identified by the CDC that require immediate attention (16). MRSA strains are also found in livestock and domestic pets, and transmissions from animals to humans have often been reported (17–19). In addition, staphylococcus infections are also associated with food poisoning (20), similar to several pathogens that belong to the order Bacillales. For example, Listeria monocytogenes and Bacillus cereus can cause gastrointestinal infections as well as food poisoning (21, 22). Though L. monocytogenes infections are relatively rare, they can lead to invasive listeriosis, cause severe symptoms, and be fatal. Persistence of this pathogen in food is a major problem and is associated with transmission to humans (23). Bacillus spp. are well known for their ability to form endospores that can persist in the environment (24). Among pathogenic Bacillus species, Bacillus anthracis can cause anthrax by contact with infected food-producing animals, as well as by direct contact with endospores when it is used as a biological weapon (25). However, S. aureus remains the most clinically prominent pathogen of this group.
Among the mechanisms that allow S. aureus to cause persistent infection is its ability to adopt a slow-growth phenotype, called small-colony variant (SCV). SCVs have been associated with chronic and persistent infections and are often recovered from lungs of patients with cystic fibrosis (CF) or from osteomyelitis, septic arthritis, bovine mastitis, and colonized orthopedic devices (26–29). SCVs possess an impaired respiratory chain, which affects their oxidative metabolism, causes slow growth, and changes the expression of virulence factors (28, 30). The reduced proton motive force (PMF) of SCVs reduces their susceptibility to aminoglycosides, which rely on an active PMF to cross the cell membrane barrier. Respiratory deficiency is often caused by mutation in genes involved in hemin or menadione biosynthesis, which are important components of the respiratory chain (28). SCVs have the ability to produce large quantities of biofilm (31–33) and also persist within nonphagocytic host cells (12, 34, 35), enhancing their ability to survive in the presence of antibiotics and host immune factors. Switching between the normal and SCV phenotypes seems to be part of S. aureus pathogenesis and is a phenomenon that may promote recurrence and chronicity of infections (36). Our laboratory extensively documented the very potent (in the nanomolar range) and selective antibacterial activities of TO against SCVs of Staphylococcus, Bacillus, and Listeria spp. (11–13, 37). We also investigated several structural analogs of TO in order to better understand its structure-activity relationship (37) and identified a promising TO derivative possessing a diamino group replacing the 3β-hydroxyl group of TO (FC04-100) (Fig. 1). In addition to its anti-SCV activity, FC04-100 interestingly gained antibacterial activity against prototypical strains (13). The mode of action of TO and its analogs was clearly associated with the respiratory chain, but their molecular target was unknown until now.
FIG 1.
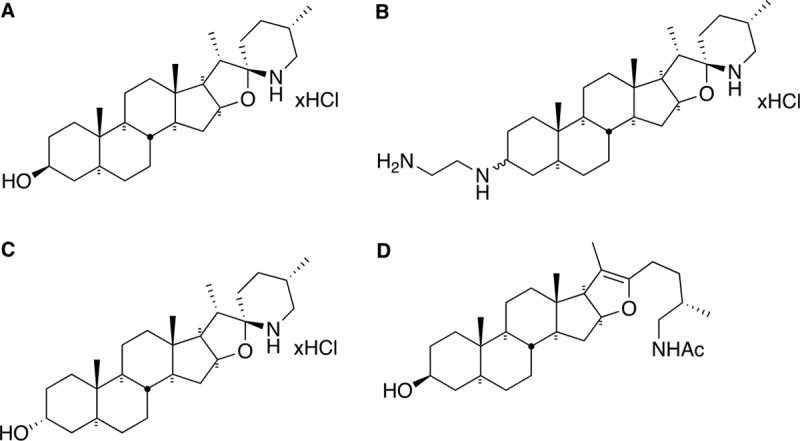
Structures of TO and analogs used in this study. (A) TO is characterized by 6 rings, 12 stereogenic centers, a 3 β-hydroxyl group, and spiro-fused rings in the form of an aminoketal. (B) FC04-100 contains a diamine in position 3. The two epimers in position 3 were separated as major (M) and minor (m) although due to the complexity of nuclear magnetic resonance signals, their respective structures could not be unambiguously assigned. (C) TO analog FC02-190 shows an α-hydroxyl group in position 3. (D) Analog FC04-116 shows an open spiroaminoketal moiety.
The aim of this study was thus to identify the molecular target of TO and its analog FC04-100. We studied drug-resistant S. aureus SCVs raised via selective pressure to TO and used whole-genome sequencing to highlight the molecular target associated with resistance. The subunit C of the bacterial ATP synthase emerged as the molecular target of TO and FC04-100.
RESULTS
Generation and selection of mutants.
In order to identify the molecular target of TO, we raised TO-resistant mutants in an S. aureus SCV strain (Newbould ΔhemB). These mutants were selected by serial passage in broth containing 2-fold dilutions of TO, and three isolated colonies were picked from passages of interest (i.e., showing an increase of the TO MIC). A low-level resistance for TO was observed at passage 7 (isolates named P07intR-1, -2, and -3; P indicates passage and intR indicates intermediate resistance) and similarly at passages 11 (isolates P11intR-1, -2, and -3) and 20 (isolates P20intR-1, -2, and -3), for which the TO MIC increased from 0.06 to 0.25 to 1 μg/ml (Table 1). Isolates with high-level resistance, i.e., a TO MIC of >64 μg/ml, were observed only after 30 passages (isolates named SaR1-1, -2, and -3). These serial passages were performed two more times, and the same stepwise increase in TO resistance was observed, with a high level of resistance reached at passage 30 (isolates named SaR3-1, -2, and -3 and SaR4-1, -2, and -3), but the low-level resistance appeared at passages 15 and 18, respectively. Serial passages were also performed another time starting with a low-level-resistant isolate from the initial passage 7 (i.e., P07intR-1), and these steps also led to high-level resistance at passage 30 (isolates named SaR2, -1, -2, and -3). The mutant descriptions and MICs of TO, FC04-100 stereoisomers (FcM, FC04-100 major stereoisomer; Fcm, FC04-100 minor stereoisomer), and gentamicin are reported in Table 1. Note that we previously reported the MICs of the FC04-100 mixture of stereoisomers to be 8 to 16 and 0.5 to 2 μg/ml for prototypic S. aureus and SCVs, respectively (13, 37). Table 1 shows that FcM is the most potent component of the FC04-100 mixture.
TABLE 1.
Susceptibility profile of the studied S. aureus strains and TO-resistant mutants selected after serial passage in the presence of TO or FC04-100
| Strain or clonesa | MIC (μg/ml)b |
|||
|---|---|---|---|---|
| TO | FcM | Fcm | GEN | |
| S. aureus Newbould | >128 | 2 | 8 | 0.25 |
| S. aureus Newbould ΔhemB | 0.06 | 0.06 | 2 | 8 |
| P07intR-1, -2, and -3* | 0.25 | 0.5–1 | 4 | 8 |
| P11intR-1, -2, and -3* | 0.25–0.5 | 0.5–1 | 4 | 8 |
| P20intR-1, -2, and -3* | 0.25–1 | 1 | 4 | 8 |
| SaR1-1, -2, and -3# | >64 | 2 | 8 | 8 |
| SaR2-1, -2, and -3§ | >64 | 2 | 8 | 8 |
| SaR3-1, -2, and -3# | >64 | 2 | 8 | 8 |
| SaR4-1, -2, and -3# | >64 | 2 | 8 | 8 |
| SaR5-1, -2, and -3† | >64 | 2 | 8–16 | 8–16 |
| SaR6-1, -2, and -3† | >64 | 2 | 8 | 8 |
Clones of S. aureus Newbould ΔhemB are indicated as follows: *, clones selected after 7, 11, or 20 passages, respectively, in the presence of TO; #, clones selected after 30 passages in the presence of TO; §, clones from a clone of S. aureus Newbould ΔhemB with low-level of resistance to TO (P07intR-1) after an additional 30 passages in the presence of TO; †, clones selected after 30 passages in the presence of FC04-100.
TO, tomatidine; FcM, FC04-100 major stereoisomer; Fcm, FC04-100 minor stereoisomer; GEN, gentamicin. For the MIC, the “>” sign denotes the highest concentration tested.
For all mutants that had a high level of resistance to TO and that were raised by selective pressure from TO (SaR1, SaR2, SaR3, and SaR4) (Table 1), cross-resistance was observed with the TO analog FC04-100 (both stereoisomers FcM and Fcm). Resistance to TO increased >1,024-fold (the MIC increased from 0.06 μg/ml to >64 μg/ml), but resistance to FcM increased only 32-fold (from 0.06 to 2 μg/ml) and only 4-fold to Fcm (from 2 to 8 μg/ml) (Table 1). Interestingly, the drug-resistant SCVs selected with FC04-100 (Table 1, mutants SaR5 and SaR6) showed the same susceptibility profiles as those selected with TO (i.e., SaR1, SaR2, SaR3, and SaR4). This strongly suggests similar modes of action for TO and its analog FC04-100 although the latter preserves some noticeable activity against TO-resistant mutants.
Since both FC04-100 stereoisomers (FcM and Fcm) possess some antibacterial activity against prototypic strains of S. aureus (MIC of 2 to 8 μg/ml against strain Newbould) (Table 1), we tried to raise mutants using selective pressure by FC04-100. Using the same approach as for TO, we failed to obtain any FC04-100-resistant mutants from prototypical S. aureus. Indeed, the MIC of FC04-100 remained the same toward strain Newman throughout the 30 passages.
Spontaneous mutation frequency for TO resistance and MPC.
The frequency of spontaneous mutations leading to low-level resistance to TO (MIC of 0.25 to 1 μg/ml; intR strains) (Table 1) was measured by spreading strain Newbould ΔhemB on agar supplemented with TO at 0.5 μg/ml. Using this selection pressure, the mutation frequency was 1.25 × 10−8 to 1.96 × 10−8. Using higher concentrations of TO in selective plates (16 and 32 μg/ml of TO), the mutation frequency dropped and ranged from <5.88 × 10−9 to 2.32 × 10−8. There were no more colonies detected on plates containing 64 μg/ml of TO, i.e., a concentration equivalent to the MIC of the high-level-resistant mutants (Table 1). The mutant prevention concentration (MPC) of TO is thus 64 μg/ml, i.e., the lowest drug concentration preventing mutant colonies, as observed on both technical and biological triplicates.
The frequency of spontaneous mutations leading to FcM resistance, as determined on plates containing 2 and 4 μg/ml FcM, ranged from <5.88 × 10−9 to 1.79 × 10−8. The MPC of FcM was 8 μg/ml. For comparison, we used rifampin as a reference antibiotic. The frequency of spontaneous mutations leading to rifampin resistance on plates containing 8 μg/ml of rifampin (initial MIC of 0.015 μg/ml) was 0.48 × 10−7 to 1.04 × 10−7. The mutation frequency for high-level resistance to rifampin was greater than that observed for low- and high-level resistance to TO or FcM.
Identification of mutations associated with resistance.
Whole-genome sequences of the first series of resistant mutants (P07intR-1, -2, and -3; P11intR-1, -2, and -3; P20intR-1, -2, and -3; and SaR1-1, -2, and -3) (Table 1) were compared to the sequence of the parental SCV strain. For each of these 12 isolates, between 22 and 87 single nucleotide variants (SNVs) and indels were identified. Only the SNVs and indels common to each of three isolates of a given passage were considered to be associated with resistance. This way, we identified only two mutations shared by these independently selected resistant isolates (see Fig. S1 and S2 in the supplemental material). A base substitution (G to T in position 446) was found in the low-level TO-resistant strains (represented by isolates from passages 7, 11, and 20) in the ccpA gene, which encodes the catabolite control protein A, a key metabolic regulator in low-GC Gram-positive bacteria. This base substitution led to an amino acid change (G149V) (Fig. S1). The second mutation, found in high-level TO-resistant isolates (i.e., SaR1-1, -2, and -3 from passage 30), is a base substitution (G to T in position 49) in the atpE gene which encodes the ATP synthase subunit C. Subunit C forms the rotor which turns when a proton flux enters the ATP synthase to produce the energy necessary to synthesize ATP (38). This mutation also leads to an amino acid change (A17S) (isolates SaR1-1, -2, and -3) (Fig. S2). Mutations in ccpA and/or atpE were confirmed by PCR amplification and Sanger sequencing for all isolates submitted to whole-genome sequencing (i.e., P07intR-1, -2, and -3; P11intR-1, -2, and -3; P20intR-1, -2, and -3; and SaR1-1, -2, and -3).
Following these results, other independent serial passages in the presence of TO were performed with S. aureus Newbould ΔhemB, also starting with an isolate with low-level TO resistance (i.e., P07int-R-1, an isolate from the initial passage 17), as described in Materials and Methods. These serial passages yielded high-level TO-resistant isolates at passage 30. PCR amplification and Sanger sequencing for the ccpA and atpE target genes were performed on these isolates. The same base substitution in the ccpA gene was found in all new high-level TO-resistant isolates selected in the presence of TO (SaR2, SaR3, and SaR4; three isolates each) (Fig. S1), which confirmed the involvement of this gene in TO resistance. For the atpE gene, a different base substitution was found in high-level TO-resistant isolates (C77T) which led to a different amino acid change (S26L) than that initially found in the SaR1 series of isolates (SaR2, SaR3, and SaR4; three isolates each) (Fig. S2).
Genes ccpA and atpE were also examined for drug-resistant isolates selected by serial passages with the TO analog FC04-100. For this series of isolates (SaR5 and SaR6; three isolates each), no mutation was found in the ccpA gene (Fig. S1), consistent with the fact that only one level of resistance was found for the analog (Table 1). For the atpE gene, two additional and different base substitutions were found (G52T and T139C). These two mutations also led to different amino acid changes (respectively, G18C and F47L) (Fig. S2). Overall, low-level resistance to TO (MIC of 0.25 to 1 μg/ml) was associated with a specific substitution in CcpA (G149V), whereas high-level resistance to TO (MIC of >64 μg/ml) was associated with both the CcpA substitution and a substitution in AtpE (either A17S or S26L). In contrast, the G18C and F47L substitutions in AtpE were associated only with the highest level of resistance to FC04-100 (MIC of 2 μg/ml for FcM).
The atpE gene is conserved among Bacillales.
TO and its analog possess an activity spectrum specific to the order Bacillales (13), which includes pathogens like S. aureus, Bacillus anthracis, and Listeria monocytogenes. We thus compared the ATP synthase subunit C (AtpE) amino acid sequences among Bacillales and nontargeted species (Fig. 2).
FIG 2.

Amino acid sequence alignments of the ATP synthase subunit C for selected species. First, the alignments for several species of Bacillales present a consensus sequence, highlighted in green. Amino acids additionally identical to those of S. aureus are highlighted in yellow. Amino acids mutated in TO/FC04-100-resistant mutants are in red and bold characters in the S. aureus NCTC 8325 sequence. Also shown below the Bacillales consensus sequence are the alignments for some bacterial species not targeted by TO. The changes in amino acids found in TO/FC04-100-resistant S. aureus (SaR1 to SaR6) are indicated below the alignments. The essential ion-binding glutamate (aspartate in E. coli) is indicated in bold black. Changes in amino acids reported for the bedaquiline-resistant strains of Mycobacterium tuberculosis or Mycobacterium smegmatis (MyR denotes a mixture of these two species) are also indicated below the alignments (40).
Additionally, using blastp and a comparison to the reference strain S. aureus NCTC 8325, we found that AtpE sequences are highly similar, with 100% identity for most of the Staphylococcus spp. (S. aureus, Staphylococcus epidermidis, Staphylococcus pasteuri, and Staphylococcus warneri), 96% for Staphylococcus saprophyticus, 94% for two other coagulase-negative staphylococci (Staphylococcus haemolyticus and Staphylococcus lugdunensis), and 83% for pathogenic bacilli (B. anthracis and B. cereus). The lowest identity was found to be 75 to 79% for Bacillus subtilis, Bacillus coagulans, and Listeria spp. (L. monocytogenes and Listeria ivanovii). The exception was Bacillus pseudofirmus, which shared only 52% identity (data not shown). Additionally, comparison of >1,000 strains of S. aureus and S. epidermidis using blastp revealed 100% identity in their ATP synthase subunit C amino acid sequences (data not shown). Interestingly, amino acids found to be mutated in high-level TO/FcM-resistant S. aureus mutants were conserved in all Bacillales, except S26 in B. coagulans (Fig. 2). Some sequences conserved around these amino acids (Fig. 2, green boxes) are not conserved in nontarget species (e.g., Streptococcus pneumoniae, Escherichia coli, Mycobacterium spp., and Homo sapiens), as shown in Fig. 2. The conserved consensus sequence (in boldface) for Bacillales (B. pseudofirmus not included) is LXXXAAAIAXGLXALGAGIGNGLIVXXTXEGXARQPEXXXXLXXXMFXGXXLVEALPIIXVVIAF. BLAST analysis of that consensus sequence identified all Bacillales. This consensus sequence, where mutations associated with high-level resistance to TO and its analog are found, could explain the specificity of the antibacterial activity of these drugs for Bacillales and may point to the interaction site(s) between these molecules and subunit C.
In contrast, nontarget species presented low identity for the AtpE sequence in comparison with that of S. aureus NCTC 8325. The highest similarity was found with Mycobacterium tuberculosis, having only 55% identity, while the lowest was with S. pneumoniae (31%). There was no significant identity match with the human sequence (Fig. 2). Again, the absence of the Bacillales conserved consensus sequence in the nontarget species may explain the absence of activity of TO against these species. Bedaquiline, now approved for the treatment of Mycobacterium tuberculosis infections (39), is an ATP synthase subunit C inhibitor specific to Mycobacterium (10). Also shown in Fig. 2 for comparison are mutation sites (not all presented here) in Mycobacterium isolates which provide resistance to bedaquiline (40). Interestingly, similar to the role of mutations in TO resistance, any of these distinct mutations in Mycobacterium can provide resistance to bedaquiline (see MyR amino acid changes illustrated in Fig. 2 compared to the SaR mutations recorded in the present study).
Genetic manipulation of atpE.
Since the target species of TO (i.e., the Bacillales) share a consensus sequence in the AtpE protein (Fig. 2, green boxes), we performed genetic manipulations in S. aureus and B. subtilis to validate the role of AtpE in TO resistance. First, overexpression of the atpE gene in the TO- and FcM-susceptible S. aureus ΔhemB background provided resistance to these agents up to the level seen in the non-SCV strain Newbould (Table 2, Newbould ΔhemB atpE versus the empty vector control). This further suggests that TO and FcM share a target and that overexpression of that target reverses susceptibility of S. aureus ΔhemB.
TABLE 2.
Susceptibility profile of genetically manipulated S. aureus and B. subtilis strains
| Strain | MIC (μg/ml)a |
||
|---|---|---|---|
| TO | FcM | TO (+HQNO)b | |
| S. aureus Newbould | >64 | 2 | 0.12 |
| S. aureus Newbould ΔhemB/empty vector | 0.06 | 0.06 | 0.06 |
| S. aureus Newbould ΔhemB atpE | >64 | 8 | ND |
| S. aureus Newbould ΔhemB SaR5 | >64 | 2 | >64 |
| B. subtilis 168 | >64 | ND | 0.12 |
| B. subtilis 168 with SaR5 atpE mutation | >64 | ND | >64 |
TO, tomatidine; FcM, FC04-100 major stereoisomer; ND, not determined. For the MIC, the “>” sign denotes the highest concentration tested.
HQNO was added at a fixed concentration of 10 μg/ml during the susceptibility test.
Furthermore, introducing the mutation found in the atpE gene of one of the high-level TO-resistant S. aureus mutants (e.g., SaR5) in the Bacillus atpE gene provided resistance to TO mixed with the electron transport chain inhibitor 4-hydroxy-2-heptylquinoline-N-oxide (HQNO) (Table 2). Indeed, we used HQNO to reveal the resistance phenotype in the non-SCV strain B. subtilis 168. We have shown previously that wild-type S. aureus strains exposed to HQNO behave like SCVs and become hypersensitive to TO (12). Here, we show that B. subtilis, possessing the mutation of S. aureus SaR5 in atpE, remains resistant to TO in the presence of HQNO (MIC > 64 μg/ml), as opposed to the result seen with B. subtilis carrying the wild-type atpE sequence (MIC of 0.12 μg/ml). This shows that the sole transfer of the SaR5 mutation from S. aureus to B. subtilis provides TO resistance.
ATP synthase subunit C model.
The impact of the S. aureus mutations was investigated by building a model structure of the monomeric subunit C of strain Newbould ΔhemB and of a dodecameric assembly of subunit C using SWISS-MODEL (41), built on homology with PDB accession numbers 3ZO6 and 1WU0. Figure 3A shows the positions of the four amino acids mutated in TO/FcM-resistant isolates (Ala17, Gly18, Ser26, and Phe47) (see also Fig. S2). Each of these amino acids is a putative binding/interaction site for TO and/or its analog and is near the essential ion-binding site amino acid Glu54, suggesting that compound binding could interfere with the function of amino acid Glu54. Figure 3B to E show the configuration of the mutated amino acids, which are all individually associated with high-level resistance. It can be hypothesized that each mutated amino acid affects TO/FcM binding or interaction with the ATP synthase subunit C or allows proton transfer even if TO/FcM is bound. Figure 3F and G present the dodecameric assembly of subunit C of a nonresistant strain, and Fig. 3H to K present mutated amino acids Ser17 (SaR1), Cys18 (SaR5), Leu26 (SaR2, -R3, and -R4), and Leu47 (SaR6) in the dodecameric assembly, respectively. Amino acid Leu26, in comparison to Ser26, clearly appears exposed at the surface of the subunit C in the internal portion of the assembly, and Leu47, in comparison to Phe47, appears exposed in the external portion. On the other hand, Ser17 and Cys18 are located between the subunits, which may affect assembly integrity although Ser17 in mutant SaR1 had the lowest impact on the multimeric model.
FIG 3.

Monomeric and multimeric models of ATP synthase subunit C built, respectively, on homology with templates of PDB accession numbers 1WU0 and 3ZO6, using the SWISS-MODEL server. (A) Position of amino acids (in white) implicated in high-level TO resistance in the wild-type polypeptide. Essential amino acid Glu54 is in yellow. (B) Position of Ser17 mutation in SaR1. (C) Position of Cys18 mutation in SaR5. (D) Position of Leu26 mutation in SaR2, SaR3, and SaR4. (E) Position of Leu47 mutation in SaR6. (F) Overview of the multimeric assembly of ATP synthase subunit C. (G) Position of amino acids (Ala17, red; Gly18, green; Ser26, cyan; Phe47, magenta) implicated in resistance in the wild-type multimeric assembly. Glu54 is also shown in yellow. (H) Position of the Ser17 mutation in the multimeric assembly in SaR1. (I) Position of the Cys18 mutation in the multimeric assembly in SaR5. (J) Position of the Leu26 mutation in the multimeric assembly in SaR2, SaR3, and SaR4. (K) Position of the Leu47 mutation in the multimeric assembly in SaR6. The models were drawn using PyMOL software (version 1.8.7.0; DeLano Scientific, San Francisco, CA).
Inhibition of ATP synthesis in prototypic and SCV strains.
Following genomic investigation, we hypothesized that TO and FC04-100 would target the C subunit of the ATP synthase and predicted that those molecules would inhibit ATP synthesis by S. aureus Newbould ΔhemB. To test this hypothesis, we established an ATP synthesis assay using inverted membrane vesicles derived from either strain Newbould or its SCV counterpart. In this assay, the 50% inhibitory concentrations (IC50s) of known ATP synthase poisons (N,N′-dicyclohexylcarbodiimide [DCCD], cyanide m-chlorophenylhydrazone [CCCP], and oligomycin) were similar to those reported in the scientific literature (9, 38) and ranged from 0.82 ± 0.17 μg/ml to 8.67 ± 1.9 μg/ml for membrane vesicles from S. aureus (prototypical or SCV strains), as well as for isolated mitochondria collected from pulmonary Calu-3 cells (Table 2).
As shown in Fig. 4A (and in more detail in Table 3), TO and FcM possess similar IC50s for the S. aureus SCV ATP synthase (18.5 ± 1.9 and 18.9 ± 3.6 μg/ml, respectively). Accordingly, the less potent FC04-100 enantiomer (Fcm; MIC of 2 μg/ml for the SCV strain) demonstrated a higher IC50 in the assay (47.0 ± 9.6 μg/ml). Other TO analogs with low or no antibiotic activity were also tested as additional comparators in the assay. Hence, the 3α-hydroxyl enantiomer of TO, FC02-190 (TO is 3β), which possesses weaker antibacterial activity against S. aureus SCV (MIC of 8 μg/ml), had a higher IC50 (85.1 ± 7.0 μg/ml). Furthermore, no inhibitory activity against whole cells (MIC > 128 μg/ml) or inhibition of ATP synthesis was observed (IC50 of >1,024 μg/ml) for a TO analog possessing an open spiroaminoketal moiety, FC04-116 (Fig. 4A). A correlation between structure, MIC, and IC50 thus emerges for the S. aureus SCV (Fig. 4C), demonstrating that simply having a steroidal backbone is not sufficient to inhibit ATP synthesis. The orientation of the position 3 group and an intact spiroaminoketal moiety in TO and its analogs are important for ATP synthase inhibition and whole-cell inhibitory activity (MIC). Also noteworthy, tomatine (the 3β-glycosylated form of TO), which is devoid of inhibitory activity against S. aureus SCVs (12), does not inhibit ATP production in this assay (Table 3), showing that extensive modification of the 3β-hydroxyl group of TO is deleterious for antibacterial effect against both whole bacterial cells and isolated membrane vesicles. Other structurally unrelated antibiotics had no inhibitory activity against staphylococcal ATP synthase (e.g., levofloxacin, a fluoroquinolone, or gentamicin, an aminoglycoside antibiotic) (Table 3), including bedaquiline (a diarylquinoline), a specific inhibitor of the Mycobacterium ATP synthase also targeting subunit C (IC50 of >1,028 μg/ml) (Fig. 4A and Table 3). This reinforces the idea that bacterial ATP synthases are sufficiently distinct from each other to respond to specific inhibitors, as suggested by the analysis of their amino acid sequences (Fig. 2). Similarly, neither bedaquiline (9) nor TO and its analogs (Table 3 and Fig. 4B) display any inhibitory activity toward ATP production by mitochondria, an important feature for ultimate use in higher species. Extrapolation of IC50 data points for mitochondria (Table 3) suggests a selectivity index (SI; IC50 for mitochondria/IC50 for bacterial ATP synthase) of >105 for TO analogs. In contrast, typical ATP synthase inhibitors, such as DCCD and oligomycin, are nonselective (SI of ≤1).
FIG 4.

Effect of TO and analogs on the ATP synthase activity of S. aureus. (A) Relative ATP synthase activity of the SCV ΔhemB strain in the presence of various inhibitors. The control (CTRL) represents the maximal ATP production in the absence of ATP synthase inhibitor whereas the assay performed without addition of NADH represents the minimal value. nd, not determined. (B) Relative ATP synthase activity of the SCV ΔhemB atpE mutants (SaR1, SaR4, SaR5, and SaR6) in the presence of TO. The effects of TO on the parental ΔhemB strain and the prototypical strain Newbould (WT) and on human mitochondria are also shown for comparison. (C) Correlation between TO and analogs (FcM, Fcm, FC02-190, and FC04-116). Log2 MICs and the log10 IC50s were determined in the ATP synthase assay using S. aureusΔhemB membrane vesicles. (D) ATP production (relative light units, RLU) by membrane vesicles prepared from the SCV ΔhemB atpE mutants. ATP production by the parental ΔhemB strain and the prototypical strain Newbould (WT) is also shown. In panel D, letters shared between or among the groups indicate no significant difference.
TABLE 3.
MIC and inhibition of ATP synthesis (IC50) by TO, TO analogs, and reference compounds for different bacterial strains and mitochondriaa
| Compound | ΔhemB strain |
Newbould |
E. coli |
IC50 (μg/ml) for mitochondria | |||
|---|---|---|---|---|---|---|---|
| MIC (μg/ml) | IC50 (μg/ml) | MIC (μg/ml) | IC50 (μg/ml) | MIC (μg/ml) | IC50 (μg/ml) | ||
| TO | 0.06 | 18.5 ± 1.9 | >128 | 95.5 ± 13.7 | >128 | 264.73 ± 30.8 | >1,024 |
| FcM | 0.06 | 18.9 ± 3.6 | 2 | 40.2 ± 13.8 | 32 | 102.42 ± 16.6 | >1,024 |
| Fcm | 2 | 47.0 ± 9.6 | 8 | 111.0 ± 11.3 | 64 | 223.09 ± 45.9 | >1,024 |
| FC02-190 | 8 | 85.1 ± 7.0 | >128 | >1,024 | >128 | >1,024 | >1,024 |
| FC04-116 | >128 | >1,024 | >128 | >1,024 | >128 | >1,024 | >1,024 |
| DCCD | NDb | 1.44 ± 0.54 | ND | 1.32 ± 0.32 | ND | 1.62 ± 0.54 | 0.82 ± 0.17 |
| CCCP | ND | 1.46 ± 0.27 | ND | 6.11 ± 1.1 | ND | 2.80 ± 1.01 | 7.19 ± 2.23 |
| Oligomycin | ND | 8.67 ± 1.9 | ND | 9.41 ± 1.5 | ND | 5.63 ± 1.23 | 5.91 ± 0.82 |
| Tomatine | >128 | >1,024 | >128 | >1,024 | >128 | >1,024 | >1,024 |
| Bedaquiline | >128 | >1,024 | >128 | >1,024 | >128 | >1,024 | >128 |
| Gentamicin | 8 | >1,024 | 0.25 | >1,024 | 1 | >1,024 | >1,024 |
| Levofloxacin | 0.25 | >1,024 | 0.25 | >1,024 | 0.03 | >1,024 | >1,024 |
The “>” sign denotes the highest concentration tested.
ND, not determined.
Additionally, we investigated wild-type S. aureus membrane vesicles to examine the influence of a fully functional respiratory chain on TO and TO analog activities. Similarly, we used wild-type E. coli membrane vesicles as a comparator. Table 3 shows that even though TO does not possess noticeable antibacterial activity against either wild-type S. aureus or E. coli, some inhibition of ATP synthesis is observed in this cell-free assay. The IC50 of TO was, however, much higher and demonstrated 5 to 14 times less inhibitory activity in the ATP synthesis assay against wild-type S. aureus and E. coli, respectively, than S. aureus SCVs. The most active TO analog was FcM, with a MIC of 2 μg/ml and IC50 of 40.2 ± 13.8 μg/ml against wild-type S. aureus Newbould and a MIC of 32 μg/ml (IC50 of 102.4 ± 16.6 μg/ml) against E. coli (Table 3).
ATP synthesis by S. aureus SCV mutants.
Next, we investigated whether mutations resulting in TO resistance had an impact on ATP synthesis. As expected because of an impaired respiratory chain in the SCV ΔhemB strain, ATP production by this strain in the membrane vesicle assay was significantly reduced compared to that produced by the prototypic strain Newbould (Fig. 4D). Interestingly, ATP production by the mutants SaR1, SaR4, SaR5, and SaR6 (previously described in Fig. S2) was further reduced compared to that of the ΔhemB parental strain (Fig. 4D). These results indicate that the mutations in atpE leading to high-level TO resistance further impair the ability of the SCV ΔhemB strain to produce ATP.
As mentioned earlier, the MIC of TO increased from 0.06 μg/ml to >64 μg/ml for the high-level-resistant mutants (Table 1). As expected for an altered drug target, the IC50s of TO, its 3α-hydroxyl analog (FC02-190), and Fcm increased drastically using the mutant inverted membrane vesicles in this assay (IC50 of >512 μg/ml against all mutants) (Fig. 4B and Table 4). Overall, the mutations in the AtpE subunit clearly had an impact on the resistance of whole cells and tolerance to ATP synthesis inhibition in the membrane vesicle assay. However, FcM, which possesses a residual MIC of 2 μg/ml against all mutants (initial MIC of 0.06 μg/ml against the parental ΔhemB strain), also demonstrated some inhibitory activity in the ATP synthesis assay against SaR1 and SaR5, with IC50s of 238 and 110 μg/ml, respectively. SaR1 and SaR5 were the most fit mutants in terms of ATP production (Fig. 4D). Interestingly, the multimeric model of subunit C (Fig. 3) showed that, in contrast to the mutations in SaR1 (Ala17Ser) and SaR5 (Gly18Cys), mutations in SaR4 (Ser26Leu; identical to SaR2 and SaR3) and R6 (Phe47Leu) were those protruding from the internal or external portion of the assembly, respectively, possibly better preventing binding of TO and analogs to the multimeric structure of subunit C and further reducing ATP production (Fig. 4D).
TABLE 4.
MIC and selective inhibition of ATP synthesis (IC50) by TO and TO analogs for different TO-resistant mutantsa
| Compound | SaR1-1 |
SaR4-1 |
SaR5-1 |
SaR6-1 |
||||
|---|---|---|---|---|---|---|---|---|
| MIC (μg/ml) | IC50 (μg/ml) | MIC (μg/ml) | IC50 (μg/ml) | MIC (μg/ml) | IC50 (μg/ml) | MIC (μg/ml) | IC50 (μg/ml) | |
| TO | >64 | >512 | >64 | >512 | >64 | >512 | >64 | >512 |
| FC02-190 | >64 | >512 | >64 | >512 | >64 | >512 | >64 | >512 |
| FcM | 2 | 238 ± 22.1 | 2 | >512 | 2 | 110 ± 9.8 | 2 | >512 |
| Fcm | 8 | >512 | 8 | >512 | 8 | >512 | 8 | >512 |
The “>” sign denotes the highest concentration tested.
DISCUSSION
The phytoanticipin α-tomatine, a sterol glycol-alkaloid, possesses antifungal activity and protects solanaceous plants against phytopathogens (42). It was reported that α-tomatine disrupts eukaryote cell membranes by forming complexes with ergosterol and cholesterol (42, 43). On the other hand, TO, the aglycone version of α-tomatine, loses inhibitory activity against most phytopathogens (42) and does not possess sterol binding activity (43). In Saccharomyces cerevisiae and several protozoans, however, it was reported that TO perturbs ergosterol biosynthesis by targeting Erg6 (C-24 sterol methyltransferase) and also of Erg4 (C-24 sterol reductase), two enzymes absent in mammals and most bacteria (44–47). Such findings in fungi and protozoans suggest a different mode of action for the potent and specific antibacterial activity of TO against SCVs of the Bacillales (13). Noteworthy, TO activity is equivalent for SCVs that are auxotrophs for either hemin or menadione (12, 13) or even thymidine (unpublished data). This suggests that TO targets a common weakness among SCVs. In the present study, we examined the genomes of in vitro-generated TO-resistant S. aureus SCVs to identify mutations in genes associated with resistance and identified the bacterial ATP synthase subunit C (encoded by atpE) as their cellular target.
ATP synthase is a ubiquitous enzyme that utilizes energy stored in the transmembrane electrochemical gradient to synthesize ATP (48). However, it essentially differs across species (49, 50). Little is known about the S. aureus ATP synthase; however, it was shown to be important for growth and survival (38, 51), reinforcing the hypothesis that mutations in atpE need to allow some level of functionality. For other Bacillales like L. monocytogenes, it was reported that ATP synthase also plays a role in pH homeostasis (52) and that deletion of ATP synthase in B. subtilis greatly affects growth (53). Our previous work showed that TO and its analog FC04-100 possess a narrow yet specific spectrum of activity against the SCVs of Bacillales (13). We show here that specificity could be explained by the presence of conserved amino acid sequences in the ATP synthase subunit C across species of the Bacillales order. This also indicates that the cellular function(s) of ATP synthase is of primary importance for survival in Bacillales, at least in the SCV background which possesses an altered respiratory chain. Previous studies revealed that extensive metabolic remodeling occurs in persistent bacteria such as SCVs (54–56), during sporulation in Bacillus spp. (57), and in slow-growing bacteria and biofilm-producing bacteria (33, 58, 59). It has been hypothesized that targeting energy metabolism is of interest for the development of antibiotics (60). Also, Balemans et al. (38) showed that targeting the Gram-positive ATP synthase is a possible approach toward the discovery of new antibacterial agents.
The subunit C of bacterial ATP synthase is a clinically validated antimicrobial target since the novel antibiotic bedaquiline, which targets subunit C of the ATP synthase of M. tuberculosis, was recently approved for the treatment of tuberculosis (10, 39). Hards et al. (61) proposed that bedaquiline binds to subunit C and perturbs the interface between subunits A and C, leading to a dysfunctional proton cycle that stops ATP synthase function. In order to allow the translocation of protons, subunits A and C must move relative to each other. The proton transfer chain, elucidated in E. coli (62) and in M. tuberculosis (63), possesses two conserved amino acids of interest: Glu61 of subunit C in M. tuberculosis (Asp61 in E. coli and Glu54 in S. aureus) that transfers the proton it receives to R186 of subunit A (R210 in E. coli and presumably R174 in S. aureus). Further functional assays and structural models showed that bedaquiline interacts with the ATP synthase subunit C by several means and covers the c-rotor ion-binding site, preventing the proton cycle and ATP synthase activity (64). The ATP synthase subunit C model presented in Fig. 3 indicates the position of mutated amino acids that give high-level resistance to TO and some degree of resistance to its analog FcM. It also represents putative binding/interaction sites that could impact the nearby essential Glu54 amino acid. TO could also interact with the essential glutamate, similarly to the mechanism of action of bedaquiline. In that case, similarly to bedaquiline (40, 65), any of the mutated amino acids found in TO-resistant strains could modify the structure of ATP synthase subunit C in a manner that prevents TO binding or proton transfer even in the presence of bound TO. Models of the ATP synthase subunit C (Fig. 3) also showed that two of the four identified mutations leading to high-level TO resistance (in SaR2, -R3, and -R4, Ser26Leu; and in SaR6, Phe47Leu) showed protruding amino acids internal or external to the assembly, whereas the other two mutated amino acids reside between the subunits (in SaR1, Ala17Ser; in SaR5, Gly18Cys), while sustaining a higher level of ATP production and keeping some degree of sensitivity to FcM (i.e., measurable IC50) compared to that seen with mutations in SaR4 and SaR6. It is possible that mutations in SaR4 and SaR6 block binding of the inhibitors, whereas those in SaR1 and SaR5 compensate to allow better proton translocation despite binding. Further structural studies are required to elucidate the resistance mechanism.
The proton motive force is the weakness of SCVs (28). The proton motive force normally generated by a strong electron transport chain is used by the bacterial ATP synthase to generate ATP (66). Interestingly here, all low-level TO-resistant SCVs showed a mutation in the ccpA gene (which encodes the catabolite control protein A), and this mutation is conserved in sequential clones possessing high-level resistance to TO with further mutation in atpE. CcpA is a highly conserved carbon catabolite regulator in low-GC Gram-positive bacteria and has been intensively studied in B. subtilis (67–69), S. xylosus (70), and S. aureus (71, 72). It functions as a catabolite activator or repressor that allows bacteria to use preferential carbon sources over secondary carbon sources, as in the presence of glucose. CcpA can also enhance glycolysis and inhibit the tricarboxylic acid (TCA) cycle. As such, a mutation in ccpA would be expected to affect the formation of substrates for the respiratory chain and, consequently, the proton gradient and could potentially moderately compensate for the consequences of the ATP synthase inhibition by TO.
The ability of TO and semisynthetic derivatives to target S. aureus SCVs may lead to important clinical applications. Indeed, it is well known that cystic fibrosis (CF) patients are highly susceptible to colonization by this bacterial phenotype. Indeed, Pseudomonas aeruginosa and Staphylococcus aureus very often cocolonize the lungs of CF patients (73), and it was previously demonstrated that the P. aeruginosa exoproduct 4-hydroxy-2-heptylquinoline-N-oxide (HQNO) promotes the emergence of S. aureus SCVs and also increases biofilm formation by prototypical S. aureus (31). Consequently, S. aureus SCVs have been independently associated with worsening lung disease in CF children (27). Since CF patients are often subjected to inhaled aminoglycoside (tobramycin) therapy to control P. aeruginosa infections, a combination treatment with steroidal alkaloids such as TO or FC04-100 appears attractive to tackle all three pathogens: P. aeruginosa (with the aminoglycoside), prototypical S. aureus (with the synergy of the aminoglycoside and steroidal alkaloid [11, 13]), and its SCV phenotype (with the steroidal alkaloid). An analog of TO such as FC04-100 that also possesses activity against prototypic S. aureus strains would certainly be advantageous, especially since it keeps high selectivity against the bacterial ATP synthase, with an SI of at least 105 (inhibition of ATP production by mitochondria versus bacterial ATP synthase).
Here, we show that TO possesses good affinity for the ATP synthase of both prototypic and SCV inverted membrane vesicles but that the relationship between IC50 and whole-cell activity (MIC) correlates well only in SCVs. This may suggest an alternate mode of action for FC04-100 or more specifically FcM, which shows activity against prototypical S. aureus (MIC 2 μg/ml), in contrast to TO, which has no activity despite a good IC50 for the prototypical ATP synthase. However, the simplest explanation could reside in the physical properties of this TO analog. FC04-100 is positively charged and less hydrophobic and more soluble than TO, which may allow FC04-100 to reach its target in prototypical cells knowing that the surface properties of SCVs and prototypical cells are indeed quite different (28). Also noteworthy, the major (FcM) and minor (Fcm) stereoisomers of FC04-100 studied here could not be clearly defined, but FcM was more potent. This was consistent with previous observations which showed that TO, which possesses a 3β-hydroxyl group, is much more potent than its 3α isomer (i.e., FC02-190) (37).
Overall, this study allowed us to identify ATP synthase subunit C as the bacterial cellular target for TO and its analog FC04-100. Specificity of antibacterial activity for Bacillales appears to be explained by the presence of conserved motif sequences in the ATP synthase subunit C. FC04-100 is a possible candidate for the development of this class of antibiotics since resistance development is more difficult and since its potency is increased against prototypic Bacillales strains.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
S. aureus strains Newbould (ATCC 29740) and Newman were used as representatives of the normal phenotype. The S. aureus strain Newbould ΔhemB (ΔhemB strain) was used as the SCV counterpart. The SCV ΔhemB strain was generated from strain Newbould by disrupting the hemB gene with the ermA cassette by homologous recombination (74). Normal-phenotype S. aureus strains were grown in 4-liter flasks at 225 rpm with 1 liter of cation-adjusted Mueller-Hinton broth at 35°C (CAMHB; BD, Mississauga, ON, Canada). Except where otherwise stated, the ΔhemB strain and SCV mutant derivatives were grown in 4-liter flasks at 225 rpm with 1 liter of brain heart infusion (BHI) broth (BD, Mississauga, ON, Canada) at 35°C. The Escherichia coli strain ATCC 25922 was also used for preparation of inverted membrane vesicles (see below).
Chemical reagents and antibiotics.
Tomatidine hydrochloride (TO) was purchased from Molekula (Shaftesbury, Dorset, United Kingdom) or Onbio (Richmond Hill, ON, Canada), and gentamicin was from Sigma (Oakville, ON, Canada). TO and FC04-100 (mixed stereoisomers at position 3) (Fig. 1) were solubilized at 2 mg/ml in dimethyl sulfoxide (DMSO). Gentamicin was solubilized at 10 mg/ml in water. Synthesis of FC04-100 was performed as previously described (37). The two diastereoisomers of FC04-100 (one major [FcM] and one minor [Fcm]) were separated by reverse-phase chromatography using a Waters preparative liquid chromatograph (LC) (Sample Manager 2767 fraction collector), a Binary Gradient Module 2545 with two 515 high-performance LC (HPLC) pumps, a System Fluidics Organizer (SFO), and Photodiode Array Detector 2998 (XSelect charged-surface hybrid [CSH] OBD Prep C18 column; particle size, 5 μm; internal diameter, 19 mm; length, 250 mm) (buffer A, 0.1% HCOOH in H2O; buffer B, 0.1% HCOOH in ACN; flow 20 ml/min). Although separation of the two epimers of FC04-100 was successful, their unambiguous structural assignment remains elusive to this day, including the production of crystals for X-ray crystallography. Mass spectrometry spectra were recorded on a Waters SQ Detector 2 (electrospray) instrument (Mississauga, ON, Canada).
Antibiotic susceptibility testing.
The MICs of drugs yielding no visible bacterial growth were determined by a broth microdilution technique by following the recommendations of the Clinical and Laboratory Standards Institute (75), except that the incubation period was extended to 48 h, and the medium used was BHI broth for SCV strains. In some experiments, the MIC of TO was determined in the presence of the electron transport inhibitor 4-hydroxy-2-heptylquinoline-N-oxide (HQNO; Axxora, San Diego, CA). HQNO was solubilized in DMSO at a concentration of 5 mg/ml and used at a fixed concentration of 10 μg/ml.
Generation of TO-resistant mutants and FC04-100-resistant mutants.
The generation of TO-resistant mutants was elicited by serial passage of S. aureus strain Newbould ΔhemB (30 passages of 48 h each) in a series of 2-fold dilutions of TO (range, 0.06 to 64 μg/ml in BHI broth) in 96-well plates. At each passage, the MIC was determined, and the well representing 0.5× MIC was diluted in fresh broth and used to inoculate (∼106 CFU/ml) a new series of TO dilutions for the next passage. At each passage, the 0.5× MIC well was also plated on BHI agar (BHIA), and the next day, three isolates were collected and frozen. Intermediate TO-resistant mutants were in this way isolated from passages 7, 11, and 20. Following 30 passages, three isolates from the highest TO concentration tested (64 μg/ml) were purified on BHIA supplemented with 32 μg/ml of TO. Such passages and isolate selection were repeated twice and also an additional time starting from an intermediate TO-resistant isolate initially selected on passage 7.
The generation of FC04-100-resistant mutants was similarly provoked by serial passage of strain Newbould ΔhemB on a series of 2-fold dilutions of FC04-100 (range, 0.06 to 64 μg/ml in both BHI broth and CAMHB) in 96-well plates. Three isolates growing at the highest concentration reached after 30 passages (4 μg/ml) were purified on BHIA supplemented with 4 μg/ml of FC04-100 (mixture of the stereoisomers).
The generation of non-SCV FC04-100-resistant mutants was also attempted by serial passage of the prototypic strain Newman on a series of 2-fold dilutions of FC04-100 (range, 0.25 to 128 μg/ml in both BHI broth and CAMHB) in 96-well plates.
Spontaneous mutation frequency and MPC.
S. aureus Newbould ΔhemB from a 20-h agar plate was suspended in 1× phosphate-buffered saline (PBS) to reach a 0.5 McFarland standard (∼1.5 × 108 CFU/ml). Ten milliliters of this suspension was added to 90 ml of BHI broth and incubated for 24 h at 35°C and 250 rpm. One hundred microliters of the culture was plated on each of a series of BHIA plates containing increasing concentrations of TO or FcM. Controls with 8 μg/ml of rifampin and with 3.2% DMSO (diluent for TO and FcM) were also used. Tests were performed three independent times in triplicate. The mutation frequency was calculated by dividing the number of mutant colonies growing after 72 h at 35°C by the initial number of CFU of the plated inoculum. The mutant prevention concentration (MPC) was defined as the lowest concentration where no mutant colonies were observed three independent times and in each of the triplicates.
Whole-genome sequencing, assembly, and annotation.
Whole-genome shotgun libraries (three isolates per passage of interest) were prepared and sequenced using Illumina technology. Briefly, genomic DNA was extracted using a Qiagen QIAamp DNA minikit and fragmented to a size of ∼200 to 400 bp by treatment with the double-stranded DNA (dsDNA) Shearase enzyme (Zymo Research) according to the manufacturer's instructions. Libraries were then prepared according to Rodrigue et al. (76). Illumina sequencing was performed on an Illumina HiSeq 2000 sequencing system at the Plateau de Biologie Moléculaire et Génomique Fonctionnelle of the Institut de Recherche Cliniques de Montréal (Montreal, QC, Canada). Samples were multiplexed in a single sequencing lane, and approximately 1 to 2 million paired-end reads of 50 bp were obtained for each library. The resulting reads were de novo assembled using a Roche gsAssembler, version 2.6. Contig sequences were aligned and annotated using the reference genome of S. aureus strain Newman with ABACAS (77) and RATT (78). Four independently sequenced S. aureus Newbould ΔhemB genomes were first compared to the reference Newman genome to identify and eliminate inherent single nucleotide variants (SNVs) from the subsequent comparison of Newbould ΔhemB to resistant isolates. TO-resistant Newbould ΔhemB sequences were then compared against the parental SCV strain Newbould ΔhemB to identify SNVs associated with resistance.
PCR and Sanger sequencing.
Mutations were confirmed by PCR amplification with specific primers to S. aureus targets genes. The primers atpE-fwd (5′-TTGATTCACCAGCGCGTATTGTC-3′) and atpE-rev (5′-AGGTATCTGCTTCAATCAGCG-3′) were used for atpE gene amplification. Two sets of primers were used for the ccpA gene: ccpAdeb-fwd (5′-CAAGCTTTTCGCTAAATTTTTCC-3′), ccpAdeb-rev (5′-CGCCTTCTTTATAACTTTCAGC-3′), ccpAfin-fwd (5′-ATGGTATTATTTTCCTTGGTGGT-3′), and ccpAfin-rev (5′-GCGAGTTGGTACGAATCTAC-3′). Sanger sequencing was performed using capillary electrophoresis at Plateforme de Séquençage et de Génotypage des Génomes, Centre de Recherche du Centre Hospitalier de l'Universitaire Laval (QC, Canada).
Overexpression of AtpE in Newbould ΔhemB.
Plasmid pCN36 (79), in which the constitutive promoter PblaZ from pCN40 (79) was inserted in front of the multiple cloning site, was used to clone and express the ATP synthase subunit C atpE gene from S. aureus strain Newbould. Plasmid constructs were generated using E. coli DH5a (Invitrogen, Burlington, ON, Canada), restriction enzymes (New England BioLabs [NEB] Inc., Pickering, ON, Canada), and the T4 DNA ligase (NEB). The recombinant plasmid purified from E. coli was then used to transform first the S. aureus RN4220 (restriction-defective) strain and then the final host strain Newbould ΔhemB by electroporation (80). Plasmid constructs were validated by restriction digestion patterns and DNA sequencing.
Transfer of the S. aureus atpE mutation and resistance to B. subtilis.
To insert the mutation found in the atpE gene of S. aureus into B. subtilis strain 168, we used the plasmid pMiniMad2, which bears an erythromycin resistance marker and a temperature-sensitive origin of replication (a kind gift from D. Rudner, Harvard Medical School, Boston, MA) (81). First, the atpE gene from B. subtilis was amplified using the primers BSatpE-fwd (5′-GGTGCTATCCAGGCATTTATC-3′) and BSatpE-rev (5′-TTGTCTCTTTTCAGCCGGCA-3′), and the amplicon was cloned into pMiniMad2 with BamHI and EcoRI (New England Biolab). Then, using a QuikChange II XL site-directed mutagenesis tool (Agilent Technologies Canada, Mississauga, ON, Canada) according to the manufacturer's instructions, oligonucleotide primers containing the mutation found in the atpE gene of one of the high-level TO-resistant S. aureus mutants (SaR5) was used to generate the sequence modification in the B. subtilis atpE gene before transforming XL-Gold Ultracompetent Escherichia coli cells (Agilent). The oligonucleotide primers used were 5′-GGTTTAGGCGCACTTGGTGCATGTATTCGTAACGGTTTGATT-3′ and 5′-CAATCAACCGTTACCAATACATGCACCAAGTGCGCCTAAACC-3′. The transformation of B. subtilis 168 with pMiniMad2 containing the mutated atpE gene was done using natural competence (82). The bacteria having integrated the plasmid were first selected at 40°C using the erythromycin resistance marker. Then, cells were grown at room temperature for 24 h and grown for several passages in LB at 37°C without antibiotic to promote excision and loss of the plasmid. Bacteria that had lost erythromycin resistance were then selected, genomic DNA was extracted (Qiagen genomic DNA extraction kit), and the atpE gene was amplified to confirm the position of the mutation by Sanger sequencing.
Preparation of inverted membrane vesicles.
Bacterial cells were grown as described above to reach an A600 of ∼0.6 to 0.8. Bacteria were harvested by centrifugation at 6,000 × g for 10 min at 4°C, washed, and suspended in KPN buffer (20 mM potassium phosphate, 140 mM NaCl [pH 7.5]). Cells of S. aureus were first treated with lysostaphin (400 μg/ml) for 1 h at 37°C before addition of 3 μg/ml of a protease inhibitor cocktail (Sigma-Aldrich), DNase (6 μg/ml), and RNase (6 μg/ml). After 30 min of treatment, cells were disrupted by a French pressure cell at 16,000 lb/in2, and the bacterial lysates were centrifuged at 6,000 × g for 30 min at 4°C to remove unbroken cells. The supernatant was then centrifuged at 150,000 × g for 40 min at 4°C using a fixed-angle rotor to collect the membranes. The membranes were suspended in a minimal volume of KPN buffer and stored at −80°C. Protein concentration was estimated by a Micro BCA protein assay kit (Thermo Fisher Scientific, Rockford, IL) using bovine serum albumin as a standard.
Measurement of ATP synthesis activity.
The ATP synthesis activity was determined in isolated membrane vesicles from S. aureus by energizing them with NADH and quantifying the amount of ATP produced using the luciferin/luciferase system, similarly to the method described by Balemans et al. (38). Briefly, S. aureus membrane vesicles were diluted to a concentration of 50 μg of protein/ml with 50 mM morpholinepropanesulfonic acid (MOPS), pH 7.5, containing 10 mM MgCl2. The membrane vesicles were preincubated with TO, FC04-100, or comparator molecules under stirring conditions at room temperature for 10 min. Subsequently, 2.5 mM NADH (final concentration) was added, and the mixture was further incubated with vigorous shaking for 1 min. The reaction was started by addition of 1 mM ADP (final concentration) and 10 mM (final concentration) potassium phosphate. After 30 min, 400 μl of stop solution (2 mM EDTA, 1% trichloroacetic acid) was added in each reaction mixture. Five milliliters of this mixture was added to 100 μl of Tris-acetate (Ac) buffer (100 mM Tris-HOAc, 2 mM EDTA, pH 7.75) in a 96-well plate. After addition of 50 μl of the luciferase reagent (ATP Bioluminescence Assay kit HS II; Roche), luminescence was measured with a luminometer (BMG Labtech, Offenburg, Germany).
Isolation of human mitochondria.
Using the method of Balemans et al. (38) with some modifications, the human lung cancer cell line Calu-3 (ATCC HTB-55; Manassas, VA, USA) was maintained in a 75-cm2 tissue culture flask in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum and 1% of an antibiotic-antimycotic solution (Wisent Inc., Saint-Jean-Baptiste, QC, Canada). Cell cultures were maintained at 37°C in 5% CO2. To isolate mitochondria, Calu-3 cells in one 75-cm2 tissue culture flask at 70% confluence were collected by centrifugation at 370 × g for 10 min and then suspended in 10 packed-cell volumes (1 ml) of NKM buffer (1 mM Tris-HCl, pH 7.4, 0.13 M NaCl, 5 mM KCl, 7.5 mM MgCl2). This step was repeated twice before cells were suspended in 6 packed-cell volumes of homogenization buffer (10 mM Tris-HCl, pH 6.7, 10 mM KCl, 0.15 mM MgCl2, 1 mM phenylmethylsulfonyl difluoride [PMSF], and 1 mM dithiothreitol [DTT], with the last two components immediately added before use). The cells were transferred to a glass homogenizer and incubated for 10 min on ice before homogenization with a tight pestle. The homogenate was poured into a conical centrifuge tube containing 1 packed-cell volume of a 2 M sucrose solution and mixed gently. Unbroken cells, nuclei, and large debris were removed by centrifugation at 1,200 × g for 5 min, and the supernatant was transferred to another tube. This process was repeated twice, transferring the supernatant to a new tube each time and discarding the pellet. The mitochondria were finally collected by centrifugation at 7,000 × g for 10 min and suspended in a 3 packed-cell volumes of mitochondrial suspension buffer (10 mM Tris-HCl, pH 6.7, 0.15 mM MgCl2, 0.25 M sucrose, 1 mM PMSF, 1 mM DTT). Mitochondria were collected again by centrifugation at 9,500 × g for 5 min, kept at 4°C, and used the same day in a mitochondrial ATP synthesis inhibition assay. Protein concentration was estimated by a Micro BCA protein assay kit.
Mitochondrial ATP synthesis inhibition assay.
Using the method of Balemans et al. (38), human mitochondria (0.25 mg/ml) were incubated in 50 mM morpholino-ethanesulfonic acid (MES; pH 6.5), 5 mM MgCl2, 20 mM KH2PO4, 100 μM P1, P5-di(adenosine-5) pentaphosphate (Ap5A), and 25 mM glucose. ATP production was then activated by addition of the oxidative phosphorylation substrates ADP (67 μM) and succinate (5 mM). The resulting concentrations of ATP were determined by using an ATP Bioluminescence Assay Kit HS II and a microplate luminometer as described above for the bacterial membrane vesicle assay.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Calcul Québec, Compute Canada, and the Centre de Calcul Scientifique of the Université de Sherbrooke for access and technical support while using the Mammouth-mp2 supercomputer. We also thank Marie-Ève Lacombe-Harvey for visualization of the model structure of subunit C.
This study was supported by a basic research grant from Cystic Fibrosis Canada to F.M. and E.M. and by a team grant from the Fonds Québécois de la Recherche sur la Nature et les Technologies (FQRNT) to F.M., K.B., and E.M. This work was also supported by Discovery grant 89758-2010 from the Natural Sciences and Engineering Research Council (NSERC) of Canada to F.M. M.L.B. received studentships from NSERC and from FQRNT during this study.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02197-17.
REFERENCES
- 1.Review on Antimicrobial Resistance 2014. Antimicrobial resistance: tackling a crisis for the health and wealth of nations. The Review on Antimicrobial Resistance, London, United Kingdom: https://amr-review.org/sites/default/files/AMR%20Review%20Paper%20-%20Tackling%20a%20crisis%20for%20the%20health%20and%20wealth%20of%20nations_1.pdf. [Google Scholar]
- 2.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 3.Coates AR, Halls G, Hu Y. 2011. Novel classes of antibiotics or more of the same? Br J Pharmacol 163:184–194. doi: 10.1111/j.1476-5381.2011.01250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen I, Christie PJ, Dubnau D. 2005. The ins and outs of DNA transfer in bacteria. Science 310:1456–1460. doi: 10.1126/science.1114021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lupo A, Coyne S, Berendonk TU. 2012. Origin and evolution of antibiotic resistance: the common mechanisms of emergence and spread in water bodies. Front Microbiol 3:18. doi: 10.3389/fmicb.2012.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perry JA, Wright GD. 2013. The antibiotic resistance “mobilome”: searching for the link between environment and clinic. Front Microbiol 4:138. doi: 10.3389/fmicb.2013.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silver LL. 2011. Challenges of antibacterial discovery. Clin Microbiol Rev 24:71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahajan R. 2013. Bedaquiline: first FDA-approved tuberculosis drug in 40 years. Int J Appl basic Med Res 3:1–2. doi: 10.4103/2229-516X.112228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haagsma AC, Abdillahi-Ibrahim R, Wagner MJ, Krab K, Vergauwen K, Guillemont J, Andries K, Lill H, Koul A, Bald D. 2009. Selectivity of TMC207 towards mycobacterial ATP synthase compared with that towards the eukaryotic homologue. Antimicrob Agents Chemother 53:1290–1292. doi: 10.1128/AAC.01393-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andries K, Verhasselt P, Guillemont J, Göhlmann HWH, Neefs J-M, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 11.Mitchell G, Lafrance M, Boulanger S, Séguin DL, Guay I, Gattuso M, Marsault É, Bouarab K, Malouin F. 2012. Tomatidine acts in synergy with aminoglycoside antibiotics against multiresistant Staphylococcus aureus and prevents virulence gene expression. J Antimicrob Chemother 67:559–568. doi: 10.1093/jac/dkr510. [DOI] [PubMed] [Google Scholar]
- 12.Mitchell G, Gattuso M, Grondin G, Marsault É, Bouarab K, Malouin F. 2011. Tomatidine inhibits replication of Staphylococcus aureus small-colony variants in cystic fibrosis airway epithelial cells. Antimicrob Agents Chemother 55:1937–1945. doi: 10.1128/AAC.01468-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guay I, Boulanger S, Isabelle C, Brouillette E, Chagnon F, Bouarab K, Marsault E, Malouin F. 2018. Tomatidine and analog FC04-100 possess bactericidal activities against Listeria, Bacillus and Staphylococcus spp. BMC Pharmacol Toxicol 19:7. doi: 10.1186/s40360-018-0197-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Otto M. 2012. Molecular insight into how MRSA is becoming increasingly dangerous. Virulence 3:521–523. doi: 10.4161/viru.21523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.David MZ, Daum RS. 2010. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin Microbiol Rev 23:616–687. doi: 10.1128/CMR.00081-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Centers for Disease Control and Prevention. 2013. Antibiotic resistance threats in the United States, 2013. Centers for Disease Control and Prevention, Atlanta, GA: https://www.cdc.gov/drugresistance/threat-report-2013/. [Google Scholar]
- 17.Leonard FC, Markey BK. 2008. Methicillin-resistant Staphylococcus aureus in animals: a review. Vet J 175:27–36. doi: 10.1016/j.tvjl.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 18.García-Álvarez L, Holden Lindsay MTG, Webb H, Brown CR, Curran DFJ, Walpole MD, Brooks E, Pickard K, Teale DJ, Parkhill C, Bentley J, Edwards SD, Girvan GF, Kearns EK, Pichon AM, Hill B, Larsen RLR, Skov AR, Peacock RL, Maskell SJ, Holmes DJ MA. 2011. Methicillin-resistant Staphylococcus aureus with a novel mecA homologue in human and bovine populations in the UK and Denmark: a descriptive study. Lancet Infect Dis 11:595–603. doi: 10.1016/S1473-3099(11)70126-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cuny C, Wieler LH, Witte W. 2015. Livestock-associated MRSA: the impact on humans. Antibiotics (Basel) 4:521–543. doi: 10.3390/antibiotics4040521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sergelidis D, Angelidis AS. 2017. Methicillin-resistant Staphylococcus aureus: a controversial food-borne pathogen. Lett Appl Microbiol 64:409–418. doi: 10.1111/lam.12735. [DOI] [PubMed] [Google Scholar]
- 21.Vázquez-Boland JA, Kuhn M, Berche P, Chakraborty T, Domínguez-Bernal G, Goebel W, González-Zorn B, Wehland J, Kreft J. 2001. Listeria pathogenesis and molecular virulence determinants. Clin Microbiol Rev 14:584–640. doi: 10.1128/CMR.14.3.584-640.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bottone EJ. 2010. Bacillus cereus, a volatile human pathogen. Clin Microbiol Rev 23:382–398. doi: 10.1128/CMR.00073-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferreira V, Wiedmann M, Teixeira P, Stasiewicz MJ. 2014. Listeria monocytogenes persistence in food-associated environments: epidemiology, strain characteristics, and implications for public health. J Food Prot 77:150–170. doi: 10.4315/0362-028X.JFP-13-150. [DOI] [PubMed] [Google Scholar]
- 24.Checinska A, Paszczynski A, Burbank M. 2015. Bacillus and other spore-forming genera: variations in responses and mechanisms for survival. Annu Rev Food Sci Technol 6:351–369. doi: 10.1146/annurev-food-030713-092332. [DOI] [PubMed] [Google Scholar]
- 25.Beierlein JM, Anderson AC. 2011. New developments in vaccines, inhibitors of anthrax toxins, and antibiotic therapeutics for Bacillus anthracis. Curr Med Chem 18:5083–5094. doi: 10.2174/092986711797636036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Atalla H, Gyles C, Jacob CL, Moisan H, Malouin F, Mallard B. 2008. Characterization of a Staphylococcus aureus small colony variant (SCV) associated with persistent bovine mastitis. Foodborne Pathog Dis 5:785–799. doi: 10.1089/fpd.2008.0110. [DOI] [PubMed] [Google Scholar]
- 27.Wolter DJ, Emerson JC, McNamara S, Buccat AM, Qin X, Cochrane E, Houston LS, Rogers GB, Marsh P, Prehar K, Pope CE, Blackledge M, Déziel E, Bruce KD, Ramsey BW, Gibson RL, Burns JL, Hoffman LR. 2013. Staphylococcus aureus small-colony variants are independently associated with worse lung disease in children with cystic fibrosis. Clin Infect Dis 57:384–391. doi: 10.1093/cid/cit270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Proctor RA, von Eiff C, Kahl BC, Becker K, McNamara P, Herrmann M, Peters G. 2006. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat Rev Microbiol 4:295–305. doi: 10.1038/nrmicro1384. [DOI] [PubMed] [Google Scholar]
- 29.Kahl BC, Becker K, Löffler B. 2016. Clinical significance and pathogenesis of staphylococcal small-colony variants in persistent infections. Clin Microbiol Rev 29:401–427. doi: 10.1128/CMR.00069-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moisan H, Brouillette E, Jacob CL, Langlois-Begin P, Michaud S, Malouin F. 2006. Transcription of virulence factors in Staphylococcus aureus small-colony variants isolated from cystic fibrosis patients is influenced by SigB. J Bacteriol 188:64–76. doi: 10.1128/JB.188.1.64-76.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mitchell G, Séguin DL, Asselin A-E, Déziel E, Cantin AM, Frost EH, Michaud S, Malouin F. 2010. Staphylococcus aureus sigma B-dependent emergence of small-colony variants and biofilm production following exposure to Pseudomonas aeruginosa 4-hydroxy-2-heptylquinoline-N-oxide. BMC Microbiol 10:33. doi: 10.1186/1471-2180-10-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh R, Ray P, Das A, Sharma M. 2009. Role of persisters and small-colony variants in antibiotic resistance of planktonic and biofilm-associated Staphylococcus aureus: an in vitro study. J Med Microbiol 58:1067–1073. doi: 10.1099/jmm.0.009720-0. [DOI] [PubMed] [Google Scholar]
- 33.Mitchell G, Brouillette E, Séguin DL, Asselin A-E, Jacob CL, Malouin F. 2010. A role for sigma factor B in the emergence of Staphylococcus aureus small-colony variants and elevated biofilm production resulting from an exposure to aminoglycosides. Microb Pathog 48:18–27. doi: 10.1016/j.micpath.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 34.Sendi P, Proctor RA. 2009. Staphylococcus aureus as an intracellular pathogen: the role of small colony variants. Trends Microbiol 17:54–58. doi: 10.1016/j.tim.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 35.Mitchell G, Grondin G, Bilodeau G, Cantin AM, Malouin F. 2011. Infection of polarized airway epithelial cells by normal and small-colony variant strains of Staphylococcus aureus is increased in cells with abnormal cystic fibrosis transmembrane conductance regulator function and is influenced by NF-κB. Infect Immun 79:3541–3551. doi: 10.1128/IAI.00078-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tuchscherr L, Medina E, Hussain M, Völker W, Heitmann V, Niemann S, Holzinger D, Roth J, Proctor RA, Becker K, Peters G, Löffler B. 2011. Staphylococcus aureus phenotype switching: an effective bacterial strategy to escape host immune response and establish a chronic infection. EMBO Mol Med 3:129–141. doi: 10.1002/emmm.201000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chagnon F, Guay I, Bonin M-A, Mitchell G, Bouarab K, Malouin F, Marsault É. 2014. Unraveling the structure-activity relationship of tomatidine, a steroid alkaloid with unique antibiotic properties against persistent forms of Staphylococcus aureus. Eur J Med Chem 80:605–620. doi: 10.1016/j.ejmech.2013.11.019. [DOI] [PubMed] [Google Scholar]
- 38.Balemans W, Vranckx L, Lounis N, Pop O, Guillemont J, Vergauwen K, Mol S, Gilissen R, Motte M, Lançois D, De Bolle M, Bonroy K, Lill H, Andries K, Bald D, Koul A. 2012. Novel antibiotics targeting respiratory ATP synthesis in Gram-positive pathogenic bacteria. Antimicrob Agents Chemother 56:4131–4139. doi: 10.1128/AAC.00273-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cohen J. 2013. Infectious disease. Approval of novel TB drug celebrated—with restraint. Science 339:130. doi: 10.1126/science.339.6116.130. [DOI] [PubMed] [Google Scholar]
- 40.Segala E, Sougakoff W, Nevejans-Chauffour A, Jarlier V, Petrella S. 2012. New mutations in the mycobacterial ATP synthase: new insights into the binding of the diarylquinoline TMC207 to the ATP synthase C-ring structure. Antimicrob Agents Chemother 56:2326–2334. doi: 10.1128/AAC.06154-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guex N, Peitsch MC. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 42.Sandrock RW, VanEtten HD. 1998. Fungal sensitivity to and enzymatic degradation of the phytoanticipin α-tomatine. Phytopathology 88:137–143. doi: 10.1094/PHYTO.1998.88.2.137. [DOI] [PubMed] [Google Scholar]
- 43.Roddick JG. 1979. Complex formation between solanaceous steroidal glycoalkaloids and free sterols in vitro. Phytochemistry 18:1467–1470. doi: 10.1016/S0031-9422(00)98476-0. [DOI] [Google Scholar]
- 44.Dorsaz S, Snäka T, Favre-Godal Q, Maudens P, Boulens N, Furrer P, Ebrahimi SN, Hamburger M, Allémann E, Gindro K, Queiroz EF, Riezman H, Wolfender J-L, Sanglard D, Snäkä T, Favre-Godal Q, Maudens P, Boulens N, Furrer P, Ebrahimi SN, Hamburger M, Allémann E, Gindro K, Ferreira Queiroz E, Riezman H, Wolfender J-L, Sanglard D. 2017. Identification and mode of action of a plant natural product targeting human fungal pathogens. Antimicrob Agents Chemother 61:e00829-17. doi: 10.1128/AAC.00829-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Medina JM, Rodrigues JCF, De Souza W, Atella GC, Barrabin H. 2012. Tomatidine promotes the inhibition of 24-alkylated sterol biosynthesis and mitochondrial dysfunction in Leishmania amazonensis promastigotes. Parasitology 139:1253–1265. doi: 10.1017/S0031182012000522. [DOI] [PubMed] [Google Scholar]
- 46.Desmond E, Gribaldo S. 2009. Phylogenomics of sterol synthesis: insights into the origin, evolution, and diversity of a key eukaryotic feature. Genome Biol Evol 1:364–381. doi: 10.1093/gbe/evp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simons V, Morrissey JP, Latijnhouwers M, Csukai M, Cleaver A, Yarrow C, Osbourn A. 2006. Dual effects of plant steroidal alkaloids on Saccharomyces cerevisiae. Antimicrob Agents Chemother 50:2732–2740. doi: 10.1128/AAC.00289-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Capaldi RA, Aggeler R. 2002. Mechanism of the F1F0-type ATP synthase, a biological rotary motor. Trends Biochem Sci 27:154–160. doi: 10.1016/S0968-0004(01)02051-5. [DOI] [PubMed] [Google Scholar]
- 49.von Ballmoos C, Cook GM, Dimroth P. 2008. Unique rotary ATP synthase and its biological diversity. Annu Rev Biophys 37:43–64. doi: 10.1146/annurev.biophys.37.032807.130018. [DOI] [PubMed] [Google Scholar]
- 50.von Ballmoos C, Wiedenmann A, Dimroth P. 2009. Essentials for ATP synthesis by F1F0 ATP synthases. Annu Rev Biochem 78:649–672. doi: 10.1146/annurev.biochem.78.081307.104803. [DOI] [PubMed] [Google Scholar]
- 51.Ko KS, Lee JY, Song JH, Baek JY, Oh WS, Chun JS, Yoon HS. 2006. Screening of essential genes in Staphylococcus aureus N315 using comparative genomics and allelic replacement mutagenesis. J Microbiol Biotechnol 16:623–632. [Google Scholar]
- 52.Cotter PD, Gahan CG, Hill C. 2000. Analysis of the role of the Listeria monocytogenes F0F1-ATPase operon in the acid tolerance response. Int J Food Microbiol 60:137–146. doi: 10.1016/S0168-1605(00)00305-6. [DOI] [PubMed] [Google Scholar]
- 53.Santana M, Ionescu MS, Vertes A, Longin R, Kunst F, Danchin A, Glaser P. 1994. Bacillus subtilis F0F1 ATPase: DNA sequence of the atp operon and characterization of atp mutants. J Bacteriol 176:6802–6811. doi: 10.1128/jb.176.22.6802-6811.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kriegeskorte A, König S, Sander G, Pirkl A, Mahabir E, Proctor RA, von Eiff C, Peters G, Becker K. 2011. Small colony variants of Staphylococcus aureus reveal distinct protein profiles. Proteomics 11:2476–2490. doi: 10.1002/pmic.201000796. [DOI] [PubMed] [Google Scholar]
- 55.Seggewiss J, Becker K, Kotte O, Eisenacher M, Yazdi MR, Fischer A, McNamara P, Al Laham N, Proctor R, Peters G, Heinemann M, von Eiff C. 2006. Reporter metabolite analysis of transcriptional profiles of a Staphylococcus aureus strain with normal phenotype and its isogenic hemB mutant displaying the small-colony-variant phenotype. J Bacteriol 188:7765–7777. doi: 10.1128/JB.00774-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Proctor RA, Kriegeskorte A, Kahl BC, Becker K, Löffler B, Peters G. 2014. Staphylococcus aureus small colony variants (SCVs): a road map for the metabolic pathways involved in persistent infections. Front Cell Infect Microbiol 4:99. doi: 10.3389/fcimb.2014.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoch JA. 1993. Regulation of the phosphorelay and the initiation of sporulation in Bacillus subtilis. Annu Rev Microbiol 47:441–465. doi: 10.1146/annurev.mi.47.100193.002301. [DOI] [PubMed] [Google Scholar]
- 58.Lewis K. 2007. Persister cells, dormancy and infectious disease. Nat Rev Microbiol 5:48–56. doi: 10.1038/nrmicro1557. [DOI] [PubMed] [Google Scholar]
- 59.Høiby N, Bjarnsholt T, Givskov M, Molin S, Ciofu O. 2010. Antibiotic resistance of bacterial biofilms. Int J Antimicrob Agents 35:322–332. doi: 10.1016/j.ijantimicag.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 60.Hurdle JG, O'Neill AJ, Chopra I, Lee RE. 2011. Targeting bacterial membrane function: an underexploited mechanism for treating persistent infections. Nat Rev Microbiol 9:62–75. doi: 10.1038/nrmicro2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hards K, Robson JR, Berney M, Shaw L, Bald D, Koul A, Andries K, Cook GM. 2015. Bactericidal mode of action of bedaquiline. J Antimicrob Chemother 70:2028–2037. doi: 10.1093/jac/dkv054. [DOI] [PubMed] [Google Scholar]
- 62.Rastogi VK, Girvin ME. 1999. Structural changes linked to proton translocation by subunit c of the ATP synthase. Nature 402:263–268. doi: 10.1038/46224. [DOI] [PubMed] [Google Scholar]
- 63.de Jonge MR, Koymans LHM, Guillemont JEG, Koul A, Andries K. 2007. A computational model of the inhibition of Mycobacterium tuberculosis ATPase by a new drug candidate R207910. Proteins 67:971–980. doi: 10.1002/prot.21376. [DOI] [PubMed] [Google Scholar]
- 64.Preiss L, Langer JD, Yildiz Ö Eckhardt-Strelau L, Guillemont JEG, Koul A, Meier T, Yildiz O, Eckhardt-Strelau L, Guillemont JEG, Koul A, Meier T. 2015. Structure of the mycobacterial ATP synthase Fo rotor ring in complex with the anti-TB drug bedaquiline. Sci Adv 1:e1500106. doi: 10.1126/sciadv.1500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Petrella S, Cambau E, Chauffour A, Andries K, Jarlier V, Sougakoff W. 2006. Genetic basis for natural and acquired resistance to the diarylquinoline R207910 in mycobacteria. Antimicrob Agents Chemother 50:2853–2856. doi: 10.1128/AAC.00244-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hong S, Pedersen PL. 2008. ATP synthase and the actions of inhibitors utilized to study its roles in human health, disease, and other scientific areas. Microbiol Mol Biol Rev 72:590–641. doi: 10.1128/MMBR.00016-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Henkin TM. 1996. The role of the CcpA transcriptional regulator in carbon metabolism in Bacillus subtilis. FEMS Microbiol Lett 135:9–15. doi: 10.1111/j.1574-6968.1996.tb07959.x. [DOI] [PubMed] [Google Scholar]
- 68.Tobisch S, Zühlke D, Bernhardt J, Stülke J, Hecker M. 1999. Role of CcpA in regulation of the central pathways of carbon catabolism in Bacillus subtilis. J Bacteriol 181:6996–7004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim H-J, Roux A, Sonenshein AL. 2002. Direct and indirect roles of CcpA in regulation of Bacillus subtilis Krebs cycle genes. Mol Microbiol 45:179–190. doi: 10.1046/j.1365-2958.2002.03003.x. [DOI] [PubMed] [Google Scholar]
- 70.Jankovic I, Egeter O, Brückner R. 2001. Analysis of catabolite control protein A-dependent repression in Staphylococcus xylosus by a genomic reporter gene system. J Bacteriol 183:580–586. doi: 10.1128/JB.183.2.580-586.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seidl K, Stucki M, Ruegg M, Goerke C, Wolz C, Harris L, Berger-Bächi B, Bischoff M. 2006. Staphylococcus aureus CcpA affects virulence determinant production and antibiotic resistance. Antimicrob Agents Chemother 50:1183–1194. doi: 10.1128/AAC.50.4.1183-1194.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seidl K, Müller S, François P, Kriebitzsch C, Schrenzel J, Engelmann S, Bischoff M, Berger-Bächi B. 2009. Effect of a glucose impulse on the CcpA regulon in Staphylococcus aureus. BMC Microbiol 9:95. doi: 10.1186/1471-2180-9-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fugère A, Lalonde Séguin D, Mitchell G, Déziel E, Dekimpe V, Cantin AM, Frost E, Malouin F. 2014. Interspecific small molecule interactions between clinical isolates of Pseudomonas aeruginosa and Staphylococcus aureus from adult cystic fibrosis patients. PLoS One 9:e86705. doi: 10.1371/journal.pone.0086705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brouillette E, Martinez A, Boyll BJ, Allen NE, Malouin F. 2004. Persistence of a Staphylococcus aureus small-colony variant under antibiotic pressure in vivo. FEMS Immunol Med Microbiol 41:35–41. doi: 10.1016/j.femsim.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 75.Clinical and Laboratory Standards Institute. 2013. Performance standards for antimicrobial susceptibility testing; twenty-second informational supplement. CLSI document M100-S22. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 76.Rodrigue S, Materna AC, Timberlake SC, Blackburn MC, Malmstrom RR, Alm EJ, Chisholm SW. 2010. Unlocking short read sequencing for metagenomics. PLoS One 5:e11840. doi: 10.1371/journal.pone.0011840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Assefa S, Keane TM, Otto TD, Newbold C, Berriman M. 2009. ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics 25:1968–1969. doi: 10.1093/bioinformatics/btp347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Otto TD, Dillon GP, Degrave WS, Berriman M. 2011. RATT: Rapid Annotation Transfer Tool. Nucleic Acids Res 39:e57. doi: 10.1093/nar/gkq1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Charpentier E, Anton AI, Barry P, Alfonso B, Fang Y, Novick RP. 2004. Novel cassette-based shuttle vector system for Gram-positive bacteria. Appl Environ Microbiol 70:6076–6085. doi: 10.1128/AEM.70.10.6076-6085.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Löfblom J, Kronqvist N, Uhlén M, Ståhl S, Wernérus H. 2007. Optimization of electroporation-mediated transformation: Staphylococcus carnosus as model organism. J Appl Microbiol 102:736–747. doi: 10.1111/j.1365-2672.2006.03127.x. [DOI] [PubMed] [Google Scholar]
- 81.Patrick JE, Kearns DB. 2008. MinJ (YvjD) is a topological determinant of cell division in Bacillus subtilis. Mol Microbiol 70:1166–1179. doi: 10.1111/j.1365-2958.2008.06469.x. [DOI] [PubMed] [Google Scholar]
- 82.Harwood CR, Cutting SM. 1990. Molecular biological methods for Bacillus. John Wiley & Sons, Chichester, United Kingdom. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.