

ABSTRACT
Avibactam is a novel non-β-lactam β-lactamase inhibitor that has been approved in the United States and Europe for use in combination with ceftazidime. Combinations of avibactam with aztreonam or ceftaroline fosamil have also been clinically evaluated. Until recently, there has been very little precedence of which pharmacokinetic/pharmacodynamic (PK/PD) indices and magnitudes are appropriate to use for β-lactamase inhibitors in population PK modeling for analyzing potential doses and susceptibility breakpoints. For avibactam, several preclinical studies using different in vitro and in vivo models have been conducted to identify the PK/PD index of avibactam and the magnitude of exposure necessary for effect in combination with ceftazidime, aztreonam, or ceftaroline fosamil. The PD driver of avibactam critical for restoring the activity of all three partner β-lactams was found to be time dependent rather than concentration dependent and was defined as the time that the concentration of avibactam exceeded a critical concentration threshold (%fT>CT). The magnitude of the CT and the time that this threshold needed to be exceeded to elicit particular PD endpoints varied depending on the model and the partner β-lactam. This review describes the preclinical studies used to determine the avibactam PK/PD target in combination with its β-lactam partners.
KEYWORDS: avibactam, diazabicyclooctane, BL-BLI, PK/PD
INTRODUCTION
Selecting the dose of a candidate antibiotic for evaluation in clinical trials requires the translation of pharmacokinetic/pharmacodynamic (PK/PD) data derived from in vitro and in vivo experimental models to the prediction of clinical responses in patients (1–3). Data from preclinical studies are typically used to identify the PK/PD index that best describes the relationship between exposure and the antimicrobial effect for the antibiotic in question. The magnitude of this PK/PD index which produces the desired effect is termed the PK/PD target. Due to interindividual variation in human PKs, a population-based modeling approach is necessary to predict whether potential dosing regimens will result in the achievement of the PK/PD target in a substantial majority of patients (4, 5). Population PK models developed from patient PK data are used to simulate antibiotic exposures in a representative patient population and to predict the proportion of patients who would achieve a level of drug exposure that meets the prespecified PK/PD target, known as the “probability of target attainment” (PTA). Such analyses can thus guide the selection of a dosage regimen that is likely to result in a high PTA (>90%). Neglecting key considerations of these principles can lead to failures of clinical trials due to inadequate drug exposures needed to be effective against key target pathogens (6). This exposes patients to unnecessary risks and delays or prevents the availability of a drug that may address an unmet need. Thus, appropriately characterizing the PK/PD index and magnitude preclinically is a key step in dose selection.
Avibactam is a novel diazabicyclooctane non-β-lactam β-lactamase inhibitor which is active against Ambler class A, class C, and some class D β-lactamases (7). When combined with ceftazidime, an antipseudomonal cephalosporin, avibactam restores the activity of ceftazidime against most multidrug-resistant Enterobacteriaceae and Pseudomonas aeruginosa, including those producing extended-spectrum β-lactamases (ESBLs) and non-metallo-β-lactamase (non-MBL) carbapenemases (8–10). Ceftazidime-avibactam is approved in the United States for the treatment of adults with complicated intra-abdominal infections (cIAI), complicated urinary tract infections (cUTI), hospital-acquired bacterial pneumonia, and ventilator-associated bacterial pneumonia and in Europe for the treatment of adults with cIAI, cUTI, and hospital-acquired pneumonia (including ventilator-associated pneumonia) and patients with aerobic Gram-negative infections and limited treatment options (11, 12).
Avibactam has also been evaluated in combination with the β-lactams aztreonam and ceftaroline fosamil. Aztreonam is a monocyclic β-lactam (monobactam) that is stable in the presence of MBLs, but is hydrolyzed by ESBLs. In combination with avibactam, aztreonam is active in vitro and in vivo against Enterobacteriaceae that harbor an MBL with or without one or more class A or C or some class D serine β-lactamases (13–15). Ceftaroline fosamil, the prodrug of the active component ceftaroline, is a broad-spectrum cephalosporin with in vitro activity against Gram-positive bacteria and some non-ESBL-producing Gram-negative species; when combined with avibactam, ceftaroline has shown in vitro activity against Enterobacteriaceae producing various ESBLs (16). A phase 2 trial of aztreonam-avibactam in patients with cIAI is currently ongoing (NCT02655419); ceftaroline fosamil-avibactam has been evaluated in a phase 2 clinical trial in patients with cUTI (NCT01281462) (17, 18).
For β-lactam–β-lactamase inhibitor combinations, it is important to establish a PK/PD index for both components, i.e., the β-lactamase inhibitor as well as the β-lactam antibiotic (19). Until recently, there has been very little precedence for the PK/PD indices and magnitudes that are appropriate to use for β-lactamase inhibitors in population PK modeling for analyzing potential doses and susceptibility breakpoints for β-lactam–β-lactamase inhibitor combinations. A thorough characterization of the PD of β-lactamase inhibitors in combination with their partner antibiotics enables an optimization of the dosing regimen. This review describes the preclinical studies used to identify the PK/PD index of avibactam and the magnitudes of exposure necessary for effect in combination with the different β-lactams, ceftazidime, aztreonam, and ceftaroline fosamil.
PK/PD TARGETS IN ANTIBIOTIC DRUG DEVELOPMENT AND THE PRINCIPLE OF ESTABLISHING A PK/PD INDEX FOR A β-LACTAMASE INHIBITOR
Although PK/PD indices and magnitudes for antibiotics could theoretically be identified from clinical trials, it is not ethical to give doses which are so low that a proportion of patients are likely to fail therapy. If the agent is effective and the dose regimen is well designed, there are likely to be few exposure-related failures. In addition, patients with infections due to multidrug-resistant pathogens are often critically ill, and clinical outcomes may be confounded by underlying diseases and/or surgical interventions. Consequently, exposure-effect analyses from clinical trial data do not usually enable discriminatory PK/PD indices and magnitudes to be identified (6, 19). Exposure targets must therefore be derived, initially, from nonhuman experiments that measure antibacterial activity in terms of bacterial killing or from outcome measures such as morbidity or mortality in animal infection models. Fortunately, in the analysis of PK/PD targets of antibacterial agents, substantial understanding can be achieved from preclinical in vitro and in vivo laboratory studies and subsequent modeling (6, 20, 21). Antibacterial drug exposures that result in bacteriostasis or 10- to 100-fold bacterial killing in experimental models are broadly correlated with clinical efficacy (6, 22, 23).
The analysis of PK/PD targets for antibacterial agents differs in a key aspect from that for drugs directed at mammalian biology, in that the target is the infecting pathogen. The PK/PD target established preclinically in either in vitro or in vivo systems is therefore relevant in humans, although penetration into the infection site may need to be taken into consideration. The exposure (PK) component of the PK/PD relationships for antibacterial agents is generally related to one of the following parameters, based on plasma concentrations of the unbound drug: Cmax (the maximum plasma concentration of the drug during the dosing interval), AUC (the area under the plasma concentration-time curve, mostly considered over 24 h [AUC0–24]), or T>Ccrit (time above a critical concentration of the drug as a proportion of the regular dosing interval [interdose period]). In addition, the variability in the susceptibility of target pathogens to the antibiotic also needs to be considered in the PK/PD target for dose finding (24, 25). For antibacterial agents, the respective PK/PD indices have been established as the ratios fCmax/MIC, fAUC/MIC, or fT>MIC, where the f symbol indicates that the concentration is that of “free” (unbound) drug (26, 27). The critical MIC used in setting PK/PD targets should be based on analyses of MIC distributions observed in global surveillance studies of contemporary isolates of key pathogens from the intended indications.
Certain classes of antibiotics tend to share PK/PD indices. In concordance with those for other β-lactam antibiotics, the PK/PD indices for ceftazidime, ceftaroline fosamil, and aztreonam are well established as fT>MIC. The initial hypothesis of the PD of avibactam in combination with β-lactams was based on the theoretical concept that if the avibactam exposure is effective, then the PD of the combination should be the same as that of the partner β-lactam (28), i.e., related to fT>MIC of the combination. Avibactam alone does not have significant antibacterial activity and thus does not change the PD of the β-lactam partner. For avibactam, different experimental approaches have been used to characterize the PD index of exposure best related to restoring the activity of the partner β-lactam under dynamic conditions, including in in vitro hollow-fiber models and in vivo mouse thigh and lung infection models, as described in the following sections.
AVIBACTAM PK/PD TARGETS IN COMBINATION WITH CEFTAZIDIME
Ceftazidime PK/PD target.
The achievement of 50% fT>MIC for ceftazidime has been shown to be associated with the killing of Enterobacteriaceae and P. aeruginosa by ceftazidime in neutropenic mouse infection models (29–31) and with microbiological eradication in patients with Gram-negative infections (23, 32). These PK/PD correlates led to the use of 50% fT>MIC as the pivotal exposure for associating particular ceftazidime doses with PK/PD target attainment and in establishing interpretive MIC criteria (25, 33–35). A ceftazidime-avibactam MIC value of 8 mg/liter was chosen for the PK/PD target because global surveillance data had reported ceftazidime-avibactam MIC90 values of 0.5 to 1 mg/liter and ≤8 mg/liter for phenotypically and genotypically unselected clinical isolates of Enterobacteriaceae and P. aeruginosa, respectively (36–42).
Determination of the avibactam PK/PD target when combined with ceftazidime against Enterobacteriaceae.
Taking the concept that with complete β-lactamase inhibition, the PK/PD profile of ceftazidime-avibactam would revert to the PK/PD profile of ceftazidime, it was important to identify the concentration of avibactam that must be maintained to achieve sufficient β-lactamase inhibition to protect the activity of ceftazidime. Thus, a “critical” or “threshold” concentration of avibactam (CT) was defined as that which occurs during the exponential decline of the concentration of avibactam during a dose cycle, below which the inhibition of β-lactamases is inadequate to prevent growth (of ceftazidime-resistant bacteria) in the presence of ceftazidime.
A series of hollow-fiber model experiments were used to analyze the PK/PD activity of avibactam in restoring the bactericidal activity of ceftazidime against ceftazidime-resistant Enterobacteriaceae and thus determine the CT of avibactam in this setting (43). In the first set of experiments, avibactam concentrations were varied to simulate human PK profiles, while ceftazidime was held constant at a concentration higher than that of the MIC of ceftazidime-avibactam in combination but lower than that of the MIC of ceftazidime alone for all strains tested. The three ceftazidime-resistant Enterobacteriaceae isolates used in these experiments produced either derepressed AmpC (Enterobacter cloacae) or one of two ESBLs, SHV-5 or CTX-M-15 (Klebsiella pneumoniae). The avibactam concentration below which adequate inhibition of β-lactamases was lost was identified as that at which bacterial numbers started to increase again. The magnitude of the CT was estimated as being equal to or less than the concentration of avibactam remaining in the hollow-fiber system at the time point at which growth suppression was last experimentally demonstrated.
Figure 1 shows how CT was estimated in these hollow-fiber experiments, using the E. cloacae isolate as an example. Four estimates of the CT of avibactam were obtained with the three isolates, with a mean value of ≤0.21 mg/liter (range, ≤0.15 to ≤0.28 mg/liter). Several further observations about the CT of avibactam were noteworthy from these experiments. The four values of CT were similar, with similar CT values obtained for two species: ≤0.15 and ≤0.2 mg/liter for E. cloacae and ≤0.22 and ≤0.28 mg/liter for K. pneumoniae. There was no direct correlation between the MIC of ceftazidime-avibactam (which spanned a 4-fold range of MICs studied [1 to 4 mg/liter] among the three isolates) and the CT for each isolate and no obvious dependence on the specific β-lactamase encountered. Within these experiments, while the avibactam AUC0–24 ranged between 16.4 and 126 mg · h/liter, there was no relationship between growth suppression and the AUC (43).
FIG 1.
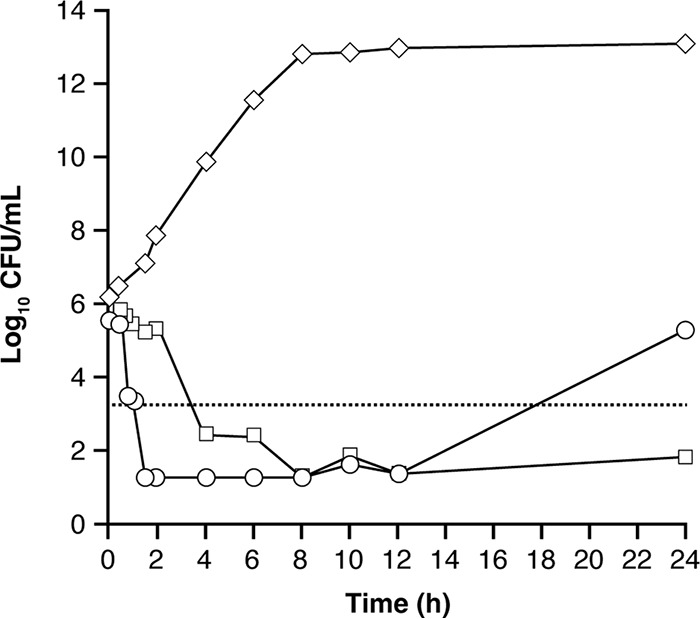
Responses of ceftazidime-resistant E. cloacae to continuous infusion of ceftazidime combined with two different concentration-time profiles of avibactam in the hollow-fiber model. Results are shown for isolate E. cloacae 293HT96 (stably derepressed AmpC; MIC ceftazidime, >128 mg/liter; MIC ceftazidime-avibactam, 4 mg/liter [with avibactam fixed at 4 mg/liter {59}]). ◇, untreated growth control; ◽, continuous infusion of ceftazidime (8.2 mg/liter) and avibactam (1.6 mg/liter); ○, continuous infusion of ceftazidime (8.2 mg/liter) with a single-dose profile of avibactam (Cmax, 31 mg/liter at 0.5 h). The dotted line represents 99.9% bacterial killing. For the single-dose profile of avibactam, growth recommenced after the 12-h time point when the concentration of avibactam on the exponentially declining concentration-time curve was 0.15 mg/liter, resulting in an estimated critical concentration threshold (CT) of ≤0.15 mg/liter. Adapted and republished from reference 43.
In a second set of hollow-fiber experiments, the “growth suppression windows” yielded by constant concentrations of avibactam infused for different periods of time were determined using a broader panel of isolates (including K. pneumoniae carbapenemase [KPC]-3-producing K. pneumoniae and stably derepressed AmpC-producing Citrobacter freundii). In these experiments, constant concentrations of avibactam ranging from 0.25 to 1.0 mg/liter were infused for different time periods in the background of ceftazidime concentrations varied to simulate human PK profiles. This enabled a CT of avibactam to be determined on the basis of a period of infusion of a constant concentration of the compound rather than following a peak of avibactam as described in the previous set of experiments. This was termed CTQ8 to indicate that it was determined in a background of 8 hourly (q8h) cycling of ceftazidime concentrations. The CTQ8 yielded in the background of ceftazidime exposures equivalent to a dose of 2 g q8h was ≤0.5 mg/liter. When comparing this value of CTQ8 to the CT to be used in setting a PK/PD target, it should be regarded a relatively conservative magnitude. This is because the avibactam concentration was constant and did not account for the pharmacologic effect of the higher avibactam concentrations yielded in vivo at early time points after dosing.
Taking together the results from these hollow-fiber experiments, a minimum avibactam CT of 0.5 mg/liter was considered appropriate for estimating the PTA for ceftazidime-avibactam against Enterobacteriaceae (43). As there was no relationship between the CT and MIC and no dependence on the different β-lactamases expressed, it can be considered that this CT of 0.5 mg/liter was sufficient to fully inhibit the β-lactamases in each of the isolates.
Determination of the avibactam PK/PD target when combined with ceftazidime against P. aeruginosa.
A series of dose fractionation studies in neutropenic mouse thigh and lung infection models was used to define the PK/PD index of avibactam in combination with ceftazidime against ceftazidime-resistant P. aeruginosa isolates (44). In these experiments, dose fractionation was used to determine which PD index best described the PD of avibactam in combination with ceftazidime. The isolates used in these studies were tested with ceftazidime-avibactam MICs ranging from 2 to 16 mg/liter and produced AmpC β-lactamase (Tables 1, 2, and 3). Before the dose fractionation experiments were undertaken, it was necessary to establish a dose of ceftazidime monotherapy against each bacterial strain that would just allow maximal growth in the neutropenic mouse model. The concept was to establish the ceftazidime dose response of the system at a point whereby any reduction in bacterial growth was the result of β-lactamase inhibition by avibactam, restoring the activity of ceftazidime against the resistant strain. This approach enabled the determination of dose-response curves for avibactam when it was administered in the presence of ceftazidime dosed every 2 h (q2h) at the amount determined empirically for that strain, as described above.
TABLE 1.
Assessment of the hypothesis that fT>CT is the exposure variable more closely linked than fAUC to the pharmacodynamic effect of avibactam in combination with ceftazidime against ceftazidime-resistant P. aeruginosa in the neutropenic mouse lung infection model, using bacterial stasis as the pharmacodynamic endpointa
| Strainb | MIC (mg/liter) |
AVIc |
||||
|---|---|---|---|---|---|---|
| Static total daily dose (mg · kg−1 · day−1) |
%fT>CT of 1 mg/liter associated with stasisd |
|||||
| CAZe | CAZ-AVI | q2h | q8h | q2h | q8h | |
| 11 | 128 | 16 | 45.6 | 463 | 19.7 | 20.9 |
| 18 | 32 | 2 | 56.4 | 151 | 23.5 | 16.1 |
Constructed from the data of Berkhout et al. (44).
Resistance summaries for the strains used in this experiment are as follows. Strain 11: OprD−, AmpCcon, class A−, class B−; strain 18: OprD−, AmpCind?, class A−, class B−.
AVI, avibactam.
Stasis-associated exposure times as percentages of the dosing interval calculated to be yielded by the interpolated doses shown.
CAZ, ceftazidime.
TABLE 2.
Magnitudes of avibactam exposures associated with stasis and killing of P. aeruginosa in the neutropenic mouse thigh infection model in the background of q2h dosing of ceftazidimea
| Strainb | MIC (mg/liter) |
Exptf | Avibactam %fT>1 mg/literc yielding: |
||
|---|---|---|---|---|---|
| CAZd | CAZ-AVIe | Stasis | 1-log10 kill | ||
| 1 | 128 | 8 | Codosing | 37.2 | 65.7 |
| 5 | 128 | 8 | Codosing | 14.1 | 32.9 |
| 7 | 64 | 4 | AVI fractionation | 74.1 | Not reported |
| Codosing | 50.4 | 65.3 | |||
| 11 | 128 | 16 | Codosing | 29.1 | 37.5 |
| 18 | 32 | 2 | AVI fractionation | 30.2 | Not reported |
| Codosing | 24.2 | 33.2 | |||
| 19 | 64 | 4 | Codosing | 62.5 | 67.2 |
| Mean | 40.2 | 50.3 | |||
| SD | 20 | 17 | |||
Data were retabulated from Berkhout et al. (44).
Resistance summaries for the strains used in this experiment are as follows. Strain 1, nitrocefinase activity, ++; AmpC transcript, overexpressed; β-lactamase genotype, blaAmpC; class A−, class B−; strain 5: nitrocefinase activity, ++++; AmpC transcript, overexpressed; β-lactamase genotype, blaAmpC; class A−, class B−; strain 7: nitrocefinase activity, +++; AmpC transcript, overexpressed; β-lactamase genotype, blaAmpC; class A−, class B−; strain 11: OprD−, AmpCcon, class A−, class B−; strain 18: OprD−, AmpCind?, class A−, class B−; strain 19: OprD−, AmpCcon, class A−, class B−.
Times are expressed as the percentages of the dosing interval.
CAZ, ceftazidime.
AVI, avibactam.
Codosing, ceftazidime and avibactam were codosed but the amount of avibactam was varied, without fractionating any given total daily dose; AVI fractionation, avibactam dose fractionation experiments.
TABLE 3.
Magnitudes of avibactam exposures associated with stasis and bacterial killing of ceftazidime-resistant P. aeruginosa in the neutropenic mouse lung infection model in the background of q2h dosing of ceftazidimea
| Strainb | MIC (mg/liter) |
Codosing expte | Avibactam %fT>1 mg/literf associated with: |
|||
|---|---|---|---|---|---|---|
| CAZc | CAZ-AVId | Stasis | 1-log10 kill | 2-log10 kill | ||
| 5 | 128 | 8 | q2h | 19.4 | 20.6 | Not reported |
| 7 | 64 | 4 | q2h | 21.4 | 22.4 | Not reported |
| 11 | 128 | 16 | q2h | 19.7 | 34.9 | 55.3 |
| q8h | 20.9 | 21.6 | 22.5 | |||
| 18 | 32 | 2 | q2h | 23.5 | 26.7 | 31.8 |
| q8h | 16.1 | 17.8 | 20.2 | |||
| Mean | 20.2 | 24.0 | 32.4 | |||
| SD | 2.5 | 6.1 | 16 | |||
Data are from Berkhout et al. (44).
Resistance summaries for the strains used in this experiment are as follows. Strain 5: nitrocefinase activity, ++++; AmpC transcript, overexpressed; β-lactamase genotype, blaAmpC; class A−, class B−; strain 7: nitrocefinase activity, +++; AmpC transcript, overexpressed; β-lactamase genotype, blaAmpC; class A−, class B−; strain 11: OprD−, AmpCcon, class A−, class B−; strain 18: OprD−, AmpCind?, class A−, class B−; strain 19: OprD−, AmpCcon, class A−, class B−.
CAZ, ceftazidime.
AVI, avibactam.
q2h, ceftazidime and avibactam were dosed together at every administration; q8h, ceftazidime was dosed q2h but avibactam was codosed at 0, 8, and 16 h from the initiation of dosing.
Times are expressed as percentages of the dosing interval.
The doses of avibactam were fractionated in the background of this specific q2h dosing schedule of ceftazidime, with responses measured as log10 change in CFU (44). The responses were plotted as a function of the PD indices fAUC, fCmax, and fT>CT for avibactam to determine which of these best correlated with antibacterial efficacy for ceftazidime-avibactam. For fT>CT, the responses were plotted against three values of CT, covering a 16-fold range: 0.25 mg/liter, 1 mg/liter, and 4 mg/liter. An example dose fractionation experiment for one of the P. aeruginosa strains is shown in Fig. 2. There was no significant relationship between the response to ceftazidime-avibactam and the avibactam Cmax, suggesting that Cmax was not the driver of efficacy. However, both %fT>CT and AUC values of avibactam showed reasonable correlations with efficacy.
FIG 2.

Dose fractionation study of avibactam in combination with ceftazidime against a ceftazidime-resistant P. aeruginosa strain in the neutropenic mouse thigh infection model. AVI, avibactam; CAZ, ceftazidime; ΔlogCFU, change in log10 CFU compared to the initial inoculum. Republished from reference 44.
A subsequent experiment in the lung infection model tested the hypothesis that %fT>CT was a more predictive avibactam index than AUC (44). Identical daily doses of avibactam were given either q2h or q8h in the background of the q2h dosing schedule of ceftazidime described above. The two widely different dose intervals were selected to gather more data points and increase the ability to distinguish between AUC and time as PK/PD indices. The avibactam static total daily doses and %fT>CT values required for stasis in two isolates of P. aeruginosa are summarized in Table 1. The total daily dose of avibactam that resulted in a static effect was lower for the more frequent q2h dosing of avibactam than for q8h dosing in both strains of ceftazidime-resistant P. aeruginosa (by factors of 10.1 and 2.7) (Table 1) (44). Despite this difference in total daily dose, the values of %fT>CT of 1 mg/liter that yielded stasis from the two avibactam dosing frequencies were similar (16.1 and 23.5%) (Table 1). These results were consistent with the hypothesis that the PD of avibactam in restoring the antibacterial activity of ceftazidime in ceftazidime-resistant P. aeruginosa was time dependent rather than concentration dependent (i.e., linked to fT>concentration [CT] rather than fAUC). Results from similar experiments in the thigh infection model showed a significant relationship between an increased frequency of dosing and a change in log10 CFU for similar avibactam total daily doses, confirming the importance of %fT>CT in the PD of avibactam (44). A CT of 1 mg/liter was chosen as the reference concentration, because in the dose fractionation study, higher r2 coefficients were found for the association between the PD effect of avibactam (change in log CFU) and %fT>CT 1 mg/liter than for %fT>CT 0.25 mg/liter and %fT>CT 4 mg/liter (r2 0.67 versus 0.61 and 0.61, respectively).
Having chosen the reference concentration of avibactam, further ceftazidime-avibactam codosing experiments were conducted with four P. aeruginosa strains in the lung model and six strains in the thigh model to determine the relationship between the magnitude of %fT>CT 1 mg/liter and the magnitude of the PD effect. The %fT>CT 1 mg/liter values that yielded PD effects of net stasis and a 1-log10 kill in the neutropenic mouse thigh infection model for the different P. aeruginosa strains are shown in Table 2, while those derived from the lung model, including the %fT>CT 1 mg/liter that yielded a 2-log10 kill, are shown in Table 3. The mean exposure that yielded 1-log10 killing of P. aeruginosa in the neutropenic mouse thigh model (50.3%) was equivalent to the avibactam exposure target of approximately 50% fT>CT of 1 mg/liter. Moreover, this target exceeded the avibactam exposure of 40% fT>CT of 1 mg/liter that was associated with bacterial stasis (Table 2). In addition, the avibactam target of 50% fT>CT of 1 mg/liter exceeded the exposures associated with stasis, 1-log10 kill, and 2-log10 kill of P. aeruginosa in the neutropenic mouse lung infection model (Table 3). Again, in these sets of experiments where the ceftazidime-avibactam MIC ranged from 2 to 16 mg/liter, there was no noticeable correlation between the avibactam CT and MIC or the level of β-lactamase expressed.
On the basis of these experiments, %fT>CT was determined as the PK/PD index that was best associated with the restoration of ceftazidime efficacy by avibactam in the neutropenic mice thigh and lung infection models. The most appropriate CT value of avibactam associated with efficacy against ceftazidime-resistant P. aeruginosa was 1 mg/liter.
Ceftazidime-avibactam PK/PD targets used to support dosage selection.
Joint PTA analyses based on the simultaneous achievement of both the ceftazidime and avibactam PK/PD targets in ≥90% of patients have subsequently been used to support ceftazidime-avibactam dosage selection (45). The ceftazidime-avibactam dose was designed to provide adequate target attainment in ≥90% of patients in whom the infecting organism would respond in a susceptibility test with a ceftazidime-avibactam MIC of 8 mg/liter. The ceftazidime-avibactam MIC value of 8 mg/liter was chosen as the cutoff for assessing PTA on the basis of reported ceftazidime-avibactam MIC90 values from global surveillance data for Enterobacteriaceae and P. aeruginosa, as described above (36–42). Therefore, we propose that the approach to dosage selection based on ≥90% of patients attaining ceftazidime exposures adequate to treat infecting bacteria against which the ceftazidime-avibactam MIC would be 8 mg/liter is conservative, because the PTA is based on the 90th percentile MIC of the MIC frequency distribution of clinical isolates as opposed to the whole distribution. That is, in patients infected by most examples of Enterobacteriaceae or P. aeruginosa and dosed with ceftazidime-avibactam, the ceftazidime PK/PD target is predicted to be well exceeded.
The avibactam exposure target of fT>CT of 1 mg/liter for at least 50% of the dosing interval was chosen for determining the PTA for avibactam in combination with ceftazidime. This also matched the exposure target for ceftazidime (50% fT > ceftazidime-avibactam MIC of 8 mg/liter) (45–48). The CT of 1 mg/liter value was regarded as an adequate index for analyses of PK/PD target attainment against P. aeruginosa on the basis of the neutropenic mouse thigh and lung infection studies and as a conservative index for analyses of PK/PD target attainment against Enterobacteriaceae, as CT and CTQ8 were estimated to be ≤0.5 mg/liter in the in vitro hollow-fiber studies by Coleman and colleagues (43). The experiments included a range of different isolates with differing β-lactamase expressions and a clinically relevant ceftazidime-avibactam MIC range. In both the in vitro and in vivo sets of experiments, there was no relationship between CT and either MIC or the identity of the β-lactamases tested, demonstrating that the CT of 1 mg/liter sufficiently inhibits all β-lactamase expression in the isolates tested. Thus, the avibactam PK/PD target of CT of 1 mg/liter was considered appropriate to use together with the ceftazidime PK/PD target in joint PTA analyses for ceftazidime-avibactam dosage selection.
AVIBACTAM PK/PD INDEX AND TARGET IN COMBINATION WITH AZTREONAM
Singh and colleagues (49) used an in vitro hollow-fiber model and a neutropenic mouse infection model to investigate the PK/PD indices and magnitudes for aztreonam and avibactam in combination against Enterobacteriaceae (49). Six aztreonam-resistant Enterobacteriaceae isolates (three K. pneumoniae and three Escherichia coli) all coproducing an MBL and an ESBL and/or a class C β-lactamase (CMY type) were used in these experiments. The MIC values of aztreonam-avibactam against these isolates ranged from 0.125 to 8 mg/liter. Two sets of hollow-fiber experiments were performed; the first evaluated the PK/PD index of aztreonam in the presence of avibactam and the second evaluated the PK/PD index and magnitude of avibactam in the presence of a fixed dose of aztreonam. The avibactam PK/PD target derived from the hollow-fiber experiments was then validated in a neutropenic mouse thigh infection model.
The first set of hollow-fiber experiments was designed to evaluate whether the aztreonam PK/PD index changed with the addition of avibactam (49). In these experiments, avibactam at 4 mg/liter was continuously infused in combination with different dosage regimens of aztreonam, and the responses of two of the Enterobacteriaceae isolates were measured. As was inferred for ceftazidime, the PK/PD index that best correlated with the efficacy of aztreonam in combination with avibactam was %fT>MIC of the combination. The magnitude of the index associated with a 1-log10 kill over 24 h was 50 to 55%. These initial experiments demonstrated that the aztreonam PK/PD index was not changed by the presence of avibactam and that the magnitudes associated with activity against aztreonam-resistant bacteria in the presence of avibactam were as expected for a β-lactam against susceptible bacteria.
In the second set of hollow-fiber studies, a fixed dose of aztreonam was administered every 6 h to simulate a human-like PK profile in the presence of different dosage regimens of avibactam (49). As the previous experiments had shown that 50% fT>MIC was sufficient for aztreonam efficacy in the presence of avibactam against Enterobacteriaceae, the fixed aztreonam dose was therefore designed to provide 50 to 100% fT>MIC against the isolate under study. An example dose fractionation experiment of avibactam in combination with aztreonam against one of the E. coli strains is shown in Fig. 3. From these dose fractionation experiments, the effect of avibactam in restoring the antibacterial activity of aztreonam was found to correlate best with %fT>CT. The PK/PD indices fAUC and fCmax were also analyzed but did not correlate well with response. The magnitude of CT that best correlated with efficacy was evaluated for CT values ranging from 0.5 to 4 mg/liter. For five of the six isolates, a CT of 2.5 mg/liter provided the best fit; for these isolates (two K. pneumoniae and three E. coli), the mean value of %fT>CT of 2.5 mg/liter that yielded a 1-log10 kill was 47.5% (range, 40.9 to 58.2%) (Table 4). In the other K. pneumoniae isolate, a CT of 2 mg/liter provided a better correlation, with a value of 38% fT>CT yielding a 1-log10 kill. As the efficacy best correlated with a CT of 2.5 mg/liter in five of the six isolates tested, this threshold value was chosen for use in PTA analyses for dosage selection. As with the ceftazidime-avibactam experiments described above, there was no relationship between the CT and aztreonam-avibactam MIC or β-lactamase expression.
FIG 3.

PK/PD relationship between fAUC, fCmax, and %fT>CT for avibactam in the presence of fixed dosing of aztreonam against an E. coli strain in the hollow-fiber model. Symbols represent experimental observations for different dosage regimens of avibactam and continuous lines represent the predicted best-fit model. Republished from reference 49 with permission from the British Society for Antimicrobial Chemotherapy.
TABLE 4.
Magnitudes of avibactam exposures associated with stasis and killing of metallo-β-lactamase- and ESBL- and/or CMY-type β-lactamase-coproducing isolates of K. pneumoniae and E. coli in the background of 6-hourly dosing of aztreonam over 24 h in a hollow-fiber model in vitroa
| Strainb | Avibactam %fT>2.5 mg/literc yielding: |
|
|---|---|---|
| Stasis | 1-log10 kill | |
| K. pneumoniae ARC3602 | 39.3 | 46.1 |
| K. pneumoniae ARC3803 | 41.8 | 44.3 |
| E. coli ARC3600 | 36.1 | 40.9 |
| E. coli ARC3805 | 56.4 | 58.2 |
| E. coli ARC3807 | 43.2 | 48.1 |
| Mean | 43.4 | 47.5 |
| SD | 7.8 | 6.5 |
Data were retabulated from Singh et al. (49) with permission from the British Society for Antimicrobial Chemotherapy. ESBL, extended-spectrum β-lactamase.
K. pneumoniae ARC3802 omitted because only bacterial responses correlating with an fT>2 mg/liter were reported for that isolate.
Times are expressed as percentages of the 24-h period of the experiment.
The bacterial responses associated with these avibactam exposure magnitudes were confirmed in further dose fractionation experiments in the neutropenic mouse thigh (49) and lung (our unpublished data) infection models. In the thigh infection model, four-hourly codosing of aztreonam and avibactam (4:1 by weight) provided aztreonam 70 to 100% fT>MIC of the combination. With the aztreonam exposures set to >50% fT>MIC, the efficacy of aztreonam-avibactam correlated with avibactam %fT>CT of 2 to 2.5 mg/liter. Twenty-four-hour stasis was achieved against an E. coli isolate at 23% fT>CT 2.5 mg/liter and against a K. pneumoniae isolate at 25% fT>CT 2 mg/liter. The maximal effect of avibactam was achieved at 35 to 40% fT>CT 2 to 2.5 mg/liter for both isolates, which was consistent with the results observed in the hollow-fiber model experiments.
These studies showed that, as had been found for ceftazidime, the PK/PD index that best correlated with the restoration of the antibacterial activity of aztreonam by avibactam was %fT>CT. In both the hollow-fiber and neutropenic mouse infection models, a CT value of 2.5 mg/liter for avibactam correlated best with the restoration of aztreonam efficacy against aztreonam-resistant Enterobacteriaceae. On the basis of these results, PK/PD targets of aztreonam 60% fT>MIC for aztreonam-avibactam and avibactam 50% fT>CT of 2.5 mg/liter were considered appropriate for PTA analyses to guide dosage selection for aztreonam-avibactam (our unpublished data).
PK/PD STUDIES OF AVIBACTAM IN COMBINATION WITH CEFTAROLINE IN AN IN VITRO HOLLOW-FIBER MODEL
Louie and colleagues (50) used a hollow-fiber model to analyze the PK/PD index of avibactam in combination with ceftaroline against two K. pneumoniae isolates (one expressing KPC-2, SHV-27, and TEM-1 and the other expressing CTX-M-15) and against an E. cloacae isolate expressing stably derepressed AmpC. In the first set of experiments, different doses of avibactam were administered as a continuous infusion in the presence of ceftaroline concentrations simulating human PK profiles following a dose of 600 mg q8h. These dose-ranging experiments enabled an identification of the effective 24-h AUC for avibactam, which informed the design of the dose fractionation experiments to identify the avibactam PK/PD index (50).
For the dose fractionation experiments, different dosage regimens of avibactam were given together with ceftaroline 600 mg q8h (50). In these experiments, each avibactam dosage regimen was designed to generate approximately the same 24-h AUC as that provided by an avibactam continuous infusion of 8 mg/liter (24-h AUC of 192 mg · h/liter). Four regimens of avibactam in combination with ceftaroline were administered: avibactam given as a continuous infusion, the whole exposure administered once daily, half the exposure administered twice daily, and one third of the total exposure administered q8h. One major difference between the studies of avibactam in combination with ceftaroline and those of the combination with ceftazidime or aztreonam was that instead of 24-h killing and the prevention of growth, the endpoint of the ceftaroline studies was the maintenance of a reduced bacterial CFU/ml and the prevention of outgrowth of resistant variants over 10 or 13 days (50). In dose fractionation experiments with K. pneumoniae expressing KPC-2, SHV-27, and TEM-1 (Fig. 4), the administration of the avibactam total daily dose (as a single dose) once daily failed by day 2 of treatment. Moreover, giving one half of the avibactam total daily dose every 12 h failed by day 4. In contrast, the more fractionated schedules of avibactam administration (where avibactam was given q8h or as a continuous infusion over 24 h) were successful in suppressing the emergence of resistance for the duration of the experiment (Fig. 4). Similar results were achieved with dose fractionation experiments using the K. pneumoniae CTX-M-15 isolate or the E. cloacae isolate. As the more fractionated avibactam dosage regimens maximized the time above the threshold concentration, these experiments showed that the activity of avibactam in combination with ceftaroline was clearly linked to fT>CT more closely than it was to fAUC (50).
FIG 4.
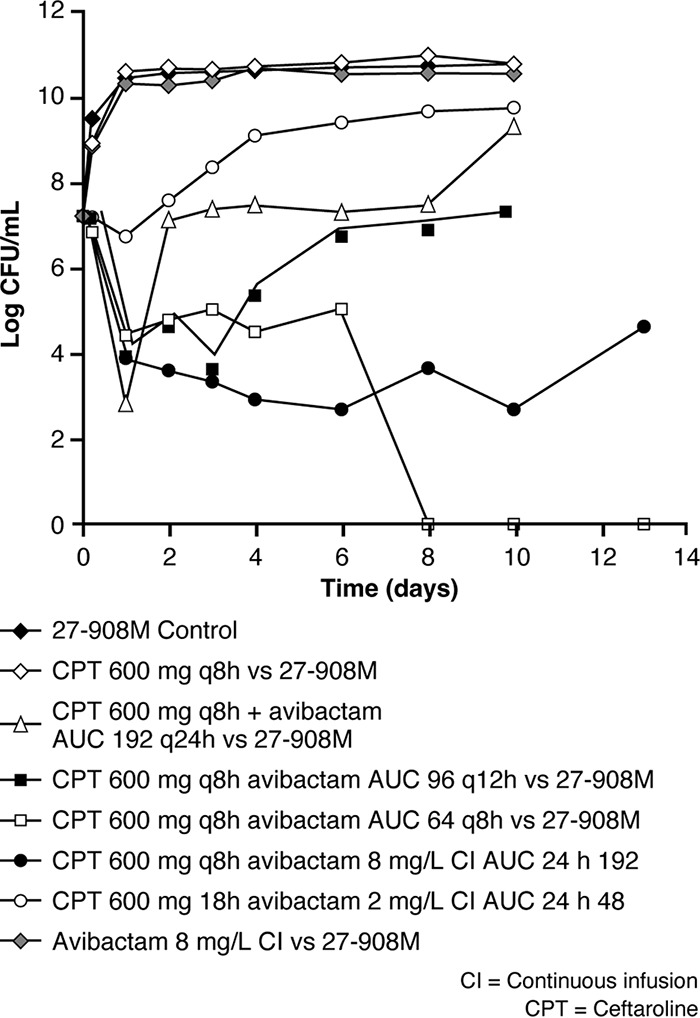
Dose fractionation study for avibactam in combination with ceftaroline 600 mg q8h against a K. pneumoniae strain in the hollow-fiber model. AUC, area under the concentration-time curve. Republished from reference 50.
PK/PD STUDIES OF AVIBACTAM IN COMBINATION WITH CEFTAROLINE OR CEFTAZIDIME IN A SINGLE-COMPARTMENT CONSTANT-VOLUME FERMENTER MODEL IN VITRO
MacGowan and colleagues (51) fractionated doses of avibactam combined with constant q8h dosing of either ceftazidime or ceftaroline in a one-compartment in vitro PK/PD model against each of three ceftazidime- and ceftaroline-resistant β-lactamase-producing isolates of Enterobacteriaceae (CTX-M-type-producing E. coli, stably derepressed AmpC-producing E. cloacae, and KPC-type-producing K. pneumoniae). Unlike the results of the hollow-fiber studies and neutropenic mouse models described above, the activity of avibactam in potentiating the bacterial killing by ceftazidime and ceftaroline over 24 h more closely fit the PK/PD indices fAUC and fCmax than the index fT>CT. The authors proposed that different experimental designs might produce differently distributed data. It was suggested that the greater number of observations at 0% fT>CT and 100% fT>CT in these experiments could have resulted in a better correlation to AUC and Cmax rather than the time above the threshold, in contrast to the other experiments described above which had fewer observations at the extremes. This potentially explains the different conclusions from this model compared with those from the other studies with avibactam combinations, all of which concluded that the avibactam fT>CT was the more closely fitting PK/PD index for avibactam than fAUC (51).
COMPARISON BETWEEN MODELS AND AVIBACTAM COMBINATIONS
Table 5 summarizes the PK/PD indices that best fit avibactam in the studies discussed above. For each combination, concentration-time profiles simulating predicted concentrations in human patients following dosing with clinically achievable regimens were studied in hollow-fiber models. Dose fractionation experiments were also conducted in neutropenic mice infection models for the ceftazidime-avibactam and aztreonam-avibactam combinations and in an in vitro single-compartment constant-volume fermenter model for ceftazidime-avibactam and ceftaroline fosamil-avibactam. Despite the different endpoints and durations of the experiments, in most of the models and across all the β-lactam combinations, there was an optimal fit between PD effect and avibactam fT>CT. The magnitude of CT varied between 0.5 mg/liter and 2.5 mg/liter depending on the model and the partner β-lactam. It is not unexpected for the CT of avibactam to be different depending on the partner β-lactam; therefore, it is important to study PK/PD indices for each different combination. The only exception has been that in the single-compartment fermenter model in which combinations of ceftazidime and ceftaroline (but not aztreonam) were studied, a better fit was obtained between the activity of avibactam and the index fAUC (and fCmax) than between the activity of avibactam and fT>CT in restoring the activities of both of these β-lactams (51). As discussed above, this could be a function of the experimental conditions in this particular analysis which resulted in a clustering of “time above” parameters at the extremes.
TABLE 5.
PK/PD indices for avibactam acting in combination with β-lactams under conditions of dynamic rising and falling concentration-time curves
| β-Lactamase inhibitor | β-Lactam | Model | Period (days) | Derived PK/PD index | Reference |
|---|---|---|---|---|---|
| Avibactam | Ceftazidime | Hollow fiber; entericsa | 1 | CT ≥0.5 mg/liter maintained β-lactamase-null phenotype | 43 |
| Avibactam | Ceftazidime | Neutropenic mouse thigh; P. aeruginosa | 1 | fT>CT 1 mg/liter | 44 |
| Avibactam | Ceftazidime | Neutropenic mouse lung; P. aeruginosa | 1 | fT>CT 1 mg/liter | 44 |
| Avibactam | Aztreonam | Hollow fiber; enterics | 1 | fT>CT 2–2.5 mg/liter | 49 |
| Avibactam | Aztreonam | Neutropenic mouse thigh; enterics | 1 | fT>CT 2–2.5 mg/liter | 49 |
| Avibactam | Ceftaroline | Hollow fiber; enterics | 10–13 | fT>CT | 50 |
| Avibactam | Ceftazidime | Constant-volume fermenter; enterics | 1 | fAUC | 51 |
| Avibactam | Ceftaroline | Constant-volume fermenter; enterics | 1 | fAUC | 51 |
Enterics, Enterobacteriaceae.
CLINICAL CONTEXT
The PKs of avibactam in combination with ceftazidime have been well characterized in population PK models developed using patient PK data from the ceftazidime-avibactam phase 3 studies (52). Joint target attainment was calculated using the models such that PTA was based on each patient achieving both the ceftazidime and avibactam targets simultaneously. From these models, the ceftazidime and avibactam PK/PD target plasma concentrations of >8 mg/liter for ceftazidime and >1 mg/liter for avibactam for more than 50% of the dosing interval have been predicted to be achieved in over 90% of patients dosed with ceftazidime-avibactam 2,000 mg + 500 mg q8h (52). These calculations of joint PTA supported ceftazidime-avibactam dosage selection and clinical breakpoint analyses (45).
The efficacy of the selected ceftazidime-avibactam dosage regimen with respect to comparators has been demonstrated across five phase 3 clinical trials, including a study which included only patients with infections caused by ceftazidime-resistant pathogens (53–57). It is important to consider whether antibiotic dosing strategies are adequate for the suppression of resistance in key target pathogens. Although the evidence for the emergence of resistance to ceftazidime-avibactam is limited, there have been isolated reports of the development of ceftazidime-avibactam resistance in some KPC-producing K. pneumoniae (58). In vitro models can be used to help identify PK/PD targets predicted to minimize the risk of selecting for resistance to the antibiotic in patients (19). This has been investigated for ceftaroline fosamil-avibactam (50) but not for ceftazidime-avibactam or aztreonam-avibactam.
CONCLUSIONS
The primary reason for conducting nonclinical PK/PD studies for antibiotics is to define quantitative PK/PD exposure targets that can be used to analyze the PTA among simulated populations of patients and thus guide the selection of appropriate doses for assessment in phase 2 and 3 clinical trials. For β-lactam–β-lactamase inhibitor combinations, it is important to determine the PK/PD index and magnitude for both components, as described here for avibactam in combination with the β-lactams ceftazidime, aztreonam, and ceftaroline fosamil. Across all combinations, with the exception of one experimental model in which combinations of ceftazidime or ceftaroline fosamil with avibactam were investigated (51), the PD of avibactam in restoring the antibacterial activity of the three partner β-lactams was found to be time dependent rather than concentration dependent (i.e., linked to fT>CT rather than fAUC). The magnitude of CT varied depending on the model and the partner β-lactam, emphasizing the importance of studying PK/PD indices for each different β-lactam–β-lactamase inhibitor combination. For ceftazidime-avibactam, PTA analyses based on the achievement of the preclinical PK/PD targets described here were used to guide the selection of ceftazidime-avibactam dosage regimens used in phase 3 clinical trials, which have demonstrated clinical efficacy similar to that of carbapenems in patients with cIAI, cUTI, and nosocomial pneumonia, including those with infections caused by ceftazidime-resistant pathogens (53–57).
ACKNOWLEDGMENTS
This review was sponsored by AstraZeneca and Pfizer.
AstraZeneca's rights to ceftazidime-avibactam, aztreonam-avibactam, and ceftaroline fosamil-avibactam were acquired by Pfizer in December 2016. W.W.N. is a former employee of and current shareholder in AstraZeneca and was a paid consultant to AstraZeneca and a principal in a partnership that was paid by Pfizer in connection with the development of the manuscript. S.D. is a former employee of and shareholder in AstraZeneca; P.N. is a former employee of AstraZeneca and still retains shares in AstraZeneca. T.R. is an employee of and shareholder in Allergan. I.A.C. is a former employee of Allergan and still retains shares in Allergan.
Editorial support was provided by Sirisha Bulusu, MBiochem at Prime, Knutsford, UK, and was funded by AstraZeneca and Pfizer. The opinions, conclusions, and interpretation of the data in this review are the responsibility of the authors.
REFERENCES
- 1.Mager DE, Woo S, Jusko WJ. 2009. Scaling pharmacodynamics from in vitro and preclinical animal studies to humans. Drug Metab Pharmacokinet 24:16–24. doi: 10.2133/dmpk.24.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bueters T, Gibson C, Visser SA. 2015. Optimization of human dose prediction by using quantitative and translational pharmacology in drug discovery. Future Med Chem 7:2351–2369. doi: 10.4155/fmc.15.143. [DOI] [PubMed] [Google Scholar]
- 3.Felmlee MA, Morris ME, Mager DE. 2012. Mechanism-based pharmacodynamic modeling. Methods Mol Biol 929:583–600. doi: 10.1007/978-1-62703-050-2_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee JY, Garnett CE, Gobburu JV, Bhattaram VA, Brar S, Earp JC, Jadhav PR, Krudys K, Lesko LJ, Li F, Liu J, Madabushi R, Marathe A, Mehrotra N, Tornoe C, Wang Y, Zhu H. 2011. Impact of pharmacometric analyses on new drug approval and labelling decisions: a review of 198 submissions between 2000 and 2008. Clin Pharmacokinet 50:627–635. doi: 10.2165/11593210-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 5.Sundqvist M, Lundahl A, Nagard MB, Bredberg U, Gennemark P. 2015. Quantifying and communicating uncertainty in preclinical human dose-prediction. CPT Pharmacometrics Syst Pharmacol 4:243–254. doi: 10.1002/psp4.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ambrose PG, Bhavnani SM, Rubino CM, Louie A, Gumbo T, Forrest A, Drusano GL. 2007. Pharmacokinetics-pharmacodynamics of antimicrobial therapy: it's not just for mice anymore. Clin Infect Dis 44:79–86. doi: 10.1086/510079. [DOI] [PubMed] [Google Scholar]
- 7.Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Durand-Reville TF, Lahiri S, Thresher J, Livchak S, Gao N, Palmer T, Walkup GK, Fisher SL. 2013. Kinetics of avibactam inhibition against class A, C, and D β-lactamases. J Biol Chem 288:27960–27971. doi: 10.1074/jbc.M113.485979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sader H, Castanheira M, Mendes RE, Flamm RK, Farrell DJ, Jones RN. 2015. Ceftazidime-avibactam activity against multidrug-resistant Pseudomonas aeruginosa isolated in U.S. medical centers in 2012 and 2013. Antimicrob Agents Chemother 59:3656–3659. doi: 10.1128/AAC.05024-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nichols WW, de Jonge BL, Kazmierczak KM, Karlowsky JA, Sahm DF. 2016. In vitro susceptibility of global surveillance isolates of Pseudomonas aeruginosa to ceftazidime-avibactam (INFORM 2012 to 2014). Antimicrob Agents Chemother 60:4743–4749. doi: 10.1128/AAC.00220-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Jonge BL, Karlowsky JA, Kazmierczak KM, Biedenbach DJ, Sahm DF, Nichols WW. 2016. In vitro susceptibility to ceftazidime-avibactam of carbapenem-nonsusceptible Enterobacteriaceae isolates collected during the INFORM global surveillance study (2012 to 2014). Antimicrob Agents Chemother 60:3163–3169. doi: 10.1128/AAC.03042-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allergan. 2018. AVYCAZ (ceftazidime-avibactam) for injection, for intravenous use. Prescribing information. http://www.allergan.com/assets/pdf/avycaz_pi Accessed 7 February 2018.
- 12.Pfizer. 2016. ZAVICEFTA. Summary of product characteristics. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/004027/WC500210234.pdf Accessed 7 February 2018.
- 13.Livermore DM, Mushtaq S, Warner M, Zhang J, Maharjan S, Doumith M, Woodford N. 2011. Activities of NXL104 combinations with ceftazidime and aztreonam against carbapenemase-producing Enterobacteriaceae. Antimicrob Agents Chemother 55:390–394. doi: 10.1128/AAC.00756-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crandon JL, Nicolau DP. 2013. Human simulated studies of aztreonam and aztreonam-avibactam to evaluate activity against challenging Gram-negative organisms, including metallo-β-lactamase producers. Antimicrob Agents Chemother 57:3299–3306. doi: 10.1128/AAC.01989-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biedenbach DJ, Kazmierczak K, Bouchillon SK, Sahm DF, Bradford PA. 2015. In vitro activity of aztreonam-avibactam against a global collection of Gram-negative pathogens from 2012 and 2013. Antimicrob Agents Chemother 59:4239–4248. doi: 10.1128/AAC.00206-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Castanheira M, Sader HS, Farrell DJ, Mendes RE, Jones RN. 2012. Activity of ceftaroline-avibactam tested against Gram-negative organism populations, including strains expressing one or more β-lactamases and methicillin-resistant Staphylococcus aureus carrying various staphylococcal cassette chromosome mec types. Antimicrob Agents Chemother 56:4779–4785. doi: 10.1128/AAC.00817-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pfizer 2017. NCT02655419. Determine the PK and safety and tolerability of ATM-AVI for the treatment of cIAIs in hospitalized adults (REJUVENATE). https://clinicaltrials.gov/ct2/show/NCT02655419 Accessed 7 February 2018.
- 18.Forest Laboratories. 2017. NCT01281462. Comparative study of coadministered ceftaroline fosamil and NXL104 vs. intravenous doripenem in adult subjects with complicated urinary tract infections. https://clinicaltrials.gov/ct2/show/NCT01281462 Accessed 7 February 2018.
- 19.European Medicines Agency. 2016. Guideline on the use of pharmacokinetics and pharmacodynamics in the development of antimicrobial medicinal products. Document EMA/CHMP/594085/2015. European Medicines Agency, London, United Kingdom: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/07/WC500210982.pdf Accessed 7 February 2018. [Google Scholar]
- 20.Nielsen EI, Friberg LE. 2013. Pharmacokinetic-pharmacodynamic modeling of antibacterial drugs. Pharmacol Rev 65:1053–1090. doi: 10.1124/pr.111.005769. [DOI] [PubMed] [Google Scholar]
- 21.Rex JH, Goldberger M, Eisenstein BI, Harney C. 2014. The evolution of the regulatory framework for antibacterial agents. Ann N Y Acad Sci 1323:11–21. doi: 10.1111/nyas.12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mouton JW, Brown DF, Apfalter P, Canton R, Giske CG, Ivanova M, MacGowan AP, Rodloff A, Soussy CJ, Steinbakk M, Kahlmeter G. 2012. The role of pharmacokinetics/pharmacodynamics in setting clinical MIC breakpoints: the EUCAST approach. Clin Microbiol Infect 18:E37–E45. doi: 10.1111/j.1469-0691.2011.03752.x. [DOI] [PubMed] [Google Scholar]
- 23.Muller AE, Punt N, Mouton JW. 2013. Optimal exposures of ceftazidime predict the probability of microbiological and clinical outcome in the treatment of nosocomial pneumonia. J Antimicrob Chemother 68:900–906. doi: 10.1093/jac/dks468. [DOI] [PubMed] [Google Scholar]
- 24.Kuti JL, Nicolau DP. 2005. Making the most of surveillance studies: summary of the OPTAMA program. Diagn Microbiol Infect Dis 53:281–287. doi: 10.1016/j.diagmicrobio.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Dudley MN, Ambrose PG, Bhavnani SM, Craig WA, Ferraro MJ, Jones RN, Antimicrobial Susceptibility Testing Subcommittee of the Clinical and Laboratory Standards Institute. 2013. Background and rationale for revised Clinical and Laboratory Standards Institute interpretive criteria (breakpoints) for Enterobacteriaceae and Pseudomonas aeruginosa: I. Cephalosporins and aztreonam. Clin Infect Dis 56:1301–1309. doi: 10.1093/cid/cit017. [DOI] [PubMed] [Google Scholar]
- 26.Drusano GL. 2004. Antimicrobial pharmacodynamics: critical interactions of ‘bug and drug’. Nat Rev Microbiol 2:289–300. doi: 10.1038/nrmicro862. [DOI] [PubMed] [Google Scholar]
- 27.Mouton JW, Dudley MN, Cars O, Derendorf H, Drusano GL. 2005. Standardization of pharmacokinetic/pharmacodynamic (PK/PD) terminology for anti-infective drugs: an update. J Antimicrob Chemother 55:601–607. doi: 10.1093/jac/dki079. [DOI] [PubMed] [Google Scholar]
- 28.Dudley MN. 1995. Combination β-lactam and β-lactamase-inhibitor therapy: pharmacokinetic and pharmacodynamic considerations. Am J Health Syst Pharm 52:S23–S28. [DOI] [PubMed] [Google Scholar]
- 29.Andes D, Craig WA. 2002. Animal model pharmacokinetics and pharmacodynamics: a critical review. Int J Antimicrob Agents 19:261–268. doi: 10.1016/S0924-8579(02)00022-5. [DOI] [PubMed] [Google Scholar]
- 30.Craig WA. 2003. Basic pharmacodynamics of antibacterials with clinical applications to the use of β-lactams, glycopeptides, and linezolid. Infect Dis Clin North Am 17:479–501. doi: 10.1016/S0891-5520(03)00065-5. [DOI] [PubMed] [Google Scholar]
- 31.Craig WA. 2007. Pharmacodynamics of antimicrobials: general concepts and applications, p 1–19. In Nightingale CH, Ambrose PG, Drusano GL, Murakawa T (ed), Antimicrobial pharmacodynamics in theory and clinical practice, 2nd ed Informa Healthcare, New York, NY. [Google Scholar]
- 32.MacVane SH, Kuti JL, Nicolau DP. 2014. Clinical pharmacodynamics of antipseudomonal cephalosporins in patients with ventilator-associated pneumonia. Antimicrob Agents Chemother 58:1359–1364. doi: 10.1128/AAC.01463-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeRyke CA, Kuti JL, Nicolau DP. 2007. Reevaluation of current susceptibility breakpoints for Gram-negative rods based on pharmacodynamic assessment. Diagn Microbiol Infect Dis 58:337–344. doi: 10.1016/j.diagmicrobio.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 34.Frei CR, Wiederhold NP, Burgess DS. 2008. Antimicrobial breakpoints for Gram-negative aerobic bacteria based on pharmacokinetic-pharmacodynamic models with Monte Carlo simulation. J Antimicrob Chemother 61:621–628. doi: 10.1093/jac/dkm536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.EUCAST. 2010. Ceftazidime. Rationale for the EUCAST clinical breakpoints, version 1.0. http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Rationale_documents/Ceftazidime_Rationale_Document_1.0_2010Nov.pdf Accessed 7 February 2018.
- 36.Flamm RK, Stone GG, Sader HS, Jones RN, Nichols WW. 2014. Avibactam reverts the ceftazidime MIC90 of European Gram-negative bacterial clinical isolates to the epidemiological cut-off value. J Chemother 26:333–338. doi: 10.1179/1973947813Y.0000000145. [DOI] [PubMed] [Google Scholar]
- 37.Sader H, Castanheira M, Flamm RK, Farrell DJ, Jones RN. 2014. Antimicrobial activity of ceftazidime-avibactam against Gram-negative organisms collected from U.S. medical centers in 2012. Antimicrob Agents Chemother 58:1684–1692. doi: 10.1128/AAC.02429-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levasseur P, Girard A-M, Claudon M, Goossens H, Black MT, Coleman K, Miossec C. 2012. In vitro antibacterial activity of the ceftazidime-avibactam (NXL104) combination against Pseudomonas aeruginosa clinical isolates. Antimicrob Agents Chemother 56:1606–1608. doi: 10.1128/AAC.06064-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walkty A, DeCorby M, Lagacé-Wiens P, Karlowsky J, Hoban D, Zhanel G. 2011. In vitro activity of ceftazidime combined with NXL104 versus Pseudomonas aeruginosa isolates obtained from patients in Canadian hospitals (CANWARD 2009 study). Antimicrob Agents Chemother 55:2992–2994. doi: 10.1128/AAC.01696-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang X, Zhang F, Zhao C, Wang Z, Nichols WW, Testa R, Li H, Chen H, He W, Wang Q. 2014. In vitro activity of ceftazidime-avibactam and aztreonam-avibactam against 372 Gram-negative bacilli collected in 2011 and 2012 from 11 teaching hospitals in China. Antimicrob Agents Chemother 58:1774–1778. doi: 10.1128/AAC.02123-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reference deleted.
- 42.Castanheira M, Farrell SE, Krause KM, Jones RN, Sader HS. 2014. Contemporary diversity of beta-lactamases among Enterobacteriaceae in the nine U.S. census regions and ceftazidime-avibactam activity tested against isolates producing the most prevalent beta-lactamase groups. Antimicrob Agents Chemother 58:833–838. doi: 10.1128/AAC.01896-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coleman K, Levasseur P, Girard AM, Borgonovi M, Miossec C, Merdjan H, Drusano G, Shlaes D, Nichols WW. 2014. Activities of ceftazidime and avibactam against β-lactamase-producing Enterobacteriaceae in a hollow-fiber pharmacodynamic model. Antimicrob Agents Chemother 58:3366–3372. doi: 10.1128/AAC.00080-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berkhout J, Melchers MJ, van Mil AC, Seyedmousavi S, Lagarde CM, Schuck VJ, Nichols WW, Mouton JW. 2016. Pharmacodynamics of ceftazidime and avibactam in neutropenic mice with thigh or lung infection. Antimicrob Agents Chemother 60:368–375. doi: 10.1128/AAC.01269-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.European Medicines Agency. 2016. Zavicefta European public assessment report. European Medicines Agency, London, United Kingdom: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004027/WC500210236.pdf Accessed 7 February 2018. [Google Scholar]
- 46.Cerexa, Inc. 2014. Briefing package, NDA 206494: ceftazidime-avibactam. Cerexa, Inc, Alameda, CA: https://www.pharmamedtechbi.com/~/media/Supporting%20Documents/The%20Pink%20Sheet%20DAILY/2014/December/12314%20FDA%20briefing%20docs.pdf. [Google Scholar]
- 47.Nicolau DP. 2015. Focus on ceftazidime-avibactam for optimizing outcomes in complicated intra-abdominal and urinary tract infections. Expert Opin Investig Drugs 24:1261–1273. doi: 10.1517/13543784.2015.1062873. [DOI] [PubMed] [Google Scholar]
- 48.Mawal Y, Critchley IA, Riccobene TA, Talley AK. 2015. Ceftazidime-avibactam for the treatment of complicated urinary tract infections and complicated intra-abdominal infections. Expert Rev Clin Pharmacol 8:691–707. doi: 10.1586/17512433.2015.1090874. [DOI] [PubMed] [Google Scholar]
- 49.Singh R, Kim A, Tanudra MA, Harris JJ, McLaughlin RE, Patey S, O'Donnell JP, Bradford PA, Eakin AE. 2015. Pharmacokinetics/pharmacodynamics of a β-lactam and β-lactamase inhibitor combination: a novel approach for aztreonam/avibactam. J Antimicrob Chemother 70:2618–2626. doi: 10.1093/jac/dkv132. [DOI] [PubMed] [Google Scholar]
- 50.Louie A, Castanheira M, Liu W, Grasso C, Jones RN, Williams G, Critchley I, Thye D, Brown D, Vanscoy B, Kulawy R, Drusano GL. 2012. Pharmacodynamics of β-lactamase inhibition by NXL104 in combination with ceftaroline: examining organisms with multiple types of β-lactamases. Antimicrob Agents Chemother 56:258–270. doi: 10.1128/AAC.05005-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.MacGowan A, Tomaselli S, Noel A, Bowker K. 2017. The pharmacodynamics of avibactam in combination with ceftaroline or ceftazidime against β-lactamase-producing Enterobacteriaceae studied in an in vitro model of infection. J Antimicrob Chemother 72:762–769. doi: 10.1093/jac/dkw480. [DOI] [PubMed] [Google Scholar]
- 52.Das S, Riccobene T, Carrothers T, Wright J, Macpherson M, Lovern M, Hing J, Xiong Y, Taylor A, Comisar C. 2017. Pharmacokinetic/pharmacodynamic validation of the ceftazidime-avibactam dose in patients with nosocomial pneumonia, including ventilator-associated pneumonia, poster P1185 27th European Congress of Clinical Microbiology and Infectious Diseases (ECCMID), Vienna, Austria, 22 to 25 April 2017. [Google Scholar]
- 53.Mazuski JE, Gasink LB, Armstrong J, Broadhurst H, Stone GG, Rank D, Llorens L, Newell P, Pachl J. 2016. Efficacy and safety of ceftazidime-avibactam plus metronidazole versus meropenem in the treatment of complicated intra-abdominal infection: results from a randomized, controlled, double-blind, phase 3 program. Clin Infect Dis 62:1380–1389. doi: 10.1093/cid/ciw133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carmeli Y, Armstrong J, Laud PJ, Newell P, Stone G, Wardman A, Gasink LB. 2016. Ceftazidime-avibactam or best available therapy in patients with ceftazidime-resistant Enterobacteriaceae and Pseudomonas aeruginosa complicated urinary tract infections or complicated intra-abdominal infections (REPRISE): a randomised, pathogen-directed, phase 3 study. Lancet Infect Dis 16:661–673. doi: 10.1016/S1473-3099(16)30004-4. [DOI] [PubMed] [Google Scholar]
- 55.Wagenlehner FM, Sobel JD, Newell P, Armstrong J, Huang X, Stone GG, Yates K, Gasink LB. 2016. Ceftazidime-avibactam versus doripenem for the treatment of complicated urinary tract infections, including acute pyelonephritis: RECAPTURE, a phase 3 randomized trial program. Clin Infect Dis 63:754–762. doi: 10.1093/cid/ciw378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qin X, Tran BG, Kim M, Wang L, Nguyen D, Chen Q, Song J, Laud PJ, Stone GG, Chow JW. 2017. A randomized, double-blind, phase 3 study comparing the efficacy and safety of ceftazidime/avibactam plus metronidazole versus meropenem for complicated intra-abdominal infections in hospitalised adults in Asia. Int J Antimicrob Agents 49:579–588. doi: 10.1016/j.ijantimicag.2017.01.010. [DOI] [PubMed] [Google Scholar]
- 57.Torres A, Zhong N, Pachl J, Timsit JF, Kollef M, Chen Z, Song J, Taylor D, Laud PJ, Stone GG, Chow JW. 2018. Ceftazidime-avibactam versus meropenem in nosocomial pneumonia, including ventilator-associated pneumonia (REPROVE): a randomised, double-blind, phase 3 non-inferiority trial. Lancet Infect Dis 18:285–295. doi: 10.1016/S1473-3099(17)30747-8. [DOI] [PubMed] [Google Scholar]
- 58.Shields RK, Chen L, Cheng S, Chavda KD, Press EG, Snyder A, Pandey R, Doi Y, Kreiswirth BN, Nguyen MH, Clancy CJ. 2017. Emergence of ceftazidime-avibactam resistance due to plasmid-borne blaKPC-3 mutations during treatment of carbapenem-resistant Klebsiella pneumoniae infections. Antimicrob Agents Chemother 61:e02097-16. doi: 10.1128/AAC.02097-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clinical and Laboratory Standards Institute. 2013. Performance standards for antimicrobial susceptibility testing; 23rd informational supplement. CLSI document M100-S23. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]