

ABSTRACT
In Toxoplasma gondii, calcium-dependent protein kinase 1 (CDPK1) is an essential protein kinase required for invasion of host cells. We have developed several hundred CDPK1 inhibitors, many of which block invasion. Inhibitors with similar 50% inhibitory concentrations (IC50s) were tested in thermal shift assays for their ability to stabilize CDPK1 in cell lysates, in intact cells, or in purified form. Compounds that inhibited parasite growth stabilized CDPK1 in all assays. In contrast, two compounds that showed poor growth inhibition stabilized CDPK1 in lysates but not in cells. Thus, cellular exclusion could explain exceptions in the correlation between the action on the target and cellular activity. We used thermal shift assays to examine CDPK1 in two clones that were independently selected by growth in the CDPK1 inhibitor RM-1-132 and that had increased 50% effective concentrations (EC50s) for the compound. The A and C clones had distinct point mutations in the CDPK1 kinase domain, H201Q and L96P, respectively, residues that lie near one another in the inactive isoform. Purified mutant proteins showed RM-1-132 IC50s and thermal shifts similar to those shown by wild-type CDPK1. Reduced inhibitor stabilization (and a presumed reduced interaction) was observed only in cellular thermal shift assays. This highlights the utility of cellular thermal shift assays in demonstrating that resistance involves reduced on-target engagement (even if biochemical assays suggest otherwise). Indeed, similar EC50s were observed upon overexpression of the mutant proteins, as in the corresponding drug-selected parasites, although high levels of CDPK1(H201Q) only modestly increased resistance compared to that achieved with high levels of wild-type enzyme.
KEYWORDS: Toxoplasma gondii, bumped kinase inhibitor, cellular thermal shift assay, inhibitor, protein kinase
INTRODUCTION
Toxoplasma gondii is an obligate intracellular parasite that causes toxoplasmic encephalitis in immunocompromised individuals, birth defects when acquired in pregnancy, and ocular damage, whether infection is congenital or acquired later in life (1). Infections have been experimentally linked to behavioral changes in animals and statistically linked to schizophrenia in humans, which may result from the parasite's predilection to reside in the brain (1, 2). After initial infection (usually asymptomatic), the parasites persist as dormant cysts and periodically reemerge, causing cell lysis followed by reinfection. Unfortunately, treatments for T. gondii infections are suboptimal (3), even though one-third of the world's population is estimated to be infected with the parasite (1).
We have taken a target-directed approach to discover new candidates to treat T. gondii infection. The chosen target is calcium-dependent protein kinase 1 (CDPK1), originally discovered for its role in invasion (4) and now known to be essential for both invasion and egress (5–7). Since both of these steps are essential to the proliferation of the parasite in the mammalian host, inhibition of CDPK1 would likely reduce or abrogate pathogenesis. Importantly, CDPK1 has an ATP-binding site that is larger than the ATP-binding sites found in most protein kinases, since the gatekeeper residue at the back of the pocket is glycine, the smallest amino acid, rather than the larger amino acids present in most other kinases. Indeed, no glycine gatekeeper protein kinases are found in humans, and CDPK1 is the only such kinase in T. gondii. Hence, we and others previously developed inhibitors that complement the enlarged ATP-binding pocket of CDPK1 (bumped kinase inhibitors [BKIs]), many of which were active against parasites (8–12). A general structure of BKIs is depicted in Fig. 1A. Several optimized inhibitors have now been used successfully in animal infection models (9, 13–15). We also demonstrated that CDPK1 with a methionine substitution for the gatekeeper residue (G128M) has higher 50% inhibitory concentrations (IC50s) for BKIs, and when parasites overexpress CDPK1(G128M), 50% effective concentrations (EC50s) are higher than those for parasites overexpressing the wild-type (WT) enzyme (5, 12, 16). Thus, mutation of the gatekeeper residue could lead to resistance to these BKIs in vivo. CDPKs also have a calcium activation domain (CAD) containing four EF hands plus a junction region. The CAD rotates with respect to the kinase domain upon binding calcium (Fig. 1B), with both relieving the autoinhibition that blocks the peptide binding site (17) and stabilizing the active form of the enzyme (18). BKIs bind both the active (when Ca2+ is present) and inactive (when Ca2+ is absent) isoforms of CDPK1 (19).
FIG 1.

TSAs assessing the BKI RM-1-132 interaction with CDPK1. (A) General structures of BKIs. The hinge region of the kinases forms hydrogen bonds with the adenine ring of ATP. The hydrophobic pocket contains the gatekeeper residue. (B) Crystal structures of CDPK1 in the inactive (without calcium [− calcium]) and active (with calcium [+ calcium]) states. (Left) The inactive-state active site contains AMP; (right) the active conformation (PDB accession number 3HX4) has four Ca2+ ions bound to the CAD and an ATP analog in the active site. The αC helix of the protein kinase domain and the activation segment are in cyan. The kinase domain is in dark gray. The CAD is in light gray, and the CAD helices discussed are labeled. (C) Thermal melt curves of purified recombinant CDPK1 in the presence or absence of the BKI RM-1-132. (Left) Data in the presence or absence of 1 mM added calcium; (right) data in the presence or absence of 1 mM EGTA. Fluorescence units (FU) are given in thousands. (D) L-CETSA. Three experiments were performed with DMSO, and two experiments were performed with RM-1-132. Each experiment used duplicate tests, the results of which were averaged; different experiments are indicated by different symbols. The lines are drawn to facilitate viewing, as temperature points were adjusted in some assays. An example of primary Western blot data is shown in Fig. S3 in the supplemental material. (E) IC-CETSA. Six experiments were performed with DMSO, and two experiments were performed with RM-1-132 (all in duplicate, except one DMSO experiment was done once). The lines are drawn to facilitate viewing, as temperature points were adjusted in some assays.
Compounds that interact with proteins often lead to stabilization of the protein in the presence of thermal stress. For purified proteins, thermal shift assays (TSAs) can be particularly useful when an enzymatic assay is not available or convenient. However, TSAs using purified protein do not reveal whether target engagement is favored in complex mixtures, such as cell lysates, in which other components may compete for the ligand, or when ligands must cross biological membranes, as with intact cells. Hence, Martinez Molina et al. (20) recently developed complementary cellular thermal shift assays (CETSAs) that utilize cell lysates (L-CETSAs) or intact cells (IC-CETSAs). While they are more cumbersome than assays using purified protein, such studies can reveal whether target engagement occurs under more relevant conditions. Subsequently, CETSAs have been used with protein kinase inhibitors both across the proteome (21) and to confirm engagement of the target(s) (22, 23). We recently used the L-CETSA to confirm the target engagement of selected BKIs with CDPK1 of the equine protozoan pathogen Sarcocystis neurona (24). Here, we report the use of these assays with T. gondii CDPK1 (TgCDPK1) to demonstrate that all BKIs tested, which had low-nanomolar IC50s, stabilized CDPK1 in cell lysates. In contrast, only the compounds that were effective in preventing parasite invasion of the mammalian host cell or inhibiting parasite growth stabilized CDPK1 in intact cells. We then selected two independent clonal lines with modest resistance to BKIs (4- to 10-fold) and found that each had a point mutation in CDPK1. Although the mutant CDPK1s in purified form still showed low-nanomolar IC50s, both L-CETSAs and IC-CETSAs revealed reduced thermal shifts compared to those for the wild-type enzyme, illustrating the value of observing target engagement in a cellular setting.
RESULTS
TSAs and CETSAs demonstrate BKI interactions with CDPK1.
Often, compounds effectively inhibit a purified target protein yet lack cellular activity and thus are noted to be exceptions to the cellular structure-activity relationship (SAR). The recently developed CETSA protocols allow answering the question of whether compound binding occurs in complex mixtures (i.e., cell lysates) and in intact cells (20). Since BKIs target the ATP-binding pocket of CDPK1, they compete in vivo with high levels of ATP for access to CDPK1. It is also possible that other protein kinases, such as mitogen-activated protein kinase-like 1 (MAPKL-1; which binds the BKI NM-PP1 [25]), bind to BKIs. We therefore implemented two variations, L-CETSAs and IC-CETSAs, which monitor target engagement in cell lysates and intact cells, respectively, to evaluate the several compounds that we had previously studied for their inhibition of purified recombinant CDPK1 and their effects on T. gondii invasion and growth.
The compounds chosen are listed in Table 1 (their structures are provided in Fig. S1 in the supplemental material). All compounds had low-nanomolar IC50s when tested against purified recombinant CDPK1 in a Kinase-Glo enzyme assay and had predominantly monophasic curves in the invasion/proliferation assays used to determine the EC50s. Three compounds (compounds 1294, 1553, and RM-1-132) showed good activity against T. gondii in the invasion/proliferation assay, while one had modest activity (compound 1568) and the last one had little activity (compound 1265). The data are summarized in Table 1.
TABLE 1.
Interaction of BKIs with CDPK1
| Compounda | IC50 (μM)b | EC50 (μM)c | Thermal shiftd (°C) by: |
||
|---|---|---|---|---|---|
| TSA | L-CETSA | IC-CETSA | |||
| 1265 | 0.004 | >6.25 | 7.4 | 6.6 | 0.7 |
| 1294 | 0.003 | 0.14 | 11.9 | 10.9 | 5.0 |
| 1553 | 0.001 | 0.06 | 12.0 | 10.1 | 3.3 |
| 1568 | 0.001 | 0.76 | 11.3 | 5.9 | 0.8 |
| RM-1-132 | 0.003 | 0.09 | 12.3 | 12.1 | 6.0 |
The EC50s and IC50s of compounds 1265, 1294, and RM-1-132 (12) and of compound 1553 (9) were published previously. The RM-1-132 EC50 shown here includes additional data from the current work.
The assay likely does not discriminate between IC50s of 1 nM or less.
EC50 in a 48-h invasion/growth assay.
Shifts in the measured Tm compared to that for the solvent (DMSO) control. CDPK1 Tms were as follows: for purified recombinant protein (no EGTA), 50.7 ± 0.9°C; for lysate, 48.0 ± 0.5°C; and for intact cells, 50.1 ± 1°C.
Figure 1C shows examples of the TSA results obtained using purified CDPK1 with the solvent control and compound RM-1-132 (20 μM). Although the TSA buffer contains no added calcium, we tested the effect of chelating any trace calcium that was present using EGTA. Surprisingly, the melting temperature (Tm) was increased in the absence of calcium (Fig. 1C), as confirmed by the titration of both calcium and EGTA (Fig. S2). This result seems counterintuitive, since calcium activates the enzyme. However, as noted above, the interactions between the CAD and the protein kinase domain are completely different in the presence and absence of calcium (Fig. 1B). Without calcium, the calcium activation domain occludes the peptide binding site and a long helix forms from the junction region (the region between the kinase domain and the first EF hand) and the first portion of EF hand 1 (5). Upon calcium binding, the calcium activation domain rotates to the opposite face of the molecule and the long helix is bent into three separate helices (26) (Fig. 1B). The purified protein showed a large shift upon addition of BKIs whether calcium was present or absent (Fig. 1C).
For L-CETSAs and IC-CETSAs, the concentration of BKIs was decreased to 2 μM to reflect a more relevant scenario that might be achieved in vivo. For example, in mice, compound 1294 yielded an average plasma level of 2 μM at 24 h (13) and compound 1553 reached a steady-state plasma concentration of 6 μM (24). After incubation with BKIs, the samples were heated at the selected temperatures and the remaining soluble CDPK1 was quantitated by Western blotting, as described in Materials and Methods (see the example results in Fig. 1D and E and Fig. S3). Comparison of Ponceau-stained blots with and without BKIs indicated that the compounds did not induce nonspecific protein aggregation (see the example results in Fig. S3). As summarized in Table 1, all compounds stabilized CDPK1 in cell lysates, with three compounds having similarly large shifts of approximately 10°C and two having more moderate shifts of approximately 6°C, despite the similarity of the IC50s (measured at 1 to 4 nM). However, it is possible that some compounds had IC50s lower than those indicated since the assay mixture contained enzyme at a 2.1 nM protein concentration (the proportion of active enzyme is unknown). Those BKIs with weaker shifts in the L-CETSA corresponded to those with poorer EC50s in the growth/invasion assay. The reason for the differential shifts in L-CETSAs is unclear, although the ligand-induced shift size for purified proteins (27, 28) and proteins in cell lysates (21) generally tracks with the Ki. When these compounds were tested for their ability to stabilize CDPK1 in intact cells via IC-CETSAs, compounds 1265 and 1568, the two compounds that had relatively high EC50s (above 0.5 μM), had negligible shifts, demonstrating a lack of target engagement in intact cells and explaining the lack of an effect on parasite invasion and growth. The three BKIs that had relatively low EC50s (below 0.2 μM) had moderate IC-CETSA shifts. These shifts were not as pronounced as those seen by L-CETSA; the smaller shift in IC-CETSA reflected both a higher Tm without drugs (50.9°C versus 48°C) and a lower Tm with drugs. The latter could reflect the limited permeation of the compound into the cell during the 60-min incubation time. The former could reflect the presence of relatively high concentrations of the natural ligand ATP in the intact parasites, which is diluted when the parasites are lysed for L-CETSA, as was previously shown for many ATP-binding proteins (21). Additionally, the conformation of CDPK1 could differ in the two assays. In the L-CETSA, CDPK1 should be fully active due to the presence of 1 mM calcium in the buffer, while inside cells (IC-CETSA), activation may be heterogeneous, although extracellular T. gondii parasites in calcium-containing medium like that used here may have increased spikes of intracellular calcium compared to parasites within host cells (29). The means of measuring stabilization is different in these CETSAs than in TSAs utilizing purified proteins. The former relies on protein aggregation as the endpoint, while the latter relies on a fluorogenic compound binding to newly exposed hydrophobic regions. This biophysical difference could lead to some divergence in assay results beyond those attributable to the complex milieu of CETSAs.
Selection of T. gondii with low-level resistance to the BKI RM-1-132.
Having established the assays for T. gondii CDPK1, we decided to apply it to examine T. gondii resistance to CDPK1 inhibitors. Previous work showed that mutation of the gatekeeper residue at the back of the ATP-binding pocket from glycine to methionine (G128M) reduces the size of the pocket, rendering the enzyme and the cells overexpressing it resistant to several BKIs (5, 6, 12). Similarly, this mutation rendered the parasites resistant to RM-1-132 (Fig. S4). We subjected the T. gondii RH strain to selection with the BKI RM-1-132 at the EC90 (0.8 μM). After 16 passages, the assays showed a shift in EC50 for the nonclonal populations from both flasks (A and C) compared to that for the parental cell line (this shift was less prominent than the shift seen with the G128M mutant; Fig. S5). At the same time, clonal cell lines were isolated from the RM-1-132-selected cell cultures and drug selection was dropped.
Multiple clones were isolated from each flask, and representatives were tested for their EC50 with RM-1-132 (Fig. 2A). These clones showed a shift similar to that for the nonclonal, selected populations, with a 4-7-fold shift for the flask A clones and a −8- to 13-fold shift for the flask C clones (Fig. S5). Preliminary data (not shown) suggested that the lines also showed modest resistance to other structurally related BKIs (unrelated BKIs were not tested). Clones A-f8 and C-d12 were selected for further study.
FIG 2.
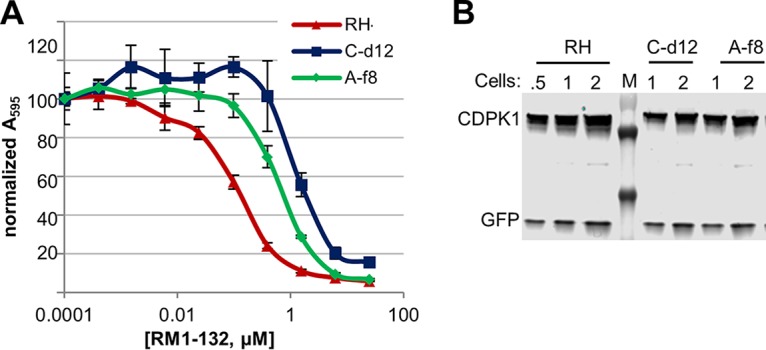
RM-1-132-resistant clones. (A) Clones C-d12 and A-f8 show higher EC50s for RM-1-132. Following selection and cloning of transfectants, 2-day invasion/growth assays were performed as described in Materials and Methods. (B) Western blot demonstrating that mutant CDPK1s are not overexpressed. GFP is expressed from another locus and was used as a loading control. Cell numbers are in millions. Markers (M) are 70 (faint), 55, and 35 kDa, from top to bottom, respectively.
Previous work has shown that CDPK1 functions at the time of invasion (4); BKI EC50s are much higher when compounds are added after invasion (5). In preliminary studies, we observed that when RM-1-132 was added after invasion, clone C-d12 was not more resistant than the wild-type parent (EC50, 2.7 μM versus 4.1 μM). This supports the contention that the resistance observed in C-d12 is at the level of the invasion pathway (which includes CDPK1) and not at the level of intracellular growth (where CDPK1 does not play a role). Western blot analysis showed no evidence of overexpression of CDPK1 in the mutant cells, and clone C-d12 had somewhat less CDPK1 than the parental RH clone (Fig. 2B).
After isolation of RNA and cDNA synthesis, PCR yielded the CDPK1 coding sequence (CDS), which in each case was sequenced. Clones from flask A had histidine 201 within the activation loop of the kinase domain replaced by glutamine (H201Q), and those from flask C had leucine 96 on helix αC (a critical regulatory element in many kinases) replaced by proline (L96P). In the active conformation, these residues are distant from each other (14.8 Å), but in the inactive conformation (without calcium), these two residues are spatially adjacent, with their respective side chains being in van der Waals contact (Fig. 1B and 3). This interaction could help stabilize the close association of the αC-helix consisting of residues 92 to 106 with the short helical segment containing residues 197 to 201. Mutation of leucine 96 to proline, effectively removing the side chain, would reduce this interaction surface and thus would be expected to destabilize the low-calcium conformation of the protein. The consequence of mutating histidine 201 to glutamine is less easy to predict. However, in neither the high- nor the low-calcium conformations were these residues particularly close to the ATP/RM-1-132 binding site, suggesting that the mutations would not directly perturb inhibitor binding.
FIG 3.

Location of amino acids mutated in CDPK1 in clones A-f8 (H201Q) and C-d12 (L96P). The images are close-ups of the region of CDPK1 where mutations are located. The kinase domain is in green, with its activation segment (ACT) in yellow, and the CAD-EF hand domain is in blue. The locations of the mutations are shown as stick residues (WT residues are shown) along with their van der Waals surfaces, based on structures in the presence or absence of calcium. The PDB accession number for the active form (with calcium) is 3HX4, and the PDB accession number for the inactive form (without calcium) is 3SX4.
Site-directed mutagenesis was used to mutate the WT CDPK1 Escherichia coli expression plasmid so that it expressed the CDPK1(H201Q) and CDPK1(L96P) mutants found in the resistant clonal lines Af-8 and C-d12, respectively. The proteins were expressed in E. coli and purified, and their activity was tested in a Kinase-Glo assay using a peptide substrate (30). Under the conditions used, both proteins showed robust activity. Surprisingly, the IC50s for RM-1-132 were similar, being 2 to 4 nM for the mutant and WT enzymes (Table 2). In preliminary studies using the Kinase-Glo assay, we estimated the Kms for ATP and the peptide substrate of the mutant proteins (for the H201Q mutant, the ATP Km was 5.5 μM and the peptide Km was 14.7 μM; for the L96P mutant, the ATP Km was 8.4 μM and the peptide Km was 18.5 μM). Comparison with WT CDPK1 (ATP Km, 12.4 μM; peptide Km, 20.5 μM [5]) did not provide an explanation for the differential EC50s observed for the selected populations. It is important to note that the assay was conducted with a small peptide substrate, and it is still possible that the affinity for protein substrates differs for the mutant proteins.
TABLE 2.
Effects of RM-1-132 on WT and mutant CDPK1 and parasites
| Cell line (CDPK1) | EC50 shifta (μM) | IC50 (μM) | TSA |
TSA + EGTA |
L-CETSAc |
IC-CETSAc |
||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tm (°C) | Thermal shift (°C) | Tm (°C) | Thermal shift (°C) | Tm (°C) | Thermal shift (°C) | Tm (°C) | Thermal shift (°C) | |||
| RH (WT) | NA | 0.003 | 50.7 ± 0.9b | 12.3 ± 0.5 | 54.8 | 11.3 | 48.0 | 12.2 | 50.9 | 6.0 |
| A-f8 (H201Q) | 4.1 | 0.002 | 47.8 ± 0.6 | 11.4 ± 0.5 | 51.0 | 11.2 | 49.4 | 9.0 | 50.1 | 3.0 |
| C-d12 (L96P) | 9.7 | 0.004 | 43.4 ± 0.8 | 9.2 ± 0.5 | 48.1 | 9.2 | 49.9 | 6.0 | 48.5 | 2.7 |
EC50 fold shift compared to that for the RH parental strain. NA, not applicable.
The standard deviation is provided when at least three separate assays were done.
Data points for the cell lysate and intact cell assays are shown in Fig. 5B.
Use of TSAs and CETSAs to characterize resistant clones.
We used TSAs to compare the mutant proteins with WT CDPK1. Both proteins had lower Tms than WT CDPK in the presence of calcium (Table 2), as well as in its absence. Comparison of the results in Fig. 1C and 4A shows that the thermal melt curves for CDPK1(L96P) were dramatically different from those for the WT, being quite flat in the presence of calcium. The curve for CDPK1(H201Q) was more similar to that for the WT (not shown). Addition of EGTA at a final concentration of 200 μM (or more) to the reaction mixtures normalized the thermal melt plots for the mutants, in addition to increasing the thermal stability of all the enzymes (including the WT) by 4 to 5°C. As with WT CDPK1, the addition of RM-1-132 dramatically enhanced the stability of both mutant proteins in buffer containing calcium, as well as when EGTA was present.
FIG 4.

TSAs and CETSAs of CDPK1 mutant clones. (A) TSAs using purified recombinant CDPK1(L96P). Comparison of the top and bottom panels shows that the chelation of calcium or the addition of RM-1-132 stabilizes the mutant protein, although the stability remains lower than that of the WT (Fig. 1C). Fluorescence units (FU) are given in thousands. (B) CETSAs using each clonal line. (Left) L-CETSAs; (right) IC-CETSAs. CDPK1 clone A-f8 has the H201Q mutation, while clone C-d12 has the L96P mutation. Each symbol indicates the result of a separate duplicate assay.
In L-CETSA studies, CDPK1 in the mutant lines showed clear evidence of binding to RM-1-132, although the shift in Tm was smaller than that for the WT parent (Table 2 and Fig. 4B). This contrasts with the findings of preliminary L-CETSA studies of parasites expressing the gatekeeper mutant CDPK1(G128M), where the thermal shift was reduced to ∼1.4°C (not shown). The CETSAs showed some differences from the TSAs when purified protein was used. For example, the decreased stability of CDPK1(L96P) observed using purified protein was not obvious in lysates and was only hinted at in whole-cell CETSAs (Fig. 4B). Additionally, in contrast to the large shift in Tm observed for the purified mutant proteins treated with RM-1-132, in IC-CETSAs the shift for the mutant cell lines was only slightly above the 2°C cutoff that is often taken as indicative of target engagement. Thus, despite the similar IC50s and thermal shifts observed in TSAs and L-CETSAs for the WT and mutant CDPK1s, target engagement by the mutant CDPKs in intact cells was near the limit of detection by IC-CETSAs.
We attempted to recapitulate the cellular phenotype by overexpressing hemagglutinin (HA)-tagged WT and the CDPK1(H201Q) and CDPK1(L96P) mutants. Clones overexpressing CDPK1 and CDPK1(H201Q) were easily obtained, but despite numerous stable CDPK1(L96P) transfectants being isolated, only one clone that expressed the CDPK1(L96P) protein was found. This could suggest that overexpression of CDPK1(L96P) is deleterious. Western blot assays showed that HA-tagged WT CDPK1 and the CDPK1(H201Q) mutant were both expressed to about 10-fold higher levels than endogenous CDPK1 (Fig. 5A). In contrast, CDPK1(L96P) was expressed at lower levels, being approximately equal in abundance to the endogenous WT protein (Fig. 5A). Figure 5B compares the EC50s of the parental strain, resistant clones, and transfectants. Expression of CDPK1(L96P) at endogenous levels recapitulated the C-clone resistance phenotype, with a 6.4-fold increase in EC50 over that for the parental RH clone. High-level expression of CDPK1(H201Q) caused a 5.3-fold shift in the EC50 compared to that for the parental RH clone, similar to what was seen with the A clones (6.5-fold). This compares with a 3.9-fold shift upon high-level expression of the WT protein.
FIG 5.
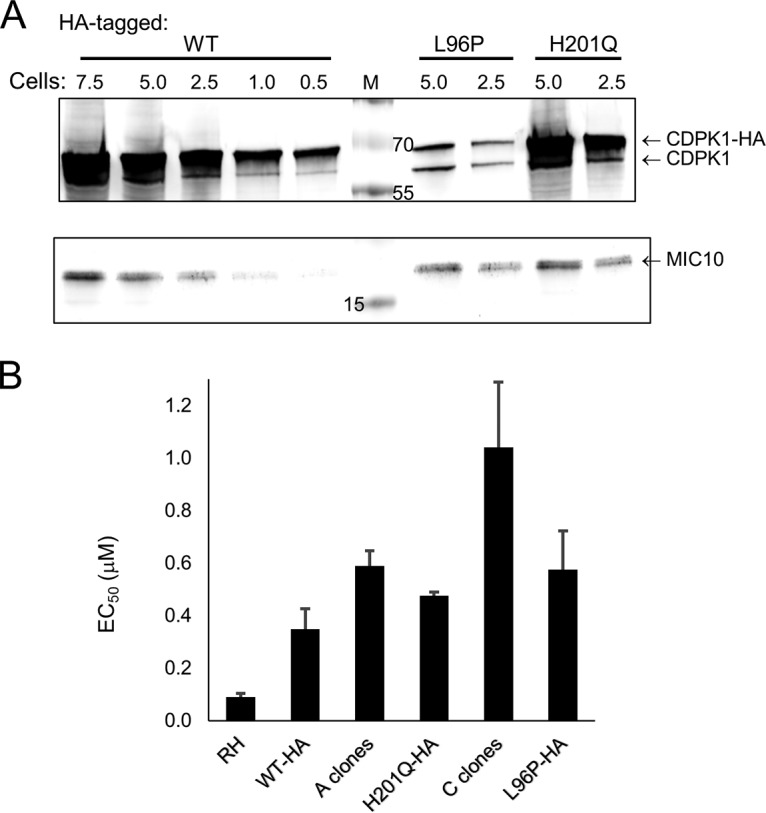
Transfectants expressing HA-tagged CDPK1 proteins. (A) Western blot of parasites overexpressing WT and the indicated mutant CDPK1-HA proteins. Endogenous CDPK1 was also expressed. Lysates bearing the specified number of parasites (in millions) were separated by SDS-PAGE and blotted with rabbit anti-CDPK1 and rabbit anti-MIC10 (a gift of Vern Carruthers) (37). MIC10 was used as a loading control on the same gel and indicates that the H201Q mutant lanes were somewhat overloaded. Mouse anti-HA.11 (clone 16B12; Covance) was used to demonstrate that the upper band revealed with anti-CDPK1 in each lane represents the HA-tagged recombinant protein (not shown). Lane M, molecular markers (in kilodaltons). (B) RM-1-132 EC50s of T. gondii expressing CDPK1 mutant proteins. The clones expressing HA-tagged proteins also expressed endogenous CDPK1. Data were collected in triplicate within each assay, and assays were repeated four or more times [except for the assay with CDPK1(H201Q)-HA, which was done twice]. The bars represent the standard error of the mean.
DISCUSSION
CDPK1 is an established drug target for T. gondii with genetic and chemical validation. Multiple CDPK1 inhibitors have been developed, showing a range of IC50s and EC50s in vitro, and some of them, including compounds 1294 and 1553, have been shown to be efficacious in animal models (9, 13, 16, 31). Compounds inhibiting CDPK1 orthologues of several other pathogens, such as Cryptosporidium spp., Babesia spp., and Sarcocystis neurona, also showed potential in animal models (19, 24, 32). Given the potential of the compounds targeting CDPK1, we undertook studies to assess target engagement in complex environments, such as cell lysates and intact cells. Those studies showed that compounds with similar, low-nanomolar IC50s stabilized CDPK1 in cell lysates but not necessarily to the same degree. The two compounds with the poorest EC50s stabilized CDPK1 in lysates to a lesser degree. Furthermore, in IC-CETSAs, those compounds did not show evidence of engaging the target in whole cells. This suggests that some molecules in our series were excluded from the cellular environment, explaining why some potent inhibitors of the TgCDPK1 enzyme lack cellular potency and, thus, are exceptions to cellular structure-activity relationship (SAR) trends. Therefore, TSAs using purified protein combined with L-CETSA and IC-CETSA data can be useful in exploring exceptions to cellular SAR, especially in systems where target validation is challenging or when phenotypes are not apparent outside the cellular setting.
CDPK1 is probably one of the more complex molecules that have been studied by TSAs and CETSAs: it has two natural intracellular ligands, ATP and calcium, which bind two distinctly folded domains that interact with one another. Thus, in the intact cell one can anticipate at least four isoforms with respect to ligand binding: isoforms that occur with and without ATP and isoforms that occur with and without calcium. Incomplete titration of the protein's four distinct calcium binding sites at nonsaturating calcium concentrations may generate additional intermediate isoforms. While binding of calcium to EF-hand proteins, such as centrins, results in greater thermal stability, such an effect in CDPK1 is apparently swamped by the large reorganization of the protein via movement of the CAD to the opposite face of the kinase domain, since decreased thermal stability was observed. Additionally, there is likely heterogeneity in phosphorylation. One phosphorylation site has been reproducibly observed on CDPK1: serine 61 in the ATP-binding site (33). This phosphorylation is likely inhibitory since examination of the structure shows that the phosphoserine would clash with the phosphate tail of ATP. It also could reduce interactions of CDPK1 with inhibitors in CETSAs. However, the physiological relevance of this modification and the kinase and phosphatase involved remain unknown.
We were able to select two independent lines of parasites with modest resistance to the BKI RM-1-132. Both had a single mutation in CDPK1, L96P or H201Q, residues that appear to interact with one another in the inactive form of the target enzyme. The close juxtaposition of the two residues that were mutated in separate resistant cell lines in the inactive conformation of CDPK1 raises the possibility that this region may contribute to maintaining the inactive conformation, analogous to the allosteric effect described for the junction region and the active conformation (18). Possibly, these mutations destabilize the inactive version, making it easier for the enzyme to transition to the active state, effectively raising the concentration of active enzyme under certain conditions.
Surprisingly, purified recombinant CDPK1 with these mutations did not show detectable changes in the RM-1-132 IC50. However, the thermal shifts induced by BKIs in CDPK1 of the mutant cell lines were reduced in intact cells compared to the parental parasites, and expression of CDPK1(L96P) at endogenous levels produced a resistance phenotype that largely recapitulated the RM-1-132 resistance phenotype seen in the in vitro-selected C clones. Furthermore, the resistance phenotype of the C-d12 clone appeared to be related to invasion (the pathway in which CDPK1 functions). Overexpression of CDPK1(H201Q) also caused a shift in the EC50 similar to that seen in the A clones. However, it conferred only a modest shift in EC50 compared to that for the similarly overexpressed WT CDPK1. Two explanations could be posed for this finding. The first is that overexpression of WT CDPK1 increases the abundance of active CDPK1 to a threshold level, while CDPK1(H201Q) has already reached this threshold at a lower abundance. This proposal fits with the hypothesis that the H201Q mutation destabilizes the inactive form and increases the proportion of active enzyme. The second possibility is that the H2012Q CDPK1 mutation in the A clones may act in concert with other genetic or epigenetic changes, such as the modulation of pumps or transporters or the pathway modulation/rewiring that occurred during the 52-day selection process. Future whole-genome sequencing, transcriptomics, and clustered regularly interspaced short palindromic repeat/Cas9 correction of the H201Q mutation in the selected clone could reveal the basis of this interesting finding.
Our experiments were not designed to measure the probability of resistance to BKIs emerging under conditions likely to be used clinically. Nonetheless, resistant mutants emerged in both flasks by 52 days. Each flask was initially inoculated with 3.3 × 107 parasites, well below the numbers expected in an infected person, and selection was performed with 0.8 μM (EC90) RM-1-132. At least for some compounds, pharmacokinetic studies in mice have revealed plasma levels above this concentration without toxicity to the host (e.g., BKI 1553 levels were at 6 μM for 24 h following an oral dose of 10 mg/kg of body weight [9]). In addition, with higher levels of inhibitors, other targets, such as MAPKL-1 may be engaged (34), further reducing the chance for the emergence of acquired resistance. Whether we would have been able to isolate resistant parasites in the lab using concentrations that high is open to question. Finally, it is also important to point out that since T. gondii is only rarely transmitted from person to person (most exposure is from parasites derived from animal infections), drug resistance is unlikely to spread in the human population. Hence, the challenge of drug resistance is focused on curing the individual with little fear of spreading resistant parasites. As a result, effective new BKIs to combat toxoplasmosis and toxoplasmic encephalitis are likely to have a long and useful life.
MATERIALS AND METHODS
Protein expression in Escherichia coli, enzymatic assay, and compounds.
CDPK1 lacking the first 29 amino acids and its mutants were N-terminally tagged with six His residues followed by a protease C cleavage site, expressed in E. coli BL21(DE3), and purified by immobilized metal-affinity chromatography followed by size exclusion chromatography as described previously (5). The enzymatic activity of the 57-kDa protein was assessed using the Kinase-Glo assay (30) with 2.1 nM CDPK1, 10 μM ATP, and 20 μM peptide substrate (PLARTLSVAGLPGKK; American Peptide Company, Inc., Sunnyvale, CA) plus 2 mM CaCl2. The reaction buffer contained 1 mM EGTA (pH 7.2), 10 mM MgCl2, 20 mM HEPES, pH 7.5 (KOH), and 0.1% bovine serum albumin.
BKIs.
Initial publication of the compounds (including their EC50s and IC50s) was in reference 12 for compounds 1265 (original name, compound 9n), 1294 (original name, compound 15o), and RM-1-132 (original name, compound 15n) and reference 9 for compound 1553 (original name, compound 32). The synthesis protocol for compound 1568, 1-(5-(4-amino-1-isobutyl-1H-pyrazolo[3,4-d]pyrimidin-3-yl)indolin-1-yl)-2-phenylethanone is as follows: 2-phenyl-1-(5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indolin-1-yl)ethanone and 3-iodo-1-isobutyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine were subjected to the Suzuki coupling procedure (9). The crude product was purified on a silica gel using a dichloromethane-methanol gradient. The spectral data were as follows: 1H nuclear magnetic resonance (300 MHz, CDCl3) δ 8.45 to 8.33 (m, 2H), 7.59 to 7.45 (m, 2H), 7.42 to 7.28 (m, 5H), 5.68 to 5.53 (br s, 2H), 4.23 (d, J = 7.46 Hz, 2H), 4.16 (t, J = 8.50 Hz, 2H), 3.86 (s, 2H), 3.26 (t, J = 8.29 Hz, 2H), 2.41 (m, 1H), 0.95 (d, J = 6.84 Hz, 6H); mass spectrometry (electrospray ionization), 427.4 m/z [MH+] (C25H27N6O; predicted molecular mass, 427.2); high-performance liquid chromatography purity, >97%.
Thermal shift assays.
The thermal stability of recombinant CDPK1 in the presence or absence of BKIs was determined as previously described (35). Each assay well contained a reaction mixture of recombinant CDPK1 enzyme (4.4 μM), 20 μM inhibitor, and 5% dimethyl sulfoxide (DMSO) in TSA buffer (5% glycerol, 25 mM HEPES, 500 mM NaCl, 0.025% NaN3, 2 mM dithiothreitol [DTT] with the pH adjusted to 7.25, with or without CaCl2 or EGTA, as indicated). A negative control lacking inhibitor was present on each assay plate. Melting was measured using SYPRO Orange (at a final dilution of 4.5× from the 5,000× stock; Invitrogen), which fluoresces when it binds to exposed hydrophobic regions of the protein. All assays were performed in duplicate (or triplicate) independently at least two times. Preliminary experiments established that DMSO concentrations from 0 to 5% did not alter the Tms of purified recombinant proteins (not shown).
L-CETSAs (20) were performed using the previously described modifications (24). Briefly, a mixture of T. gondii cells that had spontaneously egressed and those from the same culture that were mechanically released from host fibroblasts was washed in phosphate-buffered saline and the cells were counted. Parasites were resuspended in CETSA buffer (25 mM Tris HCl, pH 7.4, 2 mM DTT, 10 mM MgCl2, 1 mM CaCl2) plus phosphatase and protease inhibitor cocktail (Halt protease phosphatase inhibitor; Thermo Scientific) at 5 × 108/ml. After three freeze-thaw cycles, lysates were clarified by centrifugation at 16,100 × g for 25 min at 4°C. Aliquots were incubated with 2 μM BKI (0.5% DMSO) for 10 min at 37°C. Thermal stabilization of CDPK1 was tested at 7 temperature points (66°C, 62°C, 58°C, 54°C, 50°C, 46°C, and 42°C in duplicate per compound at 2 μM) in a reaction volume of 20 μl with 107 cell equivalents. Samples were heated for 3 min at the desired temperatures in the thermocycler and again centrifuged as described above to remove aggregated proteins. Taking care not to disturb the pellet, supernatants were removed and 2 × 106 cell equivalents (∼4 μl) mixed with sample buffer was analyzed by SDS-PAGE, with the entire duplicate series for each compound in the experiment being run on a single gel. The gels were blotted and incubated with anti-TgCDPK1 (1:50,000) (24) plus goat anti-rabbit IgG IRDye 800 and imaged on a LI-COR Odyssey instrument. The blots were stained with Ponceau to detect potential artifacts, such as nonspecific protein precipitation or gel bubbles. Each compound was tested in at least two experiments, and the solvent control was replicated three times to yield an average value used for the calculations, as described previously (24). For repeat experiments, temperatures outside the range of interest were typically not included.
For IC-CETSAs, we followed the procedure described for mammalian cells (20, 36). Briefly, mechanically harvested parasites were pelleted and rinsed in Dulbecco modified Eagle medium (DMEM) (lacking serum) and then resuspended at 4 × 108/ml in DMEM (which contains 1 mM CaCl2) prewarmed to 37°C. Compounds were added in duplicate to yield a final concentration of 2 μM BKI and 0.5% DMSO. After thorough mixing, samples were incubated for 1 h at 37°C in a CO2 incubator. The treated cells were centrifuged, rinsed in phosphate-buffered saline, and resuspended in TSA buffer containing protease and phosphatase inhibitors, as described above. Aliquots (25 μl) containing 107 cells were then heated for 3 min at the desired temperatures and placed on ice until the range of heat treatments was complete. They were then subjected to three freeze-thaw cycles, and the clarified lysate was prepared and analyzed by Western blotting as described above. Preliminary experiments tested whether parasites remained viable during the 1-h incubation in the presence or absence of drug via microscopic analysis following staining with ethidium homodimer (0.5 μM) (not shown). The viability of parasites incubated with the BKIs used in this study remained above 94%. For IC-CETSAs, each compound was tested in at least two experiments, and the solvent control was tested in six experiments. The Tms for each condition (compound and cell line) were averaged and used for the calculation of the thermal shift from the solvent control.
Invasion/proliferation assay.
Invasion/proliferation assays used T. gondii expressing the reporter enzyme β-galactosidase and followed the procedure described previously (12). Unless otherwise noted, the parasites were mixed with the BKI and then added to fibroblast monolayers in 96-well plates. After 44 h, β-galactosidase activity was measured using the substrate chlorophenol red β-galactopyranose (Sigma).
Selection of T. gondii with partial resistance to the BKI RM-1-132.
Immediately prior to initiating selection, we recloned the starting cell line RH Δhxgprt Gra2-GFP Tub-βgal. For the selections, two experimental flasks (flasks A and C) with 3.3 × 107 parasites were grown in the presence of the BKI RM-1-132 at the EC90 (0.8 μM), which had been previously determined from 2-day assays. As a control, 10 parasites expressing both green fluorescent protein (GFP) and the RM-1-132 resistance allele CDPK1(G128M) were added to 3.3 × 107 WT RH parasites and subjected to the same selection protocol. This allowed us to use flow cytometry to follow the enrichment of the resistant (green) parasites treated in the same way and, hence, to gauge when we could expect to have enriched for mutants in the experimental sample. By passage 12, ∼85% of the spiked control selection was GFP positive. We continued selection in the experimental flasks for four more passages. Then, at day 52 (passage 16), parasites from the two flasks were cloned and selection was dropped. mRNA was isolated from the selected, cloned lines and converted to cDNA. Following PCR, the entire CDS was sequenced for at least two clones per selection and the sequence was compared to the starting sequence.
Transfections.
The CDPK1-coding region (derived from cDNA), tagged with four HA epitopes and cloned into pHXGPRT for expression driven by the GRA1 promoter (5), was subjected to site-directed mutagenesis to create separate L96P and H201Q mutants, as previously described (5). The linearized plasmids were transfected by electroporation into T. gondii Δhxgprt Gra2-GFP Tub-βgal for integration into the genome. Stable transfectants were selected with mycophenolic acid plus xanthine. Clonal lines were generated by limiting dilution and checked for expression of the mutant protein using anti-HA antibodies.
Supplementary Material
ACKNOWLEDGMENTS
We thank Mira Kim for technical assistance and Stone Doggett for useful discussions.
This work was supported by the Public Health Service, National Institutes of Health, Bethesda, MD (grants R01 AI111341 and R01 HD080670 and grant NIH-R01 AI089441 [principal investigator, W.C.V.V.]), the U.S. Department of Agriculture (grant 2014-67015-22106 [principal investigator, W.C.V.V.]), and a company to develop and market CDPK1 inhibitors for animal health purposes.
W. C. Van Voorhis did not perform or interpret the data from the experiments performed in this study.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00051-18.
REFERENCES
- 1.Halonen SK, Weiss LM. 2013. Toxoplasmosis. Handb Clin Neurol 114:125–145. doi: 10.1016/B978-0-444-53490-3.00008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vyas A. 2015. Mechanisms of host behavioral change in Toxoplasma gondii rodent association. PLoS Pathog 11:e1004935. doi: 10.1371/journal.ppat.1004935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wei HX, Wei SS, Lindsay DS, Peng HJ. 2015. A systematic review and meta-analysis of the efficacy of anti-Toxoplasma gondii medicines in humans. PLoS One 10:e0138204. doi: 10.1371/journal.pone.0138204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kieschnick H, Wakefield T, Narducci CA, Beckers C. 2001. Toxoplasma gondii attachment to host cells is regulated by a calmodulin-like domain protein kinase. J Biol Chem 276:12369–12377. doi: 10.1074/jbc.M011045200. [DOI] [PubMed] [Google Scholar]
- 5.Ojo KK, Larson ET, Keyloun KR, Castaneda LJ, DeRocher AE, Inampudi KK, Kim JE, Arakaki TL, Murphy R, Zhang L, Napuli AJ, Maly DJ, Verlinde CL, Buckner FS, Parsons M, Hol WG, Merritt EA, Van Voorhis WC. 2010. A unique variation of the ATP binding site makes Toxoplasma gondii calcium-dependent protein kinase 1 a drug target for selective kinase inhibitors. Nat Struct Mol Biol 17:602–607. doi: 10.1038/nsmb.1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lourido S, Tang K, Sibley LD. 2012. Distinct signalling pathways control Toxoplasma egress and host-cell invasion. EMBO J 24:4524–4534. doi: 10.1038/emboj.2012.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lourido S, Shuman J, Zhang C, Shokat KM, Hui R, Sibley LD. 2010. Calcium-dependent protein kinase 1 is an essential regulator of exocytosis in Toxoplasma. Nature 465:359–362. doi: 10.1038/nature09022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moine E, Dimier-Poisson I, Enguehard-Gueiffier C, Loge C, Penichon M, Moire N, Delehouze C, Foll-Josselin B, Ruchaud S, Bach S, Gueiffier A, Debierre-Grockiego F, Denevault-Sabourin C. 2015. Development of new highly potent imidazo[1,2-b]pyridazines targeting Toxoplasma gondii calcium-dependent protein kinase 1. Eur J Med Chem 105:80–105. doi: 10.1016/j.ejmech.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 9.Vidadala RS, Rivas KL, Ojo KK, Hulverson MA, Zambriski JA, Bruzual I, Schultz TL, Huang W, Zhang Z, Scheele S, DeRocher AE, Choi R, Barrett LK, Siddaramaiah LK, Hol WG, Fan E, Merritt EA, Parsons M, Freiberg G, Marsh K, Kempf DJ, Carruthers VB, Isoherranen N, Doggett JS, Van Voorhis WC, Maly DJ. 2016. Development of an orally available and central nervous system (CNS) penetrant Toxoplasma gondii calcium-dependent protein kinase 1 (TgCDPK1) inhibitor with minimal human ether-a-go-go-related gene (hERG) activity for the treatment of toxoplasmosis. J Med Chem 59:6531–6546. doi: 10.1021/acs.jmedchem.6b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Z, Ojo KK, Vidadala R, Huang W, Geiger JA, Scheele S, Choi R, Reid MC, Keyloun KR, Rivas K, Siddaramaiah LK, Comess KM, Robinson KP, Merta PJ, Kifle L, Hol WG, Parsons M, Merritt EA, Maly DJ, Verlinde CL, Van Voorhis WC, Fan E. 2014. Potent and selective inhibitors of CDPK1 from and based on a 5-aminopyrazole-4-carboxamide scaffold. ACS Med Chem Lett 5:40–44. doi: 10.1021/ml400315s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z, Ojo KK, Johnson SM, Larson ET, He P, Geiger JA, Castellanos-Gonzalez A, White AC Jr, Parsons M, Merritt EA, Maly DJ, Verlinde CL, Van Voorhis WC, Fan E. 2012. Benzoylbenzimidazole-based selective inhibitors targeting Cryptosporidium parvum and Toxoplasma gondii calcium-dependent protein kinase-1. Bioorg Med Chem Lett 22:5264–5267. doi: 10.1016/j.bmcl.2012.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson SM, Murphy RC, Geiger JA, DeRocher AE, Zhang Z, Ojo KK, Larson ET, Perera BG, Dale EJ, He P, Reid MC, Fox AM, Mueller NR, Merritt EA, Fan E, Parsons M, Van Voorhis WC, Maly DJ. 2012. Development of Toxoplasma gondii calcium-dependent protein kinase 1 (TgCDPK1) inhibitors with potent anti-Toxoplasma activity. J Med Chem 55:2416–2426. doi: 10.1021/jm201713h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doggett JS, Ojo KK, Fan E, Maly DJ, Van Voorhis WC. 2014. Bumped kinase inhibitor 1294 treats established Toxoplasma gondii infection. Antimicrob Agents Chemother 58:3547–3549. doi: 10.1128/AAC.01823-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lourido S, Zhang C, Lopez M, Tang K, Barks J, Wang Q, Wildman SA, Shokat K, Sibley LD. 2013. Optimizing small molecule inhibitors of calcium-dependent protein kinase 1 to prevent infection by Toxoplasma gondii. J Med Chem 56:3068–3077. doi: 10.1021/jm4001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rutaganira FU, Barks J, Dhason MS, Wang Q, Lopez MS, Long S, Radke JB, Jones NG, Maddirala AR, Janetka JW, El BM, Hui R, Shokat KM, Sibley LD. 2017. Inhibition of calcium dependent protein kinase 1 (CDPK1) by pyrazolopyrimidine analogs decreases establishment and reoccurrence of central nervous system disease by Toxoplasma gondii. J Med Chem 60:9976–9989. doi: 10.1021/acs.jmedchem.7b01192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang W, Ojo KK, Zhang Z, Rivas K, Vidadala RS, Scheele S, DeRocher AE, Choi R, Hulverson MA, Barrett LK, Bruzual I, Siddaramaiah LK, Kerchner KM, Kurnick MD, Freiberg GM, Kempf D, Hol WG, Merritt EA, Neckermann G, de Hostos EL, Isoherranen N, Maly DJ, Parsons M, Doggett JS, Van Voorhis WC, Fan E. 2015. SAR studies of 5-aminopyrazole-4-carboxamide analogues as potent and selective inhibitors of CDPK1. ACS Med Chem Lett 6:1184–1189. doi: 10.1021/acsmedchemlett.5b00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wernimont AK, Amani M, Qiu W, Pizarro JC, Artz JD, Lin YH, Lew J, Hutchinson A, Hui R. 2011. Structures of parasitic CDPK domains point to a common mechanism of activation. Proteins 79:803–820. doi: 10.1002/prot.22919. [DOI] [PubMed] [Google Scholar]
- 18.Ingram JR, Knockenhauer KE, Markus BM, Mandelbaum J, Ramek A, Shan Y, Shaw DE, Schwartz TU, Ploegh HL, Lourido S. 2015. Allosteric activation of apicomplexan calcium-dependent protein kinases. Proc Natl Acad Sci U S A 112:E4975–E4984. doi: 10.1073/pnas.1505914112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murphy RC, Ojo KK, Larson ET, Castellanos-Gonzalez A, Perera BG, Keyloun KR, Kim JE, Bhandari JG, Muller NR, Verlinde CL, White AC, Merritt EA, Van Voorhis WC, Maly DJ. 2010. Discovery of potent and selective inhibitors of calcium-dependent protein kinase 1 (CDPK1) from C. parvum and T. gondii. ACS Med Chem Lett 1:331–335. doi: 10.1021/ml100096t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, Nordlund P. 2013. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341:84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 21.Savitski MM, Reinhard FB, Franken H, Werner T, Savitski MF, Eberhard D, Martinez MD, Jafari R, Dovega RB, Klaeger S, Kuster B, Nordlund P, Bantscheff M, Drewes G. 2014. Proteomics tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346:1255784. doi: 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- 22.Zovko A, Novak M, Haag P, Kovalerchick D, Holmlund T, Farnegardh K, Ilan M, Carmeli S, Lewensohn R, Viktorsson K. 2016. Compounds from the marine sponge Cribrochalina vasculum offer a way to target IGF-1R mediated signaling in tumor cells. Oncotarget 7:50258–50276. doi: 10.18632/oncotarget.10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fauster A, Rebsamen M, Huber KV, Bigenzahn JW, Stukalov A, Lardeau CH, Scorzoni S, Bruckner M, Gridling M, Parapatics K, Colinge J, Bennett KL, Kubicek S, Krautwald S, Linkermann A, Superti-Furga G. 2015. A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell Death Dis 6:e1767. doi: 10.1038/cddis.2015.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ojo KK, Dangoudoubiyam S, Verma SK, Scheele S, DeRocher AE, Yeargan M, Choi R, Smith TR, Rivas KL, Hulverson MA, Barrett LK, Fan E, Maly DJ, Parsons M, Dubey JP, Howe DK, Van Voorhis WC. 2016. Selective inhibition of Sarcocystis neurona calcium-dependent protein kinase 1 for equine protozoal myeloencephalitis therapy. Int J Parasitol 46:871–880. doi: 10.1016/j.ijpara.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sugi T, Kobayashi K, Takemae H, Gong H, Ishiwa A, Murakoshi F, Recuenco FC, Iwanaga T, Horimoto T, Akashi H, Kato K. 2013. Identification of mutations in TgMAPK1 of Toxoplasma gondii conferring resistance to 1NM-PP1. Int J Parasitol Drugs Drug Resist 3:93–101. doi: 10.1016/j.ijpddr.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wernimont AK, Artz JD, Finerty P Jr, Lin YH, Amani M, Allali-Hassani A, Senisterra G, Vedadi M, Tempel W, Mackenzie F, Chau I, Lourido S, Sibley LD, Hui R. 2010. Structures of apicomplexan calcium-dependent protein kinases reveal mechanism of activation by calcium. Nat Struct Mol Biol 17:596–601. doi: 10.1038/nsmb.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larsson EA, Jansson A, Ng FM, Then SW, Panicker R, Liu B, Sangthongpitag K, Pendharkar V, Tai SJ, Hill J, Dan C, Ho SY, Cheong WW, Poulsen A, Blanchard S, Lin GR, Alam J, Keller TH, Nordlund P. 2013. Fragment-based ligand design of novel potent inhibitors of tankyrases. J Med Chem 56:4497–4508. doi: 10.1021/jm400211f. [DOI] [PubMed] [Google Scholar]
- 28.Fedorov O, Marsden B, Pogacic V, Rellos P, Muller S, Bullock AN, Schwaller J, Sundstrom M, Knapp S. 2007. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc Natl Acad Sci U S A 104:20523–20528. doi: 10.1073/pnas.0708800104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borges-Pereira L, Budu A, McKnight CA, Moore CA, Vella SA, Hortua Triana MA, Liu J, Garcia CR, Pace DA, Moreno SN. 2015. Calcium signaling throughout the Toxoplasma gondii lytic cycle. A study using genetically encoded calcium indicators. J Biol Chem 290:26914–26926. doi: 10.1074/jbc.M115.652511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keyloun KR, Reid MC, Choi R, Song Y, Fox AM, Hillesland HK, Zhang Z, Vidadala R, Merritt EA, Lau AO, Maly DJ, Fan E, Barrett LK, Van Voorhis WC, Ojo KK. 2014. The gatekeeper residue and beyond: homologous calcium-dependent protein kinases as drug development targets for veterinarian Apicomplexa parasites. Parasitology 141:1499–1509. doi: 10.1017/S0031182014000857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Voorhis WC, Doggett JS, Parsons M, Hulverson MA, Choi R, Arnold SLM, Riggs MW, Hemphill A, Howe DK, Mealey RH, Lau AOT, Merritt EA, Maly DJ, Fan E, Ojo KK. 2017. Extended-spectrum antiprotozoal bumped kinase inhibitors: a review. Exp Parasitol 180:71–83. doi: 10.1016/j.exppara.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pedroni MJ, Vidadala RS, Choi R, Keyloun KR, Reid MC, Murphy RC, Barrett LK, Van Voorhis WC, Maly DJ, Ojo KK, Lau AO. 2016. Bumped kinase inhibitor prohibits egression in Babesia bovis. Vet Parasitol 215:22–28. doi: 10.1016/j.vetpar.2015.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Treeck M, Sanders JL, Elias JE, Boothroyd JC. 2011. The phosphoproteomes of Plasmodium falciparum and Toxoplasma gondii reveal unusual adaptations within and beyond the parasites' boundaries. Cell Host Microbe 10:410–419. doi: 10.1016/j.chom.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sugi T, Kawazu S, Horimoto T, Kato K. 2015. A single mutation in the gatekeeper residue in TgMAPKL-1 restores the inhibitory effect of a bumped kinase inhibitor on the cell cycle. Int J Parasitol Drugs Drug Resist 5:1–8. doi: 10.1016/j.ijpddr.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crowther GJ, Napuli AJ, Thomas AP, Chung DJ, Kovzun KV, Leibly DJ, Castaneda LJ, Bhandari J, Damman CJ, Hui R, Hol WG, Buckner FS, Verlinde CL, Zhang Z, Fan E, Van Voorhis WC. 2009. Buffer optimization of thermal melt assays of Plasmodium proteins for detection of small-molecule ligands. J Biomol Screen 14:700–707. doi: 10.1177/1087057109335749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundback T, Nordlund P, Martinez Molina D. 2014. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc 9:2100–2122. doi: 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- 37.Hoff EF, Cook SH, Sherman GD, Harper JM, Ferguson DJ, Dubremetz JF, Carruthers VB. 2001. Toxoplasma gondii: molecular cloning and characterization of a novel 18-kDa secretory antigen, TgMIC10. Exp Parasitol 97:77–88. doi: 10.1006/expr.2000.4585. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.