

ABSTRACT
The pUM505 plasmid, isolated from a clinical Pseudomonas aeruginosa isolate, confers resistance to ciprofloxacin (CIP) when transferred into the standard P. aeruginosa strain PAO1. CIP is an antibiotic of the quinolone family that is used to treat P. aeruginosa infections. In silico analysis, performed to identify CIP resistance genes, revealed that the 65-amino-acid product encoded by the orf131 gene in pUM505 displays 40% amino acid identity to the Mycobacterium smegmatis aminoglycoside phosphotransferase (an enzyme that phosphorylates and inactivates aminoglycoside antibiotics). We cloned orf131 (renamed crpP, for ciprofloxacin resistance protein, plasmid encoded) into the pUCP20 shuttle vector. The resulting recombinant plasmid, pUC-crpP, conferred resistance to CIP on Escherichia coli strain J53-3, suggesting that this gene encodes a protein involved in CIP resistance. Using coupled enzymatic analysis, we determined that the activity of CrpP on CIP is ATP dependent, while little activity against norfloxacin was detected, suggesting that CIP may undergo phosphorylation. Using a recombinant His-tagged CrpP protein and liquid chromatography–tandem mass spectrometry, we also showed that CIP was phosphorylated prior to its degradation. Thus, our findings demonstrate that CrpP, encoded on the pUM505 plasmid, represents a new mechanism of CIP resistance in P. aeruginosa, which involves phosphorylation of the antibiotic.
KEYWORDS: Pseudomonas aeruginosa, ciprofloxacin, enzyme, phosphorylation, plasmid-mediated resistance, quinolones
INTRODUCTION
Quinolones are synthetic antibiotics that constitute an important class of biologically active, broad-spectrum antibacterial drugs (1). These antibiotics are used in humans to treat a variety of bacterial infections and represent one of the most commonly prescribed classes of antibacterial drugs in the world (2). Ciprofloxacin (CIP), a fluoroquinolone, was the first quinolone to display considerable activity outside the urinary tract (3). Due to the pharmacological characteristics of CIP, as well as its potency, activity spectrum, oral bioavailability, and good safety profile, this antibiotic has been extensively used clinically worldwide (4). Importantly, CIP is the most active quinolone against Pseudomonas aeruginosa and is used to treat infections generated by this bacterium. CIP is also used to treat infections with various Gram-negative and Gram-positive bacteria and is the most commonly prescribed antibacterial drug to date according to guidelines (4, 5).
The mechanism of action of quinolone antibiotics consists of the inhibition of DNA gyrase and topoisomerase IV (Topo IV), enzymes that modulate the topological state of the DNA and are essential for transcription, DNA replication, and the repair/recombination of DNA (1). These two enzymes display structural and functional similarities; however, their specific mechanisms of action during DNA replication differ (6). The quinolones stabilize the cleavage complexes by binding the enzyme-DNA interface in the cleavage-ligation active site, resulting in the inhibition of DNA ligation (1). Therefore, the intercalation of quinolones into DNA results in increased cell death (1).
Resistance to quinolones can involve chromosomal gene mutations or may be plasmid mediated. A correlation between CIP resistance and the number of resistance-associated alterations in GyrA, GyrB subunits of topoisomerase II, and ParC and ParE subunits of topoisomerase IV in P. aeruginosa isolates has been reported previously (7). In Escherichia coli, mutations in marR (a marA repressor) lead to the MarA-mediated activation of genes encoding the AcrAB-TolC efflux pump, which plays a major role in quinolone efflux. The marR mutations also decrease the translation of ompF, a gene encoding the OmpF outer membrane porin, thus decreasing quinolone influx (4). Plasmid-mediated quinolone resistance (PMQR) confers decreased susceptibility to these antibiotics. Some examples of PMQR systems include the Qnr protein, which prevents quinolone binding to target proteins, and QepA, an efflux pump that decreases susceptibility to CIP and norfloxacin (NOR) (4, 6). Moreover, mutations in the gene encoding the aminoglycoside N-6′-acetyltransferase [AAC(6′)-Ib] enzyme, which confers resistance to aminoglycoside antibiotics, results in a variant [AAC(6′)-Ib-cr] that promotes CIP and NOR resistance (8).
pUM505 is a self-conjugating 123-kbp plasmid isolated from a clinical Pseudomonas aeruginosa isolate (9). This plasmid carries several adaptive genes, including the umuD gene (encoding a transcriptional regulator of the SOS response) (10), genes involved in Pseudomonas virulence (11, 12), and genes that increase plasmid stability (12). The aim of this work was to identify and study the product of the orf131 (renamed crpP) gene on pUM505, which confers resistance to CIP. We show that the mechanism underlying this resistance to CIP involves CIP phosphorylation.
RESULTS
Quinolone resistance is conferred by the pUM505 plasmid.
The pUM505 plasmid, originally isolated from a clinical isolate of P. aeruginosa (9), confers resistance to CIP when transferred into P. aeruginosa strain PU21. This observation suggested that pUM505 encodes a factor involved in susceptibility to this antibiotic (13). To determine whether pUM505 confers resistance to other quinolones, the pUM505 plasmid was transferred into P. aeruginosa strain PAO1, and the antimicrobial susceptibilities to CIP, NOR, levofloxacin (LVX), moxifloxacin (MXF), and nalidixic acid (NAL) were investigated. Our results showed that CIP, NOR, and MXF displayed 4-fold higher MICs for P. aeruginosa PAO1(pUM505) than for the P. aeruginosa receptor strain PAO1. In addition, the MIC values for strain PAO1 were similar to those measured for the P. aeruginosa reference strain, ATCC 27853 (Table 1). These findings indicated that the pUM505 plasmid encodes a protein involved in quinolone resistance.
TABLE 1.
Results of susceptibility tests
| Antibiotic | MIC (μg/ml) for: |
||||||
|---|---|---|---|---|---|---|---|
|
P. aeruginosa |
E. coli |
||||||
| ATCC 27853 | PAO1(pUCP20) | PAO1(pUC_crpP) | PAO1(pUM505) | ATCC 25922 | J53-3(pUCP20) | J53-3(pUC_crpP) | |
| Ciprofloxacin | 0.5 | 0.5 | 0.5 | 2.0 | 0.004 | 0.008 | 0.06 |
| Levofloxacin | 0.5 | 1.0 | 1.0 | 1.0 | 0.008 | 0.008 | 0.008 |
| Norfloxacin | 0.5 | 0.5 | 0.5 | 2.0 | 0.03 | 0.03 | 0.03 |
| Moxifloxacin | 1.0 | 2.0 | 2.0 | 8.0 | 0.008 | 0.03 | 0.03 |
| Nalidixic acid | >256 | >256 | >256 | >256 | 2.0 | 8.0 | 8.0 |
| Kanamycin | >256 | 32 | 32 | 32 | 4.0 | 4.0 | 2.0 |
| Streptomycin | 8.0 | 8.0 | 4.0 | 4.0 | 8.0 | 4.0 | 4.0 |
| Amikacin | 1.0 | 0.5 | 0.5 | 0.5 | 2.0 | 1.0 | 1.0 |
| Gentamicin | 0.5 | 0.5 | 0.5 | 0.5 | 1.0 | 0.5 | 0.5 |
Identification of the crpP gene.
In silico analysis of pUM505 failed to identify a PMQR element (13). Moreover, a BLAST analysis revealed that the protein encoded by the orf131 gene (RefSeq accession number NC_016138; region, nucleotides 119072 to 119269; renamed crpP, for ciprofloxacin resistance protein, plasmid encoded) shares 40% identity with a 42-amino-acid (42-aa) region of the aminoglycoside phosphotransferase (APH) from Mycobacterium smegmatis (Fig. 1A). The crpP gene possesses a putative sigma-70 promoter in the 5′ region and is predicted to encode a 65-aa protein (Fig. 1A; see also Fig. S1A in the supplemental material). In contrast, the APH protein from M. smegmatis contains 225 aa and is much longer than the protein encoded by crpP; however, several members of this protein family are smaller. For example, the 79-aa aminoglycoside phosphotransferase [APH(3)-IIb] from P. aeruginosa M18 confers kanamycin (KAN), gentamicin (GEN), and streptomycin (STR) resistance (14). CrpP and APH(3)-IIb display a low degree of identity (18%) and 26% similarity (Fig. 1A); however, CrpP contains two residues conserved in APH enzymes, 7-Gly (involved in catalysis) and 26-Ile (involved in ATP binding) (Fig. 1B). Hence, these analyses suggest the product encoded by the crpP gene may confer resistance to antibiotics.
FIG 1.

Comparison of CrpP to aminoglycoside phosphotransferases. (A) The coding regions and transcriptional directions of the aph, crpP, and aph(3)-IIb genes from M. smegmatis, the pUM505 plasmid, and P. aeruginosa M18, respectively, are indicated by open arrows, with the sizes of predicted polypeptides (in amino acids) indicated. Percentages of amino acid identity/similarity for the proteins compared are given in parentheses; stippled areas of the aph gene from M. smegmatis were not considered for the alignments. Shaded arrows represent adjacent genes, and the locations of the putative promoters' sequences are marked by bent arrows. (B) Alignment of the putative protein encoded by the crpP gene with the APH(3)-IIb protein from P. aeruginosa M18. Asterisks denote conserved catalytic residues in the APH(3)-IIb gene from P. aeruginosa M18. Filled squares indicate conserved catalytic residues identified in the protein encoded by the crpP gene.
The protein encoded by crpP confers resistance to CIP.
To test whether crpP, including the 5′ putative regulatory region, is involved in resistance to CIP, the regions were amplified by PCR and were cloned into the high-copy-number vector pUCP20. The pUC_crpP recombinant plasmid was transferred into P. aeruginosa strain PAO1, and susceptibility to CIP was determined. Our results showed that P. aeruginosa strain PAO1 expressing the pUC_crpP plasmid had a level of susceptibility to CIP similar to that observed with strain PAO1 (Table 1). These results suggest that the crpP product does not confer significant resistance to quinolones in the genetic background of P. aeruginosa PAO1, a strain known for its intrinsic high resistance to many antibiotics (15). However, another explanation may be that in PAO1, the crpP gene is not expressed under the control of its own promoter in the pUC20 vector. To distinguish between these possibilities, crpP gene expression in cultures of P. aeruginosa PAO1 containing the recombinant pUC-crpP plasmid was determined by quantitative reverse transcription-PCR (RT-qPCR) assays. The results showed that the crpP gene was expressed in P. aeruginosa strain PAO1 (Fig. S1B in the supplemental material). This finding suggested that this gene is functional in this strain. To determine whether crpP confers susceptibility to quinolones, the more-susceptible E. coli J53-3 bacterial background was used. MIC assays with E. coli J53-3(pUC_crpP) showed a 7.5-fold decrease in susceptibility to CIP, while susceptibilities to the other four quinolones tested (LVX, NOR, MXF, and NAL) did not differ from those of E. coli strain J53-3 (Table 1). Additionally, RT-qPCR analysis of expression showed that the crpP gene was expressed in this strain (Fig. S1B). These results indicated that crpP encodes a protein that confers decreased susceptibility to CIP on E. coli. In addition, the susceptibilities of E. coli J53-3(pUC_crpP) to KAN, STR, amikacin (AMK), and GEN were similar to those of the E. coli control strain (Table 1), suggesting that the crpP gene does not confer resistance to these aminoglycosides. These results indicated that the crpP gene on the pUM505 plasmid encodes a protein that confers selective resistance to CIP alone.
Enzymatic activity of the CrpP protein.
Because our sequence analysis suggested that crpP encodes a protein that is similar to APH antibiotic-modifying enzymes, we next determined the in vitro activity of the purified CrpP enzyme. To purify CrpP, the coding region of crpP was cloned into an overexpression vector, and a recombinant His-tagged CrpP protein was isolated, as described in Materials and Methods. We first tested whether the recombinant His-CrpP protein was active in cells in vivo. To this end, we used isopropyl β-d-1-thiogalactopyranoside (IPTG) to induce overexpression of the His-CrpP protein in E. coli BL21(pTrcHis-crpP) grown in the presence or absence of CIP. Upon His-CrpP induction, this strain grew better than the noninduced strain in the presence of CIP (see Fig. S2 in the supplemental material). This finding suggested that the N-terminal 6×His tag did not alter the activity of the enzyme, and therefore the His tag was retained as part of the recombinant protein for all subsequent experiments.
To determine the activity of CrpP on CIP, we used a coupled enzymatic assay involving NADH oxidation (16). In the presence of CrpP, NADH oxidation increases proportionally with the reaction time (Fig. 2A). Moreover, NADH oxidation also increased proportionally with the amount of protein in the assay (see Fig. S3 in the supplemental material), indicating that CIP is a substrate of CrpP. Additionally, we determined the activity of CrpP on CIP following heat-induced CrpP denaturation. This resulted in a reduced level of CrpP activity from that of the native protein at the same concentration (Fig. S3), suggesting that the activity was specific to CrpP and that the correct structure of the protein is required for its function. Additionally, we analyzed the effects of pH and Mg2+ concentration on CrpP activity. The optimal pH was 7.0, and optimal CrpP activity was observed when 10 mM MgCl2 was used (see Fig. S4 in the supplemental material).
FIG 2.

CrpP enzyme kinetics. (A) NADH oxidation rate comparisons for CIP (○) and NOR (●); 2.0 mM each antibiotic was incubated in the presence of 5.0 μg/ml His-CrpP protein at 37°C for 15 min. NADH oxidation was measured spectrophotometrically at 390 nm. (B) Michaelis-Menten plots for CIP (○) and NOR (●). The plot shows the NADH oxidation data. Data are means from four independent duplicate assays, with standard errors.
The MICs indicated that crpP confers resistance to CIP only, suggesting an enzymatic activity that is specific to this antibiotic; thus, we analyzed the catalytic activity of CrpP against NOR, LVX, MXF, and NAL. We observed a lower level of CrpP activity when NOR was used as a substrate than when CIP was used (Fig. 2A), and no activity against LVX, MXF, or NAL was observed (see Fig. S5 in the supplemental material). These results are consistent with the susceptibility to quinolones observed by measuring MICs (Table 1) and suggest that the low level of CrpP activity on NOR does not enable detection of a decreased susceptibility to this antibiotic in E. coli. Moreover, CrpP cannot use aminoglycosides, such as KAN and STR, as substrates, suggesting that CrpP is specific for CIP and NOR (Fig. S5).
We also estimated the apparent kinetic constants of CrpP for CIP and NOR (Fig. 2B). We detected a Km of 0.5483 ± 0.2762 mM and a Vmax of 2.071 ± 0.3745 μmol/min for CIP and a Km of 0.4192 ± 0.2208 mM and a Vmax of 0.2989 ± 0.4965 μmol/min for NOR. The Vmax/Km ratio was approximately 5-fold higher for CIP than for NOR (3.77 for CIP versus 0.713 for NOR). These in vitro results demonstrated that CrpP is less efficient at utilizing NOR than it is at utilizing CIP as a substrate. In addition, when ATP was used as the variable cosubstrate, CrpP activity changed as a function of the ATP concentration (Fig. 3), indicating that the activity of CrpP was ATP dependent. This further suggested that the mechanism underlying CrpP function involves phosphorylation, considering that APHs catalyze the transfer of a phosphate group to the aminoglycoside molecule, and several of these enzymes use ATP as the phosphate donor group (17). Taken together, these results indicated that CrpP confers a selective resistance to CIP, probably through a mechanism involving phosphorylation.
FIG 3.
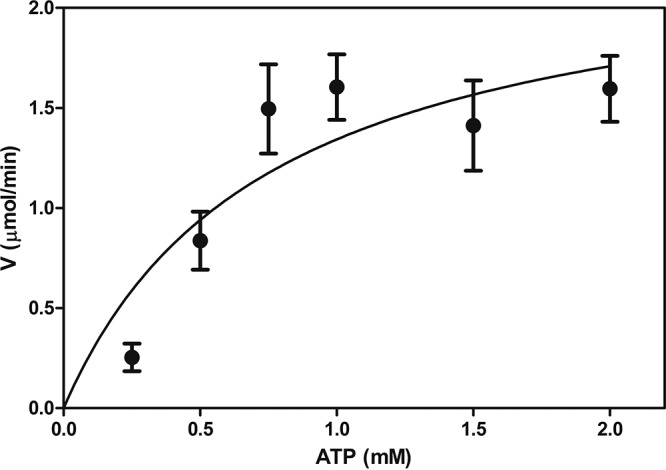
Michaelis-Menten plot of His-CrpP activity on ATP. ATP was incubated at the indicated concentrations in the presence of 5 μg/ml His-CrpP protein at 37°C for 15 min, as described in Materials and Methods. The Michaelis-Menten plot shows the data for NADH oxidation, which was measured spectrophotometrically at 390 nm. Data are means and standard errors from four independent duplicate assays.
To elucidate the mechanism of resistance to CIP, metabolite analysis was performed in vitro using liquid chromatography–tandem mass spectrometry (LC–MS-MS). When CIP was incubated with the His-CrpP protein, three peaks of interest, with retention times of 0.7, 2.18, and 3.84 min, were observed. These peaks were not observed in the chromatogram of the reaction in the absence of His-CrpP or ATP (Fig. 4A and B), suggesting a modification of the CIP molecule. Analysis of the peak with a retention time of 0.7 min clearly indicated the presence of a molecular ion corresponding to CIP-ATP (m/z 820) (Fig. 4C). In addition, several intermediary molecular ions were also identified, such as those with m/z 507, m/z 427, m/z 411, m/z 347, and m/z 331; the last ion corresponds to the CIP molecule (Fig. 4D). To better understand the mechanism of CIP inactivation, we analyzed the peaks showing retention times of 2.18 and 3.84 min (Fig. 4A). We detected mass ions corresponding to metabolites produced following fragmentation of the molecule. We propose that CIP is first activated by the binding of the ATP molecule (m/z 820), followed by phosphorylation of the molecule, confirmed by the presence of CIP monophosphate (m/z 411) (Fig. 4E). In addition, infrared spectroscopy assays showed that His-CrpP induced changes in the intensities of the characteristic absorption bands of the carboxyl group in the 2,500-to-3,300 cm−1, 1,200-to-1,420 cm−1, and 1,781 cm−1 regions, as well as in the 1,675 cm−1 absorption band, which corresponds to the vibration of the ketone carbonyl group at C-3 of the CIP molecule (see Fig. S6 in the supplemental material). This result agrees with the modification of CIP determined by LC–MS-MS, suggesting that CIP is phosphorylated on the carboxyl group. Additionally, these results suggest that the entire structure of CIP is important for CrpP function, since the carboxyl group is part of the central ring system in the quinolones, but CrpP did not show activity against other quinolones. A probable explanation for these results is that LVX and MXF contain oxygenated groups at position 8 of the central ring system in the quinolones, which can provide activation on the aromatic moiety, resulting in a higher electronic density in this part of the molecule. Meanwhile, the ethyl group of position 1 of the central ring of the quinolones in NOR could inject less electron density on the nitrogen atom than the cyclopropyl group present in CIP, producing, as a result, greater reactivity on the carboxyl group in CIP than in the other quinolones. In addition, metabolite analysis when CIP was incubated with crude extracts of an E. coli strain overexpressing the His-CrpP protein and ATP showed peaks corresponding to several degradation intermediaries of the phosphorylated CIP (CIP-P) molecule. These included molecular ions such as those with m/z 305, m/z 290, m/z 265, m/z 244, m/z 229, m/z 203, and m/z 131 (Fig. 4E; see also Fig. S7 in the supplemental material), suggesting that after CIP phosphorylation, the molecule is degraded.
FIG 4.
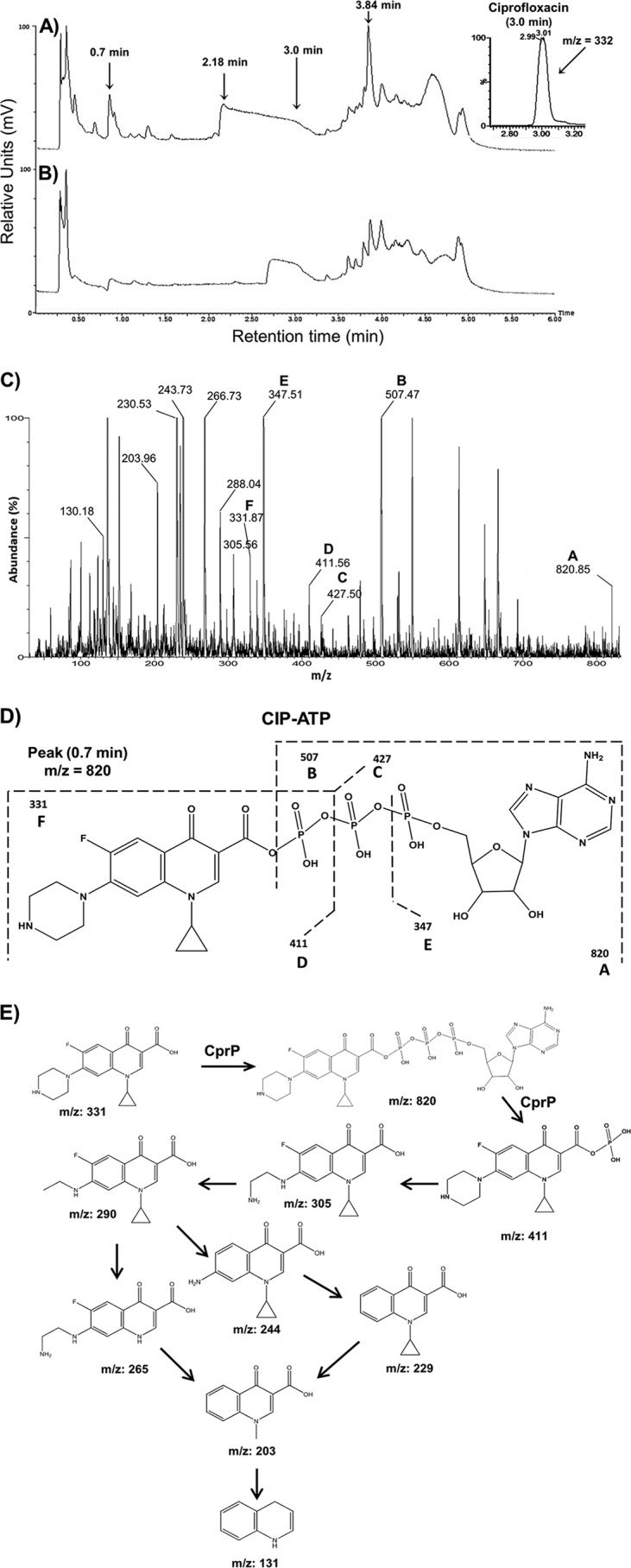
LC–MS-MS analysis of metabolites following CIP inactivation. (A and B) Representative chromatograms correspond to reaction mixtures containing the His-CrpP protein in the presence of CIP and ATP (A) or in the absence of ATP (B). The reactions were conducted as described in Materials and Methods. Arrows indicate peaks showing CIP structures (retention times of 0.7 min, 2.18 min, 3.0 min, and 3.84 min). (C) Representative mass fragmentation profile corresponding to the peak with a 0.7-min retention time. Capital letters above the peaks indicate the molecular ions that correspond to fragmented molecules derived from the ciprofloxacin-ATP molecule. Peak A (m/z 820) corresponds to the CrpP-ATP molecular ion. (D) Structure analysis of the mass fragmentation profile from panel C. (E) Reaction mechanism of CIP inactivation/fragmentation by the His-CrpP protein. The structures and m/z ratios were subtracted from the mass fragmentation profiles of peaks with retention times of 0.7, 2.18, and 3.84 min shown in panel A. Mass fragmentation profiles are shown in Fig. S7 in the supplemental material.
DISCUSSION
The pUM505 plasmid is a conjugative plasmid isolated from a clinical isolate of P. aeruginosa (9). pUM505 carries different genes, which encode proteins involved in metal resistance, DNA repair, virulence, and plasmid stability (10–13). Additionally, pUM505 conferred CIP resistance on P. aeruginosa PU21 (13), suggesting that pUM505 contains genes responsible for quinolone resistance. Because in silico analysis of the pUM505 genes did not initially identify genes previously implicated in quinolone resistance (13), these data suggested that the CIP resistance transferred by pUM505 could be conferred by genes not previously reported. The bacterial resistance to quinolones reported to date may involve chromosomal gene mutations or may be plasmid mediated (4, 6). In addition, these systems could provide resistance against several quinolones. For example, the QepA efflux pump decreases susceptibility to CIP and NOR (4, 6). For this reason, we determined whether pUM505 could confer resistance to other quinolones. The results showed that pUM505 confers decreased susceptibility to the fluoroquinolones CIP, NOR, and MXF in P. aeruginosa strain PAO1. To elucidate the molecular elements involved in fluoroquinolone resistance by pUM505, a new in silico analysis of the genes carried by this plasmid was conducted. We identified the 65-aa protein encoded by the crpP gene as a unique protein that shares sequence similarity with other proteins involved in resistance to antibiotics. CrpP displayed low identity (18%) to the APH-IIb enzyme from P. aeruginosa M18. The APH-IIb enzyme confers KAN, GEN, and STR resistance by transferring the phosphoryl group of ATP to the 3-hydroxyl groups of these antibiotics (14, 18). However, while APH enzymes share low (20 to 40%) sequence identity (18), they contain important conserved motifs, particularly at the C terminus, which is primarily responsible for catalysis and aminoglycoside binding. In addition, the ATP-binding domain is mainly located at the N terminus (18). The CrpP protein does not possess the catalytic motifs reported for APH enzymes, although it contains two conserved catalytic residues, 7-Gly (involved in catalysis) and 26-Ile (involved in ATP binding). In addition, it has been reported that the sequences of the catalytic and ATP-binding domains differ between individual APH enzymes and between APH subfamilies (18). Thus, the CrpP protein may confer resistance to antibiotics, while variability in the protein sequence (relative to those of APH enzymes) may promote the recognition of specific substrates.
The presence of the crpP gene did not decrease the quinolone susceptibility of P. aeruginosa strain PAO1, possibly because this strain has intrinsic high resistance to many antibiotics (15). However, the transfer of pUM505 to P. aeruginosa PAO1 was able to confer decreased susceptibility to CIP, NOR, and MXF. These results suggested that pUM505 might encode, in addition to the crpP gene, several mechanisms of resistance that, together, could increase Pseudomonas resistance to quinolones. It has been reported that PMQR elements [Qnr proteins, the AAC(6′)-Ib-cr enzyme, or the OqxAB, QepA1, and QepA2 efflux pump proteins] could confer reduced susceptibility to quinolones; for example, the AAC(6′)-Ib-cr enzyme increased MIC levels against E. coli 4-fold (19). However, PMQR systems are not mutually exclusive and together can confer lower levels of susceptibility to quinolones (1). Transferring crpP to E. coli J53-3 decreased the susceptibility of this strain to CIP, but not to NOR, MXF, LVX, NAL, or the aminoglycosides KAN, STR, AMK, and GEN. These results indicate that crpP encodes a protein that selectively confers resistance to CIP on E. coli. The capacity to confer selective resistance to an antibiotic is not uncommon; it is well known that APH enzymes from the same family display variable aminoglycoside resistance profiles (18). APH(3′) enzymes phosphorylate KAN and neomycin, but they tend to differ in their specificities for other aminoglycosides (18). Furthermore, AAC(6′) enzymes differ in their activities against AMK and GEN C1 (17).
The in vitro activity of the purified CrpP enzyme on CIP was determined in order to understand the mechanistic involvement of this enzyme in CIP resistance. CrpP showed enzymatic activity toward CIP and NOR when they were used as substrates. No activity toward LVX, MXF, NAL, or the aminoglycosides KAN and STR was observed, suggesting that CrpP is specific for CIP and NOR. Determination of kinetic parameters indicated that the Km values for CIP and NOR were similar, although the Vmax for CIP was >7 times higher than that for NOR. The Vmax/Km ratio was approximately 5-fold higher for CIP than for NOR, which could explain why E. coli transformed with the P. aeruginosa crpP gene did not show decreased NOR susceptibility. The APH family includes several members that are differentiated based on their substrate specificities or resistance phenotypes. Thus, members of this family confer resistance to specific antibiotics (20). Similar results were obtained by using the AAC(6′)-Ib-cr enzyme, which was more efficient at modifying KAN than CIP, suggesting its different specificities for different antibiotics (8). Moreover, CrpP used ATP as a variable cosubstrate, indicating that the activity of CrpP was ATP dependent. This further suggested that the mechanism underlying CrpP function involves phosphorylation, since APHs catalyze the transfer of a phosphate group to the aminoglycoside molecule, and several of these enzymes use ATP as the donor of the phosphate group (17).
In addition, in vitro CIP metabolite analysis using LC–MS-MS suggested that a modification of the CIP molecule occurred, in agreement with the infrared spectroscopy results. Our analysis revealed the presence of a molecular ion corresponding to CIP-ATP (m/z 820) and several intermediaries of the fragmented CIP-ATP molecule. These analyses enable us to propose that CIP is first activated by binding to the ATP molecule, followed by phosphorylation and degradation. The AAC(6′)-Ib-cr protein from E. coli is the only CIP-modifying enzyme described to date. This enzyme is a variant of the aminoglycoside acetyltransferase enzyme AAC(6′)-Ib, an acetyltransferase that confers resistance to aminoglycosides such as KAN, AMK, and tobramycin (8). The AAC(6′)-Ib-cr protein confers reduced susceptibility to CIP by decreasing the activity of this antibiotic via N-acetylation at the amino nitrogen on its piperazinyl substituent (8). Therefore, CrpP may represent a novel protein with specific enzymatic activity against CIP.
In summary, our results show that crpP encodes a novel protein capable of specifically conferring resistance to CIP in E. coli through an ATP-dependent mechanism that involves phosphorylation of the antibiotic.
MATERIALS AND METHODS
Bacterial strains and culture media.
P. aeruginosa strain PAO1 (21) and E. coli strains JM101 (22) and J53-3 (F− Met− Pro− Rifr) (23) were used as recipient strains for recombinant plasmids or susceptibility tests. The P. aeruginosa(pUM505) strain, expressing the pUM505 plasmid (Hgr Crr) (24), was used to evaluate quinolone resistance, as well as representing a DNA donor of pUM505. The E. coli BL21-CodonPlus(DE3)-RP strain (Stratagene, San Diego, CA, USA) was used for protein overexpression. The following culture media were used: nutrient broth (NB), Luria-Bertani (LB) broth (for solid media, 1.5% agar was added) (25), and M9 minimal salts medium (Sigma, St. Louis, MO, USA) supplemented with 20 mM glucose, 0.1 mM CaCl2, 2.0 mM MgSO4, and 0.1% casein peptone. When necessary, ampicillin (AMP) (100 μg/ml) or carbenicillin (CAR) (400 μg/ml) was added to agar plates.
Bacterial growth and susceptibility tests.
Bacterial cultures were obtained by diluting overnight cultures 1:50 in tubes containing 4 ml of fresh medium. The MICs for CIP, LVX, NOR, MXF, and aminoglycosides were obtained by broth microdilution in Mueller-Hinton medium according to the CLSI recommendations (26). The P. aeruginosa ATCC 27853 and E. coli ATCC 25922 strains were used as reference strains for antibiotic susceptibility testing.
Cloning of crpP.
The crpP gene was PCR amplified from the pUM505 plasmid using the following oligonucleotides: forward, 5′-CAACATGATGAATTCTACCGGAAAC-3′; reverse, 5′-GAGAAATGAAGCTTGCGTTGTT-3′ (designed with EcoRI and HindIII endonuclease sites [underlined], respectively). The PCR fragment was purified, cloned into the pJET1.2/blunt vector (Thermo Fisher Scientific, Waltham, MA, USA), and transferred to E. coli strain JM101 by electroporation; the transformants were selected on LB agar plates containing AMP. The DNA fragment containing crpP was obtained by digestion with EcoRI and HindIII and was subcloned into the corresponding sites of the pUCP20 shuttle vector (27). The resulting recombinant pUC_crpP plasmid was transferred to the P. aeruginosa PAO1 and E. coli J53-3 strains by electroporation, and transformants were selected on LB agar plates using CAR or AMP, respectively.
RT-qPCR assays.
Total RNA from bacterial cells grown in LB medium was isolated by utilizing the Tri reagent (Molecular Research Center, Inc., Cincinnati, OH, USA). After treatment with RQ1 RNase-free DNase (Promega, Madison, WI, USA), RNA was quantified by spectrophotometric analysis at 260 nm. Oligonucleotide primers and hydrolysis probes for RT-qPCR of the crpP gene and the 16S rRNA reference genes (listed in Table S1 in the supplemental material) were designed employing Biosearch Technologies software and were purchased from Biosearch Technologies (Novato, CA, USA). The crpP and 16S rRNA genes were amplified using the 5′ exonuclease probe RT-qPCR method. RT-qPCR was performed with total-RNA samples (500 ng) and the SuperScript III Platinum One-Step RT-qPCR reagent kit (Thermo Fisher Scientific, Waltham, MA, USA) on the LightCycler 480 II system (Roche Molecular Diagnostics, Pleasanton, CA, USA). Amplification signal curves were analyzed at absorption wavelengths of 530 nm. Appropriate positive and nontemplate controls were included in each test run. The relative expression of the crpP gene was normalized to expression values obtained from 16S rRNA genes from P. aeruginosa (GenBank accession number AE004091; region, nucleotides 722096 to 723631) or E. coli (GenBank accession number AJ605115). Relative gene expression was estimated via the classical calibration dilution curve and slope calculation. A 5-fold dilution series (500 to 0.05 ng total RNA) was prepared and was utilized as samples in the RT-qPCR. Efficiency (E) was obtained from standard curves utilizing the formula E = (1−1/slope − 1) × 100. Relative expression levels were determined with the efficiency correction method, which takes into account amplification efficiencies between target and reference genes (28).
Genetic techniques and sequence analysis.
General molecular genetics techniques were used according to standard protocols (25). The cloning process was verified by DNA sequencing and was conducted at Elim Biopharmaceuticals, Inc. (Hayward, CA, USA). Protein sequences were aligned using the ClustalW2 package (http://www.ebi.ac.uk/Tools/msa/clustalw2/). Sequence similarities in protein and DNA databases were searched using the blastp and blastx programs (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Potential promoter sequences were searched using Neural Network Promoter Prediction software (http://www.fruitfly.org/seq_tools/promoter.html).
Overexpression and purification of CrpP.
The crpP coding region was PCR amplified using pUM505 plasmid DNA as a template and the 5′-ACGAGGGATCCTGTGTCAAAG-3′ forward and 5′-AGCGGGATCAAGCTTAAATCGA-3′ reverse oligonucleotides, designed with BamHI and HindIII endonuclease sites (underlined), respectively. The amplified fragments were cloned into the pJET1.2/blunt vector, and the recombinant plasmids were transferred by electroporation into E. coli strain JM101; transformants were selected on LB agar plates using AMP. The crpP coding region was obtained by digestion with the BamHI and HindIII enzymes, purified, and subcloned into the corresponding restriction sites of the pTrcHisC vector (Thermo Fisher Scientific). The recombinant pTrcHis-crpP plasmid encodes the His-CrpP recombinant protein of ≈11 kDa, possessing a 6×His tag at the N terminus. The clones were validated by sequencing with the pTrcHis forward primer (Thermo Fisher Scientific). The recombinant plasmids were transferred by electroporation into the E. coli BL21-CodonPlus(DE3)-RP strain, and transformants were selected on LB agar plates using ampicillin. Overnight cultures of E. coli BL21(pTrcHis-crpP) were diluted at a 1:50 ratio into 250 ml of fresh NB medium and were cultured at 37°C with shaking until the optical density at 590 nm (OD590) reached 0.5. Overexpression of the His-CrpP protein was induced with 0.5 mM IPTG, and the culture was incubated for 6 additional hours. Cells were harvested by centrifugation, and the pellets were suspended in a loading buffer composed of 25 mM Tris (hydroxymethyl) amino methane-HCl (pH 6.8), 1% sodium dodecyl sulfate (SDS), 0.35 mM β-mercaptoethanol, 12.5% glycerol, and 0.005% bromophenol blue, after which they were heated for 5 min at 95°C. His-CrpP overexpression was verified on a 17% SDS-PAGE gel using a Tris-glycine buffer.
His-CrpP protein was purified as described by Watanabe and Takada (29). Cells were disrupted by sonication until the solution was clarified, and the debris was removed by centrifugation. The supernatant was loaded onto a nickel-nitrilotriacetic acid (NTA) resin (Qiagen, Venlo, The Netherlands) and was packed into a column. His-CrpP was recovered by elution with 200 mM imidazole, as described by Aranda et al. (30). The purification was monitored by 17% SDS-PAGE.
Phosphorylation assays.
Phosphorylation of quinolones (CIP, NOR, LVX, MXF, and NAL) or aminoglycosides (KAN and STR) by CrpP was monitored with a coupled assay using pyruvate kinase-lactate dehydrogenase, as described by Kramer and Matsumura (16), with the following modifications. We incubated 0.25, 0.5, 0.75, 1.0, 1.5, or 2.0 mM quinolone with 5.0 μg/ml of His-CrpP protein in a total volume of 1 ml of assay buffer (50 mM Tris [pH 7.6], 40 mM KCl, 10 mM MgCl2, 0.25 mg/ml NADH, 2.5 mM phosphoenolpyruvate, and 2.0 mM ATP) and 5.0 μl of pyruvate kinase-lactate dehydrogenase (600 to 1,000 μm/ml pyruvate kinase or 900 to 1,400 μm/ml lactate dehydrogenase [Sigma]) in a quartz cuvette (catalog no. 14-385-914A; Thermo Fisher). The mixtures were incubated at 37°C for 15 min. The oxidation of NADH was determined by monitoring the absorbance at 340 nm with an Amersham Biosciences Ultrospec 4300 pro UV/visible spectrophotometer (Amersham, UK). In this coupled assay system, the amount of ADP released is monitored at 340 nm through coupling with NADH oxidation. The data were plotted using nonlinear regression, the Michaelis-Menten model, and the least-squares method. GraphPad Prism software, version 5.01 for Windows, was used. Additionally, the activity of the His-CrpP protein on CIP was analyzed after its denaturation by heating at 95°C for 15 min, and the oxidation of NADH was determined as described above. To determine the optimal pH for CrpP activity, the pH of the reaction mixture was adjusted to 6.0, 6.5, 7.0, 7.5, or 8.0. Moreover, to analyze the effect of the Mg2+ concentration on CrpP activity, the MgCl2 concentration in the reaction mixture was modified to 5.0, 10.0, or 15.0 mM. Next, 0.5 mM fluoroquinolone was incubated with 5.0 μg/ml of His-CrpP protein in a total volume of 1 ml for each reaction mixture, and the oxidation of NADH was determined. Six independent assays with two replicates per experiment were conducted.
Analytical methods.
To determine whether CIP is modified by CrpP, LC–MS-MS analysis was conducted. Thus, 5.0 mM CIP was incubated with 5.0 μg/ml of His-CrpP protein for 1 h at 37°C, in the presence or absence of 2.0 mM Mg-ATP (Sigma), in buffer R (50 mM HEPES [pH 7.0], 40 mM KCl, 10 mM MgCl2) in a total volume of 1 ml. Next, the samples were centrifuged at 13,700 × g and were filtered using a 0.2-μm Millipore (Billerica, MA, USA) membrane. The LC–MS-MS system consisted of an Acquity ultraperformance liquid chromatography (UPLC) column and a Xevo TQ-S instrument (Waters, Milford, MA, USA).
LC–MS-MS measurement was carried out as follows. One microliter of the standard solution or previously filtered samples was injected into a reversed-phase LC column (Acquity UPLC BEH C18 column; particle size, 1.7 μm; inside diameter, 2.1 mm; length, 50 mm). CIP was eluted from the column using the following conditions: water containing 0.01% formic acid (A) and 100% acetonitrile (B). Elution was begun with 100% A–0% B followed by a linear gradient to 90% A–10% B (until 0.25 min), followed by 40% A–60% B (until 3.50 min), and finally by 100% A–0% B (until 4.50 min) plus 1 min for equilibration. A flow rate of 0.4 ml/min was used. A column temperature of 40°C was maintained, while the temperature of the samples in the autosampler was 10°C. For detection, a Xevo TQ-S instrument was used in the scan and multiple-reaction monitoring (MRM) modes. Scanning was carried out from 30 to 1,000 m/z with a 0.2-s scan time, a 40-V cone voltage, in the positive-ionization mode. For the MRM mode, the specific parameters for the MS-MS transition were as follows. For CIP transition, there was a precursor ion of m/z 332.2 and product ions of m/z 245.2, 288.3, and 314.2 with collision energies of 25, 15, and 20 V, respectively. The cone voltage (V) was 5 V, and the dwell time was 0.020 s. All three transitions were measured using a capillary voltage of 2.20 kV, a desolvation temperature of 500°C, a desolvation gas flow (N2) of 800 liters/h, a cone gas flow of 50 liters/h, and a nebulizer gas flow of 7.0 × 105 Pa. The calculations were performed using the ratios of the peak areas of the quantified transition components of CIP.
Additionally, samples obtained under the conditions described above were analyzed by infrared spectroscopy, and the spectra were recorded using a Nicolet iS10 spectrophotometer (Thermo Scientific) and the attenuated total reflection (ATR) technique (31).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Coordinación de la Investigación Científica (UMSNH grants 2.6 and 2.35) and the Consejo Nacional de Ciencia y Tecnología (CONACYT; grants 181747, 167071, and 256927). K.C.H.-R. and V.M.C.-J. were supported by postgraduate fellowships from CONACYT.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02629-17.
REFERENCES
- 1.Naeem A, Badshah SL, Muska M, Ahmad N, Khan K. 2016. The current case of quinolones: synthetic approaches and antibacterial activity. Molecules 21:268. doi: 10.3390/molecules21040268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aldred KJ, Kerns RJ, Osheroff N. 2014. Mechanism of quinolone action and resistance. Biochemistry 53:1565–1574. doi: 10.1021/bi5000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Emmerson AM, Jones AM. 2003. The quinolones: decades of development and use. J Antimicrob Chemother 51(Suppl 1):13–20. doi: 10.1093/jac/dkg208. [DOI] [PubMed] [Google Scholar]
- 4.Hooper DC, Jacoby GA. 2015. Mechanisms of drug resistance: quinolone resistance. Ann N Y Acad Sci 1354:12–31. doi: 10.1111/nyas.12830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Redgrave L, Sutton SB, Webber MA, Piddock JV. 2014. Fluoroquinolone resistance: mechanism, impact on bacteria, and role in evolutionary success. Trends Microbiol 22:438–445. doi: 10.1016/j.tim.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 6.Fàbrega A, Madurga S, Giralt E, Vila J. 2009. Mechanism of action of and resistance to quinolones. Microb Biotechnol 2:40–61. doi: 10.1111/j.1751-7915.2008.00063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee JK, Lee YS, Park YK, Kim BS. 2005. Alterations in the GyrA and GyrB subunits of topoisomerase II and the ParC and ParE subunits of topoisomerase IV in ciprofloxacin-resistant clinical isolates of Pseudomonas aeruginosa. Int J Antimicrob Agents 25:290–295. doi: 10.1016/j.ijantimicag.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 8.Robicsek A, Strahilevitz J, Jacoby GA, Macielag M, Abbanat D, Park CH, Buch K, Hooper DC. 2006. Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside-acetyltransferase. Nat Med 12:83–88. doi: 10.1038/nm1347. [DOI] [PubMed] [Google Scholar]
- 9.Cervantes-Vega C, Chávez J, Córdova NA, De la Mora P, Velasco JA. 1986. Resistance to metals by Pseudomonas aeruginosa clinical isolates. Microbios 48:159–163. [PubMed] [Google Scholar]
- 10.Díaz-Magaña A, Alva-Murillo N, Chávez-Moctezuma MP, López-Meza JE, Ramírez-Díaz MI, Cervantes C. 2015. A plasmid-encoded UmuD homologue regulates expression of Pseudomonas aeruginosa SOS genes. Microbiology 161:1516–1523. doi: 10.1099/mic.0.000103. [DOI] [PubMed] [Google Scholar]
- 11.Rodríguez-Andrade E, Hernández-Ramírez KC, Díaz-Peréz SP, Díaz-Magaña A, Chávez-Moctezuma MP, Meza-Carmen V, Ortíz-Alvarado R, Cervantes C, Ramírez-Díaz MI. 2016. Genes from pUM505 plasmid contribute to Pseudomonas aeruginosa virulence. Antonie Van Leeuwenhoek 109:389–396. doi: 10.1007/s10482-015-0642-9. [DOI] [PubMed] [Google Scholar]
- 12.Hernández-Ramírez KC, Chávez-Jacobo VM, Valle-Maldonado MI, Patiño-Medina JA, Díaz-Pérez SP, Jácome-Galarza IE, Ortiz-Alvarado R, Meza-Carmen V, Ramírez-Díaz MI. 2017. Plasmid pUM505 encodes a toxin-antitoxin system conferring plasmid stability and increased Pseudomonas aeruginosa virulence. Microb Pathog 112:259–268. doi: 10.1016/j.micpath.2017.09.060. [DOI] [PubMed] [Google Scholar]
- 13.Ramírez-Díaz MI, Díaz-Magaña A, Meza-Carmen V, Johnstone L, Cervantes C, Rensing C. 2011. Nucleotide sequence of Pseudomonas aeruginosa conjugative plasmid pUM505 containing virulence and heavy-metal genes. Plasmid 66:7–18. doi: 10.1016/j.plasmid.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 14.Wu DQ, Ye J, Ou HY, Wei X, Huang X, He YW, Xu Y. 2011. Genomic analysis and temperature-dependent transcriptome profiles of the rhizosphere originating strain Pseudomonas aeruginosa M18. BMC Genomics 12:438. doi: 10.1186/1471-2164-12-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hancock RE. 1998. Resistance mechanisms in Pseudomonas aeruginosa and other nonfermentative gram-negative bacteria. Clin Infect Dis 27:S93–S99. doi: 10.1086/514909. [DOI] [PubMed] [Google Scholar]
- 16.Kramer JR, Matsumura I. 2013. Directed evolution of aminoglycoside phosphotransferase (3′) type IIIa variants that inactivate amikacin but impose significant fitness costs. PLoS One 8:e76687. doi: 10.1371/journal.pone.0076687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramirez MS, Tolmasky ME. 2010. Aminoglycoside modifying enzymes. Drug Resist Updat 13:151–171. doi: 10.1016/j.drup.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith AC, Baker NE. 2002. Aminoglycoside antibiotic resistance by enzymatic deactivation. Curr Drug Targets Infect Disord 2:143–160. doi: 10.2174/1568005023342533. [DOI] [PubMed] [Google Scholar]
- 19.Strahilevitz J, Jacoby GA, Hooper DC, Robicsek A. 2009. Plasmid-mediated quinolone resistance: a multifaceted threat. Clin Microbiol Rev 22:664–689. doi: 10.1128/CMR.00016-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wright GD, Thompson PR. 1999. Aminoglycoside phosphotransferases: proteins, structure, and mechanism. Front Biosci 4:D9–D21. doi: 10.2741/A408. [DOI] [PubMed] [Google Scholar]
- 21.Holloway BW, Krishnapillai V, Morgan AF. 1979. Chromosomal genetics of Pseudomonas. Microbiol Rev 43:73–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 23.Garza-Ramos U, Barrios H, Hernández-Vargas MJ, Rojas-Moreno T, Reyna-Flores F, Tinoco P, Othon V, Poirel L, Nordmann P, Cattoir V, Ruiz-Palacios G, Fernández JL, Santamaria RI, Bustos P, Castro N, Silva-Sánchez J. 2012. Transfer of quinolone resistance gene qnrA1 to Escherichia coli through a 50 kb conjugative plasmid resulting from the splitting of a 300 kb plasmid. J Antimicrob Chemother 67:1627–1634. doi: 10.1093/jac/dks123. [DOI] [PubMed] [Google Scholar]
- 24.Cervantes C, Ohtake H, Chu L, Misra TK, Silver S. 1990. Cloning, nucleotide sequence, and expression of the chromate resistance determinant of Pseudomonas aeruginosa plasmid pUM505. J Bacteriol 172:287–291. doi: 10.1128/jb.172.1.287-291.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Green MR, Sambrook J. 2012. Molecular cloning: a laboratory manual, 4th ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 26.Clinical and Laboratory Standards Institute. 2008. Performance standards for antimicrobial susceptibility testing. Document M100–S18 CLSI, Wayne, PA. [Google Scholar]
- 27.West SE, Schweizer HP, Hricová K, Uvízl R, Neiser J, Blahut L, Urbánek K. 1994. Construction of improved Escherichia-Pseudomonas shuttle vectors derived from pUC18/19 and sequence of the region required for their replication in Pseudomonas aeruginosa. Gene 148:81–86. doi: 10.1016/0378-1119(94)90237-2. [DOI] [PubMed] [Google Scholar]
- 28.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29(9):e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watanabe S, Takada Y. 2004. Amino acid residues involved in cold adaptation of isocitrate lyase from a psychrophilic bacterium, Colwellia maris. Microbiology 150:3393–3403. doi: 10.1099/mic.0.27201-0. [DOI] [PubMed] [Google Scholar]
- 30.Aranda J, Garrido ME, Cortés P, Llagostera M, Barbé J. 2008. Analysis of the protective capacity of three Streptococcus suis proteins induced under divalent-cation-limited conditions. Infect Immun 76:1590–1598. doi: 10.1128/IAI.00987-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramer G, Lendl B. 15 March 2013. Attenuated total reflection Fourier transform infrared spectroscopy. In Encyclopedia of analytical chemistry: applications, theory and instrumentation. John Wiley and Sons, Inc, Hoboken, NJ. doi: 10.1002/9780470027318.a9287. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.