

ABSTRACT
AB-423 is a member of the sulfamoylbenzamide (SBA) class of hepatitis B virus (HBV) capsid inhibitors in phase 1 clinical trials. In cell culture models, AB-423 showed potent inhibition of HBV replication (50% effective concentration [EC50] = 0.08 to 0.27 μM; EC90 = 0.33 to 1.32 μM) with no significant cytotoxicity (50% cytotoxic concentration > 10 μM). Addition of 40% human serum resulted in a 5-fold increase in the EC50s. AB-423 inhibited HBV genotypes A through D and nucleos(t)ide-resistant variants in vitro. Treatment of HepDES19 cells with AB-423 resulted in capsid particles devoid of encapsidated pregenomic RNA and relaxed circular DNA (rcDNA), indicating that it is a class II capsid inhibitor. In a de novo infection model, AB-423 prevented the conversion of encapsidated rcDNA to covalently closed circular DNA, presumably by interfering with the capsid uncoating process. Molecular docking of AB-423 into crystal structures of heteroaryldihydropyrimidines and an SBA and biochemical studies suggest that AB-423 likely also binds to the dimer-dimer interface of core protein. In vitro dual combination studies with AB-423 and anti-HBV agents, such as nucleos(t)ide analogs, RNA interference agents, or interferon alpha, resulted in additive to synergistic antiviral activity. Pharmacokinetic studies with AB-423 in CD-1 mice showed significant systemic exposures and higher levels of accumulation in the liver. A 7-day twice-daily administration of AB-423 in a hydrodynamic injection mouse model of HBV infection resulted in a dose-dependent reduction in serum HBV DNA levels, and combination with entecavir or ARB-1467 resulted in a trend toward antiviral activity greater than that of either agent alone, consistent with the results of the in vitro combination studies. The overall preclinical profile of AB-423 supports its further evaluation for safety, pharmacokinetics, and antiviral activity in patients with chronic hepatitis B.
KEYWORDS: capsid inhibitor, HBV, pgRNA encapsidation, AB-423, CHB, sulfamoylbenzamide
INTRODUCTION
It is estimated that approximately 260 million people worldwide are suffering from a chronic infection with the hepatitis B virus (HBV), most of whom are in Asia, constituting a major public health problem (1, 2). A majority of the chronic hepatitis B (CHB) patients are unaware of their condition, and if untreated, long-term HBV infection can lead to cirrhosis, liver failure, or, eventually, hepatocellular carcinoma (HCC). It is estimated that chronic HBV infection is implicated in 80% of all cases of HCC in Asia and sub-Saharan Africa (3). In addition, 7.4% of the 36.5 million HIV-infected patients worldwide are coinfected with HBV (2). CHB leads to ∼900,000 deaths each year globally (2). In the United States, there are an estimated 2 million people who are chronically infected with HBV, and according to the CDC, CHB is implicated in ∼2,000 deaths annually (4, 5).
HBV is a noncytopathic, small DNA virus of the Hepadnaviridae family, with related viruses being found in woodchucks, ground/tree squirrels, Pekin ducks, and herons. On the basis of sequence diversity, there are eight known HBV genotypes, categorized from A to H, of which globally genotypes A to D are the most prevalent, while in the United States, genotypes A and C predominate, with 31% and 35% prevalences, respectively (6). The HBV genome is a 3.2-kb partially double-stranded circular DNA, and the viral polymerase is covalently attached to the 5′ end of the minus strand. Four types of viral particles can be detected in the serum from HBV-infected patients and include (i) 20-nm spherical structures, (ii) 22-nm-wide filaments of variable lengths comprised of the HBV surface antigen (HBsAg) and host-derived lipids devoid of viral nucleic acids, (iii) infectious virions (Dane particles) that are spherical, double-shelled structures 42 nm in diameter comprised of a lipid envelope containing HBsAg that surrounds an inner nucleocapsid composed of HBV core antigen (HBcAg) complexed with virally encoded polymerase and the viral DNA genome, and (iv) HBV RNA containing virus-like particles both in patient serum and in supernatants of HBV-infected hepatocytes (7–10).
During the life cycle of the hepatitis B virus, the virion enters the hepatocytes through Na+ taurocholate-cotransporting polypeptide (NTCP)-mediated endocytosis. Once inside the endocytic vesicle, the virus undergoes uncoating and is targeted to the nuclear pore complex, where the viral relaxed circular DNA (rcDNA) is delivered into the nucleus. In the nucleus, the rcDNA is converted to covalently closed circular DNA (cccDNA), which serves as the template for transcription of pregenomic RNA (pgRNA) and mRNAs for precore, envelope, and HBx proteins. Both the viral pgRNA and mRNAs are exported into the cytoplasm, where the mRNAs are translated into viral proteins by the host translation machinery and the pgRNA and newly synthesized viral proteins are used to generate new virions. In a single infected cell, cccDNA itself can be amplified only by reverse transcription of pgRNA to rcDNA in the cytoplasm and conversion of that rcDNA into cccDNA (11).
The current standard of care (SOC) for treating CHB patients falls into two classes: (i) nucleoside(t)ide analogs (NAs), which are direct inhibitors of the viral reverse transcriptase and DNA polymerase, and (ii) pegylated interferon alpha (PEG-IFN-α) (12, 13). While these therapies suppress active viral replication, reduce cccDNA levels, and prevent disease progression, they do not eliminate the nuclear pool of cccDNA (14–16). Due to the persistence of cccDNA, lifelong treatments with the antiviral therapies are required for a majority of patients to continuously suppress viral replication. Only a small percentage (4 to 11%) of chronic HBV patients treated for a year with PEG-IFN-α show HBsAg loss, which is similar to achieving a cure (17–20). Furthermore, some nucleoside inhibitors, such as lamivudine (LAM) and entecavir (ETV), are prone to resistance development, which could lead to treatment failures, while interferon therapy is poorly tolerated.
The goal for novel CHB therapies is to increase cure rates and reduce the treatment duration over that for the current SOC with treatment regimens that are safe and better tolerated and that do not require lifelong treatment. Clinically, these therapies should reduce a patient's risk of death due to liver disease by bringing it down, ideally, to the levels for individuals who have never been infected with HBV or to the levels for individuals who have resolved their HBV infection (21). It is hoped that the next wave of anti-HBV antivirals being developed will include direct-acting antivirals targeting viral replication (e.g., capsid inhibitors), agents that reduce s-antigen levels (e.g., small-molecule inhibitors and RNA interference [RNAi] agents), and immune modulators that stimulate host immune responses and that when these are administered in combination with current SOC agents they will increase the cure rates for CHB patients. Such a therapeutic transformation has been witnessed for hepatitis C virus infection, against which novel treatment regimens now have >90% cure rates, with all-oral interferon and ribavirin-free regimens achieving cures in 8 to 24 weeks (22).
The replication of the HBV genome is strictly dependent upon the assembly of the capsid structure around pregenomic RNA (pgRNA), and capsid assembly is emerging as a novel target for therapeutic intervention for CHB patients (23–25). The process of HBV capsid formation is shown schematically in Fig. 1. It starts with the binding of HBV DNA polymerase to the epsilon stem-loop structure of pgRNA through a protein-based priming mechanism. This initial binding step is then accompanied by the recruitment of core protein dimers that associate with the priming complex of the viral polymerase and pgRNA template, initiating the process of encapsidation. Capsid assembly is then followed by synthesis of the HBV genomic rcDNA through reverse transcription of the pgRNA, eventually leading to a fully encapsidated and enveloped infectious virion decorated with s antigen and secreted into the bloodstream (26). In addition to the role of core protein in forming the capsid structure, it has also been reported to interact with cccDNA and regulate viral gene expression (27). A role for the nucleus-localized, cccDNA-associated core protein in recruiting APOBEC3A, a cytidine deaminase, to cccDNA, mediating its selective deamination, and promoting its degradation has been suggested as the mechanism of specific and nonhepatotoxic degradation observed in IFN-α-treated HBV-infected hepatocytes (28).
FIG 1.

Intracellular events of capsid biogenesis and examples of distinct classes of capsid inhibitors. Heteroaryldihydropyrimidines (HAPs) are represented by BAY-41-4109 and GLS-4; pgRNA encapsidation inhibitors are represented by phenylpropenamides (PP), such as AT-130; and sulfamoylbenzamides (SBAs) are represented by AB-423. pol, polymerase.
A number of small-molecule compounds that inhibit capsid function have been reported (Fig. 1). These compounds mainly fall into two categories: class I capsid inhibitors, represented by heteroaryldihydropyrimidines (HAPs), such as BAY-41-4109 (29) and GLS-4 (30), which lead to misassembled noncapsid core polymers, and class II capsid inhibitors, such as the phenylpropenamides, represented by AT-130 (31, 32), which form morphologically normal capsids that are devoid of viral nucleic acid. Capsid inhibitors of both classes have shown antiviral activity in vitro in cell culture models as well as in vivo in animal models of HBV infection. The first clinical proof of concept for a capsid inhibitor was demonstrated by NVR-3-778, which produced a 1.7-log10 reduction in day 28 plasma HBV DNA levels when administered at 600 mg twice daily (BID) for 28 days in CHB patients (33). The chemical structure of NVR-3-778 has not been fully disclosed, but the core chemical structure resembles that of published class II capsid inhibitors belonging to the sulfamoylbenzamide (SBA) chemical class (34). More recently, the antiviral activities of two other capsid inhibitors, JNJ-379 and GLS4JHS, were reported in CHB patients (35, 36). Here we present the preclinical antiviral characterization of AB-423, an HBV capsid assembly inhibitor belonging to the SBA class (Fig. 1) currently in phase 1 clinical trials. We also demonstrate the potential for combining AB-423 with other mechanistic classes of HBV inhibitors, such as nucleos(t)ide analogs, RNAi agents, and IFN-α, using in vitro and in vivo HBV models.
RESULTS
AB-423 is an inhibitor of HBV replication in HBV cell culture models.
AB-423 belongs to the SBA class of capsid inhibitors, which have been described previously (34). The antiviral activity of AB-423 was evaluated in several different cell culture models of HBV infection, including models involving AML12-HBV10 (34), an immortalized murine hepatocyte cell line that inducibly expresses genotype D HBV; HepDE19 (37) and HepBHAe82 (38), two human hepatoma cell lines that also inducibly express genotype D HBV under the control of a cytomegalovirus (CMV) tetracycline (Tet)-off promoter; HepG 2.2.15 (39), a cell line that constitutively expresses genotype D HBV; and HBV-infected primary human hepatocytes (PHH). AB-423 showed inhibition of rcDNA production in a concentration-dependent manner. The antiviral activity of AB-423, as measured by rcDNA production, was comparable in the AML12-HBV10 and HepDE19 cell culture systems, with 50% effective concentrations (EC50s) of ∼0.260 μM (Table 1). AB-423 showed comparable inhibition of rcDNA production in the HepDE19 cell assay during incubation with AB-423 for 2, 3, 6, or 7 days (data not shown). Consistent with these data, AB-423 also demonstrated inhibition of cccDNA formation-dependent HBeAg production in the HepBHAe82 cell assay with an EC50 of 0.267 μM and inhibited HBV DNA levels in culture supernatants of HepG 2.2.15 cells with an EC50 of 0.134 μM. No cytotoxicity was detected in any of the three cell lines, and thus, 50% cytotoxic concentration (CC50) values are expressed in terms of the highest compound concentrations tested. Assessing antiviral activity in an authentic primary cell infection system is an important validation for potency, especially as the metabolic activity of HBV cell culture models may differ from that of PHH. Therefore, the anti-HBV potency of AB-423 was also determined in a PHH culture infection system. Treatment of infected PHH with AB-423 reduced the amount of HBV DNA in the supernatant by 1 log10 with an EC50 of 0.078 μM (Table 1). The addition of up to 40% human serum to the cell culture medium caused a modest (5-fold) decrease in the potency of AB-423 in AML12-HBV10 cells (Table 2).
TABLE 1.
Potency evaluation of AB-423 in various in vitro HBV assay systemsa
| Assay | Readout | EC50 (μM) | EC90 (μM) | CC50 (μM) |
|---|---|---|---|---|
| AML12-HBV10 cells | rcDNA | 0.263 ± 0.177 | 1.319 ± 1.076 | >10 |
| HepDE19 cells | rcDNA | 0.262 ± 0.127 | 0.905 ± 0.332 | >10 |
| HepBHAe82 cells (ELISA) | HBeAg | 0.267 ± 0.135 | 1.246 ± 0.466 | >10 |
| HepG 2.2.15 cells (qPCR) | HBV DNA | 0.134 ± 0.022 | 0.348 ± 0.080 | >10 |
| HBV-infected PHH | HBV DNA | 0.078 ± 0.031 | 0.332 ± 0.235 | ND |
The EC50 and EC90 values are the averages and standard deviations derived from at least three independent determinations. ND, not determined.
TABLE 2.
Effect of human serum on in vitro potency of AB-423
| Human seruma (%) | AB-423 |
ETV |
||
|---|---|---|---|---|
| EC50 (μM) | Serum shift ratiob | EC50 (μM) | Serum shift ratio | |
| 0 | 0.363 | 1.0 | 0.007 | 1.0 |
| 10 | 0.606 | 1.7 | 0.007 | 1.0 |
| 20 | 1.231 | 3.4 | 0.024 | 3.4 |
| 40 | 1.813 | 5.0 | 0.029 | 4.0 |
Human serum was used with standard medium, which contains 10% fetal bovine serum.
The serum shift ratio was calculated by dividing the EC50 observed in the presence of human serum by the EC50 observed in the absence of human serum. This evaluation was performed using the AML12-HBV10 cell culture system described in the Materials and Methods section.
AB-423 inhibits capsid assembly at the pgRNA encapsidation stage in an HBV cell culture model.
It has been shown that compounds belonging to the SBA class of encapsidation inhibitors allow the production of normal amounts of capsid in cells but that these capsids are predominantly empty (i.e., they lack capsid-associated pgRNA and HBV DNA) (34). In contrast, treatment with the capsid assembly modulator BAY 41-4109, a member of the HAP class of compounds, results in a strong reduction in capsid particle formation. To pinpoint the specific stage of capsid assembly that is inhibited by AB-423, HepDES19 cells were treated with AB-423 at a concentration of 3 μM for 6 days and various viral replication intermediates were extracted and analyzed (Fig. 2) by comparison to the untreated controls (i.e., cells treated with the dimethyl sulfoxide [DMSO] vehicle). Treatment of HepDES19 cells with AB-423 showed a strong inhibition of capsid-associated RNA and DNA (Fig. 2E and F, lanes 2) but no effect on the total amount of pgRNA (Fig. 2A, lane 2) or capsid particles (Fig. 2D, lane 2) in comparison to that in untreated control cells, indicating that AB-423 inhibited HBV replication at the step of pgRNA encapsidation, thus impacting the levels of downstream replication intermediates (capsid-associated RNA, capsid-associated DNA, rcDNA, and single-stranded DNA [ssDNA]). Consistent with this mechanism, there was little to no reduction in the amount of core protein or capsid particles produced in the cells (Fig. 2C and D, lanes 2). In comparison, GLS-4, a HAP related to BAY 41-4109, showed a strong reduction of capsid particle formation (Fig. 2D, lane 3), while the nucleoside inhibitor lamivudine (3TC) showed no significant effect on core protein or the levels of capsid (Fig. 2C and D, lanes 4). Moreover, 3TC enhanced the levels of encapsidated pgRNA (Fig. 2E, lane 4), in contrast to AB-423 and GLS-4, as it prevented the conversion of encapsidated pgRNA to rcDNA (Fig. 2F, lane 4). These data demonstrate that AB-423 is a specific inhibitor of viral pgRNA encapsidation in an HBV cell culture system and distinguish its mechanism of action from that of capsid assembly modulators, such as BAY 41-4109 and GLS-4, which are class I capsid inhibitors, and the nucleos(t)ide class of inhibitors, represented by 3TC.
FIG 2.
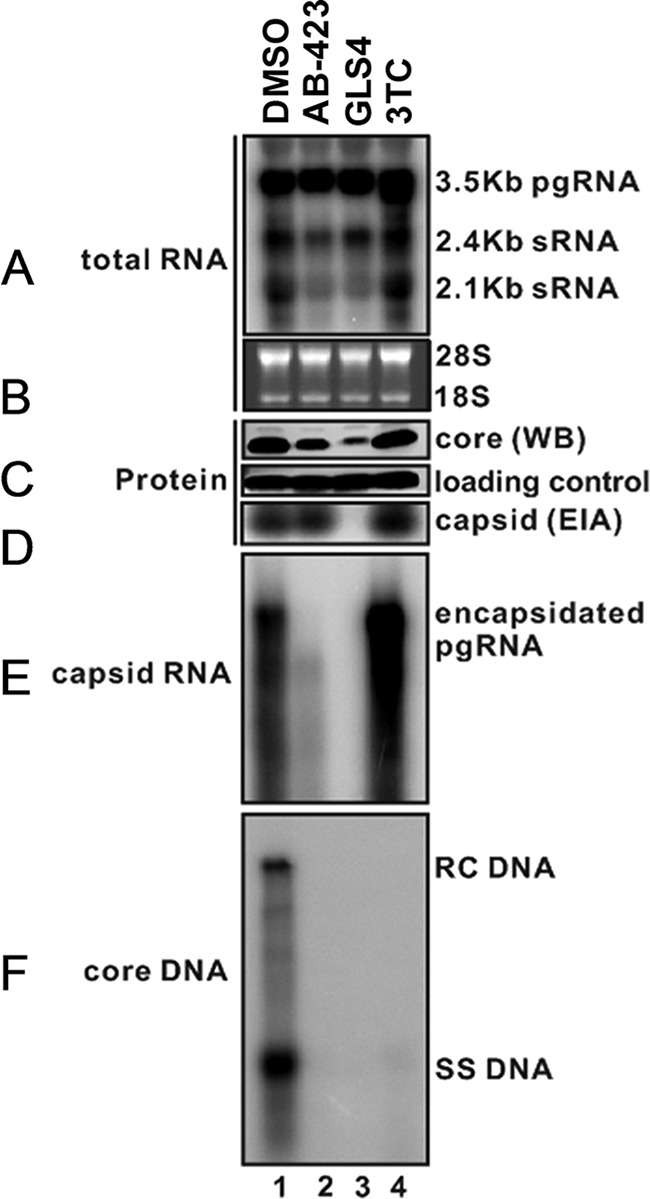
AB-423 inhibits pgRNA encapsidation in HepDES19 cells. HepDES19 cells were treated with AB-423 (3 μM), the vehicle control (DMSO), GLS-4 (a member of the HAP class of capsid inhibitors), or 3TC (a nucleoside inhibitor) for 6 days, and various viral replication intermediates were extracted and visualized by HBV RNA analysis and particle gel assays, which were performed as described in Materials and Methods. Intracellular viral RNA (A) and encapsidated pgRNA (E) were extracted and detected by Northern blotting hybridization with an HBV-specific 32P-labeled riboprobe. (B) rRNAs (18S and 28S) served as loading controls. (C) Intracellular HBV core protein was detected by Western blotting (WB) using specific antibodies. (D) The total amounts of nucleocapsids were determined by a particle gel assay. (F) HBV DNA replication intermediates (rcDNA and ssDNA) were extracted and detected by Southern blotting hybridizations.
AB-423 modulates HBV capsid assembly in a biochemical assay.
An in vitro biochemical assay of HBV capsid assembly (40) was used to demonstrate the direct interaction of AB-423 with HBV core protein and its activity on capsid assembly. The assay utilizes a mutant capsid protein that comprises the first 149 amino acids (the assembly domain) of the HBV core protein (lacking the RNA binding domain) and that is modified by the addition of a terminal cysteine and the fluorescent dye Bodipy-FL to form C150Bo. In solution, the capsid protein spontaneously reassembles as a function of protein concentration, temperature, and ionic strength. In nonassembled capsid dimer form, the C150Bo dye is fluorescent, whereas the assembly of capsid dimers quenches the fluorescence (Fig. 3). In this assay, AB-423 accelerated capsid assembly in a dose-responsive manner with a 50% inhibitory concentration (IC50) of 2.859 ± 0.430 μM (average ± standard deviation [SD]). These results support a direct role of AB-423 on capsid assembly, in agreement with the results observed in HBV tissue culture models.
FIG 3.
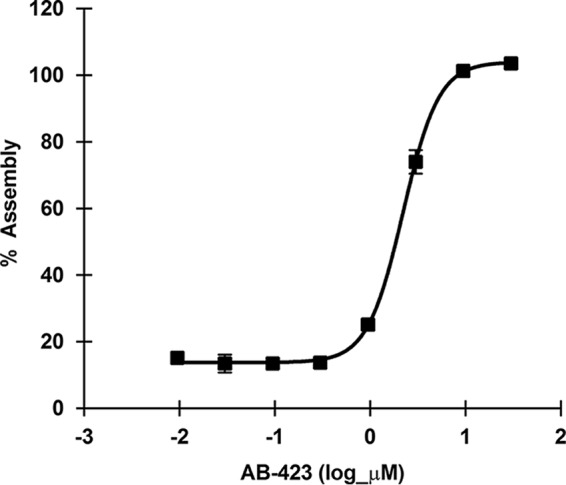
AB-423 modulates HBV capsid assembly in a biochemical assay. The activity of AB-423 was tested in an HBV capsid assembly assay using a protocol described in Materials and Methods. AB-423 was tested in a dose range format starting at the highest concentration of 30 μM and then at a serial, 1/2-log, 8-point dilution series in triplicate. The average dose-response curve and standard deviations for triplicate samples from one representative experiment are shown.
AB-423 inhibits conversion of encapsidated rcDNA to cccDNA in an infectious virus system.
In addition to preventing rcDNA synthesis through capsid inhibition, both class I and class II HBV capsid inhibitors can alter the stability of the preassembled rcDNA-containing nucleocapsid, exerting an additional activity on cccDNA formation that is distinct from inhibition of pgRNA encapsidation (41). This is likely due to interference with the normal uncoating of the nucleocapsid before rcDNA can be delivered to the nucleus for conversion to cccDNA and results in the reduction of cccDNA levels when compound exposure occurs after pgRNA encapsidation and during de novo infection. To confirm that AB-423 exhibits this activity, cells of the HBV infection-permissive cell line C3AhNTCP were treated with AB-423, the HBV entry inhibitor myrcludex B, and IFN-α starting 24 h before HBV infection was initiated and continuing to 6 days postinfection. At that time, cell monolayers were harvested and analyzed for pgRNA and cccDNA levels. As expected, by acting to prevent viral entry and cccDNA establishment from the infecting rcDNA, myrcludex B effectively reduced the levels of both cccDNA and its transcription product, pgRNA. IFN-α reduced the levels of cccDNA only slightly but exhibited a stronger effect on pgRNA, owing to its recognized activity in specific inhibition of HBV transcription but not cccDNA formation (42). In contrast, AB-423 exhibited a measurable and reproducible inhibition of cccDNA levels and the resulting expression of pgRNA. These data are consistent with inhibition of cccDNA formation by AB-423 at a step distinct from rcDNA synthesis (Fig. 4). While we cannot exclude the possibility that the inhibition of cccDNA levels after infection may have been at least partly due to the inhibition of rcDNA synthesis, which may contribute to the cccDNA pool in these cells postinfection, in both NTCP-expressing cells and primary human hepatocytes, the postinfection levels of cccDNA did not change after the initial establishment of cccDNA with or without treatment with a nucleoside analog, and thus, it is generally accepted that rcDNA synthesis and the subsequent conversion to cccDNA do not contribute to the cccDNA pool within the first several days after infection (41, 43). Based on these observations, we conclude that the inhibition of AB-423 on cccDNA formation during initial infection occurs independently of pgRNA encapsidation and rcDNA synthesis. It should be noted that the apparent potency of AB-423 on the reduction of the level of cccDNA is lower than that on the inhibition of pgRNA encapsidation (Fig. 2) and may represent a true difference in the potency between inhibition of uncoating of the nucleocapsid structure, such as during a de novo infection, and inhibition of pgRNA encapsidation by core protein dimers during nucleocapsid biogenesis. This appears to be a property common to different classes of HBV capsid inhibitors (43).
FIG 4.
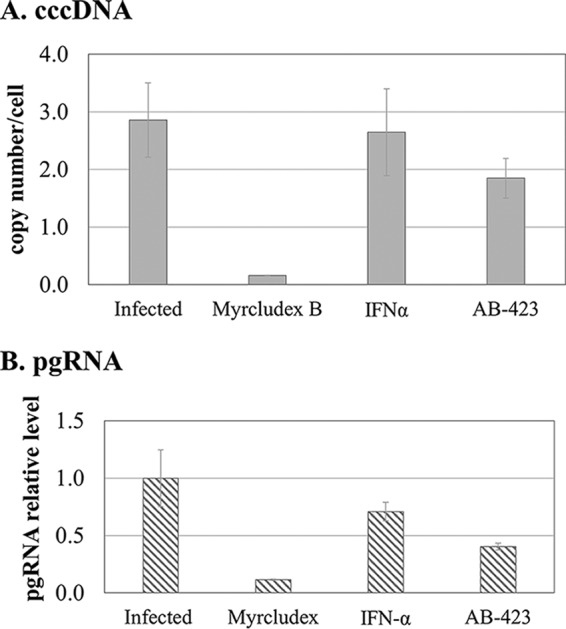
AB-423 inhibits conversion of encapsidated rcDNA to cccDNA in an infectious virus system. Inhibition of cccDNA formation by AB-423 in de novo infection was tested in C3AhNTCP cells through treatment starting at 24 h preinfection and continuing to day 6 postinfection. On day 6, cells were harvested and processed for cccDNA and pgRNA analysis by qPCR and quantitative RT-PCR, respectively, as described in Materials and Methods. Treatment was with DMSO only, myrcludex B (100 nM), IFN-α (1000 IU/ml), and AB-423 (3 μM). (A) The cccDNA copy number per cell compared to that in DMSO-treated infected samples. (B) pgRNA levels relative to those in DMSO-treated infected samples. The experiment was conducted with triplicate samples (n = 2). The bars represent the averages for triplicate samples from one trial, and the error bars represent the standard deviations.
AB-423 possesses pan-genotypic activity against HBV.
Hepatitis B virus is classified into genotypes A through H, of which genotypes A through D are the most prevalent genotypes globally. It is important that any new therapy for the treatment of CHB have broad genotype coverage. Since the HBV cell culture model systems used are based on genotype D, the activity of AB-423 against representatives of the other most prevalent HBV genotypes was evaluated. Using an HepG2 cell transient-transfection system, the antiviral activity of AB-423 against two cloned sequences each of genotypes A, B, and C and one sequence of genotype D was evaluated (Table 3). In this system, AB-423 showed inhibition of replication of HBV genotypes A, B, C, and D with EC50s of between 0.044 μM and 0.185 μM, which were comparable to or better than those observed in the standard laboratory HBV cell culture models (the HepDE19, HepBHAe82, AML12-HBV10, and HepG 2.2.15 cell models) described above. These data indicate that AB-423 has equivalent activity against representative clones of the most prevalent genotypes of HBV. While these results are consistent with the high level of conservation of the core protein dimer-dimer interface, we cannot exclude the possibility that there may be changes in the larger population of sequences that might impact the susceptibility to AB-423. The antiviral activity of AB-423 against HBV genotypes E through H has not been evaluated. Consistent with the understood mechanism of action of AB-423 on pgRNA encapsidation, AB-423 did not show any effect on the levels of HBsAg (data not shown), which in this transfection system is independent of viral encapsidation and replication.
TABLE 3.
Activity of AB-423 against HBV by genotypea
| HBV genotype | Avg EC50 ± SD (μM)b | GenBank accession no. |
|---|---|---|
| A | 0.050 ± 0.015 | AP007263 |
| A2 | 0.092 ± 0.017 | HE974371 |
| C | 0.044 ± 0.010 | AB246345 |
| C | 0.046 ± 0.012 | AB246346 |
| B | 0.056 ± 0.015 | JN406371 |
| B | 0.098 ± 0.006 | AB033554 |
| D | 0.185 ± 0.011 | U95551 |
This evaluation was performed using the HepG2 transient-transfection cell culture system, as described in the Materials and Methods section.
Average EC50s and standard deviations from 3 independent determinations are shown.
AB-423 inhibits replication of HBV nucleoside-resistant variants.
Chronic hepatitis B patients undergoing nucleos(t)ide therapy may develop resistance leading to treatment failure. Significant activity against HBV polymerase variants is needed to avoid cross-resistance with the new therapies. We therefore evaluated the activity of AB-423 against HBV polymerase variants known to confer resistance to nucleoside inhibitors (Table 4). AB-423 maintained activity against wild-type and single, double, and triple mutant HBV polymerase variants known to confer resistance to ETV or lamivudine with EC50s that were within 2-fold of the EC50 for the wild-type virus. These data are consistent with AB-423 having a mechanism of action distinct from that of the nucleos(t)ide class of HBV inhibitors.
TABLE 4.
Activity of AB-423 against nucleoside-resistant variants of HBVa
| HBV variant | Avg EC50 ± SD (μM)b |
||
|---|---|---|---|
| AB-423 | ETV | LAM | |
| rtM204I | 0.226 ± 0.031 | ND | >100 |
| rtM204I + V173L | 0.149 ± 0.004 | ND | >100 |
| rtM204I + S202G | 0.223 ± 0.032 | 10.236 ± 1.172 | ND |
| rtM204V + L180M | 0.197 ± 0.019 | ND | >100 |
| rtM204I + S202G + M250V | 0.239 ± 0.006 | 13.940 ± 4.493 | ND |
| U95551c | 0.168 ± 0.054 | 0.002 ± 0.0002 | 0.079 ± 0.056 |
ETV, entecavir; ND, not determined; LAM, lamivudine; rt, reverse transcriptase (HBV polymerase). This evaluation was performed using the HepG2 cell transient-transfection cell culture system, as described in the Materials and Methods section.
Average EC50s and standard deviations from 3 independent determinations are shown.
U95551 is the wild-type genotype D strain used as the vector background for all variants.
AB-423 shows additive to synergistic antiviral activity in vitro with nucleoside analogs and experimental RNAi agents.
The use of multiple antiviral agents in clinical combination regimens is often beneficial when the net effect of inhibiting several targets simultaneously is enhanced viral suppression, which can prevent the onset of resistance to any one agent or even hasten clearance of the infection. Conversely, combination use may be precluded due to antagonistic effects that reduce viral suppression. To guide clinical combination studies, the possibility of mechanistic antagonism may be assessed using in vitro viral culture models. In vitro combination studies were conducted using the method of Prichard and Shipman (44), wherein a checkerboard design was used to generate a 3-dimensional model of drug-drug interactions. In several cell culture models of HBV replication, cells were treated with AB-423, the nucleoside inhibitors ETV and tenofovir disoproxil fumarate (TDF), and an anti-HBV RNAi agent, ARB-1467 or ARB-1740, at concentration ranges spanning doses both above and below their respective EC50s. The two inhibitors were tested both singly and in combinations such that each concentration of inhibitor A was combined with each concentration of inhibitor B to determine the pharmacologic effect of the combination on HBV replication. Based on the MacSynergy II program and the established guidelines of Prichard and Shipman (44), all combinations tested were found to be additive to strongly synergistic (Fig. 5; see also Tables S1 to S6 in the supplemental material). Importantly, no significant antagonism was observed. These results underscore the mechanistic distinction between AB-423 and these other agents and confirm the potential utility of AB-423 for use in combination treatment regimens with varied classes of inhibitors.
FIG 5.

In vitro combination of AB-423 with nucleos(t)ide analogs and RNAi agents. Panels I, checkerboard synergy plots based on MacSynergy II analysis; panels II, antiviral dose-responses of each agent tested singly and in combination. (A) In vitro combination of AB-423 and ETV (AML12-HBV10 cell rcDNA/bDNA assay); (B) in vitro combination of AB-423 with the RNAi agent ARB-1467 (AML12-HBV10 cell rcDNA/bDNA assay); (C) in vitro combination of AB-423 with the RNAi agent ARB-1740 (AML12-HBV10 cell rcDNA/bDNA assay); (D) in vitro combination of AB-423 with TDF (HepDE19 cell rcDNA/bDNA assay); (E) in vitro combination of AB-423 with ETV (HepBHAe82 cell pcRNA/cccDNA assay); (F) in vitro combination of AB-423 with ARB-1740 (HepBHAe82 cell pcRNA/cccDNA assay). The results shown are the averages for quadruplicate samples.
Pharmacokinetic profile of AB-423 in mice.
The plasma concentration of AB-423 over time was profiled in CD-1 mice to capture its pharmacokinetic (PK) properties in vivo (Fig. 6). Following intravenous (i.v.) administration, AB-423 displayed a systemic clearance of 31 ml/min/kg with a volume of distribution of 2 liters/kg (Table 5). When AB-423 was dosed orally (p.o.) at 10 mg/kg of body weight in mice, it reached a maximum concentration (Cmax) of between 0.25 to 0.5 h and produced a Cmax and a level of exposure (area under the concentration-time curve) of 490 ng/ml and 2,238 ng · h/ml, respectively (Table 5). The oral bioavailability of AB-423 was estimated to be approximately 40% in mice. A dose-dependent gain in AB-423 exposure could be observed up to a dose of 100 mg/kg, as evident by the dose-proportional increase in exposure among the 10-, 30-, and 100-mg/kg doses. Following a single oral dose at 100 mg/kg, the liver concentration/plasma concentration ratio of AB-423 at 24 h was measured to be 2.9, while a 7-day multidosing regimen in the mouse hydrodynamic injection (HDI) study gave higher liver concentration/plasma concentration ratio of 11 (see below), suggesting favorable drug accumulation in the target organ to potentially achieve the desired pharmacological effect.
FIG 6.
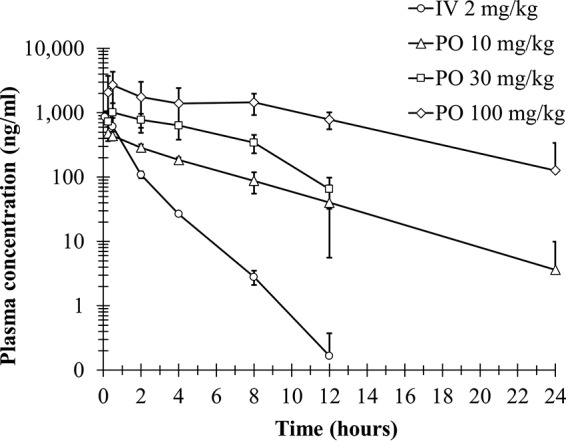
Pharmacokinetic profile of AB-423 in CD-1 mice. The plasma concentrations of AB-423 following a single i.v. or p.o. administration of AB-423 in mice are shown as the mean ± SD (n = 3 per group).
TABLE 5.
Pharmacokinetic properties of AB-423 in CD-1 micea
| Route of administration | Dose (mg/kg) | CLobs (ml/min/kg) | t1/2 (h) | Tmax (h) | Cmax (ng/ml) | AUClast (ng · h/ml) | AUCinf (ng · h/ml) | AUCextrap (%) | Vss,obs (liters/kg) | F (%) | 24-h liver concn/plasma concn ratio |
|---|---|---|---|---|---|---|---|---|---|---|---|
| i.v. | 2 | 31 | 1.1 | NA | 930 | 1,120 | 1,122 | 0 | 2 | <LLOQ | |
| p.o. | 10 | 3.4 | 0.33 | 490 | 1,994 | 2,238 | 10 | 40 | <LLOQ | ||
| p.o. | 30 | 3.2 | 1.67 | 1,028 | 5,832 | 6,174 | 5 | 37 | <LLOQ | ||
| p.o. | 100 | 5.9 | 0.50 | 2,709 | 23,024 | 25,232 | 8 | 45 | 2.9 |
Values are means (n = 3 per group). CLobs, observed clearance; t1/2, half-life; Tmax, time to maximum concentration; Cmax, maximum concentration; AUClast, area under the concentration-time curve at 1 h after the last dose; AUCinf, area under the concentration-time curve from 0 h to infinity; AUCextrap, extrapolated area under the concentration-time curve; Vss,obs, observed volume of distribution at steady state; F, bioavailability; NA, not applicable; LLOQ, lower limit of quantitation.
AB-423 inhibits HBV replication in a mouse model of HBV.
A hydrodynamic injection (HDI) model in immunodeficient NOD-SCID mice was used to determine whether AB-423 is effectively delivered via oral administration to the relevant target organ of HBV infection, the liver, to exert an antiviral effect on HBV-expressing hepatocytes in vivo. A 7-day treatment regimen with AB-423 in this model resulted in a dose-responsive reduction of the serum HBV DNA level of up to 1.4 log10 compared to that in the vehicle-treated control mice (Fig. 7). After 7 days of twice daily oral administration, the preferential accumulation of AB-423 occurred in the liver, the target organ for therapy, with a liver concentration-to-plasma concentration ratio of 11. At the dosages of 30 and 100 mg/kg BID tested, the plasma and liver concentrations of AB-423 at 1 h after the last dose were also found to be dose responsive, consistent with the observed effects on serum HBV DNA levels. Examination of other HBV markers in blood and liver showed AB-423 effects consistent with its known mechanism of action. Formation of cccDNA is understood not to occur in mouse cells (45), and in this animal model of HBV infection, the expression of HBsAg is solely driven from the incoming plasmid; therefore, AB-423 is not expected to impact serum HBsAg levels in this system. At 100 mg/kg, an HBV DNA reduction of 1.4 log10 in serum correlated with a 0.92-log10 reduction in the liver, the site of drug action where AB-423 inhibits functional capsid particle formation and the production of intact viral particles containing rcDNA. In total, these data demonstrate that oral administration of AB-423 results in anti-HBV activity in vivo and in a manner consistent with its mechanism of action.
FIG 7.
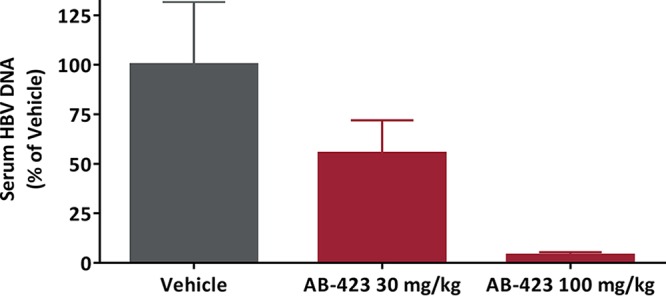
In vivo antiviral activity of AB-423 in a mouse model of HBV infection. HDI mice were administered AB-423 twice daily for 7 consecutive days. Antiviral effects are expressed as a percentage of those for the vehicle-only control group on day 7. Data are shown as the mean ± SEM. The number of animals per group for data analysis was 5 or 6.
In vivo combination studies.
While it is not feasible to evaluate many dose levels or treatment variants of compound combinations in vivo, even limited testing of dose combinations is of value, in that it allows evaluation of many more factors of drug-drug interactions than the number of factors that may be evaluated in cell culture models. In an HDI model of HBV infection in immunodeficient NOD-SCID mice, 100 mg/kg of AB-423 given orally BID for 7 days in combination with 100 ng/kg of ETV given orally once a day (QD) for 7 days resulted in a trend toward the inhibition of serum HBV DNA greater than that by either monotherapy arm alone (Fig. 8). While the serum HBV DNA reductions as a percentage of the day 0 baseline values at day 7 were statistically significant for all compound treatment groups compared to the values for the saline-treated controls (one-way analysis of variance [ANOVA], P < 0.05), the reductions for the compound treatment groups did not reach statistical significance compared to those for the vehicle-treated control group. A combination of 100 mg/kg of AB-423 given orally for 7 days with a single 0.1 mg/kg dose of ARB-1467 (an investigational RNAi agent) given i.v. resulted in a trend toward the inhibition of serum HBV DNA greater than that by either monotherapy arm alone (Fig. 8). In this case, the serum HBV DNA reductions as a percentage of the day 0 baseline values were statistically significant for all compound treatment groups versus the saline- and vehicle-treated control groups (one-way ANOVA, P < 0.05). In agreement with the in vitro findings, greater levels of inhibition were observed from liver HBV DNA assessment for both drug combinations (data not shown). As the primary function of ARB-1467 is intended to be inhibition of HBsAg, it was also verified that the addition of AB-423 did not affect ARB-1467's reduction of HBsAg in a whole-body system (Fig. 8). In summary, no antagonism was detected from in vivo investigations of AB-423 in combination with drugs from two other anti-HBV drug classes. These data were in agreement with the in vitro findings described above.
FIG 8.

In vivo antiviral activity of combination treatment with AB-423 and a nucleoside analog or an RNA interference agent. (A) Serum HBV DNA level after treatment with the AB-423 and ETV combination; (B) serum HBV DNA level after treatment with the AB-423 and ARB-1467 combination; (C) serum HBsAg level after treatment with the AB-423 and ARB-1467 combination. AB-423 was administered via oral gavage twice daily for 7 consecutive days, starting on day 0. ETV was administered via oral gavage once daily for 7 consecutive days, starting on day 0. ARB-1467 was administered as a single intravenous bolus tail vein injection on day 0. The antiviral treatment effect in HDI mice was expressed as a percentage of the individual animal's day 0 (predose) value. Data are shown as the mean ± standard error of the mean (SEM). The number of animals per group for data analysis ranged from 4 to 8.
DISCUSSION
Hepatitis B virus replication is closely linked to the assembly of 120 core protein dimers to form the virion capsid structure around the pgRNA (26). Mutations in the core protein sequence or small-molecule modulators of the capsid assembly process can be detrimental to the proper assembly of the nucleocapsid structure and have a negative impact on viral replication and infectious virus production, providing an attractive target for the development of anti-HBV therapies (23–25). Capsid inhibitors are an emerging class of anti-HBV agents for which a demonstrated proof of concept has been shown in early-stage clinical trials in chronic hepatitis B patients (33, 35, 36, 46, 47).
AB-423, a member of the SBA class of HBV capsid inhibitors, was evaluated in multiple preclinical studies to determine its in vitro anti-HBV potency, genotype spectrum, selectivity, mode of action, and in vitro and in vivo activity both as a single agent and in combination with anti-HBV agents belonging to other mechanistic classes, such as the nucleos(t)ide analogs and RNAi agents. In HBV cell culture models, AB-423 inhibited HBV replication (reduced rcDNA levels) and HBeAg production in HepBHAe82 cells, which suggests inhibition of cccDNA formation in this cell culture system (Table 1) (38). Furthermore, in two infectious virus systems, HBV-infected PHH and HBV-infected NTCP-expressing HepG2 cells, AB-423 inhibited viral replication and reduced cccDNA levels presumably through the interaction with nucleocapsid particles, disrupting the process of uncoating and release of rcDNA. These data indicate that AB-423 has the potential to reduce the production of infectious virus particles, inhibit the formation of new cccDNA through the intracellular replenishment pathway, and prevent cccDNA establishment during infection of a naive hepatocyte. Such a dual mode of action of AB-423 appears to be consistent among capsid inhibitors and differentiates them from the nucleos(t)ide class of inhibitors (43, 48). In a therapeutic setting, a drug's pharmacological activity can be impacted due to its sequestration by proteins present in human serum. Therefore, it is imperative to assess the potency of an antiviral in the presence of human serum or serum proteins. The presence of 40% human serum caused a modest 5-fold decrease in the in vitro potency of AB-423.
Mechanism-of-action studies in an HBV cell culture system showed that AB-423 is a class II capsid inhibitor. Treatment of HepDES19 cells with AB-423 resulted in the formation of normal amounts of capsid particles, but these capsids were devoid of pgRNA or rcDNA (Fig. 2). In a biochemical assay, AB-423 directly interacted with HBV core protein and accelerated capsid assembly, a property shared by both classes of capsid inhibitors. Further insight into the binding and antiviral mechanism of capsid inhibitors has been obtained through the availability of several high-resolution X-ray structures of core protein bound to capsid inhibitors, including several HAPs and, recently, an SBA (49–51). In all these published X-ray structures, the capsid inhibitors bound to the core protein dimer-dimer interface. Making the assumption that AB-423 also bound to the same site, we used a computational approach to predict the mode of AB-423 binding to core protein Cp-Y132A. The docked model of the AB-423 chemical structure was superimposed with the X-ray structures of SBA_R01 (51), representing the binding mode of a member of the SBA class of capsid inhibitors (Fig. 9A), and NVR10-001E2 (49), representing the HAP binding mode (Fig. 9B). Docking studies showed a closed binding interaction pattern between AB-423 and SBA_R01 (Fig. 9A). The central amide of the class II capsid inhibitors AB-423 and SBA_R01 forms essential hydrogen bonds with the side chains of Trp102 (chain B) and Thr128 (chain C), while the class I HAP capsid inhibitor NVR10-001E2 forms a hydrogen bond only with Trp102 (chain B) through one of its dihydropyrimidine nitrogens. While one of the sulfonamide oxygens of both class II inhibitors hydrogen bonded to the backbone NH of Ser121 of chain C through a water bridge, the sulfonamide NH of AB-423 could form an additional hydrogen bond interaction with the backbone of Leu140 of chain B (Fig. 9A). It is worth noting the two regions in the binding site occupied only by NVR10-001E2, a class I HAP capsid inhibitor: (i) a hydrophobic pocket created by aromatic residues like Trp102, Phe23, and Tyr118 of chain B (Fig. 9B) and (ii) the solvent-exposed region near Thr33 of chain B. The former pocket is occupied by the thiazole moiety of NVR10-001E2, while the latter is where the ester moiety sits. The structural difference between class I and class II capsid inhibitor-bound complexes could further aid our understanding and suggest potential differences in terms of the mechanism of action. By binding to the dimer-dimer interface of the core protein, the capsid inhibitors create additional interaction sites that lead to accelerated assembly kinetics, which can lead to the formation of noncapsid polymers (class I inhibitors) or the formation of morphologically normal capsid structures that are devoid of pgRNA or rcDNA (class II inhibitors).
FIG 9.

In silico docking of AB-423 into the X-ray structure of core protein. (A) Comparison of the predicted binding model of AB-423 and the X-ray structure of SBA_R01 (PDB accession number 5T2P; resolution, 1.69 Å) bound to the dimer-dimer interface of the Cp-Y132A viral core protein. Key residues in the vicinity of the ligand binding site are shown (the carbon of the capsid protein is in orange for chain B and pink for chain C, the carbon of the ligands is in magenta for AB-423 and green for SBA_R01, with nitrogen in blue, sulfur in yellow, oxygen in red, and fluorine in light green). The other residues within contact distance were not displayed for clarity. (B) Comparison of the predicted binding model of AB-423 and the X-ray structure of NVR10-001E2 (PDB accession number 5E0I; resolution, 1.95 Å) bound to the dimer-dimer interface of the Cp-Y132A viral core protein. Key residues in the vicinity of the ligand binding site are shown (the carbon of the capsid protein is in orange for chain B and pink for chain C, the carbon of the ligands is in magenta for AB-423 and cyan for NVR10-001E2, with nitrogen in blue, sulfur in yellow, oxygen in red, fluorine in light green, and bromine in dark brown). The other residues within contact distance are not displayed for clarity. (C to E) Chemical structures of AB-423 {(R)-5-[N-(sec-butyl)sulfamoyl]-N-(3,4-difluorophenyl)-2-fluorobenzamide} (C), SBA_R01 {4-fluoro-3-[(4-hydroxypiperidin-1-yl)sulfonyl]-N-(3,4,5-trifluorophenyl)benzamide} (D), and NVR10-001E2 [methyl 6-(2-bromo-4-fluorophenyl)-4-(morpholinomethyl)-2-(thiazol-2-yl)-1,6-dihydropyrimidine-5-carboxylate] (E).
Hepatitis B virus has eight recognized genotypes (genotypes A to H) that are geographically widely distributed, and genotypes A to D are the most prevalent genotypes globally (52). Our analysis of the core protein binding site within 4 Å of a bound SBA (51), including amino acids that form hydrogen bonds with the compound, using publicly available sequences (https://hbvdb.ibcp.fr/HBVdb/HBVdbIndex) revealed a high level of conservation of these amino acids both within and across HBV genotypes A to H (>99% for wild-type strains), with one exception being I105 (94.1% to 100% conservation). With the amino acids involved in hydrogen bond interactions, the level of conservation was >99.2%. Consistent with the high level of conservation of the binding site, AB-423 showed pan-genotypic activity in vitro against HBV genotypes A through D. Furthermore, because of its distinct mechanism of action from the nucleos(t)ide class of inhibitors, AB-423 maintained potency against nucleos(t)ide-resistant variants of HBV. The antiviral activity of AB-423 was specific for HBV, with AB-423 showing little to no specific activity against woodchuck hepatitis virus or a diverse panel of viruses belonging to other virus families (data not shown).
Hepatitis B virus has a highly limited host range, and a complete infective replication cycle has been documented to occur only in the livers of humans, chimpanzees, and tree shrews (53, 54). As the nonhuman models are relatively inaccessible, candidate therapeutics are often evaluated in a variety of other in vivo models that are artificial in one or more regards, such as having a liver engraftment with human hepatocytes. Murine cells are uninfectable due to the presence of the nonpermissive mouse sodium taurocholate cotransporting polypeptide (NTCP); in contrast, the human receptor is permissive (55–57). Mice, however, can express viral markers upon in vivo hepatocyte transfection following high-volume hydrodynamic injection (HDI) of a plasmid expressing a 1.3-fold-overlength HBV genome (58, 59). Pharmacokinetic studies in mice suggested that oral administration of AB-423 resulted in plasma and liver exposures that were in the range (concentrations greater than the EC90) required for antiviral activity. In an HDI mouse model of HBV infection, oral administration of AB-423 resulted in higher drug concentrations in the liver than in plasma and reduced serum HBV DNA levels.
The presence of cccDNA in the nucleus of HBV-infected hepatocytes in chronically infected patients is the underlying factor responsible for the persistence of HBV, and current therapies do not significantly reduce or eliminate cccDNA. Members of the nucleoside class of approved therapies are administered for prolonged periods and often lifelong to keep the patient virally suppressed. Moreover, they are leaky in their ability to completely shut down viral replication, which could lead to the replenishment of the nuclear cccDNA pool. Future combination regimens that are aimed at improving cure rates in CHB patients would comprise agents with different properties that would work together to bring about a comprehensive assault on different aspects of the HBV life cycle. These would likely involve a combination of agents that have more pronounced effects on blocking viral replication, reducing HBsAg levels, eliminating/reducing cccDNA levels, and reactivating the patient's immune system by breaking immune tolerance. It is therefore important that new agents, when combined, do not interfere/diminish each other's activity when coadministered. The capsid inhibitors are an emerging class of directly acting antiviral agents that could complement the current therapies by bringing about a stronger inhibition of viral replication in a combination regimen. Furthermore, the ability of AB-423 to interact with fully formed capsid particles and prevent the uncoating of the capsid and cccDNA establishment in the nucleus is a key differentiating property that could play a role in protecting naive hepatocytes from getting infected or hepatocytes that have been cleared of cccDNA from getting reinfected. We therefore evaluated the potential for combining AB-423 with members of the nucleos(t)ide class of anti-HBV compounds (ETV, TDF, and tenofovir alafenamide [TAF]) and with experimental RNAi agents (ARB-1467 and ARB-1740) in vitro and subsequently obtained in vivo confirmation of the potential for combination in the mouse HDI model of HBV. The results from in vitro and in vivo combination studies with nucleos(t)ide analogs and RNAi agents showed additive to synergistic inhibition of HBV replication with no antagonism, confirming that neither the NAs nor the RNAi agents tested interfered with the functionality of the other. The overall preclinical profile of AB-423 supports further evaluation for safety, pharmacokinetics, and antiviral activity in CHB patients.
MATERIALS AND METHODS
Dulbecco modified Eagle medium (DMEM)–F-12 medium was purchased from Lonza (Allendale, NJ). Fetal bovine serum (FBS) and penicillin-streptomycin were purchased from Gemini Bio-Products (West Sacramento, CA). The ETV and tenofovir disoproxil fumarate (TDF) used for the in vitro studies were purchased from SelleckChem (Houston, TX). The ETV used for the in vivo studies was purchased from LKT Laboratories (St. Paul, MN). Tetracycline, G418, and DMSO were purchased from Sigma-Aldrich (St. Louis, MO). AB-423 and reference compound GLS-4 were synthesized at Arbutus Biopharma, dissolved in 100% DMSO at a stock concentration of 10 mM or 50 mM, and stored at −20°C until use. The RNAi agents ARB-1467 and ARB-1740 were synthesized at Arbutus Biopharma as lipid nanoparticle (LNP) formulations and diluted serially in cell culture medium. AB-423, ETV, and TDF were serially diluted in 100% DMSO and added to assay plates. IFN-α was purchased from either PBL Assay Science (Piscataway, NJ) or BPS Bioscience (San Diego, CA). Myrcludex B was purchased from GenScript (Piscataway, NJ). Plateable, cryopreserved primary human hepatocytes (PHH) were purchased from BioreclamationIVT (Westbury, NY). Hepatocyte thawing medium (HTM), Williams medium E, an hepatocyte plating supplement pack, an hepatocyte maintenance supplement pack, and 96-well collagen-coated plates were purchased from Thermo Fisher Scientific (Waltham, MA). PHH plating medium (HPM) and PHH maintenance medium (HMM) were reconstituted per the manufacturer's instruction prior to use. Phosphate-buffered saline (PBS) without calcium and magnesium was purchased from Corning (Corning, NY). For the infection of PHH, polyethylene glycol 8000 was purchased from Promega (Madison, WI). Concentrated HBV particles (genotype D) isolated from HepG2-AD38 cells were used for infection of PHH and were purchased from ImQuest BioSciences (Frederick, MD). For in vitro combination studies with PHH, animals were purchased from Yecuris (Tualatin, OR). PEG-IFN-α-2a (360 μg/ml) was purchased from Roche (Shanghai, China). TAF was purchased from SelleckChem and solubilized in 100% DMSO. Genotype D HBV was concentrated from HepG 2.2.15 cell culture supernatants.
Capsid assembly assay.
The activity of AB-423 was tested in an HBV capsid assembly assay under contract by Wuxi Apptec (Shanghai, China) using a protocol described previously (40). The assay utilizes a mutant capsid protein, expressed and purified from Escherichia coli, comprising the first 149 amino acids of the HBV core protein (lacking the RNA binding domain) in which the native cysteines are mutated to alanine and a C-terminal cysteine has been added and modified by conjugation with the self-quenching fluorescent dye Bodipy-FL (C150Bo). In vitro, the capsid protein spontaneously assembles as a function of protein concentration, temperature, and ionic strength. In its nonassembled dimer form, the Bodipy-FL dye is fluorescent, and in its assembled form, the fluorescence is quenched. Capsid assembly is initiated by the addition of NaCl (0.15 M), which results in the quenching of the Bodipy-FL fluorescence signal. The no-NaCl condition was used as a no-assembly control, and 1 M NaCl was used as the 100% assembly control. If a compound binds to the HBV core protein and alters assembly kinetics, it can be detected by measuring the fluorescence changes of C150Bo in solution. AB-423 was serially diluted in DMSO and tested in this assay to determine its IC50 (the concentration of compound that results in 50% assembly). The final concentration of the C150Bo-labeled protein in the reaction was 1.5 μM.
AML12-HBV10 cell assay with bDNA quantitation of HBV rcDNA.
AML12-HBV10 is a stably transfected cell line of a mouse hepatocyte lineage which can inducibly express HBV (genotype D, serotype ayw) pgRNA and support HBV rcDNA synthesis under the control of a CMV Tet-off promoter system (34). The cell line was maintained in the presence of tetracycline to shut off HBV expression. Cells were plated in 96-well tissue culture-treated microtiter plates (20,000 cells/well) in DMEM–F-12 medium supplemented with 10% fetal bovine serum (FBS) plus 1% penicillin-streptomycin without tetracycline and incubated in a humidified incubator at 37°C with 5% CO2 overnight. On the next day, the cells were switched to fresh medium and treated with AB-423 at concentrations starting at 10 μM and then at a serial, 1/2-log, 8-point titration series in duplicate. Each plate included wells of mock-treated (0.5% DMSO) cells that served as controls. The plates were incubated for 48 h in a humidified incubator at 37°C with 5% CO2, after which the level of rcDNA present in the untreated and inhibitor-treated wells was measured using a QuantiGene (version 2.0) branched DNA (bDNA) assay kit (Affymetrix, Santa Clara, CA) with an HBV-specific custom probe set (genotype D, serotype ayw) according to the manufacturer's instructions. Concurrently, the effect of AB-423 on cell viability was assessed on replicate plates by measuring the intracellular ATP content using the Cell-Titer Glo reagent (CTG; Promega, Madison, WI) per the manufacturer's instruction. The plates were read using a Victor luminescence plate reader (PerkinElmer model 1420 multilabel counter), and the relative luminescence unit (RLU) data generated from each well were calculated to determine the percent inhibition of viral replication relative to that of the untreated control wells and analyzed using the XL-Fit module (IDBS, Boston, MA) in Microsoft Excel software to determine the 50% effective concentration (EC50) and EC90 (bDNA) as well as the 50% cytotoxic concentration (CC50) (CTG) values using a 4-parameter curve-fitting algorithm.
Serum shift assay.
The effect of human serum on the potency of AB-423 was assessed using the AML12-HBV10 cell (rcDNA/bDNA) assay described above with the following modifications in the growth medium. AML12-HBV10 cells were plated in 96-well, tissue culture-treated microtiter plates (20,000 cells/well) in DMEM–F-12 medium supplemented with 10% FBS plus 1% penicillin-streptomycin without tetracycline and incubated in a humidified incubator at 37°C with 5% CO2 overnight. On the next day, the cells were switched to fresh medium supplemented with increasing amounts of heat-inactivated normal human serum (0%, 10%, 20%, and 40%) and treated with AB-423 at concentrations starting at 10 μM and then at a serial, 1/2-log, 8-point titration series in duplicate. The final DMSO concentration in the assay was 0.5%. The plates were incubated for 48 h in a humidified incubator at 37°C with 5% CO2, after which the level of rcDNA present in the cells was measured using a bDNA assay, as described above.
HepDE19 cell assay with bDNA quantitation of HBV rcDNA.
The HepDE19 cell culture system uses an HepG2 (human hepatoma)-derived cell line that supports HBV (genotype D, serotype ayw) DNA replication and cccDNA formation under the control of a CMV Tet-off promoter system (37). HepDE19 cells were plated in 96-well collagen-coated tissue culture-treated microtiter plates (50,000 cells/well) in DMEM–F-12 medium supplemented with 10% fetal bovine serum plus 1% penicillin-streptomycin with tetracycline (1 μg/ml) and incubated in a humidified incubator at 37°C with 5% CO2 overnight. On the next day, the cells were switched to fresh medium without tetracycline and incubated for 4 h at 37°C with 5% CO2. The cells were switched to fresh Tet-free medium containing AB-423 at concentrations starting at 10 μM and then at a serial, 1/2-log, 8-point titration series in duplicate. The plates also included wells of untreated (DMSO) cells that served as controls. The final DMSO concentration in the assay was 0.5%. The plates were incubated for 3 to 7 days in a humidified incubator at 37°C with 5% CO2. The level of rcDNA present in the cells was measured using a QuantiGene (version 2.0) bDNA assay kit (Affymetrix, Santa Clara, CA) with an HBV-specific custom probe set (genotype D, serotype ayw), and the effect of AB-423 on cell viability was assessed as detailed above.
HepBHAe82 cell assay with cccDNA-dependent HBeAg quantitation using ELISA.
The HepBHAe82 cell line is an inducible HBV cell culture system similar to the HepDE19 system described above, but it has an in-frame hemagglutinin (HA) epitope tag in the N-terminal coding sequence of the HBV e antigen (HBeAg) in the HBV transgene (38). In this cell culture system, the reporters are the precore RNA (pcRNA) and its cognate protein product, and the secreted HA-tagged HBeAg is produced only upon the formation and expression of cccDNA and is detected by a chemiluminescence enzyme-linked immunosorbent assay (ELISA) (CLIA). The HepBHAe82 cell line exhibits high levels of cccDNA synthesis and HA-HBeAg production and secretion and provides a highly cccDNA-specific readout signal with low noise in a microtiter-format assay. To test the effect of AB-423 on cccDNA formation and expression, HepBHAe82 cells were plated in 96-well, tissue culture-treated microtiter plates (50,000 cells/well) in DMEM–F-12 medium supplemented with 10% FBS plus 1% penicillin-streptomycin and tetracycline (1 μg/ml) and incubated in a humidified incubator at 37°C with 5% CO2 overnight. On the next day, the cells were switched to fresh medium without tetracycline and treated with AB-423 at concentrations starting at 10 μM and then at a serial, 1/2-log, 8-point titration series in duplicate. The final DMSO concentration in the assay was 0.5%. The plates were incubated for 9 days in a humidified incubator at 37°C with 5% CO2. Following incubation, the supernatants were subjected to CLIA as described previously (38). The luminescence signal was read using an EnVision multilabel plate reader (PerkinElmer, Waltham, MA). Concurrently, the effect of inhibitor concentrations on cell viability and proliferation was assessed using replicate plates seeded at a 10 to 20% cell density, and after 4 days, the cells were assayed for the intracellular ATP content with the Cell-Titer Glo reagent as described above. Cell viability was calculated as a percentage of that of the untreated negative-control wells. The relative luminescence unit (RLU) data generated from each well were calculated to determine the percent inhibition of the untreated control wells and analyzed using the XL-Fit module in Microsoft Excel software to determine the EC50 and EC90 (HA-HBeAg ELISA) as well as the CC50 (CTG) values using a 4-parameter curve-fitting algorithm.
HepG 2.2.15 cell assay with HBV DNA quantitation using qPCR.
The HepG 2.2.15 cell line, which constitutively expresses HBV (genotype D, serotype ayw), was derived from the HepG2 cell line (39). Cells were seeded in 96-well tissue culture plates (40,000 cells/well) in 100 μl of DMEM–F-12 medium supplemented with FBS, penicillin-streptomycin, minimal essential medium nonessential amino acids, and l-glutamine medium supplemented with 2% FBS and incubated overnight in a humidified incubator at 37°C with 5% CO2. On the next day, the cells were switched to fresh medium and treated with AB-423 at concentrations starting at 10 μM and then at a serial, 1/2-log, 8-point titration series in duplicate. The plates also included wells of mock-treated (0.5% DMSO) cells that served as untreated controls. The plates were incubated for 3 days in a humidified incubator at 37°C with 5% CO2. On day 4, the medium and compound were replenished and the plates were incubated for an additional 3 days in a humidified incubator at 37°C with 5% CO2. On day 7, 150 μl of the culture supernatant from each well was collected for DNA isolation using a QIAamp 96 DNA blood kit per the manufacturer's instruction. The extracted DNA was quantified by a quantitative PCR (qPCR) assay with HBV-specific forward and reverse primers and a TaqMan probe, using the FastStart universal probe master mix (Roche, China) and an ABI 7500 real-time PCR system with 40 cycles under the following conditions: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. A standard curve generated using a plasmid carrying HBV DNA was used to determine the copy number of HBV DNA in the samples. Dose-response curves and the EC50 and EC90 were generated using GraphPad Prism software's nonlinear regression log(inhibitor)-versus-response variable slope (4-parameter) function. The effect of the compound on cell viability was assessed using the Cell-Titer Glo reagent. The relative luminescence unit (RLU) data generated from each well were calculated to determine the percent inhibition of viral replication relative to that of the untreated control wells, and the 50% cytotoxic concentration (CC50) was generated using GraphPad Prism software's nonlinear regression log(inhibitor)-versus-response variable slope (4-parameter) function. These studies were conducted under contract by Wuxi Apptec (Shanghai, China).
HBV-infected PHH assay.
Plateable, cryopreserved PHH (BioreclamationIVT, Westbury, NY), were thawed at 37°C, poured into HTM, and centrifuged at room temperature for 10 min at 100 × g. Cells were plated at 65,000 cells/well in collagen-coated 96-well plates in 100 μl HPM and incubated overnight in a humidified incubator at 37°C with 5% CO2. On the following day, the cells were washed 3 times with room temperature PBS, the medium was replaced with 100 μl of HMM, and the cells were incubated in a humidified incubator at 37°C with 5% CO2 while the inoculum was prepared.
Inoculation of cultured PHH with virus was done by a method modified from methods reported by Gripon et al. (60). To inoculate PHH with HBV, on day 1 the medium was gently removed, 70 μl of inoculum was added to each well, and the plate was incubated overnight in a humidified incubator at 37°C with 5% CO2. On the next day, the inoculated cells were washed 3 times with room temperature PBS and the medium was replaced with 100 μl of HMM, and the cells were incubated inside a humidified incubator at 37°C with 5% CO2. On day 3, infected cells were washed 3 times with PBS and the medium was replaced with 100 μl of HMM. A final wash was performed on day 4 and the medium was replaced with 50 μl of HMM in each well prior to compound addition. The compounds were diluted at a 2× concentration in HMM, and six 10-fold dilutions were prepared starting at 10 μM for AB-423 and 100 nM for ETV. Wells containing HMM only were used as infected controls that did not contain any compound treatments.
HBV DNA was extracted with a Qiagen DNeasy 96 blood and tissue kit (Qiagen Inc., Germantown, MD) using the animal tissues protocol per the manufacturer's instructions with some modification. For extraction of HBV DNA, 50 μl of culture supernatants was treated with 180 μl of buffer ATL plus proteinase K for 15 min at 55°C, and then the capsids were lysed with 410 μl of buffer AL mixed with 200-proof ethanol. Extracted DNA was eluted in a total of 100 μl buffer AE. The extracted DNA (5 μl) was subjected to a qPCR assay to determine the HBV DNA copy number using TaqMan Universal master mix II with UNG (Thermo Fisher Scientific, Waltham, MA) on an ABI 7500 real-time PCR system (Thermo Fisher Scientific, Waltham, MA) per the manufacturer's instruction. Primer and probe oligonucleotides with sequences annealing to the s-antigen region of the HBV DNA genome, used to quantitate HBV DNA, were purchased from Integrated DNA Technologies (Coralville, IA).
A standard curve was generated using HBV DNA with a known copy number as a control (ATCC, Manassas, VA). The copy number was determined by calculating the standard curve from known quantities of HBV DNA and raw threshold cycle (CT) values using GraphPad Prism software's linear regression function and then interpolating the copy number of the unknowns from the raw CT value. Dose-response curves and the EC50s were generated using GraphPad Prism software's nonlinear regression log(inhibitor)-versus-response variable slope (4-parameter) function.
Mechanism-of-action studies.
HBV RNA analysis and particle gel assays were performed as described previously (34, 37, 61). Intracellular viral RNA and encapsidated pgRNA were extracted and detected by Northern blotting hybridization with an HBV-specific 32P-labeled riboprobe. rRNAs (18S and 28S) served as loading controls. The total amounts of nucleocapsids were determined by a particle gel assay. Nucleocapsid-associated HBV DNA was detected by Southern blotting with alkaline treatment of the nucleocapsids on the membrane following the particle gel assay and hybridized with a 32P-labeled HBV-specific riboprobe. HBV DNA replication intermediates (rcDNA and ssDNA) were extracted and determined by Southern blotting hybridizations, and β-actin and intracellular HBV core protein were detected by Western blotting using specific antibodies.
Modeling of AB-423 at the dimer-dimer interface of the Cp-Y132A protein.
The model of AB-423 binding was constructed against a high-resolution crystal structure of core protein Cp-Y132A in complex with NVR10-001E2 (PDB accession number 5E0I; resolution, 1.95 Å) (49). Chains B and C were used in the docking procedure, carried out by the use of Glide SP in Maestro, followed by energy minimization (Schrödinger). An ensemble of 10 top-scoring poses was analyzed, and the best-scoring pose was used in this study.
Infectious C3AhNTCP cell assay.
The C3AhNTCP cell line stably expressing human NTCP (hNTCP) was established from a clonal isolate of HepG2 cells, as described previously (41). C3AhNTCP cells were seeded into collagen-coated 24-well plates at a density of 4 × 105 cells per well and cultured in complete DMEM containing 3% dimethyl sulfoxide (DMSO). One day later, the cells were infected with HBV prepared from HepAD38 cell culture medium at a multiplicity of infection of 500 genome equivalents per cell in DMEM containing 4% polyethylene glycol 8000. The inocula were removed at 24 h postinfection, and the cultures were maintained in complete DMEM containing 3% DMSO until harvesting. HBV RNA extraction from infected C3AhNTCP cells and real-time quantitative reverse transcription-PCR (RT-PCR) analysis were performed as described previously (34, 58). HBV cccDNA was extracted from HBV-infected C3AhNTCP cells and HepAD38 cells by a modified Hirt DNA extraction procedure (37). A fraction of the Hirt DNA preparation was digested with 1 unit of plasmid-safe ATP-dependent DNase (PSAD; Epicentre Technologies) in a 25-μl reaction mixture for 1 h at 37°C to remove the rcDNA. The DNase was inactivated by incubation of the reaction mixtures for 30 min at 70°C. The cccDNA in the PSAD-treated samples was quantified by a real-time quantitative PCR assay with HBV-specific oligonucleotide primers. The real-time PCR was performed using SYBR Premix Ex Taq master mix on a LightCycler 480 II instrument (Roche, Indianapolis, IN) as the following reaction procedure: 95°C for 10 min and then 45 cycles of 95°C for 30 s, 60°C for 5 s, and 72°C for 30 s. The amount of HBV cccDNA in a DNA preparation was determined by real-time PCR using a plasmid containing the HBV genotype D genome as the standard.
Antiviral activity against different HBV genotypes in vitro.
An HepG2 transient-transfection system with plasmid DNA harboring 1.1-mer-overlength sequences of two genome sequences each of HBV genotypes A, B, and C and one genome sequence of a genotype D wild-type isolate that had been synthesized and cloned into the pcDNA3.1 vector was used to evaluate the antiviral activity of AB-423 using transient transfection of HepG2 cells as described previously (62). These studies were performed under contract by Wuxi Apptec (Shanghai, China).
Antiviral activity against nucleoside-resistant HBV variants in vitro.
An HepG2 cell transient-transfection system and plasmid DNA harboring a 1.1-mer-overlength genotype D wild-type sequence (pcDNA3.1-HBV1.1) or nucleos(t)ide-resistant polymerase variants, introduced using site-directed mutagenesis, were used to evaluate the antiviral activity of AB-423 using transient transfection of HepG2 cells as described previously (62). These studies were performed under contract by Wuxi Apptec (Shanghai, China).
In vitro combination studies in AML12-HBV10 or HepDE19 cells using bDNA assay quantitation of HBV rcDNA.
In vitro combination studies were conducted using the method of Prichard and Shipman (44). The AML12-HBV10 or HepDE19 cell lines described above were used to evaluate the in vitro effects of AB-423 in combination with ETV or TDF. The cells were maintained in the presence of tetracycline to shut off transcription from the HBV transgene. Cells were plated at the cell densities described above in the central 60 wells of 96-well tissue culture plates as described above. On the next day, the cells were switched to fresh medium and treated with inhibitor A and inhibitor B over a range of concentrations in the vicinity of their respective EC50s. The inhibitors were diluted in either 100% DMSO (ETV, TDF, and AB-423) or growth medium (ARB-1467 and ARB-1740), and the final DMSO concentration in the assay was ≤0.5%. The two inhibitors were tested both singly and in combinations in a checkerboard fashion such that each concentration of inhibitor A was combined with each concentration of inhibitor B to determine the effects of the combination on inhibition of rcDNA production. There were four replicates of each concentration combination. The plates were incubated for 48 h in a humidified incubator at 37°C with 5% CO2, after which the level of rcDNA present in the untreated and inhibitor-treated wells was measured using a QuantiGene (version 2.0) bDNA assay kit (Affymetrix, Santa Clara, CA) with an HBV-specific custom probe set (genotype D, serotype ayw) according to the manufacturer's instructions. The RLU data generated from each well were calculated to determine the percent inhibition of the untreated control wells and analyzed using the MacSynergy II program to determine whether the combinations were synergistic, additive, or antagonistic using the interpretive guidelines established by Prichard and Shipman (44), as follows: at 95% confidence interval (CI), synergy volumes of 0 to <25 μM2 % (log volume, <2) indicated probably insignificant synergy, synergy volumes of 25 to 50 μM2 % (log volume, >2 and <5) indicated minor but significant synergy, synergy volumes of 50 to 100 μM2 % (log volume, >5 and <9) indicated moderate synergy that may be important in vivo, synergy volumes of over 100 μM2 % (log volume, >9) indicated strong synergy that is probably important in vivo, and synergy volumes of 1,000 μM2% (log volume, >90) were unusually high and the data were checked. Concurrently, the effect of inhibitor combinations on cell viability was assessed using replicate plates in triplicate, which were used to determine the ATP content as a measure of cell viability using the Cell-Titer Glo reagent (Promega) per the manufacturer's instructions.
In vitro combination studies using HepBHAe82 cells with HBeAg (e antigen) quantitation by ELISA.
The HepBHAe82 cell culture model of HBV described above was also used to evaluate in vitro the effects of combinations of AB-423 with other agents. In addition to the cccDNA-dependent HBeAg readout, a protocol for quantitative reverse transcription-PCR (RT-PCR) that is specific for the detection of precore RNA in HepBHAe82 cells was developed and used for the detection of the cccDNA-dependent mRNA (precore RNA) that is translated to produce HBeAg or HA-HBeAg.
To test the compound combinations, HepBHAe82 cells (50,000 cells/well) were plated in 96-well tissue culture-treated microtiter plates in DMEM–F-12 medium supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, and tetracycline (1 μg/ml) and incubated in a humidified incubator at 37°C with 5% CO2 overnight. On the next day, the cells were switched to fresh medium and treated with inhibitor A and inhibitor B over a range of concentrations in the vicinity of their respective EC50s. The inhibitors were diluted in either 100% DMSO (ETV, TDF, and AB-423) or growth medium (ARB-1467 and ARB-1740), and the final DMSO concentration in the assay was ≤0.5%. The two inhibitors were tested both singly and in combinations in a checkerboard fashion such that each concentration of inhibitor A was combined with each concentration of inhibitor B to determine the effects of the combination on inhibition of rcDNA production. The final DMSO concentration in the assay was 0.5%. The plates were incubated for 9 days in a humidified incubator at 37°C with 5% CO2. Following a 9-day incubation, the medium was removed and the cells were subjected to RNA extraction to measure the cccDNA-dependent precore mRNA level. Total cellular RNAs were extracted using a 96-well-format total RNA isolation kit (Macherey-Nagel Inc., Bethlehem, PA) by following the instructions of the manufacturer. RNA samples were eluted in RNase-free water. Quantitative RT-PCR was performed with a Roche LightCycler 480 instrument and an RNA master hydrolysis probe (Roche, Indianapolis, IN) using the primers and conditions required for the specific detection of cccDNA-dependent precore RNA. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA levels were also detected by standard methods and used to normalize the precore RNA levels. The levels of inhibition of precore RNA and, therefore, cccDNA expression was calculated as inhibition as a percentage of the growth in the untreated control wells and analyzed using the combination model of Prichard and Shipman (44) and MacSynergy II software to determine whether the combinations were synergistic, additive, or antagonistic using the interpretive guidelines described above. Concurrently, the effect of inhibitor combinations on cell viability and proliferation was assessed in two ways: (i) direct microscopic observation of test wells and (ii) using replicate plates seeded at a 10 to 20% cell density that, after 4 days, were assayed for the intracellular ATP content using the Cell-Titer Glo reagent (Promega, Madison, WI) per the manufacturer's instruction. Cell viability was calculated as a percentage of the viability of the cells in the untreated negative-control wells.
In vitro combination studies in PHH derived from hepatohumanized mice.
Studies were conducted under contract by Wuxi Apptec (Shanghai, China) using Fah−/−/Rag2−/−/Il2rg−/− mice (FRG mice) in which the livers had been repopulated with donor-derived PHH (63). Upon arrival, the mice were allowed to acclimate to the new environment for 7 days. The mice were monitored daily for general health and any signs of physiological and behavioral anomalies.
PHH were harvested from the mouse livers by perfusion. The isolated hepatocytes were further purified by the use of Percoll. The cells were resuspended with culture medium and seeded into 96-well plates (6 × 104 cell/well) or 48-well plates (1.2 × 105 cell/well). The PHH were infected with 400 genome equivalents per cell of genotype D HBV at 1 day postseeding (day 1). On day 2, the test compounds were diluted and added into the cell culture plates. The culture media containing the compounds were refreshed every other day. The cell culture supernatants were collected on day 8 for HBV DNA and antigen determinations. For initial determination of the EC50, dose ranges of 7 concentrations over 3-fold dilutions were used in triplicate. For combination testing, two compounds were tested in 5 dose concentration ranges in a 5-by-5 matrix in triplicate plates.
In all experiments, cytotoxicity for the cell monolayer was assessed on day 8 using a Cell Counting Kit-8 (Biolite, Tiajin, China). The HBV DNA in the culture supernatants harvested on day 8 was isolated with a QIAamp 96 DNA blood kit (Qiagen). For each sample, 100 μl of the culture supernatants was used to extract DNA, and the DNA was eluted with 100 μl, 150 μl, or 180 μl of Tris-acetate-EDTA. The HBV DNA in the culture supernatants was quantified by qPCR, using HBV-specific primers and probe. The HBsAg and HBeAg in the culture supernatants harvested on day 8 were measured by using HBsAg and HBeAg ELISA kits (Autobio Diagnostics, Zhengzhou City, China). The samples were diluted with PBS to get the signal in the range of the standard curve. For inhibition of HBV DNA, HBsAg, and HBeAg, combination effects were analyzed for synergy, additivity, or antagonism using the MacSynergy II software package as described above.
AB-423 pharmacokinetics in mice.
The pharmacokinetic properties of AB-423 were evaluated in age- and weight-matched female CD-1 mice (n = 3; Envigo, IN) in accordance with Canadian Council on Animal Care (CCAC) guidelines on good animal practices. AB-423 was solubilized in a cosolvent vehicle containing 5% ethanol and 40% polyethylene glycol 400 for the intravenous (i.v.) formulation and 5% ethanol and 60% polyethylene glycol 400 for the oral (p.o.) formulation. Animals were dosed with AB-423 intravenously at 2 mg/kg or orally at 10, 30, and 100 mg/kg. Blood samples were serially collected in EDTA-containing tubes for plasma preparation at specific time points up to 24 h to capture the PK profile of AB-423. At termination, a section of the left lateral lobe of the liver was also collected to evaluate AB-423 accumulation in the tissue. Plasma was collected by extracting the supernatant following centrifugation of blood samples in EDTA-containing tubes at 16,000 × g for 5 min. Liver homogenates were prepared by adding 2× the volume/weight of phosphate-buffered saline (PBS) to the liver-containing tubes. Tissue was then homogenized using an MP FastPrep-24 instrument (MP Biomedicals, Santa Ana, CA) at 5.0 meters/s for 30 s to produce 33.3% (wt/vol) liver homogenates. Protein precipitation was completed by using acetonitrile with an internal standard (tolbutamide) at a 1:9 sample-to-acetonitrile ratio. Following centrifugation at 3,000 rpm for 10 min, the supernatant was transferred to a 96-well plate for liquid chromatography-tandem mass spectrometry analysis (with a Sciex Qtrap 5500 system). The AB-423 standard curve was prepared by spiking known concentrations of AB-423 in the respective matrices for quantification. The PK parameters for AB-423 were determined using noncompartmental analysis with Phoenix WinNonlin software (Pharsight Corporation, CA, USA).
In vivo activity in an HDI mouse model of HBV infection.
All animal-related procedures were conducted according to written operating procedures, in accordance with Canadian Council on Animal Care (CCAC) guidelines on good animal practices, and were approved by the Institutional Animal Care and Use Committee (IACUC) at Arbutus Biopharma (Burnaby BC). Prior to the start of treatment, 10 μg of plasmid pHBV1.3 (constructed as described previously [58]) was administered to NOD.CB17-Prkdcscid/J mice via hydrodynamic injection (HDI; a rapid high-volume injection into the tail vein; n = 6 to 8 animals per group). This plasmid carries a 1.3-fold-overlength copy of an HBV genotype D genome which, when expressed, generates hepatitis B viral particles and other HBV products. AB-423 was administered via oral gavage at 30 or 100 mg/kg twice daily for 7 consecutive days starting on day 0. ETV was administered via oral gavage at 100 ng/kg once daily for 7 consecutive days starting on day 0. ARB-1467 was administered as a single intravenous bolus tail vein injection at 0.1 mg/kg on day 0. Blood was collected on days 0 (predose), 4, and 7 for HBV biomarker analysis. The serum HBV DNA concentration in mice was measured from total extracted DNA using a quantitative PCR assay with the primer/probe sequences described previously (64). Serum HBsAg concentrations were determined using a Bio-Rad enzyme immunoassay (EIA) GS HBsAg (version 3.0) kit according to the manufacturer's instructions.
Supplementary Material
ACKNOWLEDGMENTS
N.M., A.C., A.C.H.L., R.R., J.R.P., S.K., and T.O.H. designed and coordinated the studies; N.M., J.R.P., A.A., K.D.C., E.E., F.G., S.G.K., A.H.L.L., Q.L., K.D.M., S.A.M., R.M., N.M.S., S.P.R., H.M.M.S., K.S., S.T., X.W., and Q.Z. performed the experiments; K.F. conducted molecular docking and provided computational support; N.M. and R.R. had project oversight in biology; A.G.C. and B.D.D. had project oversight in chemistry; all authors reviewed and analyzed the data. N.M., A.G.C., J.R.P., A.C., K.F., T.O.H., A.C.H.L., R.R., and M.J.S. wrote and edited the manuscript.
H.G. and J.-T.G. were external collaborators, while the other authors either were/are employees of Arbutus Biopharma.
This work was sponsored by Arbutus Biopharma.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00082-18.
REFERENCES
- 1.Liang TJ, Block TM, McMahon BJ, Ghany MG, Urban S, Guo JT, Locarnini S, Zoulim F, Chang KM, Lok AS. 2015. Present and future therapies of hepatitis B: from discovery to cure. Hepatology 62:1893–1908. doi: 10.1002/hep.28025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. 2017. Global hepatitis report. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 3.Di Bisceglie AM. 2009. Hepatitis B and hepatocellular carcinoma. Hepatology 49:S56–S60. doi: 10.1002/hep.22962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention. 2015. Viral hepatitis surveillance United States. Centers for Disease Control and Prevention, Atlanta, GA. [Google Scholar]
- 5.Kowdley KV, Wang CC, Welch S, Roberts H, Brosgart CL. 2012. Prevalence of chronic hepatitis B among foreign-born persons living in the United States by country of origin. Hepatology 56:422–433. doi: 10.1002/hep.24804. [DOI] [PubMed] [Google Scholar]
- 6.Lok AS, McMahon BJ. 2007. Chronic hepatitis B. Hepatology 45:507–539. doi: 10.1002/hep.21513. [DOI] [PubMed] [Google Scholar]
- 7.Lam AM, Ren S, Espiritu C, Kelly M, Lau V, Zheng L, Hartman GD, Flores OA, Klumpp K. 2017. Hepatitis B virus capsid assembly modulators, but not nucleoside analogs, inhibit the production of extracellular pregenomic RNA and spliced RNA variants. Antimicrob Agents Chemother 61:e00680-17. doi: 10.1128/AAC.00680-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J, Shen T, Huang X, Kumar GR, Chen X, Zeng Z, Zhang R, Chen R, Li T, Zhang T, Yuan Q, Li PC, Huang Q, Colonno R, Jia J, Hou J, McCrae MA, Gao Z, Ren H, Xia N, Zhuang H, Lu F. 2016. Serum hepatitis B virus RNA is encapsidated pregenome RNA that may be associated with persistence of viral infection and rebound. J Hepatol 65:700–710. doi: 10.1016/j.jhep.2016.05.029. [DOI] [PubMed] [Google Scholar]
- 9.Wang J, Sheng Q, Ding Y, Chen R, Sun X, Chen X, Dou X, Lu F. 4 November 2017. HBV RNA virion-like particles produced under nucleos(t)ide analogues treatment are mainly replication-deficient. J Hepatol. doi: 10.1016/j.jhep.2017.10.030. [DOI] [PubMed] [Google Scholar]
- 10.Jansen L, Kootstra NA, van Dort KA, Takkenberg RB, Reesink HW, Zaaijer HL. 2016. Hepatitis B virus pregenomic RNA is present in virions in plasma and is associated with a response to pegylated interferon alfa-2a and nucleos(t)ide analogues. J Infect Dis 213:224–232. doi: 10.1093/infdis/jiv397. [DOI] [PubMed] [Google Scholar]
- 11.Glebe D, Bremer CM. 2013. The molecular virology of hepatitis B virus. Semin Liver Dis 33:103–112. doi: 10.1055/s-0033-1345717. [DOI] [PubMed] [Google Scholar]
- 12.Zoulim F, Perrillo R. 2008. Hepatitis B: reflections on the current approach to antiviral therapy. J Hepatol 48(Suppl 1):S2–S19. doi: 10.1016/j.jhep.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 13.Kim KH, Kim ND, Seong BL. 2010. Discovery and development of anti-HBV agents and their resistance. Molecules 15:5878–5908. doi: 10.3390/molecules15095878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doo EC, Ghany MG. 2010. Hepatitis B virology for clinicians. Clin Liver Dis 14:397–408. doi: 10.1016/j.cld.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fletcher SP, Delaney WE IV. 2013. New therapeutic targets and drugs for the treatment of chronic hepatitis B. Semin Liver Dis 33:130–137. doi: 10.1055/s-0033-1345713. [DOI] [PubMed] [Google Scholar]
- 16.Wursthorn K, Lutgehetmann M, Dandri M, Volz T, Buggisch P, Zollner B, Longerich T, Schirmacher P, Metzler F, Zankel M, Fischer C, Currie G, Brosgart C, Petersen J. 2006. Peginterferon alpha-2b plus adefovir induce strong cccDNA decline and HBsAg reduction in patients with chronic hepatitis B. Hepatology 44:675–684. doi: 10.1002/hep.21282. [DOI] [PubMed] [Google Scholar]
- 17.Marcellin P, Bonino F, Lau GK, Farci P, Yurdaydin C, Piratvisuth T, Jin R, Gurel S, Lu ZM, Wu J, Popescu M, Hadziyannis S, Peginterferon alfa-2a in HBeAg-Negative Chronic Hepatitis B Study Group. 2009. Sustained response of hepatitis B e antigen-negative patients 3 years after treatment with peginterferon alpha-2a. Gastroenterology 136:2169–2179.e1–e4. doi: 10.1053/j.gastro.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 18.Moucari R, Korevaar A, Lada O, Martinot-Peignoux M, Boyer N, Mackiewicz V, Dauvergne A, Cardoso AC, Asselah T, Nicolas-Chanoine MH, Vidaud M, Valla D, Bedossa P, Marcellin P. 2009. High rates of HBsAg seroconversion in HBeAg-positive chronic hepatitis B patients responding to interferon: a long-term follow-up study. J Hepatol 50:1084–1092. doi: 10.1016/j.jhep.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 19.Reijnders JG, Rijckborst V, Sonneveld MJ, Scherbeijn SM, Boucher CA, Hansen BE, Janssen HL. 2011. Kinetics of hepatitis B surface antigen differ between treatment with peginterferon and entecavir. J Hepatol 54:449–454. doi: 10.1016/j.jhep.2010.07.046. [DOI] [PubMed] [Google Scholar]
- 20.Terrault NA, Bzowej NH, Chang KM, Hwang JP, Jonas MM, Murad MH. 2016. AASLD guidelines for treatment of chronic hepatitis B. Hepatology 63:261–283. doi: 10.1002/hep.28156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Block TM, Alter H, Brown N, Brownstein A, Brosgart C, Chang KM, Chen PJ, Cohen C, El-Serag H, Feld J, Gish R, Glenn J, Greten TF, Guo JT, Hoshida Y, Kowdley KV, Li W, Lok AS, McMahon B, Mehta A, Perrillo R, Rice CM, Rinaudo J, Schinazi RF, Shetty K. 2018. Research priorities for the discovery of a cure for chronic hepatitis B: report of a workshop. Antiviral Res 150:93–100. doi: 10.1016/j.antiviral.2017.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zopf S, Kremer AE, Neurath MF, Siebler J. 2016. Advances in hepatitis C therapy: what is the current state—what come's next? World J Hepatol 8:139–147. doi: 10.4254/wjh.v8.i3.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cole AG. 2016. Modulators of HBV capsid assembly as an approach to treating hepatitis B virus infection. Curr Opin Pharmacol 30:131–137. doi: 10.1016/j.coph.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 24.Zlotnick A, Ceres P, Singh S, Johnson JM. 2002. A small molecule inhibits and misdirects assembly of hepatitis B virus capsids. J Virol 76:4848–4854. doi: 10.1128/JVI.76.10.4848-4854.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang L, Zhao Q, Wu S, Cheng J, Chang J, Guo JT. 2017. The current status and future directions of hepatitis B antiviral drug discovery. Expert Opin Drug Discov 12:5–15. doi: 10.1080/17460441.2017.1255195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seeger C, Mason WS. 2015. Molecular biology of hepatitis B virus infection. Virology 479-480:672–686. doi: 10.1016/j.virol.2015.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo YH, Li YN, Zhao JR, Zhang J, Yan Z. 2011. HBc binds to the CpG islands of HBV cccDNA and promotes an epigenetic permissive state. Epigenetics 6:720–726. doi: 10.4161/epi.6.6.15815. [DOI] [PubMed] [Google Scholar]
- 28.Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, Sprinzl MF, Koppensteiner H, Makowska Z, Volz T, Remouchamps C, Chou WM, Thasler WE, Hüser N, Durantel D, Liang TJ, Münk C, Heim MH, Browning JL, Dejardin E, Dandri M, Schindler M, Heikenwalder M, Protzer U. 2014. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 343:1221–1228. doi: 10.1126/science.1243462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stray SJ, Zlotnick A. 2006. BAY 41-4109 has multiple effects on hepatitis B virus capsid assembly. J Mol Recognit 19:542–548. doi: 10.1002/jmr.801. [DOI] [PubMed] [Google Scholar]
- 30.Wang XY, Wei ZM, Wu GY, Wang JH, Zhang YJ, Li J, Zhang HH, Xie XW, Wang X, Wang ZH, Wei L, Wang Y, Chen HS. 2012. In vitro inhibition of HBV replication by a novel compound, GLS4, and its efficacy against adefovir-dipivoxil-resistant HBV mutations. Antivir Ther 17:793–803. doi: 10.3851/IMP2152. [DOI] [PubMed] [Google Scholar]
- 31.Feld JJ, Colledge D, Sozzi V, Edwards R, Littlejohn M, Locarnini SA. 2007. The phenylpropenamide derivative AT-130 blocks HBV replication at the level of viral RNA packaging. Antiviral Res 76:168–177. doi: 10.1016/j.antiviral.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 32.King RW, Ladner SK, Miller TJ, Zaifert K, Perni RB, Conway SC, Otto MJ. 1998. Inhibition of human hepatitis B virus replication by AT-61, a phenylpropenamide derivative, alone and in combination with (−)beta-l-2′,3′-dideoxy-3′-thiacytidine. Antimicrob Agents Chemother 42:3179–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuen M-F, Kim D, Weilert F, Chan H-Y, Lalezari J, Hwang S, Nguyen T, Liaw S, Liaw S, Klumpp K, Flores L, Hartman G, Gane E. 2015. Phase 1b efficacy and safety of NVR 3-778, a first-in-class HBV core inhibitor, in HBeAg-positive patients with chronic HBV infection. Hepatology 62:1385A–1386A. [Google Scholar]
- 34.Campagna MR, Liu F, Mao R, Mills C, Cai D, Guo F, Zhao X, Ye H, Cuconati A, Guo H, Chang J, Xu X, Block TM, Guo JT. 2013. Sulfamoylbenzamide derivatives inhibit the assembly of hepatitis B virus nucleocapsids. J Virol 87:6931–6942. doi: 10.1128/JVI.00582-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zoulim F, Yogaratnam JZ, Vandenbossche JJ, Lenz O, Talloen W, Vistuer C, Moscalu I, Streinu-Cercel A, Bourgeois S, Buti M, Crespo J, Pascasio MJ, Blatt L, Fry J. 2017. Safety, tolerability, pharmacokinetics and antiviral activity of JNJ-56136379, a novel HBV capsid assembly modulator, in non-cirrhotic, treatment-naïve subjects with chronic hepatitis B. Hepatology 66:LB-15 (Abstract.) [Google Scholar]
- 36.Ding Y, Wange F, Lou J, Chen H, Li X, Wu M, Li C, Liu J, Liu C, Hu Y, Chen G, Zhang H, Zhang Y, Luo L, Niu J. 2017. Twenty-eight day safety, antiviral activity and pharmacokinetics of HBV capsid assembly inhibitor (GLS4JHS)/ritonavir combination for treatment of chronic hepatitis B infection. Hepatology 66:491A–492A. [Google Scholar]
- 37.Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81:12472–12484. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cai D, Wang X, Yan R, Mao R, Liu Y, Ji C, Cuconati A, Guo H. 2016. Establishment of an inducible HBV stable cell line that expresses cccDNA-dependent epitope-tagged HBeAg for screening of cccDNA modulators. Antiviral Res 132:26–37. doi: 10.1016/j.antiviral.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sells MA, Chen ML, Acs G. 1987. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci U S A 84:1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zlotnick A, Lee A, Bourne CR, Johnson JM, Domanico PL, Stray SJ. 2007. In vitro screening for molecules that affect virus capsid assembly (and other protein association reactions). Nat Protoc 2:490–498. doi: 10.1038/nprot.2007.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo F, Zhao Q, Sheraz M, Cheng J, Qi Y, Su Q, Cuconati A, Wei L, Du Y, Li W, Chang J, Guo JT. 2017. HBV core protein allosteric modulators differentially alter cccDNA biosynthesis from de novo infection and intracellular amplification pathways. PLoS Pathog 13:e1006658. doi: 10.1371/journal.ppat.1006658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu F, Campagna M, Qi Y, Zhao X, Guo F, Xu C, Li S, Li W, Block TM, Chang J, Guo JT. 2013. Alpha-interferon suppresses hepadnavirus transcription by altering epigenetic modification of cccDNA minichromosomes. PLoS Pathog 9:e1003613. doi: 10.1371/journal.ppat.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berke JM, Dehertogh P, Vergauwen K, Van Damme E, Mostmans W, Vandyck K, Pauwels F. 2017. Capsid assembly modulators have a dual mechanism of action in primary human hepatocytes infected with hepatitis B virus. Antimicrob Agents Chemother 61:e00560-17. doi: 10.1128/AAC.00560-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prichard MN, Shipman C Jr. 1990. A three-dimensional model to analyze drug-drug interactions. Antiviral Res 14:181–205. doi: 10.1016/0166-3542(90)90001-N. [DOI] [PubMed] [Google Scholar]
- 45.Lempp FA, Mutz P, Lipps C, Wirth D, Bartenschlager R, Urban S. 2016. Evidence that hepatitis B virus replication in mouse cells is limited by the lack of a host cell dependency factor. J Hepatol 64:556–564. doi: 10.1016/j.jhep.2015.10.030. [DOI] [PubMed] [Google Scholar]
- 46.Peters MG, Locarnini S. 2017. New direct-acting antiviral agents and immunomodulators for hepatitis B virus infection. Gastroenterol Hepatol (N Y) 13:348–356. [PMC free article] [PubMed] [Google Scholar]
- 47.Diab A, Foca A, Zoulim F, Durantel D, Andrisani O. 2018. The diverse functions of the hepatitis B core/capsid protein (HBc) in the viral life cycle: implications for the development of HBc-targeting antivirals. Antiviral Res 149:211–220. doi: 10.1016/j.antiviral.2017.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schlicksup CJ, Wang JC, Francis S, Venkatakrishnan B, Turner WW, VanNieuwenhze M, Zlotnick A. 2018. Hepatitis B virus core protein allosteric modulators can distort and disrupt intact capsids. Elife 7:e31473. doi: 10.7554/Elife.31473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klumpp K, Lam AM, Lukacs C, Vogel R, Ren S, Espiritu C, Baydo R, Atkins K, Abendroth J, Liao G, Efimov A, Hartman G, Flores OA. 2015. High-resolution crystal structure of a hepatitis B virus replication inhibitor bound to the viral core protein. Proc Natl Acad Sci U S A 112:15196–15201. doi: 10.1073/pnas.1513803112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qiu Z, Lin X, Zhou M, Liu Y, Zhu W, Chen W, Zhang W, Guo L, Liu H, Wu G, Huang M, Jiang M, Xu Z, Zhou Z, Qin N, Ren S, Qiu H, Zhong S, Zhang Y, Zhang Y, Wu X, Shi L, Shen F, Mao Y, Zhou X, Yang W, Wu JZ, Yang G, Mayweg AV, Shen HC, Tang G. 2016. Design and synthesis of orally bioavailable 4-methyl heteroaryldihydropyrimidine based hepatitis B virus (HBV) capsid inhibitors. J Med Chem 59:7651–7666. doi: 10.1021/acs.jmedchem.6b00879. [DOI] [PubMed] [Google Scholar]
- 51.Zhou Z, Hu T, Zhou X, Wildum S, Garcia-Alcalde F, Xu Z, Wu D, Mao Y, Tian X, Zhou Y, Shen F, Zhang Z, Tang G, Najera I, Yang G, Shen HC, Young JA, Qin N. 2017. Heteroaryldihydropyrimidine (HAP) and sulfamoylbenzamide (SBA) inhibit hepatitis B virus replication by different molecular mechanisms. Sci Rep 7:42374. doi: 10.1038/srep42374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sunbul M. 2014. Hepatitis B virus genotypes: global distribution and clinical importance. World J Gastroenterol 20:5427–5434. doi: 10.3748/wjg.v20.i18.5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dandri M, Burda MR, Zuckerman DM, Wursthorn K, Matschl U, Pollok JM, Rogiers X, Gocht A, Kock J, Blum HE, von Weizsacker F, Petersen J. 2005. Chronic infection with hepatitis B viruses and antiviral drug evaluation in uPA mice after liver repopulation with tupaia hepatocytes. J Hepatol 42:54–60. [DOI] [PubMed] [Google Scholar]
- 54.Wang Q, Schwarzenberger P, Yang F, Zhang J, Su J, Yang C, Cao J, Ou C, Liang L, Shi J, Yang F, Wang D, Wang J, Wang X, Ruan P, Li Y. 2012. Experimental chronic hepatitis B infection of neonatal tree shrews (Tupaia belangeri chinensis): a model to study molecular causes for susceptibility and disease progression to chronic hepatitis in humans. Virol J 9:170. doi: 10.1186/1743-422X-9-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H, Zhuang Q, Wang Y, Zhang T, Zhao J, Zhang Y, Zhang J, Lin Y, Yuan Q, Xia N, Han J. 2014. HBV life cycle is restricted in mouse hepatocytes expressing human NTCP. Cell Mol Immunol 11:175–183. doi: 10.1038/cmi.2013.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, Li W. 2012. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yan H, Peng B, He W, Zhong G, Qi Y, Ren B, Gao Z, Jing Z, Song M, Xu G, Sui J, Li W. 2013. Molecular determinants of hepatitis B and D virus entry restriction in mouse sodium taurocholate cotransporting polypeptide. J Virol 87:7977–7991. doi: 10.1128/JVI.03540-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guidotti LG, Matzke B, Schaller H, Chisari FV. 1995. High-level hepatitis B virus replication in transgenic mice. J Virol 69:6158–6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang PL, Althage A, Chung J, Chisari FV. 2002. Hydrodynamic injection of viral DNA: a mouse model of acute hepatitis B virus infection. Proc Natl Acad Sci U S A 99:13825–13830. doi: 10.1073/pnas.202398599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gripon P, Diot C, Guguen-Guillouzo C. 1993. Reproducible high level infection of cultured adult human hepatocytes by hepatitis B virus: effect of polyethylene glycol on adsorption and penetration. Virology 192:534–540. doi: 10.1006/viro.1993.1069. [DOI] [PubMed] [Google Scholar]
- 61.Guo JT, Pryce M, Wang X, Barrasa MI, Hu J, Seeger C. 2003. Conditional replication of duck hepatitis B virus in hepatoma cells. J Virol 77:1885–1893. doi: 10.1128/JVI.77.3.1885-1893.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou T, Block T, Liu F, Kondratowicz AS, Sun L, Rawat S, Branson J, Guo F, Steuer HM, Liang H, Bailey L, Moore C, Wang X, Cuconatti A, Gao M, Lee ACH, Harasym T, Chiu T, Gotchev D, Dorsey B, Rijnbrand R, Sofia MJ. 2018. HBsAg mRNA degradation induced by a dihydroquinolizinone compound depends on the HBV posttranscriptional regulatory element. Antiviral Res 149:191–201. doi: 10.1016/j.antiviral.2017.11.009. [DOI] [PubMed] [Google Scholar]
- 63.Azuma H, Paulk N, Ranade A, Dorrell C, Al-Dhalimy M, Ellis E, Strom S, Kay MA, Finegold M, Grompe M. 2007. Robust expansion of human hepatocytes in Fah−/−/Rag2−/−/Il2rg−/− mice. Nat Biotechnol 25:903–910. doi: 10.1038/nbt1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tanaka T, Inoue K, Hayashi Y, Abe A, Tsukiyama-Kohara K, Nuriya H, Aoki Y, Kawaguchi R, Kubota K, Yoshiba M, Koike M, Tanaka S, Kohara M. 2004. Virological significance of low-level hepatitis B virus infection in patients with hepatitis C virus associated liver disease. J Med Virol 72:223–229. doi: 10.1002/jmv.10566. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.