

Abstract
We describe the development of an arenophile-mediated, nickel-catalyzed dearomative trans-1,2-carboamination protocol. A range of readily available aromatic compounds was converted to the corresponding dienes using Grignard reagents as nucleophiles. This strategy provided products with exclusive trans-selectivity and high enantioselectivity was observed in case of benzene and naphthalene. The utility of this methodology was showcased by controlled and stereoselective preparation of small, functionalized molecules.
The preparation of amines plays a crucial role in the synthesis of natural products, polymers, and pharmaceuticals. Therefore, ongoing efforts in modern organic synthesis include the development of novel catalytic methods that could streamline their preparation, including functionalization of π-systems.1 Carboamination is one of the most powerful π-functionalization strategies for the synthesis of amines, as it results in the formation of C–C and C–N bonds with high atom- and step-economy.2 The past decade has seen significant developments in this area, and now several classes of these processes exist for alkenes,3 alkynes,4 and dienes.5 On the other hand, aromatic compounds could also be considered as viable substrates,6 especially when considering their availability and the synthetic versatility of the corresponding unsaturated products.7 However, due to their characteristic stability and reactivity, dearomative carboamination of arenes is virtually nonexistent.8 To the best of our knowledge, only transition-metal-catalyzed ring-opening of azabicyclic alkenes can provide products resembling those of formal dearomative 1,2-carboamination (Scheme 1A).9 Nevertheless, these reactions require more elaborate, benzyne-derived starting materials and cannot provide products resembling those obtained from mononuclear arenes.
Scheme 1.

Dearomative Carboaminations
To this end, we have recently disclosed a Pd-catalyzed dearomative syn-1,4-carboamination10 as well as a total synthesis of pancratistatins,11 which involved enantioselective, Ni-catalyzed dearomative trans-1,2-carboamination of benzene as the first step (Scheme 1B). The common design for both strategies involved photochemical dearomative cycloaddition12 between an arene and arenophile, N-methyl-1,2,4-triazoline-3,5-dione (MTAD, 1),13 which also served as a nitrogen source. Because the reactivities of Pd and Ni are intrinsically different, subsequent in situ transition-metal-catalyzed ring-opening of MTAD-arene cycloadduct I with carbon nucleophiles provided complementary syn-1,4- or trans-1,2-carboaminated products. Thus, the observed selectivity is likely the result of the outer-sphere attack of the enolate on Pd η3-intermediate II14 and inner-sphere delivery of the Grignard reagent in the case with cationic Ni η5-complex III.15
Considering the lack of dearomative difunctionalizations, as well as the noteworthy synthetic potential of the resultant products, we wondered if the Ni-catalyzed process could be translated into a general dearomative method. In addition to examining the scope, the motivation for this work also included the development of a more practical and glovebox-free enantioselective procedure. Herein we report these efforts, resulting in a general and efficient dearomative trans-1,2-carboamination. The enantioselective protocol uses low catalyst loadings of an air-stable Ni(II) precursor and permits the application of a series of Grignard reagents. Moreover, a range of aromatic precursors provides products with exclusive trans-selectivity. Finally, the synthetic utility of the method is demonstrated by selective elaboration of the dearomatized products into functionalized molecules.
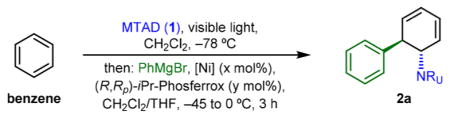
We commenced our investigations by repeating our initially reported conditions11 involving 10 mol % of [Ni(cod)2] and 20 mol % of (R,Rp)-iPr-Phosferrox, which was identified as the optimal ligand for the desymmetrization of benzene (entry 1, Table 1). Thus, using benzene and phenylmagnesium bromide, the desired product 2a was formed in 70% yield and 95:5 er. Though lowering catalyst loadings to 5/10 mol % did not result in significant erosion in efficiency (entry 2), we decided to evaluate a range of Ni(II) salts, which in combination with Grignard reagent could serve as a precursor to Ni(0).16 Gratifyingly, a variety of Ni(II) salts and complexes proved competent for this process, including NiCl2 (42%, 90:10 er, entry 3), [Ni(dmg)2] (51%, 90:10 er, entry 4), [NiCl2 glyme] (55%, 91:9 er, entry 5), and [Ni(acac)2] (59%, 93:7 er, entry 6, see also Supporting Information for full details). Interestingly, lowering the catalyst loading from 10/20 mol % resulted in beneficial effects on both yield and enantioselectivity (entries 7–10). Thus, the optimized conditions found involved application of precatalyst consisting of 1.5 mol % of [Ni(acac)2] and 2.0 mol % of (R,Rp)-iPr-Phosferrox, delivering diene product 2a in 70% yield and 97:3 er (entry 9).
Table 1.
Selected Optimization Studiesa
| |||||
|---|---|---|---|---|---|
| entry | [Ni] | x (mol%) | y (mol%) | yield (%)b | erc |
| 1 | [Ni(cod)2] | 10 | 20 | 70 | 95:5 |
| 2 | [Ni(cod)2] | 5.0 | 10 | 67 | 95:5 |
| 3 | NiCl2 | 10 | 20 | 42 | 90:10 |
| 4 | [Ni(dmg)2] | 10 | 20 | 51 | 90:10 |
| 5 | [NiCl2•glyme] | 10 | 20 | 55 | 91:9 |
| 6 | [Ni(acac)2] | 10 | 20 | 59 | 93:7 |
| 7 | [Ni(acac)2] | 10 | 12 | 51 | 95:5 |
| 8 | [Ni(acac)2] | 5.0 | 7.0 | 65 | 95:5 |
| 9 | [Ni(acac)2] | 1.5 | 2.0 | 70 | 97:3 |
| 10 | [Ni(acac)2] | 1.0 | 1.4 | 68 | 96:4 |
Standard reaction conditions: MTAD (1, 0.5 mmol, 1.0 equiv), benzene (5.0 mmol, 10 equiv), CH2Cl2 (0.20 M), visible light, −78 °C, 12 h; then PhMgBr (3 M in THF, 1.25 mmol, 2.5 equiv), solution of catalyst [Ni precursor (x mol %), (R,Rp)-iPr-Phosferrox (y mol %), CH2Cl2], −45 → 0 °C, 3 h.
Isolated yield of pure 2a after purification by flash chromatography.
Determined using HPLC analysis.
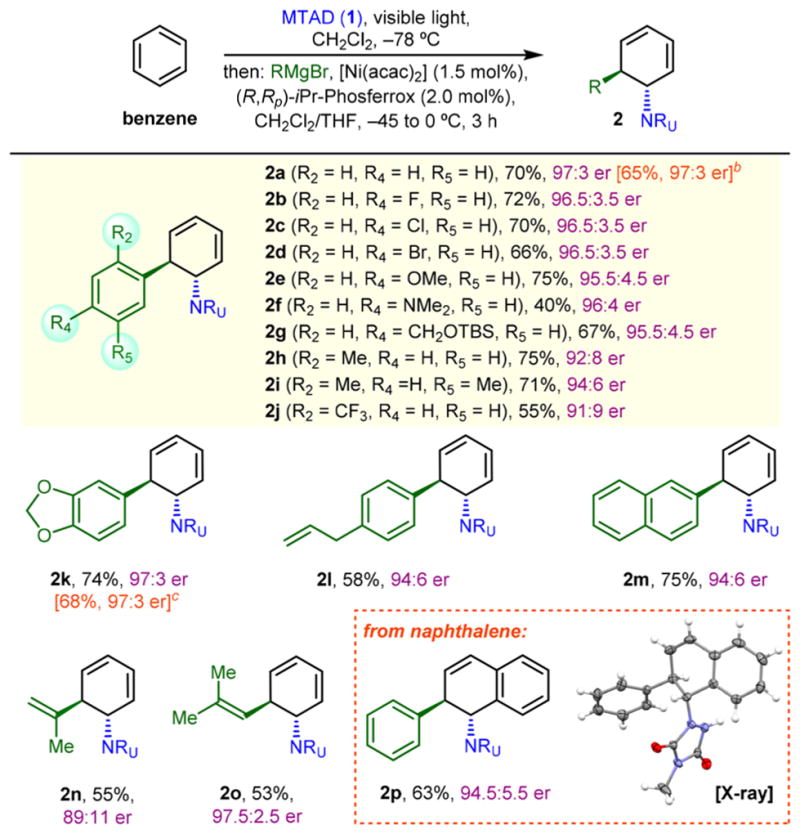
Having identified the optimal conditions for enantioselective dearomative trans-1,2-carboamination, we then examined the scope of Grignard reagents (Table 2). In addition to phenylmagnesium bromide (2a), a range of para-substituted analogues delivered products with high enantioselectivities (≥95:5 er, 2a–2g). Thus, halogens (2b–2d), electron-rich phenol and aniline (2e and 2f), as well as benzyl alcohol derivative (2g) proved compatible with this process. Noteworthy, no side products resulting from potential Ni-catalyzed Kumada-type coupling16 were observed under these conditions. Desymmetrization of benzene was also tested using more sterically demanding, ortho-substituted aryl Grignard reagents (2h–2j), which delivered products with comparable yields and selectivities. Moreover, a 3,4-methylenedioxyphenyl group, a key moiety for pancratistatins and other Amaryllidaceae alkaloids,17 was installed in 74% yield and 97:3 er (2k). This result is comparable with our previous conditions (75%, 98:2 er, see Scheme 1B, bottom) where higher loadings of [Ni(cod)2]/(R,Rp)-iPr-Phosferrox (10/20 mol %) were needed. In addition, an aryl Grignard containing an olefin (2l) and 2-naphthalenemagnesium bromide (2m) proved to be good substrates as well. Notably, this difunctionalization strategy also enables the installation of an alkene moiety, as demonstrated using terminally (2n) and internally (2o) substituted vinyl Grignard reagents.18 In addition to benzene,-naphthalene also underwent the desired asymmetric carboamination, delivering product 2p in 63% yield and 94.5:5.5 er. It is important to note that in all cases we consistently obtained products as a single diastereoisomer. Finally, the scalability of this enantioselective protocol was tested on a gram scale by examining two reactions, using normal glass media bottles surrounded with LEDs as photoreactors.19 Thus, products 2a and 2k were obtained in 65% and 68% yield on multigram scale and without any erosion in enantioselectivity.
Table 2.
Ni-Catalyzed Enantioselective Dearomative trans-1,2-Carboaminationa
|
All reactions were run on 0.5 mmol scale under the standard conditions. For 2p, 1.0 mmol of naphthalene (2.0 equiv) was used. Reported yields are of isolated products and er was determined by HPLC analysis.
Run on 3.0 g (26.5 mmol) scale.
Run on 6.0 g (53.1 mmol) scale.
Next, we sought to explore if substituted arenes could be suitable precursors for dearomative trans-1,2-carboamination (Table 3). Unfortunately, in this case, the Ni(II) precatalyst performed with notably lower efficiency compared to Ni(0). Thus, we found that application of [Ni(cod)2] and 1,1′-bis(diphenylphosphino)ferrocene (dppf) as a ligand gave consistently the best results. Though these substrates are not amenable to enantioselective desymmetrization, we were excited to find that a set of monosubstituted benzene derivatives as well as polynuclear arenes showed the desired reactivity. For example, substituted benzene derivatives containing an alkyl side chain (3a), pivaloate-protected alcohols (3b and 3c), trifluoromethyl (3d), and trimethylsilyl group (3e) were tolerated, all delivering products with high selectivity. Only in two cases, did we observe small amounts of constitutional isomers (9:1 for 3b and 11:1 for 3c). This high site-selectivity is consistent with reported examples of nucleophilic additions into stoichiometric cationic cyclohexadienyl complexes, where addition preferentially occurs at unsubstituted termini.20 Moreover, substituted polynuclear arenes were also successful substrates (3f–3k), though lower yields as well as constitutional isomers were observed with monosubstituted naphthalenes (3i–3k). Thus, electron-rich pivaloate-protected 2,3- and 1,4-dihydroxynaphthalene (3f and 3g), as well as bis-acetal protected naphthalene-1,4-dicarbaldehyde (3h) delivered the desired products with modest yields. The observed ratio of constitutional isomers in monosubstituted naphthalene series was highly dependent on the position of the substituent. As expected, 1-substituted naphthalenes, bearing more proximal substituents to the arene-MTAD cycloadduct (3i and 3k) gave higher selectivity compared to more distal, 2-substituted (3j). Finally, in addition to naphthalenes, heteroarenes such as quinoline derivative (3l) were also permitted under these conditions.
Table 3.
Arene Scope of the Dearomative trans-1,2-Carboaminationa

|
Reactions with mononuclear arenes were run on 0.5 mmol scale under the standard conditions and with 10/20 mol % of [Ni(cod)2]/dppf. For polynuclear arenes, 1 mmol (2.0 equiv) starting arene was used. Reported yields are of isolated products and the ratio of constitutional isomers (in parentheses) was determined by 1H NMR of the crude reaction mixtures.
This dearomative functionalization strategy sets the stage for further elaborations, as the corresponding products contain several modifiable regions, including olefin and urazole motifs (Figure 1). For example, benzene-derived product 2a could be converted to a fully saturated aminocyclohexane 4 through diene hydrogenation with Pt(S)/C and conversion of urazole to amine.21 Unsaturated tetrasubstituted aminocyclitol derivative 5 was obtained through singlet oxygen hetero-Diels–Alder reaction, subsequent thiourea-mediated reduction of the corresponding endoperoxide, and urazole cleavage. Similarly, diene 2a underwent [4+2]-cycloaddition with MTAD, and double urazole fragmentation/N–N-bond reduction to furnish unsaturated triamide 6. Also, naphthalene-based carboaminated product 2p was amenable to further manipulations. It was converted to saturated amine 7 using the same sequence as before. Alternatively, the arenophile moiety could serve as an oxygen surrogate, as the urazole could be readily converted to a ketone by simple oxidation with bleach,22 delivering arylketone 8. Finally, complete removal of the urazole was accomplished through Birch reduction, furnishing 2-phenyltetralin, a compound belonging to a class of potential drugs for treatment of arrhythmias and rhinoviral infections.23
Figure 1.
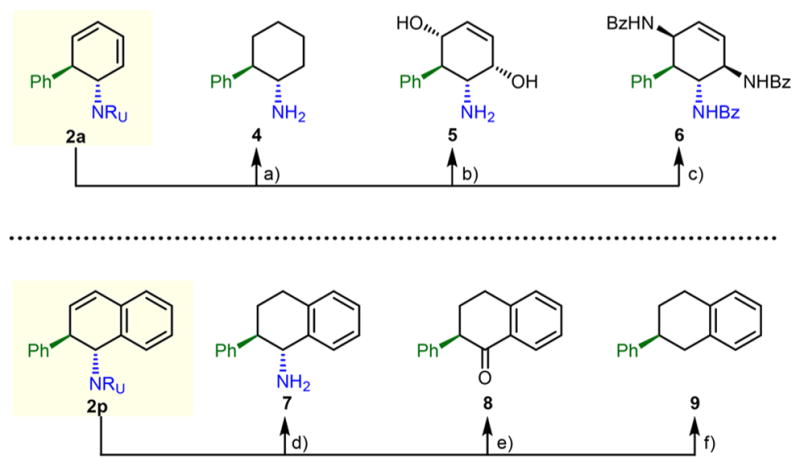
Derivatization of benzene- and naphthalene-derived products 2a and 2o. Reagents and conditions: (a) (i) α-bromoacetophenone, K2CO3, 94%; (ii) H2, Pt(S)/C (cat.), 91%; then KOH, 73%; (b) (i) α-bromoacetophenone, K2CO3, 94%; (ii) TPP, O2, visible-light, 65%; (iii) thiourea, then KOH, 73%; (c) (i) α-bromoacetophenone, K2CO3, 94% (ii) MTAD, 68%; (iii) KOH, then BzCl, then SmI2, 67%; (d) (i) α-bromoacetophenone, K2CO3, 91%; (ii) H2, Pt(S)/C (cat.), 92%; (iii) KOH, 82%; (e) (i) H2, Pd/C (cat.), 95%; (ii) NaOCl, 53%; (f) (i) H2, Pd/C (cat.), 95%; (ii) Li, NH3, 69%.
In summary, we have reported a dearomative 1,2-trans-carboamination, involving dearomative cycloaddition with an arenophile and subsequent Ni-catalyzed substitution with a Grignard reagent. A range of arenes and aryl or vinyl Grignard reagents delivered products with exclusive 1,2-trans selectivity, and high enantioselectivity when using benzene or naphthalene as substrates. In addition to expanding the currently available toolbox of dearomative transformations, this process also provides a different disconnection approach when it comes to the preparation of small, functionalized molecules. Considering no other chemical or biological equivalent for dearomative 1,2-carboamination exists, we anticipate the application of this difunctionalization strategy in the preparation of natural products and high-value added intermediates.
Supplementary Material
Acknowledgments
Financial support for this work was provided by the University of Illinois, the National Science Foundation (CAREER Award No. CHE-1654110), and the NIH/National Institute of General Medical Sciences (R01 GM122891). D.S. is an Alfred P. Sloan Fellow. L.H.W. acknowledges the NIH-NIGMS CBI Training Grant as well as the NSF for a Graduate Fellowship (GRFP). J.P. and U.K. thank the German Research Foundation (DFG) for postdoctoral fellowships. We also thank Dr. D. Olson and Dr. L. Zhu for NMR spectroscopic assistance, Dr. D. L. Gray and Dr. T. Woods for X-ray crystallographic analysis assistance, and F. Sun for mass spectrometric assistance.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b01726.
Experimental procedures and spectral data for all new compounds (PDF)
Crystallographic data for C19H17N3O2 (CIF)
Crystallographic data for C18H23N3O2Si (CIF)
References
- 1.For recent reviews, see: Pirnot MT, Wang YM, Buchwald SL. Angew Chem, Int Ed. 2016;55:48. doi: 10.1002/anie.201507594.Huang L, Arndt M, Gooßen K, Heydt H, Gooßen LJ. Chem Rev. 2015;115:2596. doi: 10.1021/cr300389u.McDonald RI, Liu G, Stahl SS. Chem Rev. 2011;111:2981. doi: 10.1021/cr100371y.Hesp KD, Stradiotto M. ChemCatChem. 2010;2:1192.Müller TE, Hultzsch KC, Yus M, Foubelo F, Tada M. Chem Rev. 2008;108:3795. doi: 10.1021/cr0306788.Hong S, Marks TJ. Acc Chem Res. 2004;37:673. doi: 10.1021/ar040051r.Müller TE, Beller M. Chem Rev. 1998;98:675. doi: 10.1021/cr960433d.
- 2.For recent reviews, see: Newhouse T, Baran PS, Hoffmann RW. Chem Soc Rev. 2009;38:3010. doi: 10.1039/b821200g.Burns NZ, Baran PS. Angew Chem, Int Ed. 2008;47:205. doi: 10.1002/anie.200704576.Wender PA, Verma VA, Paxton TJ, Pillow TH. Acc Chem Res. 2008;41:40. doi: 10.1021/ar700155p.
- 3.For recent examples, see: Gockel SN, Buchanan TL, Hull KL. J Am Chem Soc. 2018;140:58. doi: 10.1021/jacs.7b10529.Qian B, Chen S, Wang T, Zhang X, Bao H. J Am Chem Soc. 2017;139:13076. doi: 10.1021/jacs.7b06590.Liu Z, Wang Y, Wang Z, Zeng T, Liu P, Engle KM. J Am Chem Soc. 2017;139:11261. doi: 10.1021/jacs.7b06520.White DR, Hutt JT, Wolfe JP. J Am Chem Soc. 2015;137:11246. doi: 10.1021/jacs.5b07203.Piou T, Rovis T. Nature. 2015;527:86. doi: 10.1038/nature15691.Liwosz TW, Chemler SR. J Am Chem Soc. 2012;134:2020. doi: 10.1021/ja211272v.
- 4.For recent examples, see: Davis-Gilbert ZW, Yao LJ, Tonks IA. J Am Chem Soc. 2016;138:14570. doi: 10.1021/jacs.6b09939.Kajita Y, Matsubara S, Kurahashi T. J Am Chem Soc. 2008;130:6058. doi: 10.1021/ja7114426.Fürstner A, Davies PW. J Am Chem Soc. 2005;127:15024. doi: 10.1021/ja055659p.Shimada T, Nakamura I, Yamamoto Y. J Am Chem Soc. 2004;126:10546. doi: 10.1021/ja047542r.Ruck RT, Zuckerman RL, Krska SW, Bergman RG. Angew Chem, Int Ed. 2004;43:5372. doi: 10.1002/anie.200461063.
- 5.For recent examples, see: Chen SS, Wu MS, Han ZY. Angew Chem, Int Ed. 2017;56:6641. doi: 10.1002/anie.201702745.Liu Y, Xie Y, Wang H, Huang H. J Am Chem Soc. 2016;138:4314. doi: 10.1021/jacs.6b00976.Houlden CE, Bailey CD, Ford JG, Gagné MR, Lloyd-Jones GC, Booker-Milburn KI. J Am Chem Soc. 2008;130:10066. doi: 10.1021/ja803397y.
- 6.For recent reviews on dearomatizations, see: Roche SP, Porco JA. Angew Chem, Int Ed. 2011;50:4068. doi: 10.1002/anie.201006017.Pouységu L, Deffieux D, Quideau S. Tetrahedron. 2010;66:2235.Lewis SE. Chem Commun. 2014;50:2821. doi: 10.1039/c3cc49694e.Pape AR, Kaliappan KP, Kündig EP. Chem Rev. 2000;100:2917. doi: 10.1021/cr9902852.. For recent reviews on catalytic asymmetric dearomatizations, see: Zhuo CX, Zhang W, You SL. Angew Chem, Int Ed. 2012;51:12662. doi: 10.1002/anie.201204822.Zheng C, You SL. Chem. 2016;1:830.
- 7.For selected recent examples involving dearomatizations, see: Good SN, Sharpe RJ, Johnson JS. J Am Chem Soc. 2017;139:12422. doi: 10.1021/jacs.7b07745.Wilson KB, Myers JT, Nedzbala HS, Combee LA, Sabat M, Harman WD. J Am Chem Soc. 2017;139:11401. doi: 10.1021/jacs.7b05118.Nakayama H, Harada S, Kono M, Nemoto T. J Am Chem Soc. 2017;139:10188. doi: 10.1021/jacs.7b04813.Zheng J, Wang SB, Zheng C, You SL. J Am Chem Soc. 2015;137:4880. doi: 10.1021/jacs.5b01707.García-Fortanet J, Kessler F, Buchwald SL. J Am Chem Soc. 2009;131:6676. doi: 10.1021/ja9025193.Zhuo CX, You SL. Angew Chem, Int Ed. 2013;52:10056. doi: 10.1002/anie.201304591.Phipps RJ, Toste FD. J Am Chem Soc. 2013;135:1268. doi: 10.1021/ja311798q.Oguma T, Katsuki T. J Am Chem Soc. 2012;134:20017. doi: 10.1021/ja310203c.
- 8.Only certain dearomative reactions based on stoichiometric use of Cr, Mo, W, and Os can provide carbofunctionalized products. For examples, see: Liebov BK, Harman WD. Chem Rev. 2017;117:13721. doi: 10.1021/acs.chemrev.7b00480.Keane JM, Harman WD. Organometallics. 2005;24:1786.(c) See also Ref 6d.
- 9.For selected reviews involving ring-opening of unsaturated, strained heterobicycles, see: Lautens M, Fagnou K, Hiebert S. Acc Chem Res. 2003;36:48. doi: 10.1021/ar010112a.Rayabarapu DK, Cheng CH. Acc Chem Res. 2007;40:971. doi: 10.1021/ar600021z.Woo S, Keay BA. Synthesis. 1996:669.Chiu P, Lautens M. Top Curr Chem. 1997;190:190.. For selected examples of formal dearomative 1,2-carboamination, see: Zhang L, Le CM, Lautens M. Angew Chem, Int Ed. 2014;53:5951. doi: 10.1002/anie.201400218.Nishimura T, Kawamoto T, Sasaki K, Tsurumaki E, Hayashi T. J Am Chem Soc. 2007;129:1492. doi: 10.1021/ja068488c.Lautens M, Dockendorff C. Org Lett. 2003;5:3695. doi: 10.1021/ol035369i.Ogura T, Yoshida K, Yanagisawa A, Imamoto T. Org Lett. 2009;11:2245. doi: 10.1021/ol900533h.Chen CL, Martin SF. J Org Chem. 2006;71:4810. doi: 10.1021/jo060299p.Wu MS, Rayabarapu DK, Cheng CH. J Org Chem. 2004;69:8407. doi: 10.1021/jo048721u.. For an early example of Ni-catalyzed ring opening of strained oxabicyclic compounds using Grignard reagents, see: Lautens M, Ma S. J Org Chem. 1996;61:7246. doi: 10.1021/jo961615a.
- 10.Okumura M, Shved AS, Sarlah D. J Am Chem Soc. 2017;139:17787. doi: 10.1021/jacs.7b11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hernandez LW, Pospech J, Klöckner U, Bingham TW, Sarlah D. J Am Chem Soc. 2017;139:15656. doi: 10.1021/jacs.7b10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For reviews on dearomative cycloadditions, see: Remy R, Bochet CG. Chem Rev. 2016;116:9816. doi: 10.1021/acs.chemrev.6b00005.Wender PA, Ternansky R, DeLong M, Singh S, Olivero A, Rice K. Pure Appl Chem. 1990;62:1597.McCullough JJ. Chem Rev. 1987;87:811.
- 13.(a) Hamrock SJ, Sheridan RS. J Am Chem Soc. 1989;111:9247. [Google Scholar]; (b) Southgate EH, Pospech J, Fu J, Holycross DR, Sarlah D. Nat Chem. 2016;8:922. doi: 10.1038/nchem.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.For reviews involving Pdallyl intermediates, see: Weaver JD, Recio A, III, Grenning AJ, Tunge JA. Chem Rev. 2011;111:1846. doi: 10.1021/cr1002744.Lu Z, Ma S. Angew Chem, Int Ed. 2008;47:258. doi: 10.1002/anie.200605113.Trost BM, Crawley ML. Chem Rev. 2003;103:2921. doi: 10.1021/cr020027w.Trost BM, Van Vranken DL. Chem Rev. 1996;96:395. doi: 10.1021/cr9409804.
- 15.For reviews involving cationic cyclohexadienyl intermediates, see: Pike RD, Sweigart DA. Coord Chem Rev. 1999;187:183.Pearson AJ. Acc Chem Res. 1980;13:463.Davies SG, Green MLH, Mingos DMP. Tetrahedron. 1978;34:3047.
- 16.(a) Tamao K, Sumitani K, Kumada M. J Am Chem Soc. 1972;94:4374. [Google Scholar]; (b) Corriu RJP, Masse JPJ. Chem Soc, Chem Commun. 1972:144a. [Google Scholar]
- 17.For a recent review describing these natural products, see: Ghavre M, Froese J, Pour M, Hudlicky T. Angew Chem, Int Ed. 2016;55:5642. doi: 10.1002/anie.201508227.
- 18.Aliphatic, alkynyl, and heteroaromatic Grignard reagents, as well as organomagnesium reagents prepared by magnesium-halogen exchange or magnesiations, did not react under the described reaction conditions.
- 19.For full descriptions and pictures of the photochemical setups, see the Supporting Information.
- 20.For example, see: Pearson AJ, Gelormini AM, Pinkerton AA. Organometallics. 1992;11:936.Bandara BMR, Birch AJ, Kelly LF. J Org Chem. 1984;49:2496.
- 21.Adam W, Pastor A, Wirth T. Org Lett. 2000;2:1295. doi: 10.1021/ol000044c. [DOI] [PubMed] [Google Scholar]
- 22.(a) Wilson RM, Hengge AC. J Org Chem. 1990;55:197. [Google Scholar]; (b) Wilson RM, Hengge AC, Ataei A, Ho DM. J Am Chem Soc. 1991;113:7240. [Google Scholar]
- 23.For examples, see: Koskelainen T, Otsomaa L, Karjalainen A, Kotovuori P, Tenhunen J, Rasku S, Nore P, Tiainen E, Tormakangas O. 2003/006452. WO Patent. 2003Hodson HF, Batchelor JF, Selway JWT, Vinter JG, Iyer R. 19800105462. EP Patent. 1984
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.