

Abstract
After decades of rapid increase, the rate of obesity in adults in the USA is beginning to slow and the rate of childhood obesity is stabilizing. Despite these improvements, the obesity epidemic continues to be a major health and financial burden. Obesity is associated with serious negative health outcomes such as cardiovascular disease, type II diabetes, and, more recently, cognitive decline and various neurodegenerative dementias such as Alzheimer’s disease. In the past decade, major advancements have contributed to the understanding of the role of the central nervous system (CNS) in the development of obesity and how peripheral hormonal signals modulate CNS regulation of energy homeostasis. In this article, we address how obesity affects the structure and function of the blood-brain barrier (BBB), the impact of obesity on Alzheimer’s disease, the effects of obesity on circulating proteins and their transport into the brain, and how these changes can potentially be reversed by weight loss.
Keywords: blood-brain barrier, insulin, leptin, obesity
INTRODUCTION
Obesity is a condition in which excess body fat accumulates to an extent that health can be adversely affected (1). The World Health Organization has estimated that over 600 million people worldwide are obese, and obesity-attributable healthcare expenditures in the USA approach $147 billion annually (2). Obesity is caused by an imbalance of food intake and energy expenditure, but what causes this imbalance is unclear as the regulation of these processes is complex. At the least, these processes involve communication between the peripheral tissues that acquire, sense, or store nutrients with specialized nuclei in the central nervous system (CNS) that regulate feeding and metabolism (3). Brain barriers such as the vascular blood-brain barrier (BBB), the epithelial blood-cerebrospinal fluid (CSF) barrier, and tanycytic barriers are critical regulatory interfaces in such communication as many of the signals between the peripheral tissues and the CNS are blood-borne. The endothelial, epithelial, and tanycytic cells that form the various BBBs are highly specialized in that they restrict the unregulated diffusion of macromolecules between the blood and CNS and selectively regulate the blood-to-brain and brain-to-blood transport of circulating nutrients and hormonal signals that are critical in maintaining normal CNS functions. Hormones that regulate feeding and are altered in obesity including insulin, leptin, adiponectin, and ghrelin can cross the vascular BBB via specialized transport systems (4). BBB transport is necessary for these proteins to exert their functions in the CNS. Therefore, BBB transport is an important aspect of metabolic regulation. The BBB can also dynamically respond to signals from the blood and CNS, which is important for meeting the brain’s metabolic demands. However, pathological changes at the BBB occur during obesity that may ultimately exacerbate disease and can lead to additional pathological changes in the CNS such as neuroinflammation and cognitive impairment. In this review, we will discuss mainly the vascular BBB in the context of its functions in controlling the transport of regulatory proteins that affect food intake and energy expenditure, BBB dysfunctions resulting from obesity, and the prospects of the BBB as a therapeutic target for obesity.
PATHOLOGICAL CHANGES IN THE BRAIN ASSOCIATED WITH OBESITY
Obesity causes changes to a number of cell types of the neurovascular unit (NVU) that alter BBB integrity. The NVU includes capillary endothelial cells and pericytes surrounded by basal lamina, which are ensheathed by astrocytic perivascular endfeet in close proximity to neurons and microglia (5). Studies have shown that high-fat diet (HFD) feeding, containing approximately 40–45% fat, leads to neuronal loss in the arcuate nucleus and lateral hypothalamus (6). These changes are not only observed in adult mice consuming HFD but are also transferable to their offspring. In a study examining BBB disruption in the offspring of animals fed HFD or controls, offspring of HFD-fed mice showed increased BBB disruption thought to be caused by changes in the tanycyte population (a specialized ependymal cell in the brain) and expression of transporters (7). Astrocytes and microglia are important for maintaining BBB integrity, supporting neuronal metabolism, and preventing/responding to local tissue injury and have increased activation in the hypothalamus of rodents and humans with HFD consumption (8,9).
Obesity and chronic HFD consumption, especially diets rich in saturated fat, have been linked to reduced cognitive function in both humans and animals (10–13). The hippocampus, a brain region that is important for learning and memory, seems especially prone to damage. Neuronal populations within the hippocampus have a particularly high metabolic demand, making them vulnerable to a variety of environmental and biological factors (14). Dietary composition can be thought of as an environmental factor and can negatively impact hippocampal function. Several studies have shown in middle-aged rats that HFD impairs hippocampal-dependent memory processes that involve learning and utilization of stimuli in the spatial environment (15,16). However, it is still unknown whether HFD solely impairs the hippocampus or if it causes more widespread dysfunction that includes other brain regions that are important for cognition.
The neurobiological mechanisms that promote learning and memory impairment following HFD consumption are not completely understood. For the context of this review, HFD can refer to any diet with an increase in fat composition. It has been proposed that HFD consumption impacts cognitive function by influencing the integrity of the BBB (17), a proposal explored in the next section.
BBB DISRUPTION IN OBESITY
The vascular BBB is comprised of many cell types including brain microvascular endothelial cells that protect the brain from toxic substances in the circulation, influence the homeostatic environment of the brain, and regulate the transport of nutrients and endocrine signals into the CNS. In elderly humans (age 70–84 years), Gustafson et al. proposed that the BBB is disrupted in overweight or obese individuals based on an increase in the CSF/serum albumin ratio (18). Similar findings were observed in animal models. For example, a study completed in rabbits fed a cholesterol-enriched diet demonstrated increased IgG staining in the cortex, suggesting that the BBB was compromised (19). These studies support a relation linking BBB permeability with dietary factors.
To understand how BBB integrity could be influenced by dietary factors, Sprague-Dawley rats were fed a diet high in saturated fat and glucose for 90 days and then assessed by measuring permeability to sodium fluorescein. Results indicate that there is increased permeability to sodium fluorescein in the hippocampus and not in the prefrontal cortex or the striatum, other regions of the brain known to be involved in learning and memory (20). The brain regions affected in this study are different than the ones affected in the rabbit study by Ghribi et al. mentioned above. These regional differences could be due to the specific macronutrient that is enriched (saturated fat versus cholesterol), obese state and length on diet, or species differences. In addition, this increase in permeability in the rats on a HFD was accompanied by changes in tight junction proteins. HFD-fed rats had decreased expression of the tight junction proteins claudin-5 and claudin-12 in the choroid plexus and claudin-5, claudin-12, and occludin in the BBB capillaries (20). Changes in protein expression have also been observed in HFD-fed C57/BL6 mice when compared to chow-fed mice. Using a proteomic approach, Ouyang et al. isolated brain microvessels from obese and lean mice and identified 47 downregulated proteins involved in cell cycle regulation, cell metabolism, transport, cytoskeleton, chaperone, and scaffolding adaptor proteins and two upregulated proteins: heterogeneous nuclear ribonucleoproteins A1 and A2B1 (21). In contrast to other organs such as the liver (22) and the heart (23) which exhibit up- or downregulation of protein expression due to a HFD, HFD consumption causes mostly downregulation of protein expression at the BBB, which suggests that altered cellular energy metabolism might be the cause of cerebral dysfunction in obesity.
Cytoskeletal proteins play important roles in tight junction formation and function. Obesity downregulates cytoskeletal proteins at the BBB, including vimentin and tubulin (21). Vimentin is involved in nutrient transport and energy metabolism by controlling the transport of low-density lipoprotein-derived cholesterol from the lysosome to the site of esterification (24). The decreased levels of vimentin at the BBB could alter membrane fluidity preventing tight junction alignment in diet-induced obese mice. Tubulin is also involved in BBB transport and tight junction complex functions. The decreased levels of tubulin in the obese microvessels might be an indication of impaired BBB function. This is a potential explanation for changes in peptide transport as will be discussed in the following section.
Brain function requires maintenance of water and glucose homeostasis. This occurs through transporters located at the BBB, such as the water channel protein aquaporin-4 (AQP4) and the glucose transporter protein-1 (GLUT1) (25,26). While one study (20) of Sprague-Dawley rats fed a 45% HFD for 2 months showed no change in AQP4 and GLUT1, a study completed in obese Zucker rats showed downregulation of these proteins (27). The obese Zucker rat is not only a model of obesity but also an animal model of diabetes, exhibiting hyperglycemia, hyperinsulinemia, and hyperlipidemia (28). These variations in animal models may account for differences observed in gene expression. The expression of AQP4 and GLUT1 increase as the obese Zucker rat ages, implying that aging could compound the adverse effects of obesity on the BBB. It is believed that inflammation, as measured by intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion protein 1 (VCAM-1) expression in the aged obese Zucker rats, influences the expression of these transporters (27). Obesity also increases oxidative stress levels, which may contribute to BBB disruption as observed in diabetes and with systemic inflammation (29–31). Studies have shown that high-intensity workouts in individuals who are obese lead to increased serum levels of S100β, a marker of BBB disruption (32). In these obese patients, high-intensity workouts led to increased serum levels of reactive oxygen species and superoxide dismutase compared to non-obese exercised controls. Similar findings have been observed in aged HFD-fed mice which showed increased reactive oxygen species compared to controls, leading to increased BBB disruption (33).
OBESITY, BBB, AND NEUROINFLAMMATION
It has been postulated that inflammation in the CNS is a mediator of disease pathophysiology in obesity (34). Obesity increases inflammatory cytokine expression in multiple brain regions (35–37), but the predominant source of cytokines that initially trigger neuroinflammation in obesity is not completely clear (38). Blood-borne cytokines can cross the BBB in sufficient levels to elicit a CNS response, even when the BBB remains intact (39). It is likely neuroinflammation in obesity is multifaceted.
One cell type that can produce the inflammatory cytokines interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) is the adipocyte. Both IL-6 and TNF-α can be increased in obesity (40) and can cross the BBB by a saturable transport mechanism (41,42). However, it is unclear if BBB transport of these cytokines changes with obesity.
Controlled signaling of immunoregulatory cells connecting the CNS and peripheral immune system occurs via the BBB (43,44). Due to the polarized nature of brain endothelial cells, an immune activator on one side of the BBB can cause the release of an immune substance on the other side. This can occur from the blood to brain and vice versa (45). In addition, immune cells cross the BBB under normal conditions at a low rate (46) and this rate is increased with stimulation of the immune system. Immune cell trafficking across the BBB is a highly regulated multistep process that involves intercellular communication and binding of immune and endothelial cell glycoprotein receptors (47).
Immune cells are activated in obesity (40), likely due to the increased cell metabolism and subsequent inflammation. Buckman et al. showed that peripheral bone marrow-derived monocytes were able to infiltrate the CNS in obesity after 15 weeks of HFD and the number of monocyte-derived macrophages in the brain correlated with body weight and fat mass (48). However, it was unclear from this study how obesity results in infiltration of peripheral monocytes into the CNS.
Cytokines that induce BBB disruption can also activate expression of adhesion molecules on BBB endothelial cells that are important for immune cell recruitment to the brain (49). Further, immune cell engagement with endothelial cell adhesion molecules can initiate signaling events that alter tight junctions (50). Therefore, inflammation in obesity may be a cause of both BBB disruption and enhancement of diapedesis, a mechanism by which immune cells infiltrate the CNS. BBB disruption, in the context of leakiness and/or immune cell trafficking, in obese rodent models is associated with systemic inflammation and neuroinflammation (33,51). A recent study by Stranahan et al. identified protein kinase Cβ (PKCβ) as a molecular target at the BBB whose inhibition rescues obesity-induced BBB disruption and CNS infiltration of leukocytes, as well as induction of the pro-inflammatory cytokines IL-6 and TNF-α. In the same study, PKCβ inhibition also inhibited macrophage chemotaxis in an in vitro Boyden chamber assay in the absence of an intact BBB (38). Interestingly, reinstatement of the BBB integrity in obese db/db mice occurred independently of changes in body weight and food intake, supporting that treatment with a PKCβ inhibitor is sufficient to protect the BBB in obesity. These intriguing findings necessitate future studies that will provide additional insight on the BBB-protective mechanisms of PKCβ inhibition.
OBESITY, BBB, AND ALZHEIMER’S DISEASE
Given the relationship between obesity and neuronal health, it is not surprising that obesity and complications of obesity including diabetes, hypercholesterolemia, and hypertension are all risk factors for developing Alzheimer’s disease (AD) (52). In the USA, midlife obesity confers an odds ratio for developing AD of 1.6 and a 25% reduction in obesity may reduce AD prevalence by 91,000 cases (52). Several possible mechanisms exist for the obesity-AD connection ranging from changes in amyloid transport and clearance to alterations in lipid metabolism.
First, AD is characterized by age-associated progressive memory loss and cognitive decline and pathological deposition of amyloid beta and tau protein aggregates in the brain. These pathological changes are thought to be caused, in part, by decreased clearance of amyloid beta from the brain. Notably, HFD increases plasma amyloid beta and brain amyloid beta delivery, in both humans and rodent models (13,53–55). In mice, chronic HFD feeding elevated peripheral lipoprotein-associated amyloid beta which altered BBB integrity, as evidenced by perivascular leakage of IgG (56). In addition, as outlined above, obesity increases neuroinflammation and compromises BBB function, particularly in the hippocampus which is a part of the brain that atrophies early in the course of AD (57). A dysfunctional BBB as seen in obesity could serve to impede amyloid beta breakdown product clearance. For example, inflammatory conditions upregulate receptors for advanced glycation end products (RAGE) and low-density lipoprotein receptor-related protein 1 (LRP1), which are known to transport amyloid beta breakdown products into the brain in transgenic animal models, and downregulate brain-to-blood clearance (58). In support of this, RAGE protein has been found in human AD brain and RAGE levels correlate with levels of AD pathology (59). Although overall RAGE expression in the brain is increased in obesity (60), whether or not increases occur at the BBB remains to be determined. In addition, LRP1 BBB protein levels are upregulated in rodent offspring from obese mothers (7).
Another connection between obesity and AD is that obesity-related hypertension may alter the health of penetrating cerebral small vessels, leading to blood flow dysregulation and white matter ischemia which can be detected on an MRI image. Increased white matter disease burden correlates with cognitive decline, and AD patients with high white matter disease burden have worse cognitive function than those without this pathology (61).
Lastly, obesity may influence levels and functions of apolipoproteins, which are an integral part of neuronal lipid metabolism and function. The presence of the E4 allele of apolipoprotein E (apoE4) confers a significant risk for developing AD (62). In the periphery, apoE4 is associated with hyperlipidemia and hypercholesterolemia, whereas in the CNS, apoE4 is characterized by poor levels of lipidation and high amyloid burden (62,63). Variations of other apolipoproteins have also been implicated in AD and other neurodegenerative diseases (64). Some apolipoproteins cross the BBB to varying degrees, such as apoJ and apoA-I, while others are thought to be impenetrable, such as apoE (64,65). Understanding how these various apolipoproteins transport lipids in the periphery and into the brain, and how risk factors such as obesity influence these apolipoproteins, may elucidate AD pathogenesis as well as elucidate possible treatments.
BBB TRANSPORT AND TRANSPORTER CHANGES IN OBESITY
Obesity can cause changes in levels of multiple circulating factors, resulting in fluctuations in transport of regulated peptides and substrates across the BBB (Fig. 1). Obesity increases circulating triglycerides, as well as secreted adipokines including leptin, while decreasing adiponectin due to the expansion of adipocytes (40). Many adipokines can cross the BBB to act within the CNS (66). There are also indirect changes in circulating levels of other gut-hormone peptides, such as insulin and ghrelin, due to increased adipokine release (67,68).
Fig. 1.
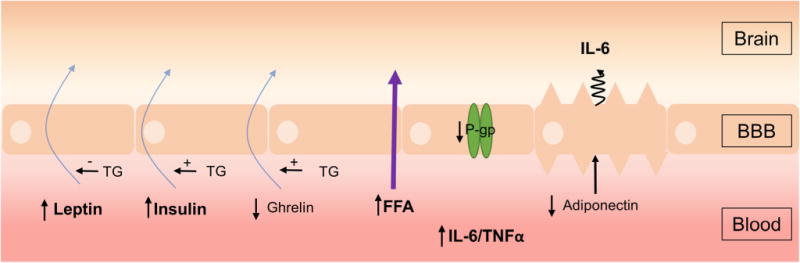
Changes at the BBB in obesity. The BBB is exposed to increased circulating levels of leptin, insulin, free fatty acids, and the cytokines IL-6 and TNF-α, whereas ghrelin and adiponectin levels are decreased in obesity (black vertical arrows). The blood-to-brain transport of leptin, insulin, and ghrelin (blue arrows) is decreased and transport of free fatty acids (FFA, purple arrow) is increased in obesity. Triglycerides (TG) can also decrease leptin transport while increasing insulin and ghrelin transport (horizontal arrows). Obesity decreases mRNA levels of P-glycoprotein (P-gp) in BBB endothelial cells. The obesity-associated decrease in circulating adiponectin stimulates the abluminal secretion of IL-6 by BBB endothelial cells, resulting in increased brain IL-6 levels
Peptides cross the BBB through saturable and unsaturable mechanisms. Saturable transport occurs through proteins that may or may not also function as the receptor. The expression or function of transport systems is often altered in disease, resulting in a perturbed exchange of information between peripheral tissues and the CNS. Obesity is one disease state in which transport of substrates across the BBB is altered as summarized in Table I. In this section, we review BBB transport systems that are known to change in obesity, and the potential consequences of these changes on CNS function.
Table I.
Summary of Changes in Peptide Transport Across the BBB in Obesity
| Peptide | BBB transport direction change due to obesity | Approximate change compared to lean | Model | Reference |
|---|---|---|---|---|
| Leptin | Decreased | 76% | Humans | (69) |
| 80% | Humans | (70) | ||
| 24% | Sheep | (71) | ||
| 69% | Mice | (72) | ||
| Insulin | Decreased | 45% | Humans | (73) |
| 45% | Dogs | (74) | ||
| 55% | Zucker rat | (75) | ||
| 54% | Mice | (76) | ||
| Adiponectin | Not investigated | No BBB transport | Humans | (77) |
| No BBB transport | Mice | (78) | ||
| Ghrelin | Decreased | 96% | Mice | (79) |
| FFA | Increased | 50% | Humans | (80) |
| Palmitate | Increased | 86% | Humans | (80) |
| Amylin | Not investigated | |||
| GLP-1 | Not investigated |
BBB blood-brain barrier, FFA free fatty acids, GLP-1 glucagon-like peptide-1
Leptin
Leptin is an adipokine that acts within the brain to control appetite (81), mediating a feedback loop between adipose tissue and the CNS. Leptin levels in blood reflect the degree of adiposity. Transport of leptin across the BBB occurs in a saturable manner (82), and leptin transport capacity is already about 50% saturated at serum levels associated with an ideal body weight (83).
During the development of obesity, obese rodents respond to centrally administered leptin via intracerebro-ventricular injections, but not to peripherally administered leptin via subcutaneous pumps or intraperitoneal injections (84,85). This suggests that impairment in CNS leptin signaling is due to resistance at the BBB transporter level. In obese mice, transport of leptin across the BBB is decreased to about 35% of that found in lean mice (72). Obese sheep also have decreased leptin BBB transport represented by decreases in the leptin CSF/plasma ratio with increasing adiposity (71). In addition, obese humans have anywhere from a 4.3–5.4-fold lower CSF/serum leptin ratio (69,70). It is important to note that the defect in leptin transport is not due to lack of functional leptin receptors at the BBB (86,87), but rather due to saturation of the transporter by leptin or reversible inhibition by other circulating factors in the blood. Circulating triglycerides can inhibit leptin transport across the BBB in a dose-dependent manner (88), whereas epinephrine, glucose, and insulin increase leptin transport (89).
Increasing delivery of leptin to the brain by overcoming transporter resistance at the BBB could significantly improve leptin therapeutic efficacy and increase weight loss. Modifications of leptin can alter the potency and efficacy, especially under conditions in which the transport system is saturated, as is the case in obesity. Analogs of leptin have been constructed to investigate if BBB transport can be improved. Indeed, PEGylated leptin and leptin modified with amphiphilic Pluronic triblock copolymers may be able to overcome leptin transport resistance at the BBB (90). There was a 33% increase in brain levels (%Inj/g) of Pluronic P85 leptin after intravenous delivery compared to unmodified leptin (90). These modifications help to not only stabilize leptin but also prevent efflux from the brain and facilitate utilization of alternative mechanisms for BBB transport, including caveola-dependent transcytosis (91).
Adiponectin
Adiponectin signals in the CNS to decrease body weight by stimulating energy expenditure (92). In humans, plasma adiponectin levels are inversely correlated to adipose tissue (93). There is controversy regarding whether or not adiponectin crosses the BBB. One study found that human CSF adiponectin levels positively correlate with systemic levels (94), but CSF adiponectin concentrations were extremely low and another human study could not detect adiponectin in the CSF of healthy human volunteers (77). Rodent studies show adiponectin does not cross the BBB (78). However, it is clear adiponectin can influence the CNS by indirect means: the low-adiponectin state due to obesity results in increased sensitivity of brain endothelial cells to inflammatory markers (95) and adiponectin can influence the polarized secretion from brain endothelial cells of inflammatory factors such as IL-6 (77).
Insulin
Insulin is secreted from the pancreas in direct proportion to body fat (68,96) and is an important negative feedback signal in the CNS to regulate body weight by decreasing food intake. Insulin is transported across the BBB in a saturable manner, which is independent of leptin (82,97). The transport rate for insulin is decreased with obesity (74,75) and CSF/serum ratios negatively correlate with body mass index (73). There is a 60% reduction in the amount of CNS insulin in obese animals that inversely correlates with the percent change in body weight (74).
There are circulating factors that are known to affect insulin transport across the BBB. For example, triglycerides increase insulin transport across the BBB by about 30% (76). In starvation, plasma triglyceride levels increase, increasing the transport of insulin across the BBB (76). Insulin can also act at the BBB to enhance amino acid transport, particularly tryptophan, across the BBB (98). As circulating insulin is increased in obesity, it is possible the transport of amino acids is increased across the BBB. However, these direct studies in obese individuals have not been done.
Amylin
Much like insulin, amylin is another peptide secreted by the pancreas after a meal. Amylin also crosses the BBB, possibly acting as a satiety agent (99). Although differences in transport due to obesity have not been investigated, peripheral administration of amylin caused weight loss in obese rats and humans (100). Roth and colleagues also propose a synergistic relationship between amylin and leptin that restores the leptin responsiveness in a model of obesity (100). These studies suggest that an integrated hormonal administration may provide a novel therapeutic approach to facilitate hormone transport across the BBB.
Ghrelin
Ghrelin is a small peptide produced primarily by the stomach. It has CNS effects primarily in the hypothalamus to regulate appetite and in the hippocampus to affect cognition. Plasma levels are decreased in obese humans and mice (67,101). Aged, obese mice lose the ability to transport circulating ghrelin across the BBB yet serum triglycerides enhance BBB transport by more than 70% (79).
Glucagon-Like Peptide-1
Glucagon-like peptide-1 (GLP-1) is secreted in response to feeding. It acts within the CNS to decrease food intake and thus has been investigated for its potential therapeutic use in weight loss. However, GLP-1 is rapidly degraded by dipeptidyl peptidase-4 (DPP-4), an enzyme increased in obesity (102,103). After peripheral injection of GLP-1, less than half of the peptide remains intact in normal-weight individuals and by 10 min, there is no GLP-1 detected (104). A more stable analog of GLP-1 with similar biological effects, [Ser8]GLP-1 (7–36), has a rapid transport rate across the BBB and crosses independently of a saturable transport system (105). The transport of GLP-1 across the BBB under obese conditions has not been examined but it is known that circulating DPP-4 levels are increased in obesity (106).
Free Fatty Acids
Even though the brain does not use free fatty acids (FFAs) as an energy source, there are data to support CNS signaling by FFAs to control feeding and energy balance (107,108). FFA levels are increased in obesity (109) and thus could potentially accumulate within the CNS.
Not surprisingly, after intravenous injection of a radiolabeled analog of FFAs, the highest levels were observed in the heart and liver, but there was radioactivity present in the brain, suggesting transport of FFA across the BBB (110). Using the same radiolabeled FFA analog as well as radiolabeled palmitate, transport into the human brain was also observed (80). The authors also found that patients with metabolic syndrome had a 50% increase in FFA and an 86% increase in palmitate uptake in the brain compared to healthy subjects. Peripheral palmitate has also been shown to accumulate in the brains of monkeys (111) and rats (112), suggestive of BBB transport.
P-glycoprotein
P-glycoprotein (P-gp) plays a key role at the BBB, acting as an efflux pump, returning its substrates back into the bloodstream, thus limiting the entry of potentially harmful substances into the CNS. The protective nature of this protein is also the bane of many pharmaceutical companies trying to design drugs to cross the BBB. Using publicly available microarray data from 145 neurologically sound adults, Vendelbo et al. found body mass index is negatively associated with expression of ABCB1 (the gene encoding P-gp) in the frontal cortex (113). It is interesting that although mRNA levels in human brain decrease with increased body mass index (113), protein levels of P-gp do not change at the BBB, at least in HFD-fed obese mice (21). Whether or not P-gp protein and/or function is altered at the BBB in humans remains to be determined. This is important as many drugs are dosed by weight and overweight subjects may therefore be at an increased sensitivity to neuropharmaceuticals.
OBESITY REVERSAL AS A THERAPEUTIC STRATEGY TO IMPROVE BBB TRANSPORT
Obesity can be a reversible disease, and when weight loss interventions are successful, profound improvements in patient health typically occur. Beneficial effects of weight loss extend to the CNS, as patients who lost weight following both dietary and surgical weight loss interventions showed improvements in cognition (114). As a link between obesity and cognitive decline has recently been established (115–117), some studies have focused on the effects of obesity on the peripheral transport of metabolic hormones into the brain.
As described in detail above, peripheral metabolic hormone resistance is commonly associated with obesity, and BBB transport of these hormones is impaired. Importantly, when obese mice were fasted, the transport of leptin (118), insulin (76), and ghrelin (79) from the blood to the brain was restored. Furthermore, obese rats switched to a low-fat diet that lost weight had higher levels of CSF insulin (119). Reversal of obesity also reduced FFA transport into the human brain by 17% (80). These findings again suggest that weight loss may restore BBB transport of peripheral metabolic mediators into the brain.
Endothelial dysfunction is prevalent among the obese population (120,121) and may be associated with changes to BBB structure and transport of peripheral hormones. In fact, a normal endothelial response to L-arginine can be restored in obese women after weight loss (121). It was also found that flow-mediated dilation, a measurement of endothelial function, was restored when severely obese patients lost weight (122). The fact that endothelial function improves with weight loss suggests that a dysfunctional mechanism of metabolite transport exists in the obese population. Taken together, a weight loss intervention may lead to the improvement of brain endothelial function in obese patients, thereby improving metabolic hormone transport and restoration of CNS metabolic activity.
Metabolic hormone resistance and vascular dysfunction may be a result of increased inflammation associated with obesity. Hotamisligil and colleagues showed that murine adipose tissue expressed pro-inflammatory factors that altered insulin receptor activity (123). Inflammation can also affect the ability of the BBB to transport peripheral metabolic hormones into the brain (124). Obesity correlates with increased basal serum levels of pro-inflammatory factors, such as TNF-α and IL-6, in rodents (125) and humans (121,126). After 10 weeks of HFD, rats that were calorically restricted for an additional 8 weeks lost weight and more importantly had reduced inflammation compared to rats that continued on HFD (125). Similar reductions in cortex inflammation, as well as decreases in oxidative stress markers, were found in mice predisposed to AD (APP/PSEN1 mice) which were fed a HFD for 7.5 months (obese) and then switched to a low-fat diet for 2.5 months (obesity reversal) (13). In addition, obese women, who lost 10% of their body weight from liposuction or from a 1-year weight loss program, also showed a reduction in serum inflammatory factors (121). While liposuction is not commonly used to treat obesity, these studies support a major role of adipocytes in the inflammatory response. In a separate study, the reduction in serum inflammatory factors also resulted in less insulin resistance (126). There are beneficial effects of reducing inflammation not only after obesity reversal but also in ameliorating obesity-associated pathologies due to obesity. Further investigation of the role of inflammation in obesity may provide novel therapeutic strategies to increase the ability of hormones crossing the BBB to trigger anorexigenic effects and prevent cognitive decline.
As highlighted above, obesity can be reversed in many ways which could potentially have a huge impact at the BBB (Fig. 2). The first and most obvious way to reverse obesity is through diet and exercise, but many patients who pursue this approach alone do not experience sufficient weight loss. When diet and exercise are not effective, other weight loss options include pharmaceutical interventions and bariatric surgery. While the studies highlighted above look at changes in BBB transport after weight loss due to dietary modifications or liposuction, BBB structure and peptide transport into the CNS have not been investigated after exercise, most pharmaceutical interventions, or bariatric surgery such as Roux-en-Y surgery. Much of the difficulty achieving weight loss in obesity may be the result of poor communication between the gut and the brain due to transport deficiencies at the BBB. As such, the view of obesity has shifted from a disease of nutritional imbalance, to a behavioral disease. To treat behavior, we must treat the brain, and to treat the brain, we must restore peripheral communication to the CNS.
Fig. 2.
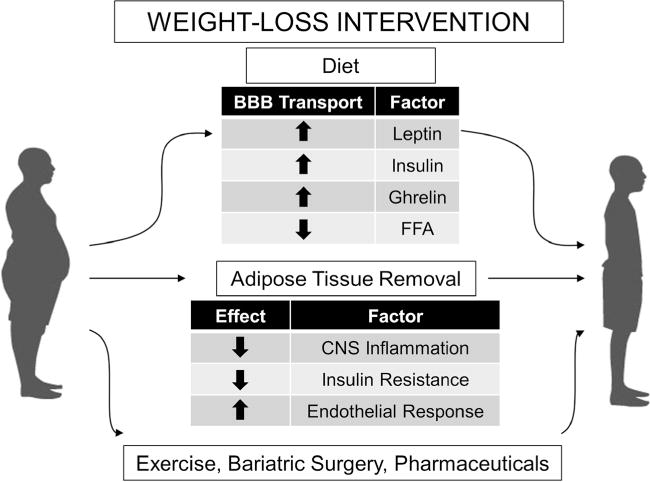
Effects of obesity reversal on BBB transport, CNS inflammation, and metabolic mediators. Obesity reversal results in many metabolic improvements including improvements in insulin resistance. These improvements extend to the CNS in which transport of peripheral peptides across the BBB is restored and inflammation is decreased, not only in the periphery but also in the CNS. Other weight loss interventions such as exercise, bariatric surgery, and pharmaceutical therapies could also exhibit similar effects on the BBB in restoring the connection between the periphery and the CNS
CONCLUSIONS
The works discussed in this review highlight that the BBB is an important interface for metabolic regulation and that BBB dysfunction is an important pathophysiological consequence of obesity and its associated CNS effects, such as cognitive impairment. Importantly, weight loss mitigates some of the BBB impairments observed in obesity such as those on leptin and insulin transport. Therefore, at least some effects of obesity on the BBB are reversible. Difficulties persist, however, in the achievement and maintenance of weight loss as an intervention for obesity. Therefore, the rigorous investigation of therapeutic strategies aimed at sensitizing the CNS to satiety signals from the periphery or enhancing their delivery to the brain continues. We recommend that future therapeutic approaches consider the BBB not only as a barrier to be overcome but also as a viable therapeutic target for the treatment of obesity.
Acknowledgments
This work was supported by numerous grants from the National Institute of Health. EMR is supported by T32-AG000057. TSS, along with WAB, is supported by R21-NS093368-01A1. WAB is also supported by RO1-AG046619, along with MAE. AFL is supported by T32-AG052354-01. AJH is supported by K23-AG047978-01. This article results from work supported by resources from the Veterans Affairs Puget Sound Health Care System, Seattle, Washington.
References
- 1.Kopelman PG. Obesity as a medical problem. Nature. 2000;404(6778):635–43. doi: 10.1038/35007508. [DOI] [PubMed] [Google Scholar]
- 2.Trogdon JG, Finkelstein EA, Feagan CW, Cohen JW. State and payer-specific estimates of annual medical expenditures attributable to obesity. Obesity. 2012;20(1):214–20. doi: 10.1038/oby.2011.169. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404(6778):661–71. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 4.Banks WA. Peptides and the blood-brain barrier. Peptides. 2015;72:16–9. doi: 10.1016/j.peptides.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7(1):41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 6.Moraes JC, Coope A, Morari J, Cintra DE, Roman EA, Pauli JR, et al. High-fat diet induces apoptosis of hypothalamic neurons. PLoS One. 2009;4(4):e5045. doi: 10.1371/journal.pone.0005045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim DW, Glendining KA, Grattan DR, Jasoni CL. Maternal obesity in the mouse compromises the blood-brain barrier in the arcuate nucleus of offspring. Endocrinology. 2016;157(6):2229–42. doi: 10.1210/en.2016-1014. [DOI] [PubMed] [Google Scholar]
- 8.Baufeld C, Osterloh A, Prokop S, Miller KR, Heppner FL. High-fat diet-induced brain region-specific phenotypic spectrum of CNS resident microglia. Acta Neuropathol. 2016;132:361–75. doi: 10.1007/s00401-016-1595-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thaler JP, Guyenet SJ, Dorfman MD, Wisse BE, Schwartz MW. Hypothalamic inflammation: marker or mechanism of obesity pathogenesis? Diabetes. 2013;62(8):2629–34. doi: 10.2337/db12-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prickett C, Brennan L, Stolwyk R. Examining the relationship between obesity and cognitive function: a systematic literature review. Obesity Research & Clinical Practice. 2015;9(2):93–113. doi: 10.1016/j.orcp.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 11.Morris MC, Evans DA, Bienias JL, Tangney CC, Wilson RS. Dietary fat intake and 6-year cognitive change in an older biracial community population. Neurology. 2004;62(9):1573–9. doi: 10.1212/01.wnl.0000123250.82849.b6. [DOI] [PubMed] [Google Scholar]
- 12.Farr SA, Yamada KA, Butterfield DA, Abdul HM, Xu L, Miller NE, et al. Obesity and hypertriglyceridemia produce cognitive impairment. Endocrinology. 2008;149(5):2628–36. doi: 10.1210/en.2007-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walker JM, Dixit S, Saulsberry AC, May JM, Harrison FE. Reversal of high fat diet-induced obesity improves glucose tolerance, inflammatory response, beta-amyloid accumulation and cognitive decline in the APP/PSEN1 mouse model of Alzheimer’s disease. Neurobiol Dis. 2017 doi: 10.1016/j.nbd.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X, Michaelis EK. Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci. 2010;2:12. doi: 10.3389/fnagi.2010.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Granholm A-C, Bimonte-Nelson HA, Moore AB, Nelson ME, Freeman LR, Sambamurti K. Effects of a saturated fat and high cholesterol diet on memory and hippocampal morphology in the middle-aged rat. Journal of Alzheimer’s disease : JAD. 2008;14(2):133–45. doi: 10.3233/jad-2008-14202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stranahan AM, Norman ED, Lee K, Cutler RG, Telljohann R, Egan JM, et al. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus. 2008;18(11):1085–8. doi: 10.1002/hipo.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood– brain barrier: structural components and function under physiologic and pathologic conditions. J NeuroImmune Pharmacol. 2006;1(3):223–36. doi: 10.1007/s11481-006-9025-3. [DOI] [PubMed] [Google Scholar]
- 18.Gustafson DR, Karlsson C, Skoog I, Rosengren L, Lissner L, Blennow K. Mid-life adiposity factors relate to blood–brain barrier integrity in late life. J Intern Med. 2007;262(6):643–50. doi: 10.1111/j.1365-2796.2007.01869.x. [DOI] [PubMed] [Google Scholar]
- 19.Ghribi O, Golovko MY, Larsen B, Schrag M, Murphy EJ. Deposition of iron and β-amyloid plaques is associated with cortical cellular damage in rabbits fed with long-term cholesterol-enriched diets. J Neurochem. 2006;99(2):438–49. doi: 10.1111/j.1471-4159.2006.04079.x. [DOI] [PubMed] [Google Scholar]
- 20.Kanoski SE, Zhang Y, Zheng W, Davidson TL. The effects of a high-energy diet on hippocampal function and blood-brain barrier integrity in the rat. J Alzheimers Dis. 2010;21(1):207–19. doi: 10.3233/JAD-2010-091414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ouyang S, Hsuchou H, Kastin AJ, Wang Y, Yu C, Pan W. Diet-induced obesity suppresses expression of many proteins at the blood–brain barrier. J Cereb Blood Flow Metab. 2014;34(1):43–51. doi: 10.1038/jcbfm.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bondia-Pons I, Boque N, Paternain L, Santamaria E, Fernandez J, Campion J, et al. Liver proteome changes induced by a short-term high-fat sucrose diet in wistar rats. J Nutrigenet Nutrigenomics. 2011;4(6):344–53. doi: 10.1159/000336075. [DOI] [PubMed] [Google Scholar]
- 23.Cruz-Topete D, List EO, Okada S, Kelder B, Kopchick JJ. Proteomic changes in the heart of diet-induced pre-diabetic mice. J Proteome. 2011;74(5):716–27. doi: 10.1016/j.jprot.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarria AJ, Panini SR, Evans RM. A functional role for vimentin intermediate filaments in the metabolism of lipoprotein-derived cholesterol in human SW-13 cells. J Biol Chem. 1992;267(27):19455–63. [PubMed] [Google Scholar]
- 25.Winkler EA, Nishida Y, Sagare AP, Rege SV, Bell RD, Perlmutter D, et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci. 2015;18(4):521–30. doi: 10.1038/nn.3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukuda AM, Badaut J. Aquaporin 4: a player in cerebral edema and neuroinflammation. J Neuroinflammation. 2012;9:279. doi: 10.1186/1742-2094-9-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tomassoni D, Tayebati SK, Cognigni MF, Amenta F. Obesity-related blood brain barrier changes in obese Zucker rats. 2015 [Google Scholar]
- 28.Lutz TA, Woods SC. Overview of animal models of obesity. In: Enna SJ, et al., editors. Current protocols in pharmacology/editorial board. 2012. CHAPTER:Unit5.61-Unit5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banks WA, Gray AM, Erickson MA, Salameh TS, Damodarasamy M, Sheibani N, et al. Lipopolysaccharide-induced blood-brain barrier disruption: roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J Neuroinflammation. 2015;12(1):223. doi: 10.1186/s12974-015-0434-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Price TO, Eranki V, Banks WA, Ercal N, Shah GN. Topiramate treatment protects blood-brain barrier pericytes from hyperglycemia-induced oxidative damage in diabetic mice. Endocrinology. 2012;153(1):362–72. doi: 10.1210/en.2011-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price TO, Farr SA, Niehoff ML, Ercal N, Morley JE, Shah GN. Protective effect of topiramate on hyperglycemia-induced cerebral oxidative stress, pericyte loss and learning behavior in diabetic mice. Int Libr Diabetes Metab. 2015;1(1):6–12. [PMC free article] [PubMed] [Google Scholar]
- 32.Roh H-T, Cho S-Y, So WY. Obesity promotes oxidative stress and exacerbates blood-brain barrier disruption after high-intensity exercise. J Sport Health Sci. 2016 doi: 10.1016/j.jshs.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tucsek Z, Toth P, Sosnowska D, Gautam T, Mitschelen M, Koller A, et al. Obesity in aging exacerbates blood-brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: effects on expression of genes involved in beta-amyloid generation and Alzheimer’s disease. J Gerontol A Biol Sci Med Sci. 2014;69(10):1212–26. doi: 10.1093/gerona/glt177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dorfman MD, Thaler JP. Hypothalamic inflammation and gliosis in obesity. Curr Opin Endocrinol Diabetes Obes. 2015;22(5):325–30. doi: 10.1097/MED.0000000000000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, et al. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology. 2005;146(10):4192–9. doi: 10.1210/en.2004-1520. [DOI] [PubMed] [Google Scholar]
- 36.Sobesky JL, Barrientos RM, De May HS, Thompson BM, Weber MD, Watkins LR, et al. High-fat diet consumption disrupts memory and primes elevations in hippocampal IL-1beta, an effect that can be prevented with dietary reversal or IL-1 receptor antagonism. Brain Behav Immun. 2014;42:22–32. doi: 10.1016/j.bbi.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang X, Dong F, Ren J, Driscoll MJ, Culver B. High dietary fat induces NADPH oxidase-associated oxidative stress and inflammation in rat cerebral cortex. Exp Neurol. 2005;191(2):318–25. doi: 10.1016/j.expneurol.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 38.Stranahan AM, Hao S, Dey A, Yu X, Baban B. Blood-brain barrier breakdown promotes macrophage infiltration and cognitive impairment in leptin receptor-deficient mice. J Cereb Blood Flow Metab. 2016;36(12):2108–21. doi: 10.1177/0271678X16642233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Banks WA, Kastin AJ, Broadwell RD. Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation. 1995;2(4):241–8. doi: 10.1159/000097202. [DOI] [PubMed] [Google Scholar]
- 40.Jung UJ, Choi MS. Obesity and its metabolic complications: the role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int J Mol Sci. 2014;15(4):6184–223. doi: 10.3390/ijms15046184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banks WA, Kastin AJ, Gutierrez EG. Penetration of interleukin-6 across the murine blood-brain barrier. Neurosci Lett. 1994;179(1–2):53–6. doi: 10.1016/0304-3940(94)90933-4. [DOI] [PubMed] [Google Scholar]
- 42.Pan W, Kastin AJ. TNFalpha transport across the blood-brain barrier is abolished in receptor knockout mice. Exp Neurol. 2002;174(2):193–200. doi: 10.1006/exnr.2002.7871. [DOI] [PubMed] [Google Scholar]
- 43.Williams K, Alvarez X, Lackner AA. Central nervous system perivascular cells are immunoregulatory cells that connect the CNS with the peripheral immune system. Glia. 2001;36(2):156–64. doi: 10.1002/glia.1105. [DOI] [PubMed] [Google Scholar]
- 44.Jais A, Solas M, Backes H, Chaurasia B, Kleinridders A, Theurich S, et al. Myeloid-cell-derived VEGF maintains brain glucose uptake and limits cognitive impairment in obesity. Cell. 2016;165(4):882–95. doi: 10.1016/j.cell.2016.03.033. [DOI] [PubMed] [Google Scholar]
- 45.Verma S, Nakaoke R, Dohgu S, Banks WA. Release of cytokines by brain endothelial cells: a polarized response to lipopolysaccharide. Brain Behav Immun. 2006;20(5):449–55. doi: 10.1016/j.bbi.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 46.Banks WA, Niehoff ML, Ponzio NM, Erickson MA, Zalcman SS. Pharmacokinetics and modeling of immune cell trafficking: quantifying differential influences of target tissues versus lymphocytes in SJL and lipopolysaccharide-treated mice. J Neuroinflammation. 2012;9:231. doi: 10.1186/1742-2094-9-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Engelhardt B. Molecular mechanisms involved in T cell migration across the blood-brain barrier. J Neural Transm (Vienna) 2006;113(4):477–85. doi: 10.1007/s00702-005-0409-y. [DOI] [PubMed] [Google Scholar]
- 48.Buckman LB, Thompson MM, Lippert RN, Blackwell TS, Yull FE, Ellacott KL. Evidence for a novel functional role of astrocytes in the acute homeostatic response to high-fat diet intake in mice. Mol Metab. 2015;4(1):58–63. doi: 10.1016/j.molmet.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Carroll SJ, Kho DT, Wiltshire R, Nelson V, Rotimi O, Johnson R, et al. Pro-inflammatory TNFalpha and IL-1beta differentially regulate the inflammatory phenotype of brain microvascular endothelial cells. J Neuroinflammation. 2015;12:131. doi: 10.1186/s12974-015-0346-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Etienne-Manneville S, Manneville JB, Adamson P, Wilbourn B, Greenwood J, Couraud PO. ICAM-1-coupled cytoskeletal rearrangements and transendothelial lymphocyte migration involve intracellular calcium signaling in brain endothelial cell lines. J Immunol. 2000;165(6):3375–83. doi: 10.4049/jimmunol.165.6.3375. [DOI] [PubMed] [Google Scholar]
- 51.Nerurkar PV, Johns LM, Buesa LM, Kipyakwai G, Volper E, Sato R, et al. Momordica charantia (bitter melon) attenuates high-fat diet-associated oxidative stress and neuroinflammation. J Neuroinflammation. 2011;8:64. doi: 10.1186/1742-2094-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barnes DE, Yaffe K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10(9):819–28. doi: 10.1016/S1474-4422(11)70072-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Julien C, Tremblay C, Phivilay A, Berthiaume L, Emond V, Julien P, et al. High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol Aging. 2010;31(9):1516–31. doi: 10.1016/j.neurobiolaging.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 54.Levin-Allerhand JA, Lominska CE, Smith JD. Increased amyloid-levels in APPSWE transgenic mice treated chronically with a physiological high-fat high-cholesterol diet. J Nutr Health Aging. 2002;6(5):315–9. [PubMed] [Google Scholar]
- 55.Hanson AJ, Bayer JL, Baker LD, Cholerton B, VanFossen B, Trittschuh E, et al. Differential effects of meal challenges on cognition, metabolism, and biomarkers for apolipoprotein E varepsilon4 carriers and adults with mild cognitive impairment. J Alzheimers Dis. 2015;48(1):205–18. doi: 10.3233/JAD-150273. [DOI] [PubMed] [Google Scholar]
- 56.Takechi R, Galloway S, Pallebage-Gamarallage MM, Lam V, Mamo JC. Dietary fats, cerebrovasculature integrity and Alzheimer’s disease risk. Prog Lipid Res. 2010;49(2):159–70. doi: 10.1016/j.plipres.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 57.Pugazhenthi S, Qin L, Reddy PH. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim Biophys Acta. 2016 doi: 10.1016/j.bbadis.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Erickson MA, Hartvigson PE, Morofuji Y, Owen JB, Butterfield DA, Banks WA. Lipopolysaccharide impairs amyloid beta efflux from brain: altered vascular sequestration, cerebrospinal fluid reabsorption, peripheral clearance and transporter function at the blood-brain barrier. J Neuroinflammation. 2012;9:150. doi: 10.1186/1742-2094-9-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miller MC, Tavares R, Johanson CE, Hovanesian V, Donahue JE, Gonzalez L, et al. Hippocampal RAGE immunoreactivity in early and advanced Alzheimer’s disease. Brain Res. 2008;1230:273–80. doi: 10.1016/j.brainres.2008.06.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Song F, Hurtado del Pozo C, Rosario R, Zou YS, Ananthakrishnan R, Xu X, et al. RAGE regulates the metabolic and inflammatory response to high-fat feeding in mice. Diabetes. 2014;63(6):1948–65. doi: 10.2337/db13-1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang M, Norman JE, Srinivasan VJ, Rutledge JC. Metabolic, inflammatory, and microvascular determinants of white matter disease and cognitive decline. American journal of neurodegenerative disease. 2016;5(5):171–7. [PMC free article] [PubMed] [Google Scholar]
- 62.Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9(2):106–18. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hanson AJ, Bayer-Carter JL, Green PS, Montine TJ, Wilkinson CW, Baker LD, et al. Effect of apolipoprotein E genotype and diet on apolipoprotein E lipidation and amyloid peptides: randomized clinical trial. JAMA Neurol. 2013:1–9. doi: 10.1001/jamaneurol.2013.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Elliott DA, Weickert CS, Garner B. Apolipoproteins in the brain: implications for neurological and psychiatric disorders. Clinical lipidology. 2010;51(4):555–73. doi: 10.2217/CLP.10.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shayo M, McLay RN, Kastin AJ, Banks WA. The putative blood-brain barrier transporter for the β-amyloid binding protein apolipoprotein J is saturated at physiological concentrations. Life Sci. 1996;60:L115–L8. doi: 10.1016/s0024-3205(96)00685-6. [DOI] [PubMed] [Google Scholar]
- 66.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–30. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tschop M, Weyer C, Tataranni PA, Devanarayan V, Ravussin E, Heiman ML. Circulating ghrelin levels are decreased in human obesity. Diabetes. 2001;50(4):707–9. doi: 10.2337/diabetes.50.4.707. [DOI] [PubMed] [Google Scholar]
- 68.Bagdade JD. Basal insulin and obesity. Lancet. 1968;2(7568):630–1. doi: 10.1016/s0140-6736(68)90712-5. [DOI] [PubMed] [Google Scholar]
- 69.Caro JF, Kolaczynski JW, Nyce MR, Ohannesian JP, Opentanova I, Goldman WH, et al. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. Lancet. 1996;348(9021):159–61. doi: 10.1016/s0140-6736(96)03173-x. [DOI] [PubMed] [Google Scholar]
- 70.Schwartz MW, Peskind E, Raskind M, Boyko EJ, Porte D., Jr Cerebrospinal fluid leptin levels: relationship to plasma levels and to adiposity in humans. Nat Med. 1996;2(5):589–93. doi: 10.1038/nm0596-589. [DOI] [PubMed] [Google Scholar]
- 71.Adam CL, Findlay PA. Decreased blood-brain leptin transfer in an ovine model of obesity and weight loss: resolving the cause of leptin resistance. Int J Obes. 2010;34(6):980–8. doi: 10.1038/ijo.2010.28. [DOI] [PubMed] [Google Scholar]
- 72.Banks WA, DiPalma CR, Farrell CL. Impaired transport of leptin across the blood-brain barrier in obesity. Peptides. 1999;20(11):1341–5. doi: 10.1016/s0196-9781(99)00139-4. [DOI] [PubMed] [Google Scholar]
- 73.Kern W, Benedict C, Schultes B, Plohr F, Moser A, Born J, et al. Low cerebrospinal fluid insulin levels in obese humans. Diabetologia. 2006;49(11):2790–2. doi: 10.1007/s00125-006-0409-y. [DOI] [PubMed] [Google Scholar]
- 74.Kaiyala KJ, Prigeon RL, Kahn SE, Woods SC, Schwartz MW. Obesity induced by a high-fat diet is associated with reduced brain insulin transport in dogs. Diabetes. 2000;49(9):1525–33. doi: 10.2337/diabetes.49.9.1525. [DOI] [PubMed] [Google Scholar]
- 75.Baskin DG, Stein LJ, Ikeda H, Woods SC, Figlewicz DP, Porte D, Jr, et al. Genetically obese Zucker rats have abnormally low brain insulin content. Life Sci. 1985;36(7):627–33. doi: 10.1016/0024-3205(85)90166-3. [DOI] [PubMed] [Google Scholar]
- 76.Urayama A, Banks WA. Starvation and triglycerides reverse the obesity-induced impairment of insulin transport at the blood-brain barrier. Endocrinology. 2008;149(7):3592–7. doi: 10.1210/en.2008-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Spranger J, Verma S, Gohring I, Bobbert T, Seifert J, Sindler AL, et al. Adiponectin does not cross the blood-brain barrier but modifies cytokine expression of brain endothelial cells. Diabetes. 2006;55(1):141–7. [PubMed] [Google Scholar]
- 78.Pan W, Tu H, Kastin AJ. Differential BBB interactions of three ingestive peptides: obestatin, ghrelin, and adiponectin. Peptides. 2006;27(4):911–6. doi: 10.1016/j.peptides.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 79.Banks WA, Burney BO, Robinson SM. Effects of triglycerides, obesity, and starvation on ghrelin transport across the blood-brain barrier. Peptides. 2008;29(11):2061–5. doi: 10.1016/j.peptides.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Karmi A, Iozzo P, Viljanen A, Hirvonen J, Fielding BA, Virtanen K, et al. Increased brain fatty acid uptake in metabolic syndrome. Diabetes. 2010;59(9):2171–7. doi: 10.2337/db09-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395(6704):763–70. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 82.Banks WA, Kastin AJ, Huang W, Jaspan JB, Maness LM. Leptin enters the brain by a saturable system independent of insulin. Peptides. 1996;17(2):305–11. doi: 10.1016/0196-9781(96)00025-3. [DOI] [PubMed] [Google Scholar]
- 83.Banks WA, Clever CM, Farrell CL. Partial saturation and regional variation in the blood-to-brain transport of leptin in normal weight mice. Am J Physiol Endocrinol Metab. 2000;278(6):E1158–65. doi: 10.1152/ajpendo.2000.278.6.E1158. [DOI] [PubMed] [Google Scholar]
- 84.Van Heek M, Compton DS, France CF, Tedesco RP, Fawzi AB, Graziano MP, et al. Diet-induced obese mice develop peripheral, but not central, resistance to leptin. J Clin Invest. 1997;99(3):385–90. doi: 10.1172/JCI119171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Halaas JL, Boozer C, Blair-West J, Fidahusein N, Denton DA, Friedman JM. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc Natl Acad Sci U S A. 1997;94(16):8878–83. doi: 10.1073/pnas.94.16.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Banks WA, Niehoff ML, Martin D, Farrell CL. Leptin transport across the blood-brain barrier of the Koletsky rat is not mediated by a product of the leptin receptor gene. Brain Res. 2002;950(1–2):130–6. doi: 10.1016/s0006-8993(02)03013-5. [DOI] [PubMed] [Google Scholar]
- 87.Maness LM, Banks WA, Kastin AJ. Persistence of blood-to-brain transport of leptin in obese leptin-deficient and leptin receptor-deficient mice. Brain Res. 2000;873(1):165–7. doi: 10.1016/s0006-8993(00)02520-8. [DOI] [PubMed] [Google Scholar]
- 88.Banks WA, Coon AB, Robinson SM, Moinuddin A, Shultz JM, Nakaoke R, et al. Triglycerides induce leptin resistance at the blood-brain barrier. Diabetes. 2004;53(5):1253–60. doi: 10.2337/diabetes.53.5.1253. [DOI] [PubMed] [Google Scholar]
- 89.Kastin AJ, Akerstrom V. Glucose and insulin increase the transport of leptin through the blood-brain barrier in normal mice but not in streptozotocin-diabetic mice. Neuroendocrinology. 2001;73(4):237–42. doi: 10.1159/000054640. [DOI] [PubMed] [Google Scholar]
- 90.Yi X, Yuan D, Farr SA, Banks WA, Poon CD, Kabanov AV. Pluronic modified leptin with increased systemic circulation, brain uptake and efficacy for treatment of obesity. J Control Release. 2014;191:34–46. doi: 10.1016/j.jconrel.2014.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Banks WA, Gertler A, Solomon G, Niv-Spector L, Shpilman M, Yi X, et al. Principles of strategic drug delivery to the brain (SDDB): development of anorectic and orexigenic analogs of leptin. Physiol Behav. 2011;105(1):145–9. doi: 10.1016/j.physbeh.2011.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Qi Y, Takahashi N, Hileman SM, Patel HR, Berg AH, Pajvani UB, et al. Adiponectin acts in the brain to decrease body weight. Nat Med. 2004;10(5):524–9. doi: 10.1038/nm1029. [DOI] [PubMed] [Google Scholar]
- 93.Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257(1):79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 94.Neumeier M, Weigert J, Buettner R, Wanninger J, Schaffler A, Muller AM, et al. Detection of adiponectin in cerebrospinal fluid in humans. Am J Physiol Endocrinol Metab. 2007;293(4):E965–9. doi: 10.1152/ajpendo.00119.2007. [DOI] [PubMed] [Google Scholar]
- 95.Vachharajani V, Cunningham C, Yoza B, Carson J, Jr, Vachharajani TJ, McCall C. Adiponectin-deficiency exaggerates sepsis-induced microvascular dysfunction in the mouse brain. Obesity (Silver Spring) 2012;20(3):498–504. doi: 10.1038/oby.2011.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stephan F, Reville P, Thierry R, Schlienger JL. Correlations between plasma insulin and body weight in obesity, anorexia nervosa and diabetes mellitus. Diabetologia. 1972;8(3):196–201. doi: 10.1007/BF01212261. [DOI] [PubMed] [Google Scholar]
- 97.Woods SC, Seeley RJ, Baskin DG, Schwartz MW. Insulin and the blood-brain barrier. Curr Pharm Des. 2003;9(10):795–800. doi: 10.2174/1381612033455323. [DOI] [PubMed] [Google Scholar]
- 98.Cangiano C, Cardelli-Cangiano P, Cascino A, Patrizi MA, Barberini F, Rossi Fanelli F, et al. On the stimulation by insulin of tryptophan transport across the blood-brain barrier. Biochem Int. 1983;7(5):617–27. [PubMed] [Google Scholar]
- 99.Banks WA, Kastin AJ. Differential permeability of the blood-brain barrier to two pancreatic peptides: insulin and amylin. Peptides. 1998;19(5):883–9. doi: 10.1016/s0196-9781(98)00018-7. [DOI] [PubMed] [Google Scholar]
- 100.Roth JD, Roland BL, Cole RL, Trevaskis JL, Weyer C, Koda JE, et al. Leptin responsiveness restored by amylin agonism in diet-induced obesity: evidence from nonclinical and clinical studies. Proc Natl Acad Sci U S A. 2008;105(20):7257–62. doi: 10.1073/pnas.0706473105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Uchida A, Zechner JF, Mani BK, Park WM, Aguirre V, Zigman JM. Altered ghrelin secretion in mice in response to diet-induced obesity and Roux-en-Y gastric bypass. Mol Metab. 2014;3(7):717–30. doi: 10.1016/j.molmet.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lamers D, Famulla S, Wronkowitz N, Hartwig S, Lehr S, Ouwens DM, et al. Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes. 2011;60(7):1917–25. doi: 10.2337/db10-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tanaka S, Kanazawa I, Notsu M, Sugimoto T. Visceral fat obesity increases serum DPP-4 levels in men with type 2 diabetes mellitus. Diabetes Res Clin Pract. 2016;116:1–6. doi: 10.1016/j.diabres.2016.04.027. [DOI] [PubMed] [Google Scholar]
- 104.Kieffer TJ, McIntosh CH, Pederson RA. Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology. 1995;136(8):3585–96. doi: 10.1210/endo.136.8.7628397. [DOI] [PubMed] [Google Scholar]
- 105.Kastin AJ, Akerstrom V, Pan W. Interactions of glucagon-like peptide-1 (GLP-1) with the blood-brain barrier. J Mol Neurosci. 2002;18(1–2):7–14. doi: 10.1385/JMN:18:1-2:07. [DOI] [PubMed] [Google Scholar]
- 106.Rohrborn D, Wronkowitz N, Eckel J. DPP4 in diabetes. Front Immunol. 2015;6:386. doi: 10.3389/fimmu.2015.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pocai A, Lam TK, Obici S, Gutierrez-Juarez R, Muse ED, Arduini A, et al. Restoration of hypothalamic lipid sensing normalizes energy and glucose homeostasis in overfed rats. J Clin Invest. 2006;116(4):1081–91. doi: 10.1172/JCI26640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Obici S, Feng Z, Morgan K, Stein D, Karkanias G, Rossetti L. Central administration of oleic acid inhibits glucose production and food intake. Diabetes. 2002;51(2):271–5. doi: 10.2337/diabetes.51.2.271. [DOI] [PubMed] [Google Scholar]
- 109.Boden G. Obesity and free fatty acids. Endocrinol Metab Clin N Am. 2008;37(3):635–46. viii–ix. doi: 10.1016/j.ecl.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Guiducci L, Gronroos T, Jarvisalo MJ, Kiss J, Viljanen A, Naum AG, et al. Biodistribution of the fatty acid analogue 18F-FTHA: plasma and tissue partitioning between lipid pools during fasting and hyperinsulinemia. J Nucl Med. 2007;48(3):455–62. [PubMed] [Google Scholar]
- 111.Arai T, Wakabayashi S, Channing MA, Dunn BB, Der MG, Bell JM, et al. Incorporation of [1-carbon-11]palmitate in monkey brain using PET. J Nucl Med. 1995;36(12):2261–7. [PubMed] [Google Scholar]
- 112.Kimes AS, Sweeney D, London ED, Rapoport SI. Palmitate incorporation into different brain regions in the awake rat. Brain Res. 1983;274(2):291–301. doi: 10.1016/0006-8993(83)90707-2. [DOI] [PubMed] [Google Scholar]
- 113.Vendelbo J, Olesen RH, Lauridsen JK, Rungby J, Kleinman JE, Hyde TM, et al. Increasing BMI is associated with reduced expression of P-glycoprotein (ABCB1 gene) in the human brain with a stronger association in African Americans than Caucasians. Pharmacogenomics J. 2016 doi: 10.1038/tpj.2016.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Veronese N, Facchini S, Stubbs B, Luchini C, Solmi M, Manzato E, et al. Weight loss is associated with improvements in cognitive function among overweight and obese people: a systematic review and meta-analysis. Neurosci Biobehav Rev. 2017;72:87–94. doi: 10.1016/j.neubiorev.2016.11.017. [DOI] [PubMed] [Google Scholar]
- 115.Jeong SK, Nam HS, Son MH, Son EJ, Cho KH. Interactive effect of obesity indexes on cognition. Dement Geriatr Cogn Disord. 2005;19(2–3):91–6. doi: 10.1159/000082659. [DOI] [PubMed] [Google Scholar]
- 116.Dahl A, Hassing LB, Fransson E, Berg S, Gatz M, Reynolds CA, et al. Being overweight in midlife is associated with lower cognitive ability and steeper cognitive decline in late life. J Gerontol A Biol Sci Med Sci. 2010;65(1):57–62. doi: 10.1093/gerona/glp035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hassing LB, Dahl AK, Pedersen NL, Johansson B. Overweight in midlife is related to lower cognitive function 30 years later: a prospective study with longitudinal assessments. Dement Geriatr Cogn Disord. 2010;29(6):543–52. doi: 10.1159/000314874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Banks WA, Farrell CL. Impaired transport of leptin across the blood-brain barrier in obesity is acquired and reversible. Am J Physiol Endocrinol Metab. 2003;285(1):E10–5. doi: 10.1152/ajpendo.00468.2002. [DOI] [PubMed] [Google Scholar]
- 119.Begg DP, Mul JD, Liu M, Reedy BM, D’Alessio DA, Seeley RJ, et al. Reversal of diet-induced obesity increases insulin transport into cerebrospinal fluid and restores sensitivity to the anorexic action of central insulin in male rats. Endocrinology. 2013;154(3):1047–54. doi: 10.1210/en.2012-1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Steinberg HO, Chaker H, Leaming R, Johnson A, Brechtel G, Baron AD. Obesity/insulin resistance is associated with endothelial dysfunction. Implications for the syndrome of insulin resistance. J Clin Invest. 1996;97(11):2601–10. doi: 10.1172/JCI118709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ziccardi P, Nappo F, Giugliano G, Esposito K, Marfella R, Cioffi M, et al. Reduction of inflammatory cytokine concentrations and improvement of endothelial functions in obese women after weight loss over one year. Circulation. 2002;105(7):804–9. doi: 10.1161/hc0702.104279. [DOI] [PubMed] [Google Scholar]
- 122.Bigornia SJ, Mott MM, Hess DT, Apovian CM, McDonnell ME, Duess MA, et al. Long-term successful weight loss improves vascular endothelial function in severely obese individuals. Obesity (Silver Spring) 2010;18(4):754–9. doi: 10.1038/oby.2009.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271(5249):665–8. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- 124.Xaio H, Banks WA, Niehoff ML, Morley JE. Effect of LPS on the permeability of the blood-brain barrier to insulin. Brain Res. 2001;896(1–2):36–42. doi: 10.1016/s0006-8993(00)03247-9. [DOI] [PubMed] [Google Scholar]
- 125.Park S, Park NY, Valacchi G, Lim Y. Calorie restriction with a high-fat diet effectively attenuated inflammatory response and oxidative stress-related markers in obese tissues of the high diet fed rats. Mediat Inflamm. 2012;2012:984643. doi: 10.1155/2012/984643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Giugliano G, Nicoletti G, Grella E, Giugliano F, Esposito K, Scuderi N, et al. Effect of liposuction on insulin resistance and vascular inflammatory markers in obese women. Br J Plast Surg. 2004;57(3):190–4. doi: 10.1016/j.bjps.2003.12.010. [DOI] [PubMed] [Google Scholar]