

Abstract
Heme, a hydrophobic and cytotoxic macrocycle, is an essential cofactor in a large number of proteins and is important for cell signaling. This must mean that heme is mobilized from its place of synthesis or entry into the cell to other parts of the cell where hemoproteins reside. However, the cellular dynamics of heme movement is not well understood, in large part due to the inability to image heme noninvasively in live biological systems. Here, using high-resolution transient absorption microscopy, we showed that heme storage and distribution is dynamic in Caenorhabditis elegans. Intracellular heme exists in concentrated granular puncta which localizes to lysosomal-related organelles. These granules are dynamic, and their breaking down into smaller granules provides a mechanism by which heme stores can be mobilized. Collectively, these direct and noninvasive dynamic imaging techniques provide new insights into heme storage and transport and open a new avenue for label-free investigation of heme function and regulation in living systems.
Graphical abstract
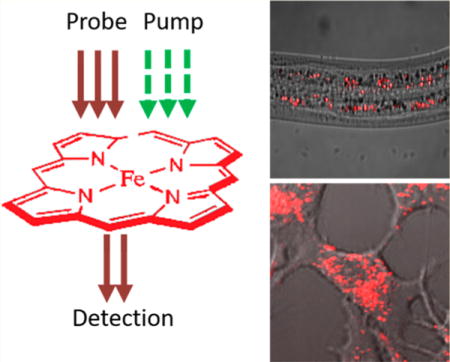
■ INTRODUCTION
Heme is an essential prosthetic group for a myriad of proteins which function in a wide spectrum of biological processes. These processes include electron transport, gas transport and sensation, circadian clock control, micro-RNA processing, and gene regulation.1–5 Heme can be exogenously acquired, primarily as a source of dietary iron uptake, and endogenously synthesized via an eight-step pathway in the eukaryote mitochondria.6 Free heme, however, is toxic because it induces oxidative stress and reacts with proteins and the lipid bilayer.7–9 Therefore, a specific molecular mechanism must exist to transport cellular heme from sites of storage or synthesis to specific hemoproteins which reside in virtually all intracellular organelles. In mammals, defects in heme synthesis lead to anemia and porphyrias.10–12 By contrast, nematodes such as the free-living Caenorhabditis elegans and related parasitic helminths lack the ability to synthesize heme but instead acquire heme from the environment.13 Helminths infect hundreds of millions of people worldwide, posing huge challenges to public health.14 The treatment of helminthiasis calls for new ways to tackle the problem even as drug resistance is on the rise.15 Therefore, studies aimed in elucidating heme transport pathways could be key in understanding human blood disorders and help develop therapeutics to curb helminthic infections.
Despite the significance of heme transport and trafficking, only a few players have been identified and characterized to date.16–19 To push this endeavor further, methods for heme imaging were developed. Zinc mesoporphyrin (ZnMP), a heme analogue that has fluorescence emission around 600 nm, has been used to follow heme uptake and distribution. Yet, the reliability of ZnMP is weakened by a lack of biological relevance, toxicity, and limited sensitivity.20,21 Another strategy for heme imaging in live cells is based on the principle that association of heme either enhances or attenuates the fluorescence resonance energy transfer (FRET) process or quenches the fluorophore.22–25 Typically, these sensors recognize only labile heme.22,23,25 Moreover, the FRET sensors could perturb normal heme homeostasis by sequestering heme and distorting labile heme.22,23,25 Recently, a horseradish peroxidase (HRP)-based sensor was engineered based on the enhancement of HRP activity upon heme-binding. The HRP protein was specifically targeted to various subcellular compartments and was sensitive enough to probe micromolar levels of labile heme.26 Yet, this sensor cannot probe heme in living cells due to the need for in situ histochemical detection. In summary, current methods either image a heme substitute or pick up indirect signals from a heme sensor.
Transient absorption (TA) microscopy27–29 allows for direct visualization and quantification of chromophores having low fluorescence quantum yield. In principle, TA microscopy uses two-pulsed laser beams, one as pump and the other as probe, to measure the excited state dynamics of a chromophore. Time-resolved TA microscopy takes a series of image of the specimen at various delays between the pump and probe pulses. The decay rate of the TA signal describes how fast the molecule returns to the ground state after excitation by the pump pulse. Because a molecule in different forms could decay at distinct rates, the decay curve serves as a spectral signature to differentiate the various forms of the molecule. The TA signal peaks when the pump and probe pulses reach the specimen at roughly the same time.
TA microscopy was first reported in 1995 by Dong et al.30 and since has been applied broadly. In the materials science field, Hartland, Vallee, Orrit, and coworkers applied TA microscopy to study carrier dynamics in a variety of metal nanomaterials.31–34 Zhang, Huang, and coworkers employed TA imaging to study semiconducting nanomaterials.35–37 Cheng and coworkers applied TA microscopy to differential semiconducting and metallic carbon nanotubes38 and to visualize graphene in biological systems.39 For biological molecules, the Warren group pioneered the development of two-color TA imaging40 to melanoma diagnosis based on the differentiation of eumelanin and pheomelanin.41 Xie group demonstrated TA imaging of nonfluorescent chromophores in living cells.42 Very recently, Wilson and coworkers reported TA measurement of heme in adipose and heart frozen tissue sections.43
Here, we exploit TA microscopy to map spatiotemporal dynamics of heme in live organisms. We first establish the sensitivity, specificity, and spatial resolution of our microscope and then deploy TA microscopy to directly visualize heme uptake and subcellular localization of heme in genetic mutants of C. elegans and live mammalian cell lines. Collectively, our results demonstrate a viable platform for label-free imaging of heme in live biological systems.
■ EXPERIMENTAL SECTION
Biological Specimens
C. elegans. WT, IQ6011, LRO marker glo-1:gfp, mrp-5 deletion strains were provided by Dr. Iqbal Hamza’s lab. The worms were maintained in a 20 °C incubator (Tritech research DT2-MP-47L) and on NGM plates as documented in Wormbook.44 To incubate worms with various concentrations of hemin (Frontier Scientific H651-9), worms were cultured on NGM plates and synchronized by bleaching and L1 arrest, as documented in Wormbook.44 Two millimolar hemin stock (dissolved in 0.3N NH4Cl, pH 8.0) was diluted in mCeHR2 medium to the desired concentration before worms were added. RNAi knockdown was performed in accordance with the protocol documented in Wormbook.44 To immobilize worms for imaging, worms were harvested and washed using M9 buffer 3 times to remove medium residues and then anesthetized in 20 mM NaN3 (Sigma S2002) for 10 min. One hundred to two hundred worms were added on predried 1% agarose (Thermo 16500500) pad sandwiched between cover glasses (VWR 16004-338).
HEK293A cell line was provided by Dr. Iqbal Hamza’s lab. Cells were cultured in DMEM medium (Thermo 11965092) with 10% fetal bovine serum (Corning 35-010-CV) and 100 U/mL penicillin/streptomycin solution (Thermo 15140122). Prior to imaging, cells were washed with phosphate buffered saline (PBS, Thermo 10010023) 3 times to remove medium residue and soaked in PBS for imaging.
Transient Absorption Microscopy
Transient absorption imaging was performed following a protocol in previous publication.45 The laser source was a femtosecond pulse laser (Spectra-Physics Insight) operating at 80 MHz with 2 synchronized outputs. Lasers at 520 and 780 nm were chosen as pump and probe beams, respectively. The pump beam was modulated at ∼2.5 MHz using acoustic-optical modulator (Isomet 1205-C); the probe beam went through a delay-tuning stage and combined with pump beam. The combined beams were directed to a 60×, 1.2 NA water immersing objective (Olympus UPlanApo/IR). After interacting with the specimen, photons were collected by a 1.4 NA oil condenser (Olympus U-AAC) and filtered to let probe beam through. The probe photons were detected using a photodiode (Hamamatsu 3994), and the modulated transient absorption signal was amplified by a lab-made resonant circuit. The signal was then further amplified and extracted by lock-in amplifier (Zurich Instrument HF2LI) with time constant setting as 7 μs. To record time-resolved TA curves, a delay-tuning stage was used to tune the delay between pump and probe pulses, with 66.7 fs per step. A whole frame was recorded at each delay position before the stage moved to the next delay position. The delay curve was processed using ImageJ software and plotted using Origin software. Decay curve was fitted with double exponential decay model as described previously.33
Quantification of heme amount inside a worm was performed by either intensity or area. In general, a constant threshold was applied to the raw images to filter out background, and then the intensity or the area of the heme-indicating pixels were calculated using ImageJ and normalized by the total pixel number of a worm imaged, which was calculated by manually defining the boundary of worm using the transmission image of the same field of view and then tallying the pixel number using “Measure” function in ImageJ software. Sensitivity of TA microscopy for hemin was obtained by finding the concentration of hemin where signal-to-noise ratio was 2.0.
SRS and TPEF Microscopy
Stimulated Raman scattering (SRS) imaging was performed in accordance with a protocol in previous publication.46 Forward-detected two-photon excitation fluorescence (TPEF) imaging was performed using the 780 nm pulsed laser as the excitation light. TA, SRS, and TPEF imaging were conducted on the same microscope platform. Quantifi-cation was done as described above.
Spontaneous Raman Spectroscopy
Spontaneous Raman spectroscopy was performed on a Horiba confocal Raman microscope (Horiba Scientific Labram HR Evolution) in accordance with the user’s manual. Worm sample was prepared as described in the Biological Specimen section. Worms were first inspected using bright field microscopy. Because heme granules typically showed as bright or dark granules under bright field microscopy, we picked those granules for spontaneous Raman spectroscopy imaging. Laser wavelength: 532 nm; laser power: 1%; pinhole size: 50 μm; dwell time: 5 s; objective: 40× air; grating: 600 l/mm.
Statistical Analysis
One-tailed Student’s t test was used for all significance tests. N is at least 5 for each group. * denotes p < 0.05; ** denotes p < 0.01; *** denotes p < 0.001.
■ RESULTS
To start, we tested the sensitivity of our TA microscope (Figure S1) in detecting heme in vitro. We used 200 fs pulses at 520 and 780 nm as pump and probe beams, respectively, and fixed the on-sample laser power to 5 and 17 mW for pump and probe. We then measured the zero delay TA intensities as a function of increasing concentrations of hemin solution (Figure 1a). TA intensity was found to be linear with respect to hemin concentrations, as predicted theoretically (Figure 1b, R = 0.998). The sensitivity of our TA microscope was found to be ∼9 μM heme based on a signal-to-noise ratio of 2.0.
Figure 1.
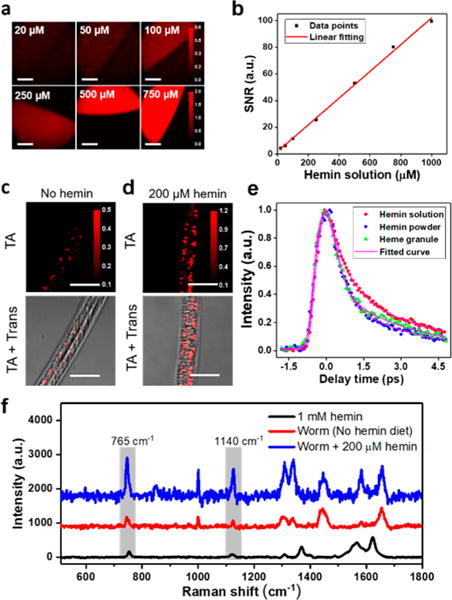
Transient absorption microscopy for in vivo imaging of heme in C. elegans. (a) TA imaging of 20, 50, 100, 250, 500, and 750 μM hemin solution (in 0.3N NH4Cl, pH 8.0). Scale bar: 70 μm. (b) TA signal-to-noise ratio plotted against concentration of hemin solution. Data were fitted linearly. (c and d) WT worms were cultured on NGM plates and synchronized to L1, which were maintained in mCeHR2 medium or mCeHR2 medium supplemented with 200 μM hemin for 16 h and then imaged using TA microscopy. Trans: transmission images. Scale bar: 20 μm. (e) Decay curves of heme granule, 1 mM hemin solution and hemin powder; about 200 pixels were selected and averaged. (f) Spontaneous Raman spectra of heme granules from worms kept on NGM plate and NGM plate supplemented with 200 μM hemin. Hemin solution (1 mM) served as reference. Pixel dwell time: 10 μs.
To determine the utility of TA microscopy for heme imaging, we exploited C. elegans as an animal model. C. elegans is a unique biological system to test instrument sensitivity and establish threshold for detection because they do not synthesize heme; endogenous heme levels are a direct function of exogenous heme supplementation. Synchronized wild-type (WT) L1 larvae were grown in liquid axenic mCeHR2 medium or mCeHR2 medium supplemented with 200 μM hemin for three days followed by zero time delay TA imaging. As revealed in Figures 1c and d, the hemin-fed worms display heme accumulation in the intestine, whereas only trace amount of heme was detected in the intestine of worms kept in medium without hemin. In addition, the hemin-fed worms displayed larger and more intense heme-containing puncta within the intestine than worms grown in low heme medium (Figures 1c and d). In addition, a trace amount of heme, which is probably from maternal passage, is observed in worms grown in medium without hemin supplementation (Figure 1c). The TA imaging reveals heme storage sites within vesicle-like granules in the intestine (Figure 1d) as well as in the pharynx and hypodermis (Figure S2).
To further characterize the heme storage sites, we recorded and compared the TA decay curve of the heme granules with that of hemin powder and of hemin solution and found the former nicely matched the decay curve of heme granule (Figure 1e). These data suggest that heme might exist in a clustered, solid-state form inside the granules. To further confirm the existence of heme inside these granules, we recorded spontaneous Raman spectra of the heme granules from worms grown on solid agar-based NGM with and without hemin (Figure 1f). Both groups show the characteristic peaks at 765 and 1140 cm−1, corresponding to vibrations of C–H bond and pyrrole ring in heme,47 also found in the control hemin solution. Taken together, these data indicate that TA microscopy has the capacity of imaging heme in live C. elegans.
We noted that the standard procedure to grow C. elegans in most biological studies is on NGM agar seeded with E. coli OP50 strain.44 Thus, we checked whether TA microscopy has the sensitivity to probe heme granules under this condition. Figure S3 shows that heme granules were observed in intestine cells of WT worms at both L2 and L4 stages. Together, these results demonstrate that TA microscopy has the sensitivity to monitor heme stores under normal growth conditions.
To examine whether TA microscopy could quantify heme uptake in worms grown under varying heme concentrations,48 we cultured WT worms in mCeHR2 medium supplemented with 5, 20, 50, 80, and 110 μM hemin for 16 h and then imaged intestinal heme stores (Figure 2a). Heme accumulation in the worms corresponds to heme supplemented in the growth medium (Figure 2b). Interestingly, the curve can be fitted linearly (Figure 2b, R2 = 0.98), indicating the uptake of heme is not saturated even at 110 μM.
Figure 2.
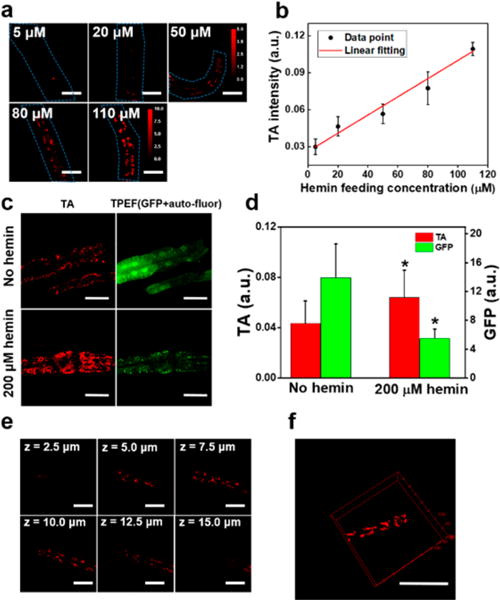
TA detection sensitivity, spatial resolution, and biocompatibility for in vivo heme imaging. (a) WT worms at L1 stage were maintained in mCeHR2 medium supplemented with 5, 20, 50, 80, and 110 μM of hemin for 16 h and then subject to TA imaging. Scale bar: 15 μm. (b) Quantification of heme level. N = 6. Linear fitting with R2 = 0.98. (c) IQ6011 worms were harvested from NGM plate and synchronized to L1, which were kept in mCeHR2 medium or mCeHR2 medium supplemented with 200 μM hemin for 11 h. Then, TA and TPEF microscopy were performed to determine heme and green fluorescence, respectively. The puncta were autofluorescent gut granules. Scale bar: 30 μm. (d) Quantification of heme level. N ∼ 8. * denotes p < 0.05. (e) TA imaging of WT C. elegans kept on NGM plate. Images at different z-depths are shown. Scale bar: 15 μm. Displaying dynamic range: 0.28–1.00. (f) 3D view of heme granules in C. elegans rendered using ImageJ. Scale bar: 30 μm. Pixel dwell time: 10 μs.
Next, we explored the accuracy of TA microscopy in monitoring heme homeostasis in C. elegans. We used IQ6011, a strain genetically engineered to express green fluorescence protein (GFP) driven by the hrg-1 promoter; intestinal GFP expression inversely correlates with heme concentrations in the growth medium.16 IQ6011 was grown either in mCeHR2 medium with or without 200 μM hemin for 11 h, and worms were imaged for heme and GFP using TA and two-photon excitation fluorescence (TPEF) microscopy, respectively. As expected, we found a negative correlation between GFP and TA intensity (Figure 2c). We also noted that in addition to the diffused GFP signal, TPEF also detected autofluorescent gut granules,49 which we filtered out. As shown in Figure 2d, when incubated with 200 μM hemin, the intestinal heme level is significantly higher, and GFP level is correspondingly down-regulated.
We noticed that planar imaging at focal plane creates a relatively large standard deviation for specimens with heterogeneity along z-direction. Three-dimensional (3D) imaging overcomes this problem by sampling frames along the z-direction. To demonstrate the 3D sectioning capability of TA microscopy for imaging heme in C. elegans, we applied a piezo scanner and sampled the worms along the axial direction with a step size of 0.5 μm. The spatial resolution along the axial direction was found to be ∼2.0 μm, and the lateral resolution was ∼0.5 μm, as determined by full-width half-maximum (Figure S4). Comparing images at different depths, we found that heme levels varied greatly, while heme levels from the planes closer to the center of the worm displayed less variation (Figure 2e, z = 7.5, 10, and 12.5 μm). These results demonstrate that TA microscopy can be adapted for the 3D imaging (Figure 2f and Supporting Video 1) and studying biological samples with axial heterogeneity.
Harnessing the high-speed capacity of TA imaging (0.5 s per frame), we characterized the dynamics of heme granules inside intestine cells of live worms. To this end, we collected WT worms at the L4 larval stage from NGM agar plates and incubated them in mCeHR2 medium supplemented with 200 μM hemin for 1 h and performed continuous TA imaging of the intestine (Supporting Video 2). Strikingly, the majority of intracellular heme granules is dynamic and mobile; one larger granule breaks down into smaller granules (Figures 3a and b). This continuous breakdown event presents a possible mechanism by which heme granules are metabolized and utilized. Collectively, these direct and noninvasive dynamic imaging provide new insights into heme storage and transport in C. elegans.
Figure 3.
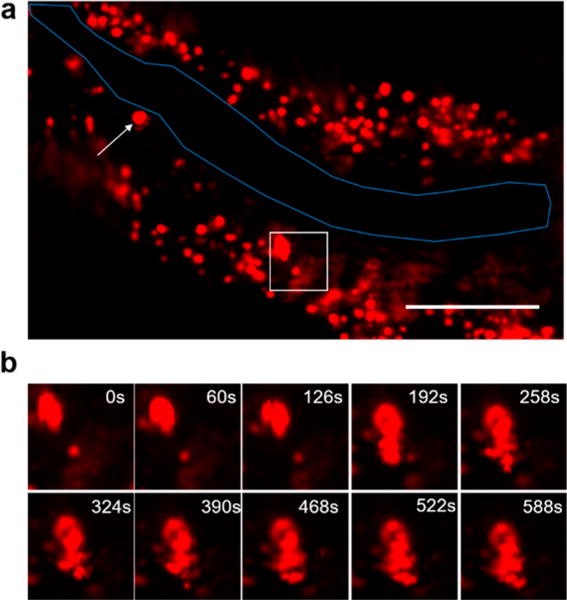
Real-time TA imaging of heme granule dynamics in live C. elegans. (a) Zero time delay TA imaging of WT worm. Overall image is shown; worm intestinal membrane is outlined in blue. Scale bar: 40 μm. (b) Field of view in the white box was cropped, and images at different time points are shown. Sample: IQ6011 strain was kept in mCeHR2 medium supplemented with 200 μM hemin for 1 h before TA imaging. Pixel dwell time: 10 μs.
We reasoned that heme could be organized within lipid droplets or enclosed by membrane structures such as lysosomal-related organelles (LRO) which contain cholesterol and lipofuscin.50–52 We used SRS microscopy (C–H vibration) to light up lipid droplets in the intestinal cells of C. elegans and imaged heme in the same field-of-view by the TA mode on the same platform. We found that heme granules and lipid droplets do not overlap (Figure 4a). We then tested whether heme granules reside in LRO which emits green autofluorescence upon excitation.50 Taking advantage of this property, we applied TPEF and TA to light up LRO and heme, respectively. Hemin-fed WT worms display solid-green fluorescence which colocalizes with heme granules (Figure 4b), indicating that heme is concentrated within LRO. To confirm this result, we used GLO-1, a lysosomal surface protein that localizes to the LRO, and GLO-1 fused GFP displays a ring-shaped morphology.53 We imaged heme-fed GLO-1::GFP transgenic worms using TPEF and TA signals. As expected, the heme granules were encircled by ring-shaped LRO membrane (Figure 4c). Collectively, these multimodal imaging data reveal that heme is stored in LROs.
Figure 4.
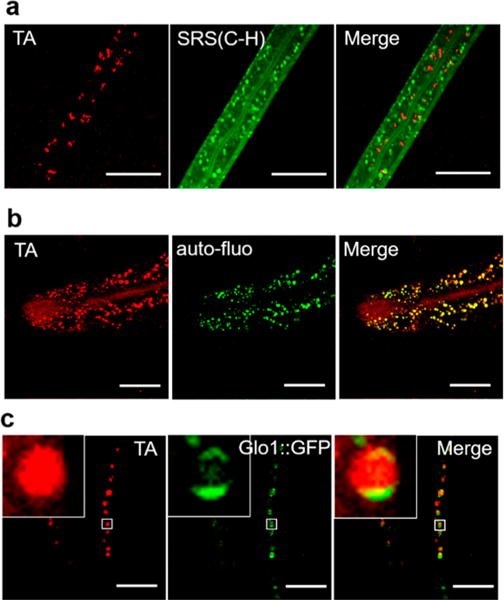
Multimodal imaging identifies subcellular heme stores in C. elegans. (a) WT worms were kept in mCeHR2 medium supplemented with 160 μM hemin for 2 days, then imaged using SRS (C–H bond) and TA microscopy. Scale bar: 20 μm. (b) WT worms were kept on NGM plates supplemented with 100 μM hemin for 24 h and then subject to TA imaging. Scale bar: 30 μm. (c) hJiS strain (Glo-1::GFP) was kept in mCeHR2 medium supplemented with 200 μM hemin for 16 h and then subject to TA and TPEF imaging for heme and green fluorescence (GFP + autofluorescence). Scale bar: 20 μm. Pixel dwell time: 10 μs.
Heme import into the worm intestine is mediated by the apical transporter HRG-4, while heme export from the intestine is mediated by the basolateral transporter MRP-5.17,19 To genetically validate heme imaging by TA microscopy, we used hrg-4 and mrp-5 mutant worms. hrg-4 depletion by RNAi resulted in significantly lower amounts of heme accumulation (Figures 5a and b), while mrp-5 mutants (ok2067) accumulated greater amounts of heme in the intestine (Figures 5c and d). In addition to storage in intestine, it has been reported that maternal heme is transported by HRG-3 to embryos.48 However, heme has not been directly visualized in either oocytes or embryos. To image heme in these tissues, we cultured WT worms in mCeHR2 medium supplemented with 20 μM heme and imaged oocytes and developing embryos by TA imaging. We observed trace amounts of heme in fertilized eggs (Figure 5e), and a moderate level in oocytes (Figure 5f). These studies further support the imaging capabilities of TA microscopy in imaging heme and screening for regulators of heme homeostasis.
Figure 5.
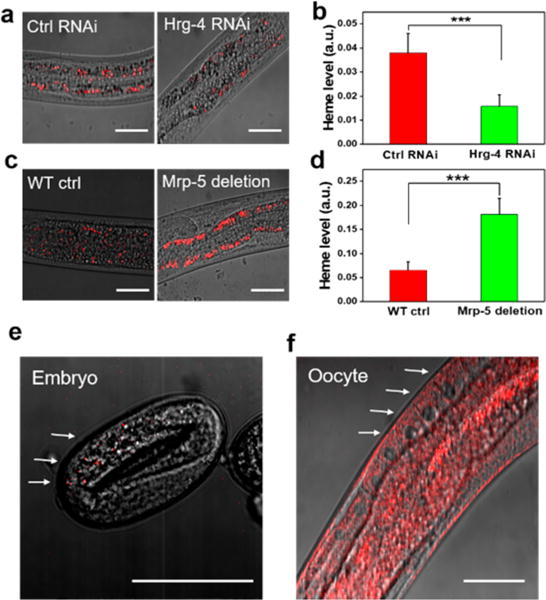
TA mapping of tissue distribution of heme inside C. elegans. (a) WT worms at L1 stage were kept on RNAi control or hrg-4 RNAi plates. L4 or adults were subject to TA imaging. Scale bar: 25 μm. (b) Quantification of heme level. *** denotes p < 0.001. N = 13 per group. (c) WT and mrp-5 deletion strains were kept on NGM plates supplemented with 200 μM hemin. L4 or adults were subject to TA imaging. Scale bar: 40 μm. (d) Quantification of heme level. *** denotes p < 0.001, N = 12 per group. (e and f) Developing egg and oocytes of WT worm were imaged using TA microscopy. The overlay of TA and transmission images are shown. Scale bar: 40 μm. In a–d, pixel dwell time: 10 μs; in e and f, pixel dwell time: 50 μs.
To visualize heme at the cellular level, we used HEK293A cells grown with or without supplementation of 100 μM hemin (Figure 6a). Under basal conditions, heme signals were observed as perinuclear, which intensified in the presence of 100 μM heme as vesicular puncta (Figure 6a). The decay curve of heme granules in mammalian cells fed with 100 μM heme resembled that of hemin solution rather than hemin powder (Figure 6b), suggesting that the puncta formed in the presence of heme are likely to be soluble heme confined within membrane-enclosed structures.
Figure 6.
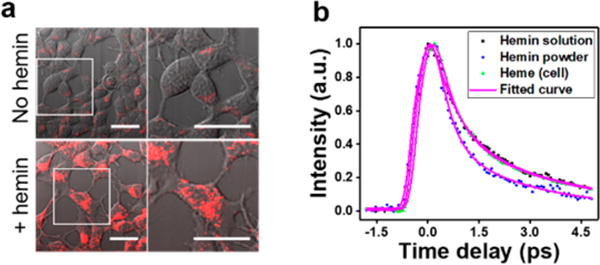
TA imaging of endogenous heme in live mammalian cells. (a) HEK293A cells were kept in DMEM medium or DMEM medium supplemented with 100 μM hemin for 2 days and subject to TA (red) and transmission (gray) imaging. Overlay of TA and transmission images are shown. Right panel: magnification view of the area in white box. Scale bar: 20 μm. (b) Time-resolved curves of hemin solution, hemin powder, and heme cluster from HEK293A cells treated with 100 μM hemin; about 200 pixels were selected and averaged. Pixel dwell time: 100 μs.
■ DISCUSSION
In this study, we demonstrated TA microscopy for imaging heme storage, distribution, and dynamics in living biological models. The on-sample pump and probe laser powers were 6 and 17 mW, respectively. We did not observe photobleaching of samples even after continuous imaging for over 10 min. The sensitivity of our TA microscope at this laser power level for hemin solution is 9.2 μM. Although the labile heme level in worms is ∼1.0 μM 17, heme also exists in a protein-bound state. We reason that the observed diffused heme in worms cultured under normal maintenance condition (Figure S3) consists of labile and protein chelated heme.54
We note that picosecond SRS microscopy has been used to image hemoglobin in red blood cells at video rate speed.46 However, the detection sensitivity of SRS microscopy for endogenous molecules is at millimolar level, which is not sufficient for imaging heme uptake and transport in biological systems. Here, we show that TA microscopy has a detection sensitivity of 9 μM based on the electronic absorption at 520 nm. Such sensitivity allowed real time imaging of heme dynamics in C. elegans.
We used the TA signal at zero time delay to quantify the heme level. This method lights up total heme in living cells, including labile and protein-associated heme. Time-resolved TA microscopy can differentiate different states of heme based on the TA signal decay rate. The challenge facing time-resolved TA imaging is the speed. Using an optical delay line, a frame-by-frame time-resolved TA image with 200 × 200 pixels takes about 1 min for 60 frames, which is not fast enough to truly capture dynamics. If a tuned amplifier array is used,54 a parallel excitation and sampling scheme could be developed to increase the imaging speed by 32 times. Another approach would be to perform volumetric pump–probe imaging of heme distribution in living organisms by using Bessel beam geometry.55 These efforts will render TA microscopy the potential to differentiate different states of heme such as oxidized and reduced heme, modified heme, and hemo-protein dynamics in living organisms.
■ CONCLUSION
We validated TA microscopy for imaging heme storage, distribution, and dynamics in living biological models. Specifically, we demonstrated 9 μM heme imaging sensitivity and established that the TA microscope was capable of detecting heme inside organs of C. elegans. We demonstrated the ability of TA microscopy to directly visualize heme dynamics within granules in C. elegans as well as heme distribution in mammalian cell lines. Integration of TA and other imaging modalities allowed label-free mapping of heme distribution to study subcellular heme localization. Our study establishes a new approach for studying heme storage, transport, and metabolism in living organisms.
Supplementary Material
Acknowledgments
This work was supported by Grant NIH R01 GM114853 to J.X.C. The authors thank Dr. Chien-Sheng Liao and Dr. Bin Liu for technical support.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.7b05046.
Rotation view of heme granules in C. elegans intestine (AVI)
Real-time imaging of heme dynamics (AVI)
Diagram of TA microscope, TA images showing that heme exists as diffused form in pharynx and hypodermis, images showing that TA microscopy has enough sensitivity to image heme in standard C. elegans culture condition, and lateral and axial intensity profiles of heme granules (PDF)
ORCID
Ji-Xin Cheng: 0000-0002-5607-6683
Author Contributions
J.X.C. and I.H. guided the project. A.J.C. performed TA and TPEF imaging of worms and TA imaging of HEK293A cells. X.Y. constructed IQ6011 and hrg-4 and mrp-5 mutants, and provided training and material in C. elegans handling. A.J.C., J.L., and P.D. performed data analysis. A.J.C. wrote the manuscript. All authors read and revised the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Ponka P. Am J Med Sci. 1999;318:241–256. doi: 10.1097/00000441-199910000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Tsiftsoglou AS, Tsamadou AI, Papadopoulou LC. Pharmacol Ther. 2006;111:327–345. doi: 10.1016/j.pharmthera.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 3.Yin L, Wu N, Curtin JC, Qatanani M, Szwergold NR, Reid RA, Waitt GM, Parks DJ, Pearce KH, Wisely GB, Lazar MA. Science. 2007;318:1786–1789. doi: 10.1126/science.1150179. [DOI] [PubMed] [Google Scholar]
- 4.Faller M, Matsunaga M, Yin S, Loo JA, Guo F. Nat Struct Mol Biol. 2007;14:23–29. doi: 10.1038/nsmb1182. [DOI] [PubMed] [Google Scholar]
- 5.Hamza I, Dailey HA. Biochim Biophys Acta, Mol Cell Res. 2012;1823:1617–1632. doi: 10.1016/j.bbamcr.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Severance S, Hamza I. Chem Rev. 2009;109:4596–4616. doi: 10.1021/cr9001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balla G, Vercellotti GM, Mullereberhard U, Eaton J, Jacob HS. Lab Invest. 1991;64:648–655. [PubMed] [Google Scholar]
- 8.Wagener FADTG, Eggert A, Boerman OC, Oyen WJG, Verhofstad A, Abraham NG, Adema G, van Kooyk Y, de Witte T, Figdor CG. Blood. 2001;98:1802–1811. doi: 10.1182/blood.v98.6.1802. [DOI] [PubMed] [Google Scholar]
- 9.Jeney V, Balla J, Yachie A, Varga Z, Vercellotti GM, Eaton JW, Balla G. Blood. 2002;100:879–887. doi: 10.1182/blood.v100.3.879. [DOI] [PubMed] [Google Scholar]
- 10.Tang N, Chen LQ, Zhuang H. Food Funct. 2014;5:390–399. doi: 10.1039/c3fo60292c. [DOI] [PubMed] [Google Scholar]
- 11.Fujiwara T, Harigae H. BioMed Res Int. 2015;2015:278536. doi: 10.1155/2015/278536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bissell DM, Anderson KE, Bonkovsky HL. N Engl J Med. 2017;377:862–872. doi: 10.1056/NEJMra1608634. [DOI] [PubMed] [Google Scholar]
- 13.Rao AU, Carta LK, Lesuisse E, Hamza I. Proc Natl Acad Sci U S A. 2005;102:4270–4275. doi: 10.1073/pnas.0500877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bethony J, Brooker S, Albonico M, Geiger SM, Loukas A, Diemert D, Hotez PJ. Lancet. 2006;367:1521–1532. doi: 10.1016/S0140-6736(06)68653-4. [DOI] [PubMed] [Google Scholar]
- 15.Albonico M, Engels D, Savioli L. Int J Parasitol. 2004;34:1205–1210. doi: 10.1016/j.ijpara.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 16.Rajagopal A, Rao AU, Amigo J, Tian M, Upadhyay SK, Hall C, Uhm S, Mathew MK, Fleming MD, Paw BH, Krause M, Hamza I. Nature. 2008;453:1127–1131. doi: 10.1038/nature06934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan XJ, Protchenko O, Philpott CC, Hamza I. J Biol Chem. 2012;287:4914–4924. doi: 10.1074/jbc.M111.326785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.White C, Yuan XJ, Schmidt PJ, Bresciani E, Samuel TK, Campagna D, Hall C, Bishop K, Calicchio ML, Lapierre A, Ward DM, Liu P, Fleming MD, Hamza I. Cell Metab. 2013;17:261–270. doi: 10.1016/j.cmet.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korolnek T, Zhang JB, Beardsley S, Scheffer GL, Hamza I. Cell Metab. 2014;19:1008–1019. doi: 10.1016/j.cmet.2014.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lutton JD, Abraham NG, Drummond GS, Levere RD, Kappas A. Proc Natl Acad Sci U S A. 1997;94:1432–1436. doi: 10.1073/pnas.94.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lutton JD, Jiang S, Drummond GS, Abraham NG, Kappas A. Pharmacology. 1999;58:44–50. doi: 10.1159/000028267. [DOI] [PubMed] [Google Scholar]
- 22.Song YQ, Yang MY, Wegner SV, Zhao JY, Zhu RF, Wu Y, He C, Chen PR. ACS Chem Biol. 2015;10:1610–1615. doi: 10.1021/cb5009734. [DOI] [PubMed] [Google Scholar]
- 23.Hanna DA, Harvey RM, Martinez-Guzman O, Yuan XJ, Chandrasekharan B, Raju G, Outten FW, Hamza I, Reddi AR. Proc Natl Acad Sci U S A. 2016;113:7539–7544. doi: 10.1073/pnas.1523802113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yuan XJ, Rietzschel N, Kwon H, Nuno ABW, Hanna DA, Phillips JD, Raven EL, Reddi AR, Hamza I. Proc Natl Acad Sci U S A. 2016;113:E5144–E5152. doi: 10.1073/pnas.1609865113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abshire JR, Rowlands CJ, Ganesan SM, So PTC, Niles JC. Proc Natl Acad Sci U S A. 2017;114:E2068–E2076. doi: 10.1073/pnas.1615195114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Min W, Freudiger CW, Lu SJ, Xie XS. Annu Rev Phys Chem. 2011;62:507–530. doi: 10.1146/annurev.physchem.012809.103512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei L, Min W. Anal Bioanal Chem. 2012;403:2197–2202. doi: 10.1007/s00216-012-5890-1. [DOI] [PubMed] [Google Scholar]
- 28.Fischer MC, Wilson JW, Robles FE, Warren WS. Rev Sci Instrum. 2016;87:031101. doi: 10.1063/1.4943211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong PT, Cheng JX. Spectroscopy-Us. 2017;32:24–36. [Google Scholar]
- 30.Dong CY, So PTC, French T, Gratton E. Biophys J. 1995;69:2234–2242. doi: 10.1016/S0006-3495(95)80148-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Dijk MA, Lippitz M, Orrit M. Phys Rev Lett. 2005;95:267406. doi: 10.1103/PhysRevLett.95.267406. [DOI] [PubMed] [Google Scholar]
- 32.Muskens OL, Del Fatti N, Vallee F. Nano Lett. 2006;6:552–556. doi: 10.1021/nl0524086. [DOI] [PubMed] [Google Scholar]
- 33.Staleva H, Hartland G. J Phys Chem C. 2008;112:7535–7539. [Google Scholar]
- 34.Muskens OL, Rivas JG, Algra RE, Bakkers EPAM, Lagendijk A. Nano Lett. 2008;8:2638–2642. doi: 10.1021/nl0808076. [DOI] [PubMed] [Google Scholar]
- 35.Gao B, Hartland GV, Huang LB. ACS Nano. 2012;6:5083–5090. doi: 10.1021/nn300753a. [DOI] [PubMed] [Google Scholar]
- 36.Gao B, Hartland GV, Huang LB. J Phys Chem Lett. 2013;4:3050–3055. [Google Scholar]
- 37.Cui Q, Ceballos F, Kumar N, Zhao H. ACS Nano. 2014;8:2970–2976. doi: 10.1021/nn500277y. [DOI] [PubMed] [Google Scholar]
- 38.Jung Y, Slipchenko MN, Liu CH, Ribbe AE, Zhong ZH, Yang C, Cheng JX. Phys Rev Lett. 2010;105:217401. doi: 10.1103/PhysRevLett.105.217401. [DOI] [PubMed] [Google Scholar]
- 39.Li JJ, Zhang WX, Chung TF, Slipchenko MN, Chen YP, Cheng JX, Yang C. Sci Rep. 2015;5:12394. doi: 10.1038/srep12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fu D, Ye T, Matthews TE, Yurtsever G, Warren WS. J Biomed Opt. 2007;12:054004. doi: 10.1117/1.2780173. [DOI] [PubMed] [Google Scholar]
- 41.Matthews TE, Piletic IR, Selim MA, Simpson MJ, Warren WS. Sci Transl Med. 2011;3:71ra15. doi: 10.1126/scitranslmed.3001604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Min W, Lu SJ, Chong SS, Roy R, Holtom GR, Xie XS. Nature. 2009;461:1105–1109. doi: 10.1038/nature08438. [DOI] [PubMed] [Google Scholar]
- 43.Domingue SR, Bartels RA, Chicco AJ, Wilson JW. Biomed Opt Express. 2017;8:2807–2821. doi: 10.1364/BOE.8.002807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Girard LR, Fiedler TJ, Harris TW, Carvalho F, Antoshechkin I, Han M, Sternberg PW, Stein LD, Chalfie M. Nucleic Acids Res. 2007;35:D472–D475. doi: 10.1093/nar/gkl894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tong L, Liu YX, Dolash BD, Jung Y, Slipchenko MN, Bergstrom DE, Cheng JX. Nat Nanotechnol. 2012;7:56–61. doi: 10.1038/nnano.2011.210. [DOI] [PubMed] [Google Scholar]
- 46.Saar BG, Freudiger CW, Reichman J, Stanley CM, Holtom GR, Xie XS. Science. 2010;330:1368–1370. doi: 10.1126/science.1197236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Webster GT, McNaughton D, Wood BR. J Phys Chem B. 2009;113:6910–6916. doi: 10.1021/jp811028a. [DOI] [PubMed] [Google Scholar]
- 48.Chen CY, Samuel TK, Sinclair J, Dailey HA, Hamza I. Cell. 2011;145:720–731. doi: 10.1016/j.cell.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guitart J, Martinez-Escala ME, Subtil A, Duvic M, Pulitzer MP, Olsen EA, Kim E, Rook AH, Samimi SS, Wood GS, Girardi M, Junkins-Hopkins J, Ivan DS, Selim MA, Sable KA, Virmani P, Pincus LB, Tetzlaff MT, Kim J, Kim YH. Mod Pathol. 2017;30:761–772. doi: 10.1038/modpathol.2016.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roh HC, Collier S, Guthrie J, Robertson JD, Kornfeld K. Cell Metab. 2012;15:88–99. doi: 10.1016/j.cmet.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reddi AR, Hamza I. Acc Chem Res. 2016;49:1104–1110. doi: 10.1021/acs.accounts.5b00553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang P, Liu B, Zhang DL, Belew MY, Tissenbaum HA, Cheng JX. Angew Chem, Int Ed. 2014;53:11787–11792. doi: 10.1002/anie.201406029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang SBO, Box AC, Xu NY, Le Men J, Yu JY, Guo FL, Trimble R, Mak HY. Proc Natl Acad Sci U S A. 2010;107:4640–4645. doi: 10.1073/pnas.0912308107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang C, Huang KC, Rajwa B, Li JJ, Yang SQ, Lin HN, Liao CS, Eakins G, Kuang SH, Patsekin V, Robinson JP, Cheng JX. Optica. 2017;4:103–109. doi: 10.1364/optica.4.000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen X, Zhang C, Lin P, Huang KC, Liang J, Tian J, Cheng JX. Nat Commun. 2017;8:15117. doi: 10.1038/ncomms15117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.