

Abstract
Extracellular vesicles (EVs) are physiological vesicles secreted from most of eukaryotes and contain cargos of their cell of origin. EVs, and particularly a subset of EV known as exosomes, are emerging as key mediators of cell to cell communication and waste management for cells both during normal organismal function and in disease. In this review, we investigate the rapidly growing field of exosome biology, their biogenesis, cargo loading, and uptake by other cells. We particularly consider the role of exosomes in Alzheimer’s disease, both as a pathogenic agent and as a disease biomarker. We also explore the emerging role of exosomes in chronic traumatic encephalopathy. Finally, we highlight open questions in these fields and the possible use of exosomes as therapeutic targets and agents.
Keywords: Alzheimer’s disease, amyloid-b peptide, chronic traumatic encephalopathy, exosomes, extracellular vesicles, microvesicles, microglia, microtubule-associated protein tau
Introduction
Extracellular vesicles
Extracellular vesicles (EVs) are membranous vesicles enclosed by a lipid bilayer and containing the cytosol of their cell of origin (reviewed in Colombo et al. 2014). The creation of EVs is an evolutionally conserved process observed in both prokaryotes and a wide variety of eukaryotes (reviewed in Yáñez-Mó et al. 2015). EVs were first observed as pro-coagulant particles in plasma, and were later termed “platelet dust” (Chargaff and West 1946; Wolf 1967). Since these early discoveries, EVs have been found in almost every cell type and bodily fluid. Our current understanding of these vesicles groups them into three major types: exosomes, microvesicles (MVs), and apoptotic bodies.
Exosomes
Exosomes are a specific subset of EV approximately 30–150 nm in size (Cobb and Gendelman 2016) that were first described in the 1980s (Trams et al. 1981; Pan and Johnstone 1983; Harding et al. 1983). They are released by almost every known cell type in the body, including cells of the central nervous system (CNS) (Perez-Gonzalez et al. 2012) such as oligodendrocytes (Krämer-Albers et al. 2007), astrocytes (Fauré et al. 2006; Taylor et al. 2007; Chiarini et al. 2017), neurons (Fauré et al. 2006), and microglia (Potolicchio et al. 2005) (reviewed in Simpson et al. 2014). Exosomes are also found in most bodily fluids, including cerebrospinal fluid (Vella et al. 2008), the blood (Caby et al. 2005), breast milk (Admyre et al. 2007), and urine (Pisitkun et al. 2004) (reviewed in Simpson et al. 2014).
Exosomes are formed when the plasma membrane undergoes invagination, creating an endosome. The endosome then undergoes internal budding, resulting in the formation of multivesicular bodies (MVBs) or late endosomes containing intraluminal vesicles (ILVs). The MVB can then fuse with the lysosome, degrading the ILVs and their contents, or instead can fuse with the plasma membrane (Raposo et al. 1996; Buschow et al. 2009). During this second type of fusion, the MVB releases the ILVs into the extracellular space, at which point they are termed exosomes (Fig. 1). Once in the extracellular space, exosomes may circulate in bodily fluids or be taken up by other cells. These two possibilities reflect the two major functions of exosomes: as a means of waste disposal for cells, or as a form of long distance cell to cell communication.
Fig 1. Schema of exosome and microvesicle synthesis and cargo molecules.
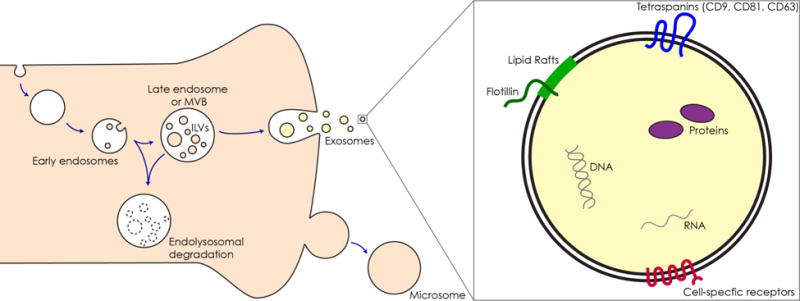
Left Schematic of exosome biogenesis. First, the plasma membrane invaginates creating the early endosome. The early endosome then buds inwardly for a second time creating intraluminal vesicles (ILVs) and becomes the late endosome or multivesicular body (MVB). The MVB can then fuse with the lysosome and ILVs along with their contents will be degraded (bottom middle). Alternately, the MVB can fuse with the plasma membrane, releasing ILVs into the extracellular space, at which point they are considered exosomes. Ectosomes or microvesicles are created by the outward budding of the plasma membrane (bottom right). Right Schema of a single exosome. Exosomes are recognized by their expression of tetraspanins (blue) such as CD9, CD81, and CD63 as well as lipid rafts and Flotilin 1 protein (green). Depending on their cell of origin, exosomes may possess cell-specific receptors (red). Internally, exosomes contain DNA (grey), RNA (grey strand), and proteins (purple) specially packaged during their assembly.
The other major type of extracellular vesicles involved in intercellular communication are the MVs, which are also called ectosomes. Biologically, MVs are distinguished from exosomes by the fact that they are generated by outward, rather than inward, budding of the plasma membrane. These other types of EV come in many sizes, but are generally, although not always, larger than exosomes (typical size 150 - 1000 nm) (reviewed in Colombo et al. 2014). Exosomes and microvesicles share many proteins markers, making it difficult to distinguish between them in the extracellular space or after vesicles have been purified (Kowal et al. 2016). Additionally, both types of EV use the Endosomal Sorting Complex Required for Transport (ESCRT) machinery in their formation (reviewed in Abels and Breakefield 2016). Of note is the fact that the ESCRT-0 proteins Hepatocyte growth factor-Regulated tyrosine kinase (HRS) and Signal Transducing Adapter Molecule 1 (STAM) are only involved in the initial phase of exosome biogenesis (reviewed in Colombo et al. 2014). Exosomes may also be synthesized via non-ESCRT dependent mechanisms, for example, ceramide-initiated biogenesis (Laulagnier et al. 2004; Trajkovic et al. 2008; Stuffers et al. 2009; Carayon et al. 2011; van Niel et al. 2011; Ghossoub et al. 2014). See Table 1 for an overview of differences between these types of EV. Due to their differing biological origins, exosomes and microvesicles may have divergent roles in the body; until this point is fully resolved, it is important to differentiate between these two classes of EV.
Table 1.
Differences observed between exosomes and microvesicles.
| Type of EV | Size | Location of Biosynthesis | Known unique markers | Biosynthesis mechanisms |
|---|---|---|---|---|
| Exosome | 30–150 nm | Endosomes | ALIX, CD9, CD81, CD 63, TSG101, Syntenin-1 | ESCRT, nSMase2, phospholipase D2, CD63 |
| Microvesicle | 100–1000 nm | Plasma membrane | Glycoprotein 1b, external phosphatidylserine, HSP90B1 | ESCRT, actin cytoskeleton depolymerization |
Apoptotic bodies are generally 800 nm – 5 μm in size, and, as the name suggests, are shed by cells undergoing apoptosis (reviewed in Budnik et al. 2016). They are created by the same mechanism as MVs (reviewed in Huang-Doran et al. 2016), and due to their larger size are not often conflated with exosomes. Furthermore, they are not involved in cell-to-cell communication, but instead are taken up by phagocytic cells and digested.
Beyond size, exosomes are distinguished from other EVs by their ubiquitous expression of the tetraspanin protein CD9. CD81 and CD63 are also common but less ubiquitously expressed in exosomes (Kowal et al. 2016). Exosomes are often characterized by their high concentration of lipid raft components, such as ceramide and sphingomyelin (de Gassart et al. 2003; Record et al. 2014). Exosomes may also carry specific cell surface proteins that reflect their cell of origin, as they are essentially composed of plasma membrane that has inwardly budded multiple times (Raposo et al. 1996; Zitvogel et al. 1998) (reviewed in Cobb and Gendelman 2016). Such cell surface proteins are thought to mediate exosomal uptake by recipient cells (Morelli et al. 2004; Nazarenko et al. 2010; Atay et al. 2011; Escrevente et al. 2011; Näslund et al. 2014). Furthermore, the cell-type specific proteins found on the surface of exosomes may play a key role in specifying what types of cell can act as recipients for a given exosome (Fitzner et al. 2011; Alvarez-Erviti et al. 2011; Hood et al. 2011; Zech et al. 2012; Chivet et al. 2014) (reviewed in Milane et al. 2015)
During the creation of ILVs within the late endosome, proteins (reviewed in Théry et al. 2002), DNA (Balaj et al. 2011; Kahlert et al. 2014), RNA (Valadi et al. 2007; Cheng et al. 2014) (reviewed in Zhang et al. 2015), and lipids (reviewed in Théry et al. 2002) are sorted into the newly forming vesicles (reviewed in Kalluri 2016; Abels and Breakefield 2016). The protein and RNA cargo of exosomes seems to be specifically curated, as it can differ from the parent cell (Valadi et al. 2007; Montecalvo et al. 2012 for RNA, Fauré et al. 2006; Saman et al. 2014; Munich et al. 2014 for protein, Nazarenko et al. 2010 for both). Additionally, the loaded cargo may change depending on the state of the parent cell (Eldh et al. 2010; Carayon et al. 2011; de Jong et al. 2012).
There are three major proposed routes for sorting protein into exosomes. The first pathway uses cytosolic machinery such as ALG-2-Interacting Protein X (ALIX) and ESCRT proteins, as well as Lysobisphosphatidic Acid (LBPA) (Matsuo et al. 2004; Géminard et al. 2004; Trajkovic et al. 2008; van Niel et al. 2011). The ESCRT pathway recruits mono-ubiquitinated proteins in ILVs for endo-lysosomal degradation (Raiborg et al. 2006; Carayon et al. 2011). The second pathway is based on sphingolipid-enriched membrane domains segregating membrane-embedded proteins, with certain proteins co-segregating with flotilins and tetraspanins in lipid rafts Wubbolts et al. 2003; de Gassart et al. 2003; Géminard et al. 2004; Trajkovic et al. 2008; Carayon et al. 2011; Perez-Hernandez et al. 2013). The third pathway for cargo loading is initiated on the luminal side of the endosomal membrane by factors such as lectins. This pathway mainly recruits glycosylated proteins to the exosomes (Barres et al. 2010; Carayon et al. 2011). In contrast, the mechanisms for loading specified DNA and RNA cargo into exosomes are only beginning to be elucidated (Villarroya-Beltri et al. 2013). See Table 2 for an overview of loading mechanisms (Wubbolts et al. 2003; de Gassart et al. 2003; Géminard et al. 2004; Trajkovic et al. 2008; Carayon et al. 2011; Perez-Hernandez et al. 2013). The third pathway for cargo loading is initiated on the luminal side of the endosomal membrane by factors such as lectins. This pathway mainly recruits glycosylated proteins to the exosomes (Barres et al. 2010; Carayon et al. 2011). In contrast, the mechanisms for loading specified DNA and RNA cargo into exosomes are only beginning to be elucidated (Villarroya-Beltri et al. 2013). See Table 2 for an overview of loading mechanisms.
Table 2.
Summary of currently known mechanisms for cargo loading into Exosomes.
| Cargo Loading Mechanisms | Involved Molecules | Type of Cargo Loaded | Shared with Microvesicles? |
|---|---|---|---|
| Cytosolic | ALIX, ESCRTs, LBPA | Proteins, ubiquitinated proteins with ESCRTs | Yes |
| Lipid Raft Microdomains | Flotillins, Tetraspanins | Membrane embedded and transmembrane proteins | Yes |
| Lectins | Galectin-5 | Glycosylated proteins | Unknown |
| Sumoylated hnRNPA2B1 | hnRNPA2B1 | microRNA | Unknown |
Exosomes may be taken up by recipient cells via membrane fusion (Parolini et al. 2009; Montecalvo et al. 2012) or endocytosis (Fitzner et al. 2011; Escrevente et al. 2011; Montecalvo et al. 2012) (reviewed in Mulcahy et al. 2014). Once taken up, exosomes can deliver their cargo to a recipient cell, thereby conveying a complex message and altering the physiological state of their host (Admyre et al. 2007; Nazarenko et al. 2010; Zech et al. 2012; Frohlich et al. 2014). Exosomes have therefore been shown to be important for cell-cell communication both during normal functions of the body (Al-Dossary et al. 2015) and during disease progression; they are especially important factors in cancer metastasis (reviewed in Milane et al. 2015; Javeed and Mukhopadhyay 2016).
In the nervous system, exosomes play roles in the maintenance of myelin (Krämer-Albers et al. 2007), intercellular communication (Potolicchio et al. 2005; Frühbeis et al. 2013; Frohlich et al. 2014), and elimination of waste (Fitzner et al. 2011). Brain tumors often produce abundant exosomes that contain tumorigenic factors that they can communicate to normal cells and may influence immune response (Graner et al. 2009). Exosomes in the brain also play roles in neurodegenerative dis-eases such as Parkinson’s (reviewed in Russo et al. 2012), Prion (Vella et al. 2008), as well as Alzheimer’s disease and Chronic Traumatic Encephalopathy (Reviewed herein). Overall, exosomes are becoming recognized as intercellular messengers able to deliver selected cargo to distinct downstream cell populations, sometimes over long distances (Hood et al. 2011). Their role in promoting or preventing pathology is just beginning to be understood.
Alzheimer’s Disease and EVs
Alzheimer’s disease is the most common form of dementia, with approximately 5.5 million Americans currently diagnosed with the disease, and an expected 13.8 million to be diagnosed with it by 2050 (Alzheimer’s Association 2017). Alzheimer’s disease (AD) is characterized by presence of amyloid plaques (sometimes called senile plaques) formed by misfolded and aggregated Aβ as well as and tau-based neurofibrillary tangles (NFTs) (Goedert et al. 1988) in the brain resulting in neuronal dysfunction and death. As AD progresses, tau becomes hyper-phosphorylated, causing it to misfold and aggregate into NFTs and neuropil threads (Hu et al. 2016) (reviewed in Geschwind 2003).
Amyloid plaque deposits are known to occur in the neocortex before appearing in the subcortex in AD, while NFT deposits appear in a characteristic and predictable spatiotemporal manner first described by Braak & Braak (Braak and Braak 1991). Braak and Braak identified six stages of AD based on tau histology. The first two are characterized by tau deposits in the transentorhinal region and then in the entorhinal cortex (EC). Therefore, these initial phases are known as the transentorhinal stages. The CA1 region of the hippocampus may also be affected at this time (Braak and Braak 1991). Later studies determined that the locus coeruleus is also one of the earliest affect areas of the brain, exhibiting pathology before the EC (Grudzien et al. 2007). During these first stages, patients are largely cognitively normal and so may be referred to as having prodromal Alzheimer’s Disease (pAD).
In stages III and IV, NFTs are more densely present in the pre-alpha region of the EC and in the CA1 region. During this time, the subiculum begins to show NFTs as well. In stage IV, the CA4 region, amygdala, claustrum, putamen and thalamic nucleus are affected. These stages are called limbic stages as they are affecting the limbic regions of the brain. Clinically, such patients appear to have mild cognitive impairment (MCI) (reviewed in Serrano-Pozo et al. 2011) but are able to carry out routine tasks.
Patients are diagnosed with stage V or VI AD when there is pathological tau in almost all components of the hippocampus. These patients also exhibit widespread neuronal loss. During stage V, the isocortex begins to exhibit NFTs. In stage VI all of the changes seen in stage V are present but are more severe in terms of their pathology, with the major difference being the involvement of the fascia dentata. These stages are considered moderate to severe forms of AD (reviewed in Serrano-Pozo et al. 2011). These phases are also known as the isocortical stages, as tau is now being deposited in the cortical regions. See Fig. 2 for a visual summary of tau deposition across the Braak & Braak stages.
Fig. 2. Schema for the staging of Alzheimer’s disease and Chronic Traumatic Encephalopathy based on tauopathy.
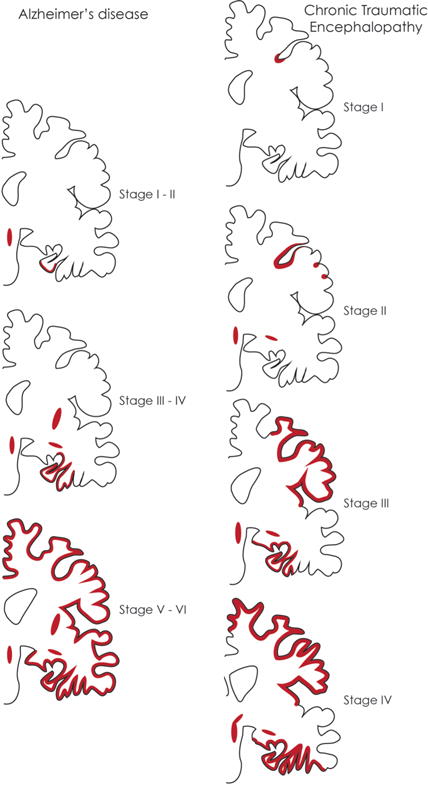
Left Schema of tau deposition (red) in Alzheimer’s disease as based on the 6 Braak & Braak stages. Top Left In stages I-II of AD, tau deposits are seen in the transentorhinal and entorhinal cortices (EC), as well as in the locus coeruleus (LC). Middle Left In AD stages III-IV, tau is additionally found in the CA1 region, subiculum, amygdala, and putamen, and stains more intensely in the EC. Bottom Left In stages V-VI, tau deposits are found throughout the hippocampus and cortex. The brain has also undergone shrinkage at this point. Right The four stages of Chronic Traumatic Encephalopathy in coronal view. Top Right CTE stage I is characterized by tau deposits in the depths cortical sulci. Middle Top Right In CTE stage II, tau has spread from the depths of the sulci to areas closer to the brain surface as well as other sulcal depths, and have begun to appear in the LC. Middle Bottom Right In CTE stage III, the brain begins to exhibit atrophy and ventricular enlargement. NFTs are now present in the hippocampus, EC, and amygdala, as well as being more widespread in the cortex. Bottom Right Stage VI of CTE is characterized by gross brain weight loss and septal defects, and widespread presence of pathological tau.
The mechanism by which misfolded protein spreads in AD and other neurodegenerative diseases is thought to occur in the manner of a prion disease, where native protein monomers become misfolded, and then act as templates for the misfolding and eventual aggregation of other monomers (Frost et al. 2009; Guo and Lee 2011; Jucker and Walker 2011) (reviewed in Guo and Lee 2014) (Fig. 3). Interestingly, oligomeric, rather than fully fibrilized forms of either Aβ or tau may be the toxic component in AD (Iba et al. 2013; Iba et al. 2015; Hu et al. 2016) (reviewed in Guo and Lee 2014). In either case, the deposition of NFTs is the best clinically correlated marker of AD progression (Nelson et al. 2012; Schöll et al. 2016). Aβ deposition does not necessarily correlate with AD stage (Arriagada et al. 1992; Jack et al. 2009). Early onset and familial forms of AD have been linked to mutations in the gene encoding amyloid precursor protein (APP) as well as in those genes that process APP into Aβ1-42, the pathological form of Aβ linked to AD (Giri et al. 2017). Intriguingly, no link has been found between AD and mutations in tau, though mutations in tau are linked to other forms of dementia and neurodegeneration (reviewed in Ballatore et al. 2007; Iqbal et al. 2015; Arendt et al. 2016).
Fig. 3. Schema of mechanisms for the spread of pathological protein seeding in the brain via direct and indirect routes.
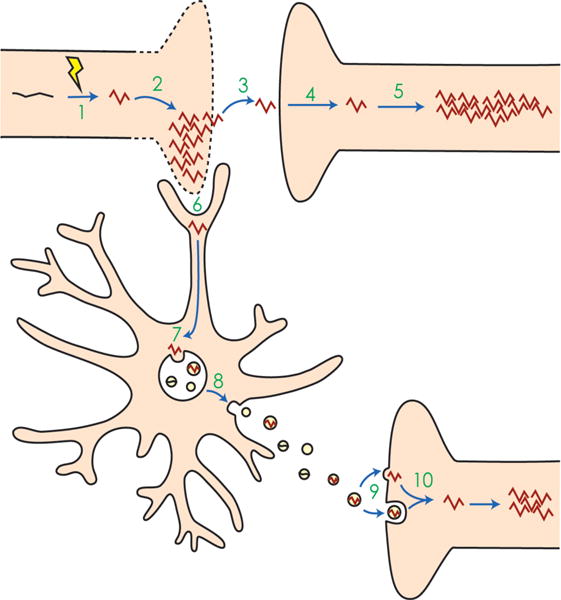
1 An event occurs that alters normal protein monomers, such as tau (black) into pathological misfolded proteins (red). In the case of tau, this event may be hyperphosphorylation. 2 The misfolded protein templates other monomers to misfold. Together, these misfolded proteins aggregate, eventually leading to cell death. 3 The misfolded protein escapes the dying cell and 4 is taken up by a nearby recipient cell. 5 In the recipient cell, the misfolded protein again templates other proteins to misfold and aggregate. 6 Alternatively, microglia phagocytose the cytopathic cells, thereby taking up the misfolded protein. 7 Misfolded proteins may be shuttled into endosomes and processed through endolysosomal degradation or incorporated into the multivesicular bodies (MVBs), where intraluminal vesicles package misfolded proteins and then 8 are released into the extracellular space as exosomes. 9 The exosomes may then be taken up by recipient cells via membrane fusion, directly releasing their cargo, or may be taken up by endocytosis. 10 The misfolded protein in the endocytosed exosome reaches the cytoplasm and templates and aggregates in the recipient cell. While in neuro-degenerative diseases such as Alzheimer’s and CTE this type of seeding is thought to occur across synapses, exosomes can also travel laterally or across long distances to be taken up by distal neurons or even other cell types, and cause pathological aggregation.
Although the propagation of tau through the brain during AD progression can largely be explained by trans-synaptic transmission (Liu et al. 2012), there is a lateral and long-distance spread of tau protein that is currently unexplained (reviewed in Guo and Lee 2014). However, this dissemination of tau could be accounted for if it is propagated through the brain by exosomes (Fig. 3). Alternately, it may be that neuron-to-neuron transmission of tau+ exosomes requires synaptic connections (Wang et al. 2017), or that MVs also account for some tau propagation (Dujardin et al. 2014). Recent work has begun to explore the possibility that exosomes act to spread misfolded proteins such as Aβ and tau in AD. With the increasing use of proteomics, researchers are beginning to understand the cellular origin of pathological exosomes. Furthermore, scientists are using exosomes as biomarkers for predicting AD diagnosis before clinical symptoms arise.
Exosomes contain AD biomarkers
It is now known that exosomes contain the classic AD biomarkers, Aβ (Rajendran et al. 2006) and tau (Saman et al. 2012). Additionally, it is now known that exosomes from the brain can cross the blood-brain barrier and are present in the blood (Graner et al. 2009; Shi et al. 2016). Recent studies have been working to understand where and how these proteins, as well as other biomarkers, appear in exosomes, and whether they can be used to track or even predict AD progression.
Extracellular vesicles present in plasma can now be reliably enriched for neuron-derived exosomes (NDEs) using antibodies against L1 Cell Adhesion Molecule (L1CAM) or Neural Cell Adhesion Molecule 1 (NCAM1), proteins known to be expressed by neurons (Kapogiannis et al. 2015; Goetzl et al. 2015a; Fiandaca et al. 2015). NDEs positive for these markers comprise up to 15% of total plasma exosomes (Kapogiannis et al. 2015).
Levels of Aβ1-42, total tau, tau phosphorylated at threonine 181 (pT181), and especially tau phosphorylated at serine 396 (pS396) in NDEs from plasma have been able to differentiate AD patients from control patients (Fiandaca et al. 2015). Using all markers except for total tau, the authors were able to generate a model that correctly segregated MCI and AD patients from controls 96.4% of the time (Fiandaca et al. 2015). There was no difference in the expression level of these markers between MCI and AD patient NDEs, suggesting that the exosomal levels of these proteins could be used as an early detection point for AD onset. A conflicting study found that there was no significant difference in total tau between control and AD NDEs (Shi et al. 2016). This group, instead, found that NDE tau levels were different between controls and patients with Parkinson’s disease, and that whole plasma levels of tau could differentiate between AD patients and controls. Interestingly, Shi et al. could only detect tau in NDEs using extremely sensitive Single Molecule Array (SiMoA) technology, while Fiandaca et al. could detect tau using conventional ELISA methods.
When examining plasma NDEs from a cohort of patients that were cognitively normal but went on to develop AD, Fiandaca et al. found that those who went on to develop AD had significantly higher levels of pT181 and pS396 even at 10 years prior to diagnosis as compared to those who did not develop the disease (Fiandaca et al. 2015). Remarkably, while pT181 and pS396 levels plateaued in the 10 years before diagnosis, Aβ levels continued to increase. Furthermore, increased levels of Aβ and pT181 were observed in patients with fronto-temporal dementia (FTD). Strikingly, pS396 was the only marker uniquely elevated in AD. A second study found that pT181, but not pS396, was significantly elevated in the NDEs of MCI patients (Winston et al. 2016), and that the markers pS396 and Aβ1-42 both increased between MCI patients and AD patients, rather than Aβ alone.
It has been shown that in AD and FTD, synaptic protein levels decrease in the brain as the disease progresses. Examining synaptic proteins found in NDEs from patient plasma, Goetzl et al. found that these proteins were largely decreased in both AD and FTD, but that growth-associated protein 43 and synapsin 1 were uniquely depleted in AD (Goetzl et al. 2016a). Similar to the tau phosphorylation levels observed in the study by Fiandaca et al., these synaptic protein levels were altered up to 10 years before the onset of clinical symptoms in patients. Interestingly, Goetzl et al. found that synaptopodin and synaptophysin levels correlated with worse mini-mental state examination (MMSE) and Alzheimer’s disease assessment scale-cognitive subscale (ADAS-cog) scores, but neither Aβ nor pT181 levels did. This last finding echoes the Fiandaca et al. finding for pT181: its level is elevated but steady from the pre-clinical state through to clinical Alzheimer’s disease diagnosis in this study as well. However, the groups diverge in their findings for Aβ, which may be due to the fact that the NDEs in the Goetzl study were L1CAM+ (as were those studied by Winston et al.), whereas the Fiandaca NDEs were NCAM1+ or sometimes double-positive with L1CAM.
Another study performed by Goetzl et al. found that neuro-protective transcription factors such as Repressor Element 1 Silencing Transcription factor (REST), Low-density Lipoprotein Receptor-related Protein 6 (LRP6), and Heat Shock Factor 1 (HSF1) were decreased in NDEs 2–10 years before the onset of clinical AD symptoms, and persisted at low levels during the clinical manifestation of the disease (Goetzl et al. 2015a). This finding suggests that exosomes may normally spread protective factors, but that in AD this ability is lost. Interestingly, REST is significantly increased in FTD over controls and AD, suggesting that it may play different roles in FTD and AD pathology.
A further study by the same group found that lysosomal proteins were altered in plasma NDEs during AD (Goetzl et al. 2015b). Two proteins were increased: Cathepsin D, which is involved in protein cleavage in the lysosomes and endosomes of neural cells, as well as Lysosomal-Associated Membrane Protein 1 (LAMP1), which maintains lysosomal integrity. Overall levels of protein ubiquitination were elevated in AD and MCI patient NDEs, which is a marker normally used to flag abnormal proteins for elimination. Finally, Heat Shock Protein 70 (HSP70), which maintains normal lysosomal permeability and prevents apoptosis, was found to be decreased in AD NDEs. These alterations in expression could be detected up to 10 years before AD diagnosis (Goetzl et al. 2015b). Together, Cathepsin D, HSP70, and ubiquitinated protein levels could differentiate AD from controls with a 100% success rate, and had 95.8% accuracy at differentiating from FTD. Interestingly, pT181, Aβ1-42, and Cathepsin D expression all increased significantly with aging, while neurogranin and REST levels decreased (Winston et al. 2016), although all of the age-related changes were smaller in magnitude than that of changes due to AD (Abner et al. 2016).
AD patient brains often show markers of insulin resistance (Kapogiannis et al. 2015). Furthermore, type 2 diabetes is associated with an increased risk of developing AD (reviewed in Suzanne and Wands 2008). Included in the markers of insulin resistance in AD brains is the protein type 1 Insulin Receptor Substrate (IRS-1). This protein shows altered patterns of phosphorylation in AD, with increased phosphorylation of serine 312 (pS312) and decreased overall phosphorylation of tyrosine residues, which can lead to decreases in insulin receptor signaling and therefore increased Aβ levels, as well as tau phosphorylation. Recently, these altered forms of IRS-1 were found in NDEs from AD patient plasma. The total level of IRS-1 was lower in AD patients as compared to control. Overall phosphorylation of tyrosine was significantly lower in AD patients than controls, as well as in those with FTD, while patients with type 2 diabetes showed an intermediate level. pS312 was most increased in AD, followed by FTD patients and then those with type 2 diabetes. Together, the altered phosphorylation markers on IRS-1 could distinguish between control and MCI/AD patients 100% of the time, and could distinguish between controls and patients with type 2 diabetes 97.5% of the time (Kapogiannis et al. 2015). pS312 alone was useful for distinguishing between FTD patients and controls, with an 84% success rate. As with some of the other markers identified in plasma NDEs, these changes were detectable up to 10 yeas before diagnosis, and there were no changes in overall IRS-1 level or its phosphorylation between MCI and AD. This discovery suggests that IRS-1 and its phosphorylation will be useful as marker to diagnose patients before they fully develop AD (Kapogiannis et al. 2015). Caution will have to be exercised in using this model in those with type 2 diabetes, however, as they often show altered levels of IRS-1 related markers, but do not always develop AD. The lack of correlation in this instance suggests that there are other factors yet to be uncovered in AD susceptibility. See Table 3 for a summary of exosomal protein markers and their relationship to AD.
Table 3.
Exosomal Protein Biomarkers for Alzheimer’s disease
| Protein | Direction of Chang e in AD | Predictive Before AD Onset? | Involvement in Other Neurodegenerative Diseases? | Studies |
|---|---|---|---|---|
| Aβ1–42 | Increased | Yes |
Fiandaca et al. 2015 Abner et al. 2016 |
|
| Cathepsin D | Increased | Yes | FTD |
Abner et al. 2016 Goetzl et al. 2015b |
| Growth associated protein 43 | Decreased | Yes | Goetzl et al. 2016 | |
| HSF1 | Decreased | Yes | Goetzl et al. 2015a | |
| HSP70 | Decreased | Yes | FTD* | Goetzl et al. 2015b |
| IRS-1, total | Decreased | No | Kapogiannis et al. 2015 | |
| IRS-1, pan tyrosine phosphorylation | Decreased | Yes | Type 2 diabetes | Kapogiannis et al. 2015 |
| IRS-1, pS312 | Increased | Yes | Type 2 diabetes, FTD | Kapogiannis et al. 2015 |
| LAMP1 | Increased | Yes | Goetzl et al. 2015b | |
| LRP6 | Decreased | Yes | Goetzl et al. 2015a | |
| Neurogranin | Decreased | No |
Winston et al. 2016 Abner et al. 2016 |
|
| REST | Decreased | Yes |
Abner et al. 2016 Winston et al. 2016 Goetzl et al. 2015a |
|
| Synapsin 1 | Decreased | Yes | Goetzl et al. 2016a | |
| Synaptophysin | Decreased | Yes | FTD | Goetzl et al. 2016a |
| Synaptopodin | Decreased | Yes | FTD | Goetzl et al. 2016a |
| Tau, total | Increased | No | PD |
Fiandaca et al. 2015 Shi et al. 2016 |
| Tau, pT181 | Increased | Yes | FTD |
Saman et al. 2012 Fiandaca et al. 2015 Abner et al. 2016 |
| Tau, pS396 | Increased | Yes |
Fiandaca et al. 2015 Abner et al. 2016 |
|
| Ubiquitination | Increased | Yes | No | Goetzl et al. 2015b |
Denotes biomarker has a more predictive effect for this disease
Exosomes within the serum also contain AD-correlating microRNAs (Cheng et al. 2015). MicroRNAs (miRs) are 18-22 nucleotides in length and are important post-transcriptional regulators of mRNA translation. MicroRNAs target mRNAs for translational repression using only 6-8 nucleotide long complementation sites, allowing them to bind many mRNA targets simultaneously and to globally sculpt the transcriptome of a cell. Exosomes are enriched for miRs, especially in comparison to the levels of free-floating miRs found in bodily fluids such as the serum (Cheng et al. 2014). The packaging of miRs into exosomes may be important to the successful delivery of miRs to recipient cells as the blood is an especially RNase rich environment (Huang et al. 2013). 13 miRs were found to be increased in AD exosomes, while 3 were decreased (Table 4). The majority of the serum exosome miRs discovered in this study had previously been identified in the brain. miR-1306-5p, one of the decreased miRs, showed the greatest combined sensitivity and specificity for predicting whether a patient had AD (Cheng et al. 2015). miR-1306-5p is involved in regulation of A Disintegrin and Metalloproteinase Domain-containing protein 10 (ADAM10), which generates secreted APP. Interestingly, one of the increased microRNAs, miR-101-3p, is known to target APP, and thereby to reduce the amount of Aβ created, while miRs -15 and -424 regulate tau phosphorylation (Cheng et al. 2015).
Table 4.
Exosomal microRNA markers for Alzheimer’s Disease.
| microRNAs upregulated in AD Exosomes | miR-361-5p, miR-30e-5p, miR-93-5p, miR-15a-5p, miR-143-3p, miR-335-5p, miR-106b-5p, miR-101-3p, miR-424-5p, miR-106a-5p, miR-18b-5p, miR-20a-5p, miR-582-5p |
| microRNAs downregulated in AD Exosomes | miR-1306-5p, miR-342-3p, miR-15b-3p |
Bold indicates miRs that were previously found to be important for AD pathogenesis in mouse or cell line models.
Exosomes containing pathological protein species are also detectable in the cerebrospinal fluid (CSF) of patients with AD. CSF is especially important to diagnosing and understanding brain diseases as it generally reflects the biochemistry of the brain and central nervous system.
Exosomes in the CSF were found to contain pT181 more often than was found in total CSF (Saman et al. 2012). Interestingly, exosomes carrying phosphorylated forms of tau were seen most often in stage III AD, with overall levels of such exosomes decreasing with disease progression. Exosomes containing phosphorylated tau were not found at all in other neurological conditions such as vascular or Lewy Body disease. The fact that this form of tau is released so early in AD pathogenesis suggests that exosomal packaging and release of tau is likely an active process of the brain rather than a passive one due to neuronal death (Saman et al. 2012).
Determining Exosomal/Pathology Source
Currently, AD research is determining which CNS cells are involved in creating, maintaining, and spreading AD pathology. Similarly, the exosome subfield is also attempting to determine the source of exosomes carrying potentially pathologic proteins for this disease. Goetzl et al. recently were able to purify a subset of astrocyte-derived exosomes (ADEs, GLAST+ exosomes) from the population of exosomes circulating in the blood (Goetzl et al. 2016b). Astrocytes have been known to act protectively in the brain during AD; they accumulate at site of Aβ deposition in order to bind, internalize, and degrade Aβ1-42 (Wyss-Coray et al. 2003; Koistinaho et al. 2004). However, some astrocytes can generate their own Aβ peptides, especially when stimulated by fibrillary forms of Aβ and inflammatory cytokines (Hartlage-Rübsamen et al. 2003; Hong et al. 2003; Zhao et al. 2011). Strikingly, exosomes isolated from cultured astrocyte media were found to accelerate Aβ aggregation in 5XFAD mice (Dinkins et al. 2016), suggesting that ADEs may drive AD pathology.
Excitingly, NDEs from the plasma of AD and MCI patients were able to propagate filamentous forms of tau to the CA1 region in normal mouse brains once injected into the hippocampus. Exosomes from AD patients were able to more extensively propagate tau into the CA1 region than were MCI NDEs (Winston et al. 2016). This difference may be due to the increased concentration of phosphorylated tau species present in the NDEs from AD patient serum, as phosphorylated tau species are considered to be more prone to aggregation.
Microglia derived exosomes can also propagate tau pathology from the EC to the dentate gyrus (Asai et al. 2015). Clearly, further work in this area is needed to identify markers for exosomes of each of the CNS cell types. Such markers will be instrumental in determining which cells are the sources of pathological exosomes, and may provide specific therapeutic targets for combating pathological propagation of proteins in AD.
Exosomes can spread pathology
As early as 2006, not only were exosomes were found to contain Aβ, but exosomal proteins were found in association with the amyloid plaques in AD patient brains (Rajendran et al. 2006). Exosomes derived from a cell culture model of AD, as well as those directly isolated from the brain, were found to contain APP proteins, and were especially enriched for C-terminal fragments of APP, the precursor of Aβ (CTF-APP, (Sharples et al. 2007; Perez-Gonzalez et al. 2012). Interestingly, the amount of CTF-APP was increased in brain-derived exosomes over brain homogenate (Perez-Gonzalez et al. 2012). Overall, inhibiting nSmase2, a key regulatory enzyme of ceramide biogenesis, reduced the level of exosomes in the brain and serum and further reduced Aβ plaque load in 5XFAD mice (Dinkins et al. 2014). Together, these data suggest that exosomes contribute to the spread of Aβ pathology.
There is an ongoing debate as to the relationship between Aβ, tau, and AD progression. Intriguingly, in cultured astrocytes, pretreatment with Aβ25-35 causes an increase in intracellular pTau levels via the Calcium-Sensing Receptor (CaSR). This in turn causes an increase in exosomal pTau levels (Chiarini et al. 2017). These findings suggest that Aβ may trigger the classic spread of tau seen in AD, and are consistent with the fact that Aβ deposits usually appear in the brain before tau does (Ingelsson et al. 2004). While Aβ appears first in AD, the presence of tau seems to induce Aβ toxicity, and may therefore be necessary for development of AD (Roberson et al. 2007; Ittner et al. 2010).
But what is it about exosomes that allows them to spread pathology so effectively in AD? Exosomes from the brain or from cell culture APP models were found to contain proteases that contribute to the biogenesis of Aβ fragments such as ADAM10, Beta-secretase 1 (BACE1), Nicastrin, and Presenilin 1 and 2 (PSEN1 and 2) (Sharples et al. 2007; Perez-Gonzalez et al. 2012; Cheng et al. 2015). These findings suggest that exosomes not only deposit Aβ, but can transmit the ability to create it between cells.
In Jurkat T cells, oligomerization of proteins was shown to be sufficient to target them to exosomes (Fang et al. 2007). Later studies performed in neuroblastoma cells investigating aggregation of a-synuclein, found that exosomes catalyzed and accelerated the nucleation and aggregation process, particularly in the presence of ganglioside lipids GM1 or 3 (Grey et al. 2015). An earlier study found that increasing GM1 in cell media was enough to accelerate Aβ assembly into insoluble forms in exosomes (Yuyama et al. 2008). The same group found that NDEs can accelerate Aβ assembly within themselves (Yuyama et al. 2012; An et al. 2013), although there are conflicting reports as to the effect of lipids on Aβ aggregation (Kakio et al. 2002; Martins et al. 2008). Together, these findings suggest that oligomerized tau and/or Aβ may be targeted to exosomes and thereby excreted from the cell. Furthermore, these data indicate that some types of exosomes may themselves promote oligomeric assembly of pathogenic proteins.
After assembly and release, Aβ oligomer-containing exosomes can then be incorporated into microglia, where Aβ is degraded (Yuyama et al. 2012). It has been suggested that this sequestration of oligomerized Aβ into exosomes and subsequent degradation can preserve synaptic plasticity (An et al. 2013). The packaging of already oligomeric proteins into exosomes may be an attempt by the cells to preserve their health that ironically leads to overall decreased health of the brain in AD. Indeed, loss of ceramide and thereby lowering of overall brain exosome load was found to reduce Aβ1-42 level in the brains of 5XFAD mice, as well as to improve their cognition (Dinkins et al. 2016).
While many studies suggest that exosomes spread Aβ in the brain, exosomes have also been shown to carry proteolytically active insulin-degrading enzyme (IDE) on their surfaces. This enzyme is known to degrade Aβ, and therefore, in this context, IDE+ exosomes may function to clear extracellular Aβ (Bulloj et al. 2015). Further work should be conducted to determine whether excretion of IDE on exosomes is impaired in AD.
As for tau, misprocessing of non-microtubule-bound tau was shown to pre-dispose it to being associated with intracellular vesicles. The vesicular association of tau was especially prevalent in areas where microtubules were disorganized and where phosphorylated tau was present (Lee et al. 2012). This finding gives clues as to some of the early mechanisms of tau accumulation into the endocytic compartment and possible packaging into exosomes. Furthermore, the tyrosine kinase Fyn, which is associated with lipid rafts and with extracellular toxicity in AD, was found to be associated with vesicular tau, allowing for the possibility of further phosphorylation of tau within vesicles (Lee et al. 2012).
Exosome-enriched vesicles from rTg4510 mouse brains (Tet-repressible P301L tau) were shown to act similarly to lysates from the same brains in that both sample types were able to increase the number of cells containing hyperphosphorylated tau in the CA1 region over wild-type injected controls (Baker et al. 2016). Furthermore, both the lysate and EVs were able to cause the endogenous tau to form oligomers in the hippocampus (Baker et al. 2016). A second study using the same type of mouse found that the initiation of tau aggregation by exosomes was concentration dependent (Polanco et al. 2016). They suggest that this threshold may be why AD develops late in life for most patients. Recent work by Asai et al. has shown that, in an inverse of the neuron to microglia exosome communication process, microglia-derived exosomes can propagate tau to neurons in the brain. Furthermore, microglia-derived exosomes are sufficient to spread tau from the entorhinal cortex (EC), one of the earliest loci of tau pathology, to the dentate gyrus, a later but still early effected area (Asai et al. 2015). Exosome biogenesis inhibition via blocking of nSmase2 activity significantly reduced tau propagation in this model, suggesting that exosomes are indeed a pathogenic agent in AD. Taken together, these studies evince that tau+ exosomes may be a major component of pathological tau propagation in the brain.
Coupled with the findings from Yuyama et al., these data suggest that it could be possible that neurons secrete potentially pathogenic forms of tau or Aβ in exosomes for uptake and degradation by microglia. The microglia may eventually reach capacity in terms of protein degradation and re-release pathological proteins in an attempt to remain healthy. These pathological protein-laden exosomes could in turn be re-uptaken by neurons, which are especially vulnerable to buildup of such proteins, resulting in their eventual death. Neurons can also transmit tau to each other via exosomes, although this seems to occur largely across synaptic connections, even when exosomes are involved (Wang et al. 2017). Exosomes created by these processes also spread pathological forms of tau laterally throughout the brain, and may eventually end up in the CSF or blood, where their detection can serve as a warning system for clinicians.
Tau overexpression in a neural cell culture experiment revealed that the abundance of tau significantly altered the composition of proteins recruited to exosomes (Saman et al. 2014), with only 35 proteins common to exosomes with or without tau overexpression. Tau overexpression alone was enough to increase the significance for the gene ontology term “Alzheimer” as curated by the Kyoto encyclopedia of genes and genomes. The resulting profile closely resembled the profile of exosomes derived from the hippocampus of stage IV AD patients. The overexpression of tau in exosomes induced enrichment of proteins associated with Aβ processing, such as APP and PSEN1. This finding is in contradiction with the general consensus that APP misprocessing occurs prior to that of tau, instead suggesting that either might be possible. Interestingly, proteins known to interact with both tau and APP were significantly overrepresented. Overexpression of tau recruited many mitochondrial proteins to the exosomes, echoing other research suggesting that autophagy and mitochondrial function may be disrupted in AD (reviewed in Cadonic et al. 2016). Finally, those proteins recruited to exosomes by tau overexpression overlapped by 80% with those proteins downregulated in familial AD. Overall, these results suggest that alterations in tau may underlie a large amount of the proteomic changes in AD, and links tau and exosomes to mitochondrial dysfunction. As the majority of recruited proteins in this model are downregulated in AD, exosomes in this case may be functioning more as a waste-disposal method rather than as a means of cell-cell communication in an attempt by cells to protect themselves.
Chronic Traumatic Encephalopathy and Exosomes
Chronic Traumatic Encephalopathy (CTE) was first identified in the 1920s as “dementia pugilistica” or punch-drunk syndrome in boxers (Martland 1928) and was first neuropathically described in 1973 (Corsellis et al. 1973). This disease consists of progressive deterioration of both motor and executive functions, generally following multiple head injuries. It is often associated with irritability, impulsivity, aggression, cognitive impairment, and depression. This disease occurs especially in athletes playing football, hockey, rugby, soccer, and wrestling (Stein et al. 2014), as well as in persons with a history of military service, headbanging, epilepsy, or who participate in other activities in which concussion or sub-concussive impacts occur (Geddes et al. 1999; McKee et al. 2009; McKee et al. 2012; Stein et al. 2014). CTE has also been reported in a case of domestic violence (Roberts et al. 1990b; McKee et al. 2009). Generally, there is a period of latency between head injuries and when symptoms first occur (Roberts et al. 1990a; McKee et al. 2009). Interestingly, not all persons subjected to repetitive head injuries develop CTE, suggesting that other factors, such as type of injury, genetics, and lifestyle, may contribute to whether an individual develops the disease (Jordan et al. 1997; McKee et al. 2012; Jeter et al. 2013; Montenigro et al. 2015; Bieniek et al. 2015; Stern 2016).
As in Alzheimer’s disease, one of the characteristics of CTE is the abnormal deposition of tau in the brain (Stein et al. 2014). However, CTE differs from AD and other tauopathies in the spatio-temporal distribution of tau present in this disease (See Fig. 2, Table 5). In fact, CTE tau deposition progresses in almost an exact inversion of that seen in AD: in CTE tau pathology first appears in the cortex before manifesting in deeper brain areas such as the CA1 region.
Table 5.
Comparison of the key features of AD and CTE
| Disease | Tau isoforms | Cell types affected by tau | Location of first tau pathology | Risk Genes | Co-depositing proteins |
|---|---|---|---|---|---|
| Alzheimer’s disease | all 6 | Neurons | LC, EC | APOE e4, APP, PSEN 1 and PSEN2 | Aβ |
| CTE | all 6 | Neurons and astrocytes | Depths of cortical sulci | APOE e4, MAPT | TDP43, infrequent Aβ |
Currently, CTE progression is divided into four stages. In CTE stage I, there are isolated foci of phosphorylated tau, NFTs, and astrocytic tangles in the sulcal depths, which are largely perivascular (Tokuda et al. 1991; Geddes et al. 1999; McKee et al. 2009; Stein et al. 2014). Approximately half of stage I pa-tients will have TAR DNA-binding protein 43 (TDP-43) inclusions (Stein et al. 2014). Clinically, patients in stage I of CTE tend to report headaches and loss of focus with some short term memory issues as well as aggression and depression (McKee et al. 2012).
Stage II CTE manifests with mild lateral ventricle enlargement. Multiple sulcal depths of the cortex begin to show phosphorylated tau, and NFTs appear in the areas of the cortex nearer to the brain surface and proximal to the initial legion sites. NFTs are also present in the LC and amygdala. TDP43 positivity is more frequent in this stage and often mimics the distribution of tau (Stein et al. 2014). Symptomatically, patients in stage II present with headaches, memory loss, and loss of focus, as well as depression and aggression.
In stage III CTE, patients exhibit mild cerebral atrophy as well as loss of white and grey matter in the mammillary bodies, thalamus, and frontal as well as temporal lobes, resulting in overall lower brain weight (Stein et al. 2014). At this time, there is more intensive enlargement of the lateral and third ventricles as well as septal abnormalities. NFTs are now found in many areas including the temporal cortex, hippocampus, entorhinal cortex, amygdala, hypothalamus and SN. TDP43 inclusions are more frequent. Aβ deposition may occur in some patients. In stage III, executive dysfunction in patients becomes more apparent, and the emergence of visuospatial difficulties begins to occur. The majority of patients with stage III of CTE are considered cognitively impaired.
The final phase of CTE, stage IV, is characterized by gross loss in brain weight, even as compared to earlier CTE stages (Stein et al. 2014). There is often cavum septum pellucidum at this stage. Neuronal loss is now observed in the CA1 regions as well as in the subiculum, which is accompanied by astrocytic tau phosphorylation. TDP43 inclusions now appear as thread-like neurites and are more widespread than in previous stages. Executive dysfunction and memory loss are common clinical symptoms at this stage, with many patients exhibiting dementia. Aggression and paranoia are often exhibited by patients, and a significant portion of them may experience suicidal tendencies. As with Alzheimer’s, definitive diagnosis of this CTE can only be made with a post-mortem neuropathological examination of the brain.
APOE ε4, a known risk factor for late-onset AD, is also associated with increased risk of developing CTE (Stern et al. 2013). This may be in part due to the fact that APOE ε4 is associated with worse clinical outcomes after traumatic brain injury (TBI) (Teasdale et al. 1997; Friedman et al. 1999), and especially chronic or repetitive TBI (Jordan et al. 1997), which often precede CTE (Shahim et al. 2016).
The fact that CTE can only be definitively diagnosed by post-mortem brain examination makes clinical recognition of the disease especially difficult in the face of two inter-related conditions: TBI and post-concussion syndrome (PCS). Both of these share many clinical symptoms with CTE and may even precede the development of CTE (Shahim et al. 2016). PCS is especially difficult to differentiate from CTE as PCS is also most often seen after an individual has suffered concussions or TBIs. Excitingly, the level of tau measured in the blood post-concussion correlated with duration of symptoms, and could potentially be useful for predicting clinical outcome as PCS symptoms are limited in duration (Shahim et al. 2014).
Little is currently understood about how tau and other proteins spread from the site of injury to the other brain areas characteristic of CTE. As with AD, trans-synaptic propagation of tau explains some, but not all, of the pathology seen in CTE. Especially as there is astrocyte involvement in CTE, it seems that mechanisms other than trans-synaptic transmission may be at work. McKee et al. proposed that tau may spread through the paravenous flow as well as via the CSF and explain the periventricular and perivascular deposits of tau and TDP43 seen in this disease (McKee et al. 2012). Exosomes may also play a role in this deposition, as they can travel along the same routes, and their capability to be preferentially uptaken by particular cell types can explain why pathology is largely restricted to neurons and astrocytes.
Due to the fact that options for diagnosing CTE are so limited, biomarkers, and especially any correlating to the earliest stages of the disease, will be essential to timely clinical intervention for patients showing signs of this disease. Therefore, researchers have been turning to plasma exosomes for potential biomarkers. As with AD, sufferers of CTE showed significantly elevated levels of tau in their blood exosomes, even when adjusted for age and body mass index (BMI). Level of tau in plasma exosomes was able to correctly discern between CTE and control patients 82% of the time (Stern et al. 2016). Interestingly, higher levels of exosomal tau were associated with worse performance on memory tests, but not with mood or behavior deficits. This correlation was not present when measuring tau in total plasma, although this measure was positively associated with cumulative head impact index (Alosco et al. 2017). However, it is yet to be seen whether tests of exosomal tau can discriminate between CTE patients and those with other types of tauopathy.
A second use of exosomes in the CTE field is the recently developed μMED device (Ko et al. 2016). This device specifically captures CD81 exosomes from serum and then measures their level of Glutamate receptor 2 (GluR2). GluR2 shows a 20% increase in release from the brain following TBI-like injury in mice. This marker is found in most neurons, as well as in some oligodendrocytes, and is inversely correlated with neuronal activity. Tests of the device showed 73% sensitivity and 71% specificity in determining TBI from control mice and was useable for up to 4 days following initial injury (Ko et al. 2016). Given the relationship of TBI to CTE, this type of technology may prove invaluable for early detection and intervention in both conditions.
Discussion/Future directions
While exosomes are clearly part of the propagation process for pathological proteins in AD, it still remains to be understood whether they are involved in tau propagation in CTE. It is also yet to be determined what triggers the misfolding of tau proteins in the first place. However, if exosomes with AD or CTE biomarkers are discovered in the blood or CSF early enough into disease progression, clinical intervention might occur in time to halt the majority of neuron loss. There is also a need for identification of markers specific to exosomes originating from the different CNS cell types in order to identify sources of pathological exosomes. Furthermore, comprehensive understanding of exosomal surface markers will aid in identification of the mechanisms by which CNS exosomes are taken up by specific cell types and potential therapeutic targets for halting exosomal uptake and pathological propagation.
Another unexplained aspect of tauopathies more generally is the variation in clinical symptoms and tau deposition observed between such diseases even though they share a known pathological agent. One explanation may be the fact that the tauopathies all have differing initial sites of injury or tau deposition (Clavaguera et al. 2009) (reviewed in Guo and Lee 2014; Arendt et al. 2016), and that these sites may determine which areas become subsequently affected. Another explanation could be that variations in tau misfolding and or in co-aggregating proteins for each disease dictate the sub-populations of neurons affected and the rate at which they become dysfunctional (Guo and Lee 2014). A currently unexplored avenue is whether there are variations in the exosomes carrying tau in each of these diseases that affect their progression and explain their overall differences.
As in AD, microglia are also activated in CTE (Cherry et al. 2016). Therefore, it may be worth exploring the origin of pathological exosomes in CTE as well to determine which may be responsible for tau propagation. Overall, markers for exosomal origin are only just beginning to be uncovered, and can provide a wealth of information as to the source of pathological proteins in neurodegenerative diseases.
Some of the studies reviewed herein have halted exosome biogenesis and seen amelioration in AD biomarkers and overall cognition (Dinkins et al. 2014; Asai et al. 2015). However, such global reduction in a highly-conserved pathway is likely to have many side effects. In looking to exosomes for therapeutic targets, it will likely be best to selectively target a subset of exosomes rather than to globally suppress them. One enticing option in this arena will be to use surface markers correlating with pathological protein cargo as targets for exosomal uptake inhibition. In this way, harmful exosomes might instead exit the brain or at least deposit their cargo extracellularly.
Another open question is how exosomes in the brain travel between cells both during normal brain function as well as when spreading pathology in disease. While trans-synaptic spreading seems to readily occur, exosomal travel between non-adjacent neurons and even to other cell types has been documented (Asai et al. 2015). The recently described glymphatic system may provide an answer to this question (Iliff et al. 2012). The glymphatic system is the lymphatic system of the brain, and consists of CSF that travels in the interstitial spaces of the brain flowing from perivascular spaces to the ventricles. Its primary function has been described as clearance of extracellular proteins, including Aβ. Interestingly, the same group found that the glymphatic system becomes disrupted after TBI (Iliff et al. 2014). If exosomes are indeed traveling through or being cleared by this system, such a disruption may reduce ability of exosomes to leave the brain, exacerbating or perhaps even causing the initial tau deposits seen in CTE.
Instead of being therapeutic targets, exosomes themselves could be used as therapeutics for neurodegenerative diseases. The therapeutic potential of exosomes is already being explored for other diseases. Naturally occurring exosomes in breast milk are known to block HIV infection by 50% (Näslund et al. 2014); and those made by dendritic cells can kill tumor cells (Munich et al. 2014) (reviewed in Andaloussi et al. 2013). On the AD front, mouse exosomes were successfully designed to target CNS cells and were loaded with small interfering RNAs (siRNAs) directed to knock down expression of BACE1, one of the proteases responsible for Aβ biogenesis (Alvarez-Erviti et al. 2011). The engineered exosomes were successful in crossing the blood brain barrier, which has been a large hurdle for small molecule-based therapies for neurodegenerative diseases, as well as in successfully reducing expression of BACE1. With the identification of other target proteins underlying AD and or CTE pathogenesis, as well as those proteins mediating exosomal propagation of pathology, engineered exosomes could open a new horizon for therapies for these diseases.
Acknowledgments
This work is funded in part by grants from the National Institute of Health (RF1 AG054199, R01AG054672), Alzheimer’s Association (DVT-14320835 and AARF-16442664), BrightFocus Foundation (A2016551S) and CurePSP Foundation.
Footnotes
Disclaimers
The authors have no conflict of interest relevant to this article.
References
- Abels ER, Breakefield XO. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell Mol Neurobiol. 2016;36:301–312. doi: 10.1007/s10571-016-0366-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abner EL, Jicha GA, Shaw LM, et al. Plasma neuronal exosomal levels of Alzheimer’s disease biomarkers in normal aging. Ann Clin Transl Neurol. 2016;3:399–403. doi: 10.1002/acn3.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Admyre C, Johansson SM, Qazi KR, et al. Exosomes with Immune Modulatory Features Are Present in Human Breast Milk. The Journal of Immunology. 2007;179:1969–1978. doi: 10.4049/jimmunol.179.3.1969. [DOI] [PubMed] [Google Scholar]
- Al-Dossary AA, Bathala P, Caplan JL, Martin-DeLeon PA. Oviductosome-Sperm Membrane Interaction in Cargo Delivery: DETECTION OF FUSION AND UNDERLYING MOLECULAR PLAYERS USING THREE-DIMENSIONAL SUPER-RESOLUTION STRUCTURED ILLUMINATION MICROSCOPY (SR-SIM) JOURNAL OF BIOLOGICAL CHEMISTRY. 2015;290:17710–17723. doi: 10.1074/jbc.M114.633156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alosco ML, Tripodis Y, Jarnagin J, et al. Repetitive head impact exposure and later-life plasma total tau in former National Football League players. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring. 2017;7:33–40. doi: 10.1016/j.dadm.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Erviti L, Seow Y, Yin H, et al. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011:1–7. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- Alzheimer’s Association. 2017 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia. 2017;13:325–373. doi: 10.1016/j.jalz.2017.02.001. [DOI] [Google Scholar]
- An K, Klyubin I, Kim Y, et al. Exosomes neutralize synaptic-plasticity-disrupting activity of Aβ assemblies in vivo. Mol Brain. 2013;6:47. doi: 10.1186/1756-6606-6-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andaloussi SE, Mäger I, Breakefield XO, Wood MJA. Extracellular vesicles: biology and emerging therapeutic opportunities. Nature Publishing Group. 2013;12:347–357. doi: 10.1038/nrd3978. [DOI] [PubMed] [Google Scholar]
- Arendt T, Stieler JT, Holzer M. Tau and Tauopathies. Brain research bulletin. 2016;126:238–292. doi: 10.1016/j.brainresbull.2016.08.018. [DOI] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–631. doi: 10.1212/WNL.42.3.631. [DOI] [PubMed] [Google Scholar]
- Asai H, Ikezu S, Tsunoda S, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18:1–14. doi: 10.1038/nn.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atay S, Gercel-Taylor C, Taylor DD. Human Trophoblast-Derived Exosomal Fibronectin Induces Pro-Inflammatory Il-1β Production by Macrophages. American Journal of Reproductive Immunology. 2011;66:259–269. doi: 10.1111/j.1600-0897.2011.00995.x. [DOI] [PubMed] [Google Scholar]
- Baker S, Polanco JC, Götz J. Extracellular Vesicles Containing P301L Mutant Tau Accelerate Pathological Tau Phosphorylation and Oligomer Formation but Do Not Seed Mature Neurofibrillary Tangles in ALZ17 Mice. JAD. 2016;54:1207–1217. doi: 10.3233/JAD-160371. [DOI] [PubMed] [Google Scholar]
- Balaj L, Lessard R, Dai L, et al. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nature Communications. 2011;2:180–19. doi: 10.1038/ncomms1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C, Lee VMY, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Barres C, Blanc L, Bette-Bobillo P, et al. Galectin-5 is bound onto the surface of rat reticulocyte exosomes and modulates vesicle uptake by macrophages. Blood. 2010;115:696–705. doi: 10.1182/blood-2009-07-231449. [DOI] [PubMed] [Google Scholar]
- Bieniek KF, Ross OA, Cormier KA, et al. Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta Neuropathol. 2015;130:877–889. doi: 10.1007/s00401-015-1502-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Budnik V, Ruiz-Cañada C, Wendler F. Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci. 2016;17:160–172. doi: 10.1038/nrn.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulloj A, Leal MC, Xu H, et al. Insulin-Degrading Enzyme Sorting in Exosomes: A Secretory Pathway for a Key Brain Amyloid-β Degrading Protease. JAD. 2015;19:79–95. doi: 10.3233/JAD-2010-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschow SI, Nolte-‘t Hoen ENM, van Niel G, et al. MHC II in Dendritic Cells is Targeted to Lysosomes or T Cell-Induced Exosomes Via Distinct Multivesicular Body Pathways. Traffic. 2009;10:1528–1542. doi: 10.1111/j.1600-0854.2009.00963.x. [DOI] [PubMed] [Google Scholar]
- Caby M-P, Lankar D, Vincendeau-Scherrer C, et al. Exosomal-like vesicles are present in human blood plasma. Int Immunol. 2005;17:879–887. doi: 10.1093/intimm/dxh267. [DOI] [PubMed] [Google Scholar]
- Cadonic C, Sabbir MG, Albensi BC. Mechanisms of Mitochondrial Dysfunction in Alzheimer’s Disease. Mol Neurobiol. 2016;53:1–13. doi: 10.1007/s12035-015-9515-5. [DOI] [PubMed] [Google Scholar]
- Carayon K, Chaoui K, Ronzier E, et al. Proteolipidic composition of exosomes changes during reticulocyte maturation. JOURNAL OF BIOLOGICAL CHEMISTRY. 2011;286:34426–34439. doi: 10.1074/jbc.M111.257444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chargaff E, West R. The biological significance of the thromboplastic protein of blood. J Biol Chem. 1946;166:189–197. [PubMed] [Google Scholar]
- Cheng L, Doecke JD, Sharples RA, et al. Prognostic serum miRNA biomarkers associated with Alzheimer’s disease shows concordance with neuropsychological and neuroimaging assessment. Mol Psychiatry. 2015;20:1188–1196. doi: 10.1038/mp.2014.127. [DOI] [PubMed] [Google Scholar]
- Cheng L, Sharples RA, Scicluna BJ, Hill AF. Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. 2014;3:263–14. doi: 10.3402/jev.v3.23743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JD, Tripodis Y, Alvarez VE, et al. Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathologica Communications. 2016:1–9. doi: 10.1186/s40478-016-0382-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarini A, Armato U, Gardenal E, et al. Amyloid β-Exposed Human Astrocytes Overproduce Phospho-Tau and Overrelease It within Exosomes, Effects Suppressed by Calcilytic NPS 2143—Further Implications for Alzheimer’s Therapy. Front Neurosci. 2017;11:711–9. doi: 10.3389/fnins.2017.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chivet M, Javalet C, Laulagnier K, et al. Exosomes secreted by cortical neurons upon glutamatergic synapse activation specifically interact with neurons. Journal of Extracellular Vesicles. 2014;3:24722–10. doi: 10.3402/jev.v3.24722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F, Bolmont T, Crowther RA, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb DA, Gendelman HE. Exosomes and Neuroregulation. In: Ikezu T, Gendelman HE, editors. Neuroimmune Pharmacology. Springer International Publishing; Cham: 2016. pp. 313–328. [Google Scholar]
- Colombo M, Raposo G, Théry C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu Rev Cell Dev Biol. 2014;30:255–289. doi: 10.1146/annurev-cellbio-101512-122326. [DOI] [PubMed] [Google Scholar]
- Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med. 1973;3:270–303. doi: 10.1017/s0033291700049588. [DOI] [PubMed] [Google Scholar]
- de Gassart A, Géminard C, Fevrier B, et al. Lipid raft-associated protein sorting in exosomes. Blood. 2003;102:4336–4344. doi: 10.1182/blood-2003-03-0871. [DOI] [PubMed] [Google Scholar]
- de Jong OG, Verhaar MC, Chen Y, et al. Cellular stress conditions are reflected in the protein and RNA content of endothelial cell-derived exosomes. Journal of Extracellular Vesicles. 2012;1:18396–13. doi: 10.3402/jev.v1i0.18396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkins MB, Dasgupta S, Wang G, et al. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiology of Aging. 2014;35:1792–1800. doi: 10.1016/j.neurobiolaging.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkins MB, Enasko J, Hernandez C, et al. Neutral Sphingomyelinase-2 Deficiency Ameliorates Alzheimer’s Disease Pathology and Improves Cognition in the 5XFAD Mouse. The Journal of neuroscience. 2016;36:8653–8667. doi: 10.1523/JNEUROSCI.1429-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujardin S, Bégard S, Caillierez R, et al. Ectosomes: A New Mechanism for Non-Exosomal Secretion of Tau Protein. PLoS ONE. 2014;9:e100760–10. doi: 10.1371/journal.pone.0100760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldh M, Ekström K, Valadi H, et al. Exosomes Communicate Protective Messages during Oxidative Stress; Possible Role of Exosomal Shuttle RNA. PLoS ONE. 2010;5:e15353–8. doi: 10.1371/journal.pone.0015353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escrevente C, Keller S, Altevogt P, Costa J. Interaction and uptake of exosomes by ovarian cancer cells. BMC Cancer. 2011;11:108. doi: 10.1186/1471-2407-11-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Wu N, Gan X, et al. Higher-Order Oligomerization Targets Plasma Membrane Proteins and HIV Gag to Exosomes. Plos Biol. 2007;5:e158–17. doi: 10.1371/journal.pbio.0050158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauré J, Lachenal G, Court M, et al. Exosomes are released by cultured cortical neurones. Molecular and Cellular Neuroscience. 2006;31:642–648. doi: 10.1016/j.mcn.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Fiandaca MS, Kapogiannis D, Mapstone M, et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015;11:600–7.e1. doi: 10.1016/j.jalz.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzner D, Schnaars M, van Rossum D, et al. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. Journal of Cell Science. 2011;124:447–458. doi: 10.1242/jcs.074088. [DOI] [PubMed] [Google Scholar]
- Friedman G, Froom P, Sazbon L, et al. Apolipoprotein E- 4 genotype predicts a poor outcome in survivors of traumatic brain injury. Neurology. 1999;52:244–244. doi: 10.1212/wnl.52.2.244. [DOI] [PubMed] [Google Scholar]
- Frohlich D, Kuo WP, Fruhbeis C, et al. Multifaceted effects of oligodendroglial exosomes on neurons: impact on neuronal firing rate, signal transduction and gene regulation. Philosophical Transactions of the Royal Society B: Biological Sciences. 2014;369:20130510–20130510. doi: 10.1098/rstb.2013.0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frühbeis C, Fröhlich D, Kuo WP, et al. Neurotransmitter-Triggered Transfer of Exosomes Mediates Oligodendrocyte–Neuron Communication. Plos Biol. 2013;11:e1001604–19. doi: 10.1371/journal.pbio.1001604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes JF, Vowles GH, Nicoll J, Revesz T. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol. 1999;98:171–178. doi: 10.1007/s004010051066. [DOI] [PubMed] [Google Scholar]
- Geschwind DH. Tau Phosphorylation, Tangles, and Neurodegeneration. Neuron. 2003;40:457–460. doi: 10.1016/s0896-6273(03)00681-0. [DOI] [PubMed] [Google Scholar]
- Géminard C, de Gassart A, Blanc L, Vidal M. Degradation of AP2 during reticulocyte maturation enhances binding of hsc70 and Alix to a common site on TFR for sorting into exosomes. Traffic. 2004;5:181–193. doi: 10.1111/j.1600-0854.2004.0167.x. [DOI] [PubMed] [Google Scholar]
- Ghossoub R, Lembo F, Rubio A, et al. Syntenin-ALIX exosome biogenesis and budding into multivesicular bodies are controlled by ARF6 and PLD2. Nature Communications. 2014;5:3477. doi: 10.1038/ncomms4477. [DOI] [PubMed] [Google Scholar]
- Giri M, Shah A, Upreti B, Rai J. Unraveling the genes implicated in Alzheimer’s disease (Review) biom rep. 2017:1–10. doi: 10.3892/br.2017.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Wischik CM, Crowther RA, et al. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci USA. 1988;85:4051–4055. doi: 10.1097/00007890-200607152-03005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetzl EJ, Boxer A, Schwartz JB, et al. Low neural exosomal levels of cellular survival factors in Alzheimer’s disease. Ann Clin Transl Neurol. 2015a;2:769–773. doi: 10.1002/acn3.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetzl EJ, Boxer A, Schwartz JB, et al. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology. 2015b;85:40–47. doi: 10.1212/WNL.0000000000001702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetzl EJ, Kapogiannis D, Schwartz JB, et al. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J. 2016a;30:4141–4148. doi: 10.1096/fj.201600816R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetzl EJ, Mustapic M, Kapogiannis D, et al. Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer’s disease. FASEB J. 2016b;30:3853–3859. doi: 10.1096/fj.201600756R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graner MW, Alzate O, Dechkovskaia AM, et al. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. 2009;23:1541–1557. doi: 10.1096/fj.08-122184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey M, Dunning CJ, Gaspar R, et al. Acceleration of alpha-Synuclein Aggregation by Exosomes. J Biol Chem. 2015;290:2969–2982. doi: 10.1074/jbc.M114.585703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudzien A, Shaw P, Weintraub S, et al. Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer’s disease. Neurobiology of Aging. 2007;28:327–335. doi: 10.1016/j.neurobiolaging.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Guo JL, Lee VMY. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. JOURNAL OF BIOLOGICAL CHEMISTRY. 2011;286:15317–15331. doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Lee VMY. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nature Publishing Group. 2014;20:130–138. doi: 10.1038/nm.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding C, Heuser J, Stahl P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. The Journal of Cell Biology. 1983;97:329–339. doi: 10.1083/jcb.97.2.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlage-Rübsamen M, Zeitschel U, Apelt J, et al. Astrocytic expression of the Alzheimer’s disease beta-secretase (BACE1) is stimulus-dependent. Glia. 2003;41:169–179. doi: 10.1002/glia.10178. [DOI] [PubMed] [Google Scholar]
- Hong HS, Hwang EM, Sim HJ, et al. Interferon γ stimulates β-secretase expression and sAPPβ production in astrocytes. Biochemical and Biophysical Research Communications. 2003;307:922–927. doi: 10.1016/S0006-291X(03)01270-1. [DOI] [PubMed] [Google Scholar]
- Hood JL, San Roman S, Wickline SA. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Research. 2011;71:3792–3801. doi: 10.1158/0008-5472.CAN-10-4455. [DOI] [PubMed] [Google Scholar]
- Hu W, Zhang X, Tung YC, et al. Hyperphosphorylation determines both the spread and the morphology of tau pathology. Alzheimers Dement. 2016;12:1066–1077. doi: 10.1016/j.jalz.2016.01.014. [DOI] [PubMed] [Google Scholar]
- Huang X, Yuan T, Tschannen M, et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genomics. 2013;14:319. doi: 10.1186/1471-2164-14-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang-Doran I, Zhang C-Y, Vidal-Puig A. Extracellular Vesicles: Novel Mediators of Cell Communication In Metabolic Disease. Trends in Endocrinology & Metabolism. 2016:1–16. doi: 10.1016/j.tem.2016.10.003. [DOI] [PubMed] [Google Scholar]
- Iba M, Guo JL, McBride JD, et al. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci. 2013;33:1024–1037. doi: 10.1523/JNEUROSCI.2642-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iba M, McBride JD, Guo JL, et al. Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC’s afferent and efferent connections. Acta Neuropathol. 2015;130:349–362. doi: 10.1007/s00401-015-1458-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Chen MJ, Plog BA, et al. Impairment of Glymphatic Pathway Function Promotes Tau Pathology after Traumatic Brain Injury. The Journal of neuroscience. 2014;34:16180–16193. doi: 10.1523/JNEUROSCI.3020-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. 2012;4:147ra111–147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelsson M, Fukumoto H, Newell KL, et al. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Liu F, Gong C-X. Tau and neurodegenerative disease: the story so far. Nature Publishing Group. 2015;12:15–27. doi: 10.1038/nrneurol.2015.225. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, et al. Dendritic Function of Tau Mediates Amyloid-β Toxicity in Alzheimer’s Disease Mouse Models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Jack CR, Lowe VJ, Weigand SD, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer”s disease: implications for sequence of pathological events in Alzheimer’s disease. Brain. 2009;132:1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javeed N, Mukhopadhyay D. Exosomes and their role in the micro-/macro-environment: a comprehensive review. J Biomed Res. 2016 doi: 10.7555/JBR.30.20150162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeter CB, Hergenroeder GW, Hylin MJ, et al. Biomarkers for the Diagnosis and Prognosis of Mild Traumatic Brain Injury/Concussion. Journal of Neurotrauma. 2013;30:657–670. doi: 10.1089/neu.2012.2439. [DOI] [PubMed] [Google Scholar]
- Jordan BD, Relkin NR, Ravdin LD, et al. Apolipoprotein E epsilon4 associated with chronic traumatic brain injury in boxing. JAMA. 1997;278:136–140. [PubMed] [Google Scholar]
- Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol. 2011;70:532–540. doi: 10.1002/ana.22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlert C, Melo SA, Protopopov A, et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. JOURNAL OF BIOLOGICAL CHEMISTRY. 2014;289:3869–3875. doi: 10.1074/jbc.C113.532267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakio A, Nishimoto S-I, Yanagisawa K, et al. Interactions of Amyloid β-Protein with Various Gangliosides in Raft-Like Membranes: Importance of GM1 Ganglioside-Bound Form as an Endogenous Seed for Alzheimer Amyloid †. Biochemistry. 2002;41:7385–7390. doi: 10.1021/bi0255874. [DOI] [PubMed] [Google Scholar]
- Kalluri R. The biology and function of exosomes in cancer. J Clin Invest. 2016;126:1208–1215. doi: 10.1172/JCI81135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapogiannis D, Boxer A, Schwartz JB, et al. Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical Alzheimer’s disease. FASEB J. 2015;29:589–596. doi: 10.1096/fj.14-262048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Hemphill MA, Gabrieli D, et al. Smartphone-enabled optofluidic exosome diagnostic for concussion recovery. Scientific Reports. 2016;6:31215. doi: 10.1038/srep31215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho M, Lin S, Wu X, et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat Med. 2004;10:719–726. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- Kowal J, Arras G, Colombo M, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci USA. 2016;113:E968–E977. doi: 10.1073/pnas.1521230113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer-Albers E-M, Bretz N, Tenzer S, et al. Oligodendrocytes secrete exosomes containing major myelin and stress-protective proteins: Trophic support for axons? Prot Clin Appl. 2007;1:1446–1461. doi: 10.1002/prca.200700522. [DOI] [PubMed] [Google Scholar]
- Laulagnier K, Grand D, Dujardin A, et al. PLD2 is enriched on exosomes and its activity is correlated to the release of exosomes. FEBS Lett. 2004;572:11–14. doi: 10.1016/j.febslet.2004.06.082. [DOI] [PubMed] [Google Scholar]
- Lee S, Kim W, Li Z, Hall GF. Accumulation of Vesicle-Associated Human Tau in Distal Dendrites Drives Degeneration and Tau Secretion in an In Situ Cellular Tauopathy Model. International Journal of Alzheimer’s Disease. 2012;2012:1–16. doi: 10.1155/2012/172837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, et al. Trans-Synaptic Spread of Tau Pathology In Vivo. PLoS ONE. 2012;7:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins IC, Kuperstein I, Wilkinson H, et al. Lipids revert inert Aβ amyloid fibrils to neurotoxic protofibrils that affect learning in mice. The EMBO Journal. 2008;27:224–233. doi: 10.1038/sj.emboj.7601953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martland HS. Punch Drunk. JAMA. 1928;91:1103–1107. doi: 10.1001/jama.1928.02700150029009. [DOI] [Google Scholar]
- Matsuo H, Chevallier J, Mayran N, et al. Role of LBPA and Alix in multivesicular liposome formation and endosome organization. Science. 2004;303:531–534. doi: 10.1126/science.1092425. [DOI] [PubMed] [Google Scholar]
- McKee AC, Cantu RC, Nowinski CJ, et al. Chronic Traumatic Encephalopathy in Athletes: Progressive Tauopathy After Repetitive Head Injury. Journal of Neuropathology and Experimental Neurology. 2009;68:709–735. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Stein TD, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2012;136:43–64. doi: 10.1093/brain/aws307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milane L, Singh A, Mattheolabakis G, et al. Exosome mediated communication within the tumor microenvironment. Journal of Controlled Release. 2015;219:278–294. doi: 10.1016/j.jconrel.2015.06.029. [DOI] [PubMed] [Google Scholar]
- Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood. 2012 doi: 10.1182/blood-2011-02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montenigro PH, Corp DT, Stein TD, et al. Chronic Traumatic Encephalopathy: Historical Origins and Current Perspective. Annu Rev Clin Psychol. 2015;11:309–330. doi: 10.1146/annurev-clinpsy-032814-112814. [DOI] [PubMed] [Google Scholar]
- Morelli AE, Larregina AT, Shufesky WJ, et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood. 2004;104:3257–3266. doi: 10.1182/blood-2004-03-0824. [DOI] [PubMed] [Google Scholar]
- Mulcahy LA, Pink RC, Carter DRF. Routes and mechanisms of extracellular vesicle uptake. Journal of Extracellular Vesicles. 2014;3:24641–15. doi: 10.3402/jev.v3.24641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munich S, Sobo-Vujanovic A, Buchser WJ, et al. Dendritic cell exosomes directly kill tumor cells and activate natural killer cells via TNF superfamily ligands. OncoImmunology. 2014;1:1074–1083. doi: 10.4161/onci.20897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarenko I, Rana S, Baumann A, et al. Cell surface tetraspanin Tspan8 contributes to molecular pathways of exosome-induced endothelial cell activation. Cancer Research. 2010;70:1668–1678. doi: 10.1158/0008-5472.CAN-09-2470. [DOI] [PubMed] [Google Scholar]
- Näslund TI, Paquin-Proulx D, Paredes PT, et al. Exosomes from breast milk inhibit HIV-1 infection of dendritic cells and subsequent viral transfer to CD4+ T cells. AIDS. 2014;28:171–180. doi: 10.1097/QAD.0000000000000159. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer Disease Neuropathologic Changes With Cognitive Status: A Review of the Literature. Journal of Neuropathology and Experimental Neurology. 2012;71:362–381. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan BT, Johnstone RM. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: selective externalization of the receptor. Cell. 1983;33:967–978. doi: 10.1016/0092-8674(83)90040-5. [DOI] [PubMed] [Google Scholar]
- Parolini I, Federici C, Raggi C, et al. Microenvironmental pH Is a Key Factor for Exosome Traffic in Tumor Cells. J Biol Chem. 2009;284:34211–34222. doi: 10.1074/jbc.M109.041152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Gonzalez R, Gauthier SA, Kumar A, Levy E. The exosome secretory pathway transports amyloid precursor protein carboxyl-terminal fragments from the cell into the brain extracellular space. JOURNAL OF BIOLOGICAL CHEMISTRY. 2012;287:43108–43115. doi: 10.1074/jbc.M112.404467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Hernandez D, Gutiérrez-Vázquez C, Jorge I, et al. The intracellular interactome of tetraspanin-enriched microdomains reveals their function as sorting machineries toward exosomes. JOURNAL OF BIOLOGICAL CHEMISTRY. 2013;288:11649–11661. doi: 10.1074/jbc.M112.445304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisitkun T, Shen R-F, Knepper MA. Identification and proteomic profiling of exosomes in human urine. Proc Natl Acad Sci USA. 2004;101:13368–13373. doi: 10.1073/pnas.0403453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanco JC, Scicluna BJ, Hill AF, Götz J. Extracellular vesicles isolated from brains of rTg4510 mice seed tau aggregation in a threshold-dependent manner. J Biol Chem. 2016 doi: 10.1074/jbc.M115.709485. jbc.M115.709485-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potolicchio I, Carven GJ, Xu X, et al. Proteomic analysis of microglia-derived exosomes: metabolic role of the aminopeptidase CD13 in neuropeptide catabolism. The Journal of Immunology. 2005;175:2237–2243. doi: 10.4049/jimmunol.175.4.2237. [DOI] [PubMed] [Google Scholar]
- Raiborg C, Wesche J, Malerød L, Stenmark H. Flat clathrin coats on endosomes mediate degradative protein sorting by scaffolding Hrs in dynamic microdomains. Journal of Cell Science. 2006;119:2414–2424. doi: 10.1242/jcs.02978. [DOI] [PubMed] [Google Scholar]
- Rajendran L, Honsho M, Zahn TR, et al. Alzheimer’s disease β-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci USA. 2006;103:11172–11177. doi: 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana S, Yue S, Stadel D, Zöller M. Toward tailored exosomes: The exosomal tetraspanin web contributes to target cell selection. International Journal of Biochemistry and Cell Biology. 2012;44:1574–1584. doi: 10.1016/j.biocel.2012.06.018. [DOI] [PubMed] [Google Scholar]
- Raposo G, Nijman HW, Stoorvogel W, et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med. 1996;183:1161–1172. doi: 10.1084/jem.183.3.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Record M, Carayon K, Poirot M, Silvente-Poirot S. Exosomes as new vesicular lipid transporters involved in cell–cell communication and various pathophysiologies. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2014;1841:108–120. doi: 10.1016/j.bbalip.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Roberts GW, ALLSOP D, BRUTON C. The Occult Aftermath of Boxing. J Neurol Neurosurg Psychiatr. 1990a;53:373–378. doi: 10.1136/jnnp.53.5.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts GW, Whitwell HL, Acland PR, Bruton CJ. Dementia in a punch-drunk wife. The Lancet. 1990b;335:918–919. doi: 10.1016/0140-6736(90)90520-f. [DOI] [PubMed] [Google Scholar]
- Russo I, Bubacco L, Greggio E. Exosomes-associated neurodegeneration and progression of Parkinson’s disease. 2012 [PMC free article] [PubMed] [Google Scholar]
- Saman S, Kim W, Raya M, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. JOURNAL OF BIOLOGICAL CHEMISTRY. 2012;287:3842–3849. doi: 10.1074/jbc.M111.277061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saman S, Lee NCY, Inoyo I, et al. Proteins recruited to exosomes by tau overexpression implicate novel cellular mechanisms linking tau secretion with Alzheimer’s disease. J Alzheimers Dis. 2014;40(Suppl 1):S47–70. doi: 10.3233/JAD-132135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöll M, Lockhart SN, Schonhaut DR, et al. PET Imaging of Tau Deposition in the Aging Human Brain. Neuron. 2016;89:971–982. doi: 10.1016/j.neuron.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harbor Perspectives in Medicine. 2011;1:a006189–a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahim P, Tegner Y, Gustafsson B, et al. Neurochemical Aftermath of Repetitive Mild Traumatic Brain Injury. JAMA Neurol. 2016;73:1308–8. doi: 10.1001/jamaneurol.2016.2038. [DOI] [PubMed] [Google Scholar]
- Shahim P, Tegner Y, Wilson DH, et al. Blood Biomarkers for Brain Injury in Concussed Professional Ice Hockey Players. JAMA Neurol. 2014;71:684–9. doi: 10.1001/jamaneurol.2014.367. [DOI] [PubMed] [Google Scholar]
- Sharples RA, Vella LJ, Nisbet RM, et al. Inhibition of -secretase causes increased secretion of amyloid precursor protein C-terminal fragments in association with exosomes. The FASEB Journal. 2007;22:1469–1478. doi: 10.1096/fj.07-9357com. [DOI] [PubMed] [Google Scholar]
- Shi M, Kovac A, Korff A, et al. CNS tau efflux via exosomes is likely increased in Parkinson’s disease but not in Alzheimer’s disease. 2016:1–7. doi: 10.1016/j.jalz.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson RJ, Lim JW, Moritz RL, Mathivanan S. Exosomes: proteomic insights and diagnostic potential. Expert Rev Proteomics. 2014;6:267–283. doi: 10.1586/epr.09.17. [DOI] [PubMed] [Google Scholar]
- Stein TD, Alvarez VE, McKee AC. Chronic traumatic encephalopathy: a spectrum of neuropathological changes following repetitive brain trauma in athletes and military personnel. Alzheimer’s Research & Therapy. 2014;6:4. doi: 10.1186/alzrt234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern RA. Cerebrospinal Fluid Biomarkers in Postconcussion Syndrome Measuring Neuronal Injury and Distinguishing Individuals at Risk for Persistent Postconcussion Syndrome or Chronic Traumatic Encephalopathy. JAMA Neurol. 2016;73:1280–1282. doi: 10.1001/jamaneurol.2016.3169. [DOI] [PubMed] [Google Scholar]
- Stern RA, Daneshvar DH, Baugh CM, et al. Clinical presentation of chronic traumatic encephalopathy. Neurology. 2013;81:1122–1129. doi: 10.1212/WNL.0b013e3182a55f7f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern RA, Tripodis Y, Baugh CM, et al. Preliminary Study of Plasma Exosomal Tau as a Potential Biomarker for Chronic Traumatic Encephalopathy. 2016;51:1099–1109. doi: 10.3233/JAD-151028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuffers S, Sem Wegner C, Stenmark H, Brech A. Multivesicular Endosome Biogenesis in the Absence of ESCRTs. Traffic. 2009;10:925–937. doi: 10.1111/j.1600-0854.2009.00920.x. [DOI] [PubMed] [Google Scholar]
- Suzanne M, Wands JR. Alzheimer’s disease is type 3 diabetes—evidence reviewed. Journal of Diabetes Science and Technology. 2008;2:1101–1113. doi: 10.1177/193229680800200619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AR, Robinson MB, Gifondorwa DJ, et al. Regulation of heat shock protein 70 release in astrocytes: Role of signaling kinases. Devel Neurobio. 2007;67:1815–1829. doi: 10.1002/dneu.20559. [DOI] [PubMed] [Google Scholar]
- Teasdale GM, Nicoll JA, Murray G, Fiddes M. Association of apolipoprotein E polymorphism with outcome after head injury. The Lancet. 1997;350:1069–1071. doi: 10.1016/S0140-6736(97)04318-3. [DOI] [PubMed] [Google Scholar]
- Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569–579. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- Tokuda T, Ikeda S, Yanagisawa N, et al. Re-examination of ex-boxers’ brains using immunohistochemistry with antibodies to amyloid beta-protein and tau protein. Acta Neuropathol. 1991;82:280–285. doi: 10.1007/BF00308813. [DOI] [PubMed] [Google Scholar]
- Trajkovic K, Hsu C, Chiantia S, et al. Ceramide Triggers Budding of Exosome Vesicles into Multivesicular Endosomes. Science. 2008;319:1244–1247. doi: 10.1126/science.1153124. [DOI] [PubMed] [Google Scholar]
- Trams EG, Lauter CJ, Salem N, Heine U. Exfoliation of membrane ecto-enzymes in the form of micro-vesicles. Biochim Biophys Acta. 1981;645:63–70. doi: 10.1016/0005-2736(81)90512-5. [DOI] [PubMed] [Google Scholar]
- Valadi H, Ekström K, Bossios A, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- van Niel G, Charrin S, Simoes S, et al. The Tetraspanin CD63 Regulates ESCRT-Independent and -Dependent Endosomal Sorting during Melanogenesis. Developmental Cell. 2011;21:708–721. doi: 10.1016/j.devcel.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella LJ, Greenwood DLV, Cappai R, et al. Enrichment of prion protein in exosomes derived from ovine cerebral spinal fluid. Veterinary Immunology and Immunopathology. 2008;124:385–393. doi: 10.1016/j.vetimm.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Villarroya-Beltri C, Gutiérrez-Vázquez C, Sánchez-Cabo F, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nature Communications. 2013;4:2980. doi: 10.1038/ncomms3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Balaji V, Kaniyappan S, et al. The release and trans-synaptic transmission of Tau via exosomes. Molecular Neurodegeneration. 2017:1–25. doi: 10.1186/s13024-016-0143-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston CN, Goetzl EJ, Akers JC, et al. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. 2016;3:63–72. doi: 10.1016/j.dadm.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf P. The nature and significance of platelet products in human plasma. British Journal of Haematology. 1967;13:269–288. doi: 10.1111/j.1365-2141.1967.tb08741.x. [DOI] [PubMed] [Google Scholar]
- Wubbolts R, Leckie RS, Veenhuizen PTM, et al. Proteomic and Biochemical Analyses of Human B Cell-derived Exosomes. J Biol Chem. 2003;278:10963–10972. doi: 10.1074/jbc.M207550200. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Loike JD, Brionne TC, et al. Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nat Med. 2003;9:453–457. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- Yáñez-Mó M, Siljander PRM, Andreu Z, et al. Biological properties of extracellular vesicles and their physiological functions. Journal of Extracellular Vesicles. 2015;4:27066–60. doi: 10.3402/jev.v4.27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuyama K, Sun H, Mitsutake S, Igarashi Y. Sphingolipid-modulated exosome secretion promotes clearance of amyloid-β by microglia. JOURNAL OF BIOLOGICAL CHEMISTRY. 2012;287:10977–10989. doi: 10.1074/jbc.M111.324616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuyama K, Yamamoto N, Yanagisawa K. Accelerated release of exosome-associated GM1 ganglioside (GM1) by endocytic pathway abnormality: another putative pathway for GM1-induced amyloid fibril formation. J Neurochem. 2008;105:217–224. doi: 10.1111/j.1471-4159.2007.05128.x. [DOI] [PubMed] [Google Scholar]
- Zech D, Rana S, Büchler MW, Zöller M. Tumor-exosomes and leukocyte activation: an ambivalent crosstalk. Cell Commun Signal. 2012;10:37. doi: 10.1186/1478-811X-10-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Li S, Li L, et al. Exosome and Exosomal MicroRNA: Trafficking, Sorting, and Function. Genomics, Proteomics & Bioinformatics. 2015;13:17–24. doi: 10.1016/j.gpb.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, O’Connor T, Vassar R. The contribution of activated astrocytes to Aβ production: implications for Alzheimer’s disease pathogenesis. Journal of Neuroinflammation. 2011;8:150. doi: 10.1186/1742-2094-8-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Regnault A, Lozier A, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med. 1998;4:594–600. doi: 10.1038/nm0598-594. [DOI] [PubMed] [Google Scholar]