

Abstract
Tumors that elude infiltration by CD8+ T lymphocytes are particularly resistant to multiple forms of treatment, including immune checkpoint blockade. Stromal TGF-β appears to play a key role in this process, potentially constituting a target for novel combinatorial regimens tackling immune-excluded neoplasms.
Keywords: atezolizumab, microsatellite instability, mutational burden, PD-1, tumor neo-antigens, tumor organoids
Immune checkpoint blockers (ICBs) have revolutionized the treatment of multiple tumors, including melanoma, lung cancer, and urothelial carcinoma. However, only a fraction of patients with these neoplasms respond to ICBs employed as standalone therapeutic interventions, and other tumors such as colorectal carcinoma (CRC) are not particularly sensitive to monotherapy with ICBs. Several features of the tumor and its microenvironment are currently employed to inform clinical decision-making with respect to ICB-based immunotherapy, including tumor mutational burden (TMB), sometimes linked to microsatellite instability; increased expression of CD274 (best known as PD-L1), a co-inhibitory ligand for programmed cell death 1 (PDCD1, best known as PD-1); and infiltration by CD8+ cytotoxic T lymphocytes (CTLs) [1]. In particular, solid neoplasms can be classified into three different phenotypes with respect to immune infiltration: (1) “inflamed” tumors; (2) “immune desert” and (3) “immune-excluded” tumors (Figure 1) [2]. Inflamed tumors display elevated numbers of CD8+ T cells that invade the tumor and are often referred as “hot” tumors. Immune deserts and immune-excluded tumors are called “cold” tumors and either exhibit little amounts of CD8+ CTLs and other immune cells (i.e. “immune desert”) or display extensive accumulation of CD8+ CTLs at tumor margins, without tumor infiltration (i.e. “immune-excluded”). While inflamed tumors generally respond to ICB-based immunotherapy, the immune-desert and immune-excluded phenotypes have been linked to limited ICB efficiency [2].
Figure 1. Blocking TGF-β enables therapeutic responses to ICBs in immune-excluded tumors.
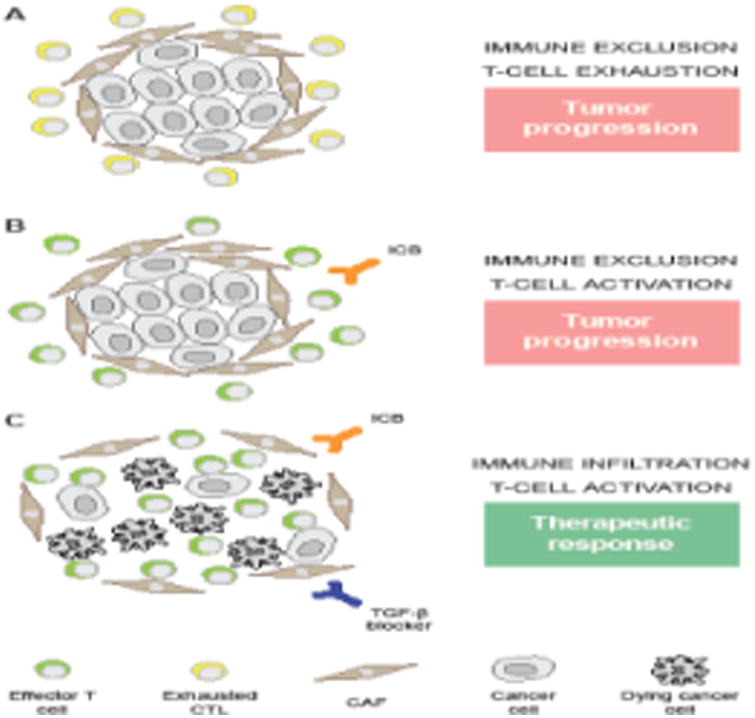
Immune-excluded tumors are characterized by the accumulation of CD8+ cytotoxic T lymphocytes (CTLs) at the tumor periphery, with limited infiltration of malignant cell nests. Some (but not all) of these tumors also exhibit a robust desmoplastic stroma characterized by high amounts of cancer-associated fibroblasts (CAFs) and a dense extracellular matrix (A). In this setting, immunotherapy with immune checkpoint blockers (ICBs) employed as standalone interventions generally provides limited clinical benefits as (re-)activated CTLs cannot get in contact with (and hence kill) malignant cells (B). Since transforming growth factor beta (TGF-β) produced by CAFs is etiologically involved in the establishment of immune exclusion, therapeutic regimens combining ICBs with an agent that inhibit TGF-β signaling, such as galunisertib or fresolimumab, may exert superior clinical benefits by simultaneously enabling T-cell activation and tumor infiltration (C).
The fact that tumor-associated T cells are frequently restricted to stromal areas rather than tumor island raises the possibility of a crosstalk between malignant cells and “normal” stromal cells that impede T cell infiltration and/or activation. Cancer-associated fibroblasts (CAFs), the most preponderant stromal component and associated with poor patient prognosis, are emerging as a suppressive intermediates within the tumor microenvironment (TME) through secretion of a myriad of chemokines and cytokines that can polarize responsive immune cell subsets [3]. Of note, transforming growth factor-beta (TGF-β), a pleiotropic cytokine involved in processes as diverse as physiological tissue repair, tumor progression, and immune regulation [4] has been described as an upstream regulator of the functions of CAFs as well as a downstream inducer of the immune suppressive status in CAFs rich tumors [5].
Preclinical data from multiple laboratories suggest that another mechanism underlying the limited sensitivity of some tumors to ICBs depends on TGF-β. However, the precise crosstalk between TGF-β, the stromal compartment and immune checkpoint blockade remains poorly characterized. Recent data from Tauriello et al. [6] and Mariathasan et al. [7] uncover the key contribution of stromal TGF-β to immune exclusion in microsatellite stable CRC and urothelial carcinoma. These findings point to TGF-β as a promising target for the development of combinatorial treatments that enable immune infiltration by CD8+ CTLs in the context of immune checkpoint blockade.
Tauriello and colleagues investigated the interplay between genetic alterations and the TME in metastatic microsatellite-stable CRCs, a tumor type classically resistant to ICB. To this end, they generated a biobank of mouse tumor organoids (MTOs) from C57BL/6J mice genetically engineered to bear the four main CRC driving mutations (i.e. Apc (A), Kras (K), Tgfbr2 (T) and Trp53 (P)) in intestinal stem cells using a Lgr5 (L) promoter. Among the eight mice strains bearing combinations of these mutations, the quadruple-mutant LAKTP genuinely recapitulated key features of advanced human microsatellite-stable CRCs, characterized by a low mutational burden, a limited amount of predicted high-affinity MHC class I-restricted neo-antigens, an immune excluded phenotype as well as increased stromal TGF-β bioavailability. Similar to clinical settings, PD-1 and PD-L1 blockades failed to induce any response in LAKTP mice whereas galunisertib, a TGF-β receptor I kinase inhibitor, unleashed CD8+ cytotoxic T-cell anti-tumor immunity that prevented metastasis. Notably, the anti-metastatic effects of galunisertib were abolished in nu/nu mice, as well as in mice depleted for CD8+ CTLs or CD4+ TH1 cells, confirming the immunological nature of the response. However, in mice with established metastatic disease in the liver, TGF-β blockade by itself was insufficient to overcome immune suppression but revealed essential to convert ICB-unresponsive into ICB-responsive tumors, therefore indicating that stromal TGF-β dictates T-cell exclusion and suppresses TH1 -effector phenotype [6].
Mariathasan et al., also studied the mechanisms behind a lack of response to immunotherapy and uncovered TGF-β signaling as a key determinant of resistance to PD-L1 blockade [7]. Using an integrated evaluation of biomarkers, tumors from a large Phase II cohort of patients with metastatic urothelial cancers were stratified according to their response to atezolimumab, a PD-L1 targeting agent. Three distinct biological processes were found to be associated with increased likelihood of response: (1) signs of an actionable tumor-targeting immune response, including high PD-L1 expression levels on immune (but not malignant) cells and a gene signature of CD8+ effector T (TEFF) cells; (2) elevated TMB; and (3) reduced transcriptional markers of cytokine-cytokine receptor interactions (including genes involved in TGF-β signaling such as TGFB1, AVCR1 and TGFBR2).
Forty-seven percent of the urothelial carcinomas included in this cohort exhibited immune exclusion. In this tumors, the gene signature for CD8+ TEFF cells per se was not statistically associated with increased likelihood for clinical responses, while a gene signature representative of TGF-β signaling in fibroblasts (F-TBRS score) was associated with stable or progressive disease. To investigate whether immune exclusion is responsible for resistance to atezolizumab and whether TGF-β has a role in this setting, Mariathasan and collaborators used the mouse mammary carcinoma EMT6 model, which generally exhibits an excluded phenotype and expresses both PD-L1 and TGF-β. In this scenario, therapeutic blockade of either PD-L1 or TGF-β (with the pan-TGF-β-targeting antibody 1D11) had limited therapeutic effects, while concurrent inhibition of both pathways enabled persistent tumor regression in 70% of mice. Accordingly, EMT6 mammary carcinoma subjected to combinatorial immunotherapy (but not tumors receiving either treatment alone) exhibited elevated CD8+ T-cell infiltration and reduced F-TBRS scores along with a significant increase in the mean distance separating CD8+ CTLs from the tumor periphery. Similar observations were obtained in the mouse MC38 CRC model [7].
One of the main difference when comparing the two above described preclinical studies relies on the score of TGF-β response signature that was found reduced in both fibroblast (F-TBRS) and T cells (T-TBRS) following galunisertib [6] but only in cancer-associated fibroblast upon 1D11 treatment [7]. However, even though the T-TBRS score was not affected, 1D11 did promote T cells infiltration into the tumor therefore improving responsiveness to ICBs. Although cautin should be employed in extrapolating preclinical data to the clinic (especially because different TGF-β neutralization agents were used), these studies converge towards the critical role of TGF-β in immune mediated exclusion and resistance to the PD-1/PD-L1 blockade (Figure 1). Two ongoing clinical trials (NCT02423343 and NCT02734160) are testing this hypothesis.
Of note, even in the context of local radiation therapy (RT) where TGF-β activation is well-known to prevent RT-induced immunogenicity [8], double blockade of TGF-β and PD-1 was required to achieve the best survival responses in a poorly immunogenic mouse breast carcinoma [9].
Importantly, a recently reported pilot trial in metastatic breast cancer patients testing TGF-β blockade by the monoclonal antibody fresolimumab with localized radiotherapy also suggests the need for dual blockade in this context. While the patients randomly assigned to receive a higher dose of fresolimumab achieved a longer survival compared to those treated at lower dose and demonstrated improved peripheral blood mononuclear cell counts and a striking boost in the CD8 central memory pool, no objective tumor responses were achieved outside the irradiated fields (abscopal responses, the main endpoint of the study). The failure to achieve abscopal responses warrants further exploration by adding ICB to localized radiotherapy and TGF-β blockade [10].
Development of combinatorial-based approaches targeting multiple ICB, with the potential inclusion of focal radiotherapy might be required to significantly enhance the proportion of patient that can benefit from immunotherapy, especially in the context of immune-excluded cancers, a common subset of tumors in patients resistant to immunotherapy. Several trials are testing TGF-β blockade (NCT02538471, NCT02581787, NCT02688712 and NCT02906397), without ICBs. Depending on the prevalence of immune-excluded tumors among the patients who accrue to these trials, the work of Tauriello et al. and Mariathasan et al. predicts for a potentially limited clinical efficacy of these studies, in the absence of ICBs.
Acknowledgments
CVB is supported by the 2017 Kellen Junior Faculty Fellowship from the Anna-Maria and Stephen Kellen Foundation (New York, NY, US). This work was supported by NIH grants to S.C.F. (S10 RR027619). S.C.F. is supported by the Breast Cancer Research Foundation (BCRF-16-054).
Footnotes
Conflicts of interest: Authors have nothing to disclose pertaining to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nishino M, et al. Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nat Rev Clin Oncol. 2017 doi: 10.1038/nrclinonc.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 3.Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582–98. doi: 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- 4.Chen W, Ten Dijke P. Immunoregulation by members of the TGFbeta superfamily. Nat Rev Immunol. 2016;16(12):723–740. doi: 10.1038/nri.2016.112. [DOI] [PubMed] [Google Scholar]
- 5.Harper J, Sainson RC. Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Semin Cancer Biol. 2014;25:69–77. doi: 10.1016/j.semcancer.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 6.Tauriello DVF, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538–543. doi: 10.1038/nature25492. [DOI] [PubMed] [Google Scholar]
- 7.Mariathasan S, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wennerberg E, et al. Barriers to Radiation-Induced In Situ Tumor Vaccination. Front Immunol. 2017;8:229. doi: 10.3389/fimmu.2017.00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vanpouille-Box C, et al. TGFbeta Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res. 2015;75(11):2232–42. doi: 10.1158/0008-5472.CAN-14-3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Formenti SC, et al. Focal Irradiation And Systemic Transforming Growth Factor-beta Blockade in Metastatic Breast Cancer. Clin Cancer Res. 2018 doi: 10.1158/1078-0432.CCR-17-3322. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]