

Abstract
Histone modification, a post-translational modification of histones and involving various covalent tags, such as methyl, phosphate and acetate groups, affects gene expression and hence modulates various cellular events, including growth and proliferation. Consequently histone-modifying proteins have become targets for the development of anti-cancer agents. Thus far, compounds that inhibit the methylation or acetylation of histones have advanced in the clinic, but inhibitors of histone phosphorylation have lagged behind. Haspin is a kinase that phosphorylates histone H3 and is a promising anticancer target. Thus far only a handful of haspin inhibitors have been reported. Using a one flask Doebner/Povarov reaction, we synthesized a library of compounds that potently inhibit haspin with IC50 values as low as 14 nM. Some of these compounds also inhibited the proliferation of cancer cell lines HCT116, HeLa and A375. The ease of synthesis of the new haspin inhibitors, coupled with their anticancer activities make these compounds interesting leads to develop into therapeutics.
Keywords: Haspin, Kinase inhibitor, Doebner/Povarov reaction
Graphical abstract
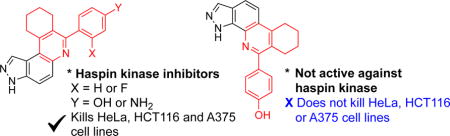
1. Introduction
Epigenetic mechanisms modulate heritable phenotypic changes that are not dictated by changes in DNA sequences. The link between epigenetic modifications and cancer development has been established[1, 2]. Histone modifications represent an important class of epigenetic mechanisms that could be targeted by drugs. The post-translational modifications of histones including methylation, acetylation, ubiquitination, phosphorylation and sumoylation regulate chromatin structure and affect the transcription of various genes[3]. As such intense efforts have been dedicated to the discovery and development of small molecule inhibitors of enzymes that control histone modifications as well as other epigenetic modification enzymes in various cancers[4–9].
Histone phosphorylation and de-phosphorylation is tightly regulated by epigenetic kinases and phosphatases[3, 10]. The phosphorylation of N-terminal tail of histone H3 at residues such as Thr3, S10, Thr11 and S28 has been well documented to regulate mitosis and meiosis in various life forms[3, 10]. Haspin (haploid germ cell-specific protein kinase), a serine/threonine kinase is involved in histone phosphorylation, particularly at Thr3 (H3T3) during mitosis[11, 12]. Haspin is expressed in several cell types and the upregulation or activation of haspin leads to H3T3 phosphorylation[3]. Dai et al demonstrated the vital role of haspin in chromosome alignment by showing that depletion of haspin via RNA interference resulted in decreased H3T3 phosphorylation, leading to the misalignment of chromosomes during metaphase[12]. Hence haspin is thought to play a role in chromosome segregation and might be important in tumor formation[11]. Despite the great potential of haspin being a target for anticancer therapy, only a handful of inhibitors have been documented[13–16]. CHR-6494, a haspin kinase inhibitor, was demonstrated to also possess antitumor properties against melanoma, colon, breast and cervical cancers[13, 17]. CHR-6494 caused a G2/M cell cycle arrest consistent with the role of haspin in mitosis[11–13, 17].
Here we report that compounds containing the 3H-pyrazolo[4,3-f]quinoline scaffold potently inhibit haspin kinase. The compounds also possess anticancer activities against colon, cervical and skin cancers.
2. Results
2.1 Pyrazolo[4,3-f]quinoline compounds inhibit haspin kinase activity
We screened our in-house chemical library using the ADP-Glo™ kinase assay system (Promega, Madison WI) for haspin inhibitors and identified compound 1, a phenolic compound that also contained a pyrazolo[4,3-f]quinoline moiety (synthesized via a Doebner/Povarov reaction[18–21], see Scheme 1), as haspin kinase inhibitors (see Figures 1 & 2).
Scheme 1.
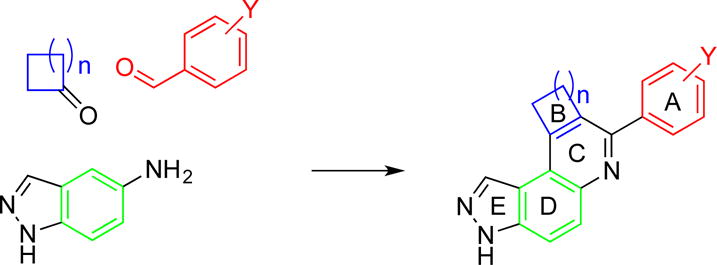
A one-flask synthesis of 3H-pyrazolo[4,3-f]quinoline-containing compounds, using HCl acid as a catalyst.
Figure 1.
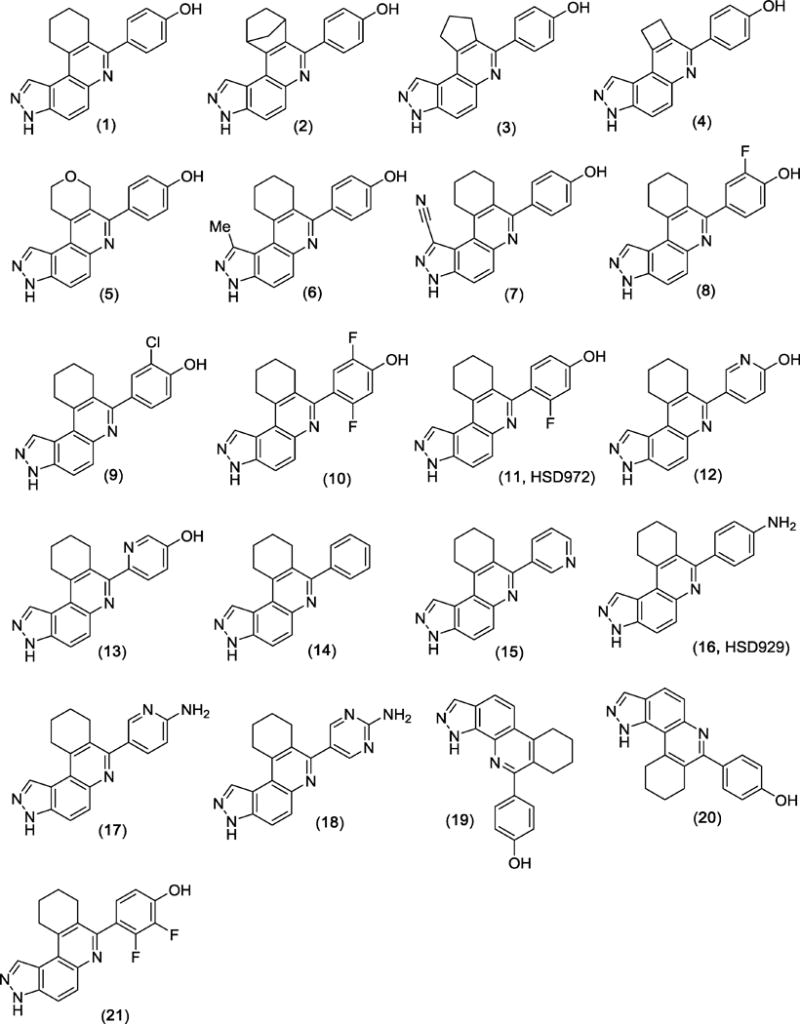
Structures of HSD972 and related compounds
Figure 2.
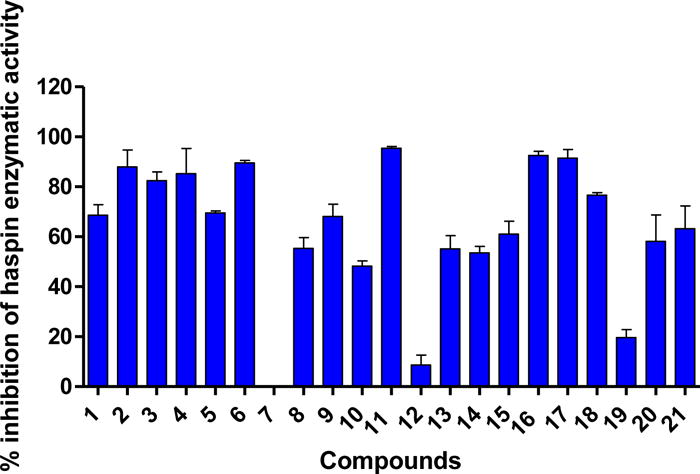
The effect of HSD972 and related compounds on haspin kinase activity. Compounds were tested at 100 nM for 3 h. Error bars represent the SEM of duplicates.
For a structure-activity-relationship (SAR) study, we proceeded to synthesize a series of compound 1 analogs. In one series of analogs, we explored the effects of changing ring B (see Scheme 1 for the naming of the rings) on haspin inhibition (compounds 2, 3, 4 and 5). Next, we explored the effects of substituting position-1 with a methyl (compound 6) or cyano (compound 7) groups. For the third series of analogs, the phenolic moiety (ring A) was substituted with halogens (compounds 8, 9, 10 and 21). We rationalized that the lower halogens (especially F) could affect the acidity of the phenolic OH without a confounding steric issue associated with higher halogen substitution (such as Br and I). The phenolic moiety was also changed to other aryl groups (such as phenyl (14), pyridine (15), hydroxypyridine (12 and 13), aniline (16), aminopyridine (17) and aminopyrimidine (18)). Finally, we designed two analogs to investigate the role that molecular architecture/geometry had on haspin inhibition. In other words, we designed analogs of compound 1, which contained the same moieties but spatially arranged differently (compound 20 containing the 1H-pyrazolo[3,4-f]quinoline moiety, compound 19 containing 1H-pyrazolo[4,3-h]quinoline moiety to compare with compound 1, which contains the 3H-pyrazolo[4,3-f]quinoline moiety). From the in vitro haspin inhibition assay, we observed varying degrees of activity from the compounds (Figure 2). Some compounds, such as HSD972, HSD929, and compound 17 were identified as potent inhibitors of haspin kinase (at 100 nM concentration, these three compounds inhibited over 90% of haspin’s enzymatic activity) whilst other compounds, like compound 7, had no inhibitory activity against haspin (Figure 2).
The SAR studies indicated that substitution at position 1 and the nature of ring B were important for haspin inhibition. For example, whereas the enzymatic inhibition profile of compound 6 (position-1 substituted with Me group) was similar to compound 1 (H at position 1), compound 7, which had a cyano group at position 1 did not inhibit haspin (see Figure 2). The nature of halogen substitution on the phenolic moiety was also important for haspin inhibition. For example, both compounds 8 and 11 (HSD972) are monosubstituted with fluorine but compound 8, which has the fluorine group ortho to the phenolic OH is less active against haspin than HSD972, which has the fluoro group meta to the phenolic OH (Figure 2). Additionally, we observed decreased potency with compounds 10 and 21, which had two fluoro substitutions on ring A. Changing the phenolic OH into amino (compound 16, HSD929) did not affect haspin inhibition. Replacement of the aniline moiety in compound 16 into aminopyridine (compound 17) did not affect haspin inhibition. However, the pyrimidine analog, compound 18, was slightly less active against haspin. compound Compounds 19 and 20 containing the 1H-pyrazolo[4,3-h]quinoline and 1H-pyrazolo[3,4-f]quinoline moieties were not as active against haspin than compound 1, which contains the 3H-pyrazolo[4,3-f]quinoline moiety (Figure 2). Therefore it appears that the 3H-pyrazolo[4,3-f]quinoline moiety is the most optimum core scaffold for developing this class of haspin inhibitors.
From the single concentration inhibition data (Figure 2), compounds HSD972, HSD929 and 17 were chosen for more detailed investigation of haspin inhibition. We determined the half maximal inhibitory concentration (IC50) of these top three active compounds against haspin (Figure 3). From the IC50 analysis, we observed that HSD972 was most potent against haspin with an IC50 value of 14 nM, compared with 45 nM and 61 nM for compounds 17 and HSD929 respectively (Figure 3).
Figure 3.
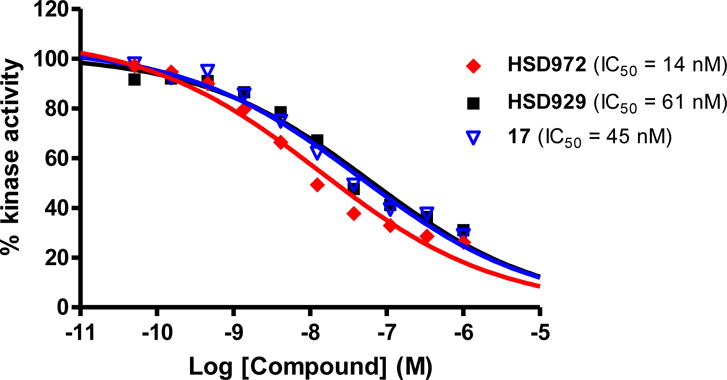
Dose-response curves showing the activity of HSD972, HSD929 and compound 17 against haspin kinase. Experiment was done as a service by Reaction Biology Corp. Data was fitted to a non-linear regression equation using GraphPad Prism 5.0 software. Each data point represents the mean of duplicates.
2.2 Antiproliferative activities of compounds
As earlier stated, haspin inhibitors have been demonstrated to have anticancer activities against various cancers[13, 17]. Following the impressive haspin inhibition activities of the compounds, particularly HSD972, HSD929 and compound 17, we proceeded to investigate whether these compounds possessed any anticancer activities. Precedent literature demonstrated the anticancer activity of CHR-6494 (a haspin inhibitor) against the colon cancer cell line HCT116, the cervical cancer cell line HeLa and as well as human melanoma cell lines RPMI-7951 and COLO-792[13, 17]. We determined the anticancer activities of the synthesized compounds (Figure 1) against, HCT116, HeLa and A375 cell lines by incubating the cell lines with 10 μM of the compounds and determining growth inhibition after 72 h. The compounds displayed varied potencies against the selected cancer cell lines. HCT116 and HeLa cell lines were more sensitive to the compounds than the melanoma A375 cell line (Figure 4). There appeared to be some correlation between haspin inhibition and antiproliferative activity of the most active compounds. For example, compounds 7 and 19, which were poor haspin inhibitors performed poorly in the cellular assays. Compound 12 on the other hand, was a poor haspin inhibitor yet it displayed a moderate antiproliferative activity against HCT116, Hela and A375 cancer cell lines. This suggests that compound 12 inhibits other targets and future studies will attempt to identify the true target of 12. HSD929 and HSD972 were both potent inhibitors of haspin and possessed potent antiproliferative activities against HCT116 and HeLa cell lines (Figure 4). An outliner was compound 1, which is a good haspin inhibitor but did not have good anticancer activities against the three tested cell lines. Various factors, such as drug permeation, stability etc. affect phenotypic outcomes and one of such factors could account for the low correlation between the high degree of haspin inhibition by compound 1 and anticancer activities.
Figure 4.
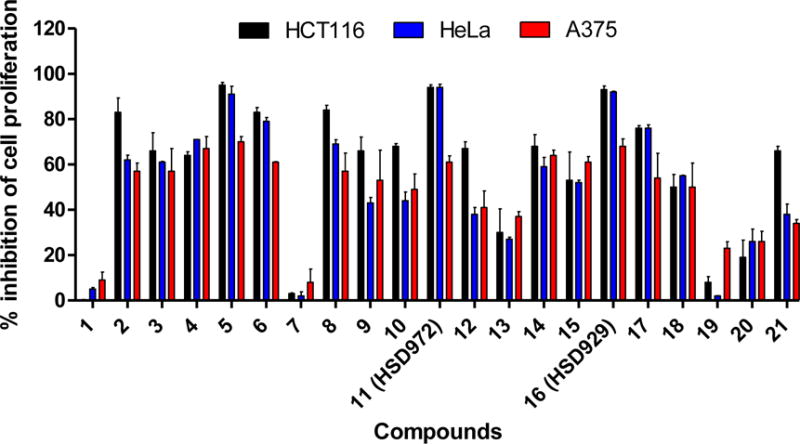
Inhibition of cell viability by HSD972 and analogs. Cells were incubated with 10 μM compounds and viability accessed after 72 h.
Since compounds HSD929 and HSD972 displayed the highest growth inhibition at 10 μM, we decided to determine the half maximum inhibitory concentration (IC50) against HCT116 and HeLa cell lines (Figure 5). HSD929 inhibited HCT116 and HeLa cell lines in a dose-dependent manner with IC50 values of 1.68 μM and 3.6 μM respectively. HSD972 also had similar potency as HSD972 with IC50 values of 2.7 μM and 4.8 μM against both cell lines.
Figure 5.
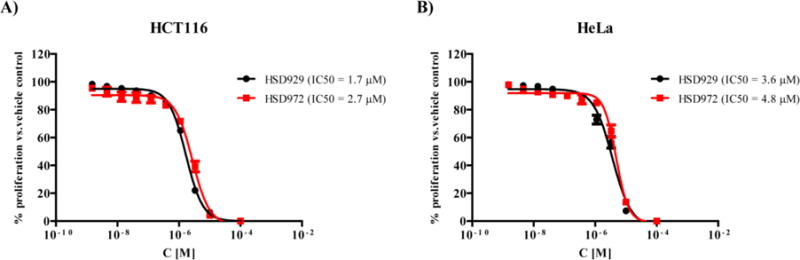
Dose-response curves showing the inhibition of proliferation of HSD929 and HSD972 on A) HCT116 and B) HeLa cell lines. Every data point represent the mean of triplicates and the error bars represent the SEM.
2.3 Haspin-HSD972 interaction at hinge region is similar to haspin-ADP interaction
To gain insights into the interaction between the active compounds and haspin, we used GLIDE in the Schrodinger software package to dock HSD972 to haspin (PDB 3DLZ)[22]. Docking results indicated that HSD972 docked to the active site (Figure 6). HSD972 formed a hydrogen bond interaction with the backbone carbonyl oxygen of Glu606, a hinge region residue[22].
Figure 6.
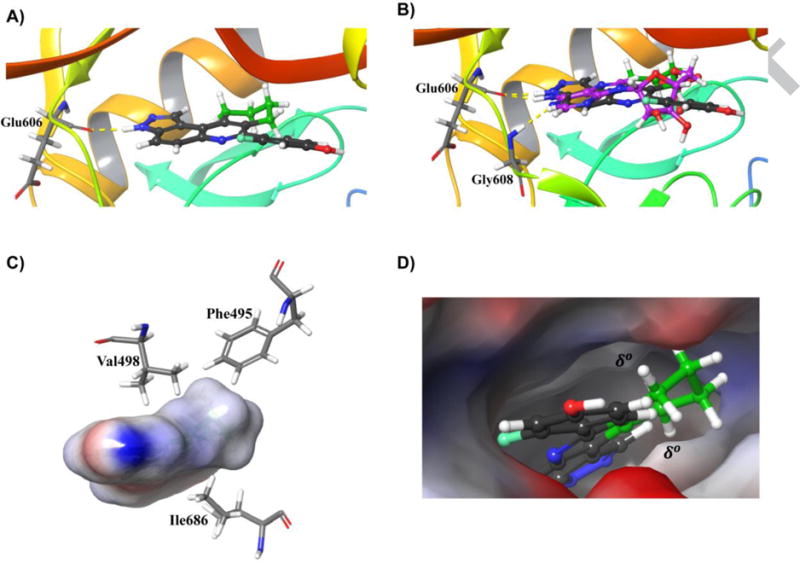
Modeling of the active site interaction between haspin (PDB 3DLZ)[22] and HSD972. A) Hydrogen bond interaction between HSD972 and hinge residue Glu606. B) Overlay of docked HSD972 with crystalized ADP (magenta) in the ATP binding pocket of haspin. Both make hydrogen bonds to Glu606. An additional hinge region interaction between ADP and Gly608 is shown. C) Hydrophobic residues around the cyclohexyl ring B of HSD972. D) Hydrophobic patches above and below the cyclohexyl ring B in the active site. The atoms of the cyclohexyl ring are colored green. Docking was performed using GLIDE in the Schrodinger software package.
The haspin Glu606 residue is known to interact with ATP in the binding pocket[22]. The N6-amino group of ADP purine ring makes a hydrogen bond contact with Glu606[22]. Interestingly, when the structures of HSD972 and crystalized ADP molecule were superimposed, the N6-amino group of the purine ring of ADP overlaid with the N3 atom of the 8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridine moiety of HSD972 (Figure 6). Both nitrogen atoms are attached to hydrogens that could serve as hydrogen bond donors to the backbone carbonyl oxygen of Glu606. Electrostatic surface potential map analysis of HSD972 revealed that the N3-H moiety had a partial positive charge (blue color, Figure S1), which allows it to be a hydrogen bond donor. Analysis of the electrostatic surface potential of haspin reveals hydrophobic patches (δ°) (Figure 6) in the active site created by residues such as Phe495, Val498 and Ile686. The hydrophobic cyclohexyl ring B of HSD972 docks in this pocket (Figure 6 C and D).
2.4 HSD972 affects cell cycle progression and apoptosis
The haspin inhibitor CHR-6494 was shown to induce a G2/M cell cycle arrest as well as apoptosis in HCT116 and HeLa cells[13]. Therefore, we wondered whether HSD972 could also induce cell cycle arrest and apoptosis in HCT116 cell line. Using the propidium iodide (PI)-staining with flow cytometry, we determined the distribution of cells in the different cell cycle phases after 24 h of treatment with HSD972 (5X IC50). It was observed that compared to the DMSO-treated cells, the population of cells in the G2/M phase was higher in the presence of HSD972 (Figure 7). A corresponding decrease in the population of cells in G1 phase was observed. A caspase 3/7 activity assay also revealed that HSD972 increased the activity of caspase 3/7 (a sign of apoptosis) after 72 hours in HCT116 cells, see Figure 8.
Figure 7.
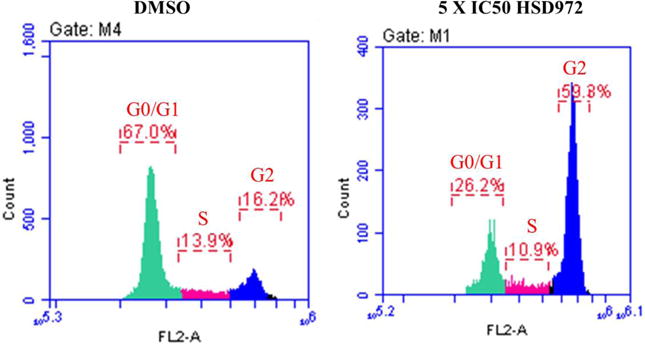
The effect of HSD972 on cell cycle progression in HCT116 cells after 24 h. An increase in cells in G2/M phase was observed with 13.5 μM HSD972 compared to DMSO [DMSO G2 (16.2%) versus HSD972 G2 (59.3%)].
Figure 8.
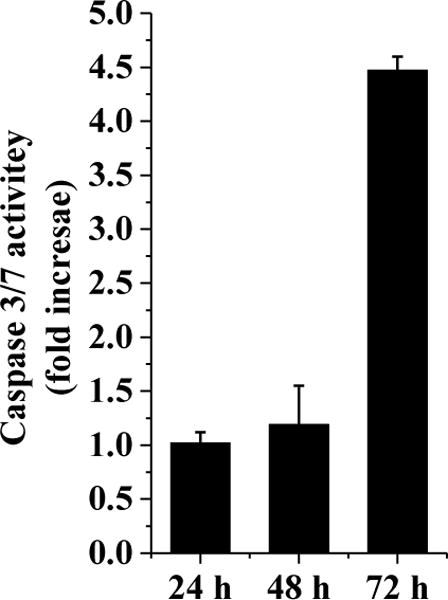
HSD972 causes an increase in caspase 3/7 activities in HCT116 cells. Data represent the normalized value to DMSO at each time point.
3. Conclusions
In this report we have identified inhibitors of haspin kinase activity and demonstrated their ability to inhibit cancer cell proliferation. HSD972 and HSD929 were the most potent inhibitors with nanomolar IC50 values for the inhibition of haspin activity. Both compounds also inhibited HCT116 and HeLa proliferation at single digit micromolar concentrations. The ease of synthesis of the reported compounds (a one flask synthesis) makes these novel pyrazolo[4,3-f]quinoline-containing kinase inhibitors ideal lead compounds to develop into anticancer therapeutics.
4. Experimental
4.1 Chemistry
4.1.1 General considerations
Unless noted otherwise, all reagents and solvents were purchased from commercial sources and used as received. All reactions were performed in a screw-cap 20 mL vials. The 1H and 13C NMR spectra were obtained in CD3OD or (CD3)2SO as solvent using a 500 MHz spectrometer with an internal standard Me4Si. Chemical shifts are reported in parts per million (δ) and are calibrated using residual undeuterated solvent as an internal reference. Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm) (multiplicity, coupling constant (Hz), integration). Multiplicities are reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, or combinations thereof. High resolution mass spectra (HRMS) were given using electron spray ionization (ESI) technique and as TOF mass analyzer. All the synthesized compounds were characterized by 1H NMR, 13C NMR, and HRMS data.
4.1.2 General procedure for the multicomponent reaction
A mixture of amine (1 mmol) and aldehyde (1 mmol) in 3 mL of absolute ethanol was refluxed for 2 h followed by addition of cyclic ketone (2.5 mmol) to the reaction mixture. A catalytic amount of conc. hydrochloric acid was added, and the reaction was continued to reflux for 6–12 h. Reaction mixture dissolved in ethyl acetate (50 mL), washed with sodium bicarbonate solution and water (10 mL × 2). The organic layer was dried (Na2SO4), concentrated under reduced pressure, and purified by silica gel chromatography [dichloromethane:methanol (99:01 to 80:20) or hexane:ethylacetate (20:80 to 05:95)] to give the desired cyclized compound. (Note: Sometime product may get precipitated out, which was filtered, washed with absolute ethanol and further purified by column chromatography).
4.1.2.1 4-(8,9,10,11-Tetrahydro-3H-pyrazolo[4,3-a]phenanthridin-7-yl)phenol (1)
Off white solid (189 mg, 60%).1H NMR (500 MHz, DMSO-d6) δ 9.66 (s, 1H), 8.54 (s, 1H), 7.83 (q, J = 9.1 Hz, 2H), 7.48 – 7.29 (m, 2H), 7.02 – 6.73 (m, 2H), 3.33-3.34 (m, 2H), 2.79 (t, J = 6.1 Hz, 2H), 2.13 – 1.94 (m, 2H), 1.85 – 1.58 (m, 2H); 13C NMR (126 MHz, DMSO) δ 157.71, 156.82, 143.40, 138.61, 136.25, 131.76, 130.89, 129.56, 121.53, 116.48, 116.04, 115.15, 114.37, 29.76, 29.18, 22.66, 22.63; HRMS (ESI) m/z calcd for C20H18N3O [M + H]+ 316.1449, found 316.1458.
4.1.2.2 4-(8,9,10,11-Tetrahydro-3H-8,11-methanopyrazolo[4,3-a]phenanthridin-7-yl)phenol (2)
A mixture of indazole (1 mmol), 4-hydroxybenzaldehyde (1 mmol) and norcamphor (2 mmol) in 6 mL tetrahydrofuran was refluxed for 6-12 h in presence of iodine (10 mol%). After completion of reaction, reaction mixture was extracted with ethyl acetate and washed with sodium thiosulphate aqueous solution (5%). The organic layer was dried (Na2SO4), concentrated under reduced pressure, purified by silica gel chromatography [ethyl acetate:hexane (60:40)] to give the desired cyclized compound.
Off white solid (82 mg, 25%).1H NMR (500 MHz, MeOD-d4) δ 8.64 (s, 1H), 7.97 – 7.90 (m, 1H), 7.80 (d, J = 9.2 Hz, 1H), 7.62 (d, J = 8.4 Hz, 2H), 6.97 (d, J = 8.4 Hz, 2H), 4.34 (s, 1H), 3.76 (s, 1H), 2.33 – 2.18 (m, 2H), 1.93 (d, J = 9.4 Hz, 1H), 1.77 (d, J = 9.0 Hz, 1H), 1.44 – 1.31 (m, 2H). 13C NMR (126 MHz, MeOD) δ 157.96, 153.18, 150.46, 144.56, 139.98, 138.03, 134.29, 130.56, 130.03, 128.51, 116.16, 114.92, 113.81, 49.47, 43.72, 42.28, 26.38, 24.64. HRMS (ESI) m/z calcd for C21H18N3O [M + H] + 328.1450, found 328.1459.
4.1.2.3 4-(3,8,9,10-Tetrahydrocyclopenta[c]pyrazolo[4,3-f]quinolin-7-yl)phenol (3)
Off-white solid (168 mg, 56%). 1H NMR (500 MHz, DMSO-d6) δ 8.42 (s, 1H), 7.85 (d, J = 9.2 Hz, 1H), 7.81 (dd, J = 9.1, 0.9 Hz, 1H), 7.74 (d, J = 8.6 Hz, 2H), 6.89 (d, J = 8.6 Hz, 2H), 3.41 (t, J = 7.6 Hz, 2H), 3.21 (t, J = 7.4 Hz, 2H), 2.20 – 2.22 (m, 2H); 13C NMR (126 MHz, DMSO) δ 158.20, 152.26, 149.32, 144.91, 135.59, 131.44, 130.38, 129.22, 118.73, 116.70, 115.46, 33.48, 33.40, 24.97; HRMS (ESI) m/z calcd for C19H16N3O [M + H]+ 302.1293, found 302.1295.
4.1.2.4 4-(8,9-Dihydro-3H-cyclobuta[c]pyrazolo[4,3-f]quinolin-7-yl)phenol (4)
Off-white solid (152 mg, 53%). 1H NMR (500 MHz, DMSO-d6) δ 9.80 (s, 1H), 8.32 (s, 1H), 8.01 (dd, J = 8.6, 1.4 Hz, 2H), 7.82 (d, J = 9.5 Hz, 1H), 7.78 (d, J = 9.1 Hz, 1H), 6.91 (dd, J = 8.6, 1.4 Hz, 2H), 3.73 – 3.64 (m, 2H), 3.62 – 3.54 (m, 2H); 13C NMR (126 MHz, DMSO) δ 159.12, 149.75, 148.04, 144.97, 137.96, 136.56, 134.42, 130.02, 129.03, 128.87, 116.88, 116.11, 115.24, 114.32, 32.33, 30.24; HRMS (ESI) m/z calcd for C18H14N3O [M + H]+ 288.1137, found 288.1139.
4.1.2.5 4-(3,8,10,11-Tetrahydropyrano[3,4-c]pyrazolo[4,3-f]quinolin-7-yl)phenol (5)
Off-white solid (206 mg, 65%). 1H NMR (500 MHz, DMSO-d6) δ 9.69 (s, 1H), 8.52 (s, 1H), 7.85 (q, J = 9.2 Hz, 2H), 7.40 (d, J = 8.5 Hz, 2H), 6.87 (d, J = 8.6 Hz, 2H), 4.77 (s, 2H), 4.11 (t, J = 5.9 Hz, 2H), 3.34 (d, J = 6.8 Hz, 2H); 13C NMR (126 MHz, DMSO) δ 158.09, 153.88, 144.20, 139.12, 138.55, 135.81, 130.66, 130.59, 129.53, 127.54, 120.80, 116.34, 115.40, 114.82, 67.17, 64.45, 28.70; HRMS (ESI) m/z calcd for C19H16N3O2 [M + H]+ 318.1243, found 318.1244.
4.1.2.6 4-(1-Methyl-8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridin-7-yl)phenol (6)
Off-white solid (132 mg, 40%). 1H NMR (500 MHz, DMSO-d6) δ 10.37 (s, 1H), 8.24 – 8.03 (m, 2H), 7.56 (d, J = 8.6 Hz, 2H), 7.04 (d, J = 8.6 Hz, 2H), 3.62 (t, J = 5.9 Hz, 2H), 2.94 (s, 3H), 2.81 (t, J = 6.2 Hz, 2H), 1.93 – 1.73 (m, 4H); 13C NMR (126 MHz, DMSO) δ 163.50, 160.27, 153.31, 150.55, 135.81, 135.11, 131.86, 131.49, 125.28, 122.45, 119.96, 116.00, 112.81, 32.79, 27.62, 21.69, 21.32; HRMS (ESI) m/z calcd for C21H20N3O [M + H]+ 330.1606, found 330.1606.
4.1.2.7 7-(4-Hydroxyphenyl)-8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridine-1-carbonitrile (7)
White solid (210 mg, 61%). 1H NMR (500 MHz, MeOD-d4) δ 8.32 (d, J = 9.3 Hz, 1H), 8.20 (d, J = 9.2 Hz, 1H), 7.62 (d, J = 8.6 Hz, 2H), 7.09 (d, J = 8.5 Hz, 2H), 3.91 (t, J = 6.3 Hz, 2H), 3.00 (t, J = 6.3 Hz, 2H), 2.24 – 2.09 (m, 2H), 2.01 – 1.90 (m, 2H). 13C NMR (126 MHz, MeOD) δ 160.62, 155.55, 153.29, 139.97, 135.54, 133.16, 130.98, 122.63, 121.71, 121.08, 119.18, 115.88, 115.65, 115.28, 33.30, 27.88, 20.92, 20.63; HRMS (ESI) m/z calcd for C21H21N4O [M + H]+ 345.1715, found 345.1720.
4.1.2.8 2-Fluoro-4-(8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridin-7-yl)phenol (8)
Off white solid (207 mg, 62%). 1H NMR (500 MHz, DMSO-d6) δ 10.81 (s, 1H), 8.83 (s, 1H), 8.26 (d, J = 9.2 Hz, 1H), 8.21 (d, J = 9.2 Hz, 1H), 7.66 (dd, J = 11.8, 2.2 Hz, 1H), 7.42 (dd, J = 8.3, 2.1 Hz, 1H), 7.26 (t, J = 8.6 Hz, 1H), 3.49 (t, J = 6.5 Hz, 2H), 2.83 (t, J = 6.2 Hz, 2H), 2.11 – 1.98 (m, 2H), 1.84 – 1.70 (m, 2H); 13C NMR (126 MHz, DMSO) δ 154.99, 151.91, 149.98, 147.90, 135.34, 132.04, 127.34, 123.18, 120.34, 118.67, 118.51, 118.31, 114.88, 40.49, 40.33, 40.25, 40.16, 40.08, 39.99, 39.83, 39.66, 39.49, 30.93, 28.11, 21.80, 21.69; HRMS (ESI) m/z calcd for C20H17FN3O [M + H]+ 334.1355, found 334.1351.
4.1.2.9 2-Chloro-4-(8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridin-7-yl)phenol (9)
Off-white solid (191 mg, 55%). 1H NMR (500 MHz, DMSO-d6) δ 11.19 (s, 1H), 8.68 (s, 1H), 8.14 (s, 2H), 7.78 (d, J = 2.2 Hz, 1H), 7.56 (dd, J = 8.4, 2.2 Hz, 1H), 7.31 (d, J = 8.5 Hz, 1H), 3.33 (d, J = 6.6 Hz, 2H), 2.80 (d, J = 6.3 Hz, 2H), 2.00 (dt, J = 12.4, 4.6 Hz, 2H), 1.80 – 1.69 (m, 2H); 13C NMR (126 MHz, DMSO) δ 155.92, 152.58, 149.60, 135.00, 131.94, 131.83, 130.50, 123.67, 122.81, 120.32, 120.19, 116.98, 114.53, 30.78, 28.06, 21.65, 21.61; HRMS (ESI) m/z calcd for C20H17ClN3O [M + H]+ 350.1060, found 350.1066.
4.1.2.10 2,5-Difluoro-4-(8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridin-7-yl)phenol (10)
Off-white solid (159 mg, 45%). 1H NMR (500 MHz, Methanol-d4) δ 8.57 (s, 1H), 7.86 (q, J = 9.1 Hz, 2H), 7.15 (dd, J = 10.8, 6.7 Hz, 1H), 6.81 (dd, J = 10.5, 7.1 Hz, 1H), 3.38 (s, 2H), 2.73 (s, 2H), 2.06 – 2.09 (m, 2H), 1.86 (d, J = 9.8 Hz, 2H); 13C NMR (126 MHz, MeOD) δ 156.74 (J = 239.16 Hz), 151.59, 148.92 (J = 238.14 Hz), 146.61, 143.45, 143.06, 138.67, 135.89, 130.94, 128.32, 122.39, 118.20, 116.93, 116.06, 113.85, 104.69, 29.48, 27.05, 22.11, 21.80; HRMS (ESI) m/z calcd for C20H218 F2N3O [M + H]+ 354.1418, found 354.1421.
4.1.2.11 3-Fluoro-4-(8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridin-7-yl)phenol (11, HSD972)
White solid (186 mg, 56%). 1H NMR (500 MHz, DMSO-d6) δ 8.57 (s, 1H), 7.84 (dd, J = 9.0, 0.9 Hz, 1H), 7.79 (d, J = 9.1 Hz, 1H), 7.22 (t, J = 8.6 Hz, 1H), 6.72 (dd, J = 8.3, 2.3 Hz, 1H), 6.66 (dd, J = 11.8, 2.3 Hz, 1H), 3.30 (t, J = 6.6 Hz, 2H), 2.61 (t, J = 6.0 Hz, 2H), 2.00 – 1.91 (m, 2H), 1.79 – 1.69 (m, 2H); 13C NMR (126 MHz, DMSO) δ 161.25 (J = 245.11 Hz), 159.59, 152.73, 143.80, 142.04, 132.20, 132.16, 130.53, 129.53, 122.03, 119.48, 119.35, 116.25, 112.08, 102.89, 102.70, 29.48, 27.50, 22.64, 22.24; HRMS (ESI) m/z calcd for C20H17FN3O [M + H]+ 334.1356, found 334.1360.
4.1.2.12 5-(8,9,10,11-Tetrahydro-3H-pyrazolo[4,3-a]phenanthridin-7-yl)pyridin-2-ol (12)
1H NMR (500 MHz, DMSO-d6) δ 11.86 (s, 1H), 8.53 (s, 1H), 7.83 (d, J = 9.0 Hz, 1H), 7.80 – 7.73 (m, 2H), 7.65 (d, J = 2.6 Hz, 1H), 6.42 (d, J = 9.4 Hz, 1H), 3.25 (t, J = 6.6 Hz, 2H), 2.84 (t, J = 6.1 Hz, 2H), 2.01 – 1.92 (m, 2H), 1.80 – 1.68 (m, 2H); 13C NMR (126 MHz, DMSO) δ 162.21, 152.68, 143.74, 142.84, 142.59, 136.37, 129.51, 129.43, 121.76, 119.38, 118.73, 116.21, 29.69, 28.78, 22.59, 22.54; HRMS (ESI) m/z calcd for C19H17N4O [M + H]+ 317.1402, found 317.1411.
4.1.2.13 6-(8,9,10,11-Tetrahydro-3H-pyrazolo[4,3-a]phenanthridin-7-yl)pyridin-3-ol (13)
Off-white solid (124 mg, 39%).1H NMR (500 MHz, MeOD-d4) δ 8.59 (s, 1H), 8.22 (dd, J = 2.8, 0.7 Hz, 1H), 7.91 (d, J = 9.7 Hz, 1H), 7.84 (d, J = 9.0 Hz, 1H), 7.56 (dd, J = 8.4, 0.7 Hz, 1H), 7.41 (dd, J = 8.5, 2.8 Hz, 1H), 3.46 – 3.35 (m, 2H), 2.83 (t, J = 6.2 Hz, 2H), 2.12 – 2.04 (m, 2H), 1.90 – 1.79 (m, 2H); 13C NMR (126 MHz, MeOD) δ 154.91, 154.11, 149.18, 143.77, 142.83, 138.73, 136.11, 135.92, 130.22, 128.53, 125.24, 123.34, 122.50, 116.13, 113.82, 29.64, 27.64, 22.11, 21.92.; HRMS (ESI) m/z calcd for C19H17N4O [M + H]+ 317.1402, found 317.1397.
4.1.2.14 7-Phenyl-8,9,10,11-tetrahydro-3H-pyrozolo[4,3-a]phenanthridine (14)
Off-white solid (235 mg, 78%).1H NMR (500 MHz, DMSO-d6) δ 8.56 (s, 1H), 7.84 (dd, J = 9.1, 0.9 Hz, 1H), 7.80 (d, J = 9.1 Hz, 1H), 7.54 – 7.51 (m, 2H), 7.48 – 7.44 (m, 2H), 7.44 – 7.39 (m, 1H), 3.29 (t, J = 6.5 Hz, 2H), 2.74 (t, J = 6.1 Hz, 2H), 2.02 – 1.91 (m, 2H), 1.77 – 1.64 (m, 2H); 13C NMR (126 MHz, DMSO) δ 156.89, 143.73, 142.34, 141.33, 139.70, 135.12, 129.62, 129.45, 129.22, 128.39, 128.15, 121.89, 116.26, 115.24, 29.64, 28.95, 22.59, 22.49; HRMS (ESI) m/z calcd for C20H18N3 [M + H]+ 300.1501, found 300.1501.
4.1.2.15 7-(Pyridin-3-yl)-8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridine (15)
Off-white solid (135 mg, 45%). 1H NMR (500 MHz, MeOD-d4) δ 9.06 (s, 2H), 8.73 (s, 1H), 8.42 (d, J = 7.3 Hz, 1H), 8.11 (d, J = 8.4 Hz, 1H), 7.96 (d, J = 8.8 Hz, 2H), 3.55 – 3.46 (m, 2H), 2.95 – 2.81 (m, 2H), 2.20 – 2.08 (m, 2H), 1.99 – 1.84 (m, 2H); 13C NMR (126 MHz, MeOD) δ 150.86, 148.85, 148.27, 147.36, 140.15, 138.36, 131.44, 123.81, 122.94, 119.15, 115.51, 30.47, 27.89, 21.51, 21.42; HRMS (ESI) m/z calcd for C19H17N4 [M + H]+ 301.1453, found 301.1465.
4.1.2.16 4-(8,9,10,11-Tetrahydro-3H-pyrazolo[4,3-a]phenanthridin-7-yl)aniline (16)
Orange solid (192 mg, 61%). 1H NMR (500 MHz, DMSO-d6) δ 8.60 (s, 1H), 8.03 – 7.90 (m, 2H), 7.38 (d, J = 8.2 Hz, 2H), 6.74 (d, J = 8.2 Hz, 2H), 3.30 (t, J = 6.4 Hz, 2H), 2.85 – 2.77 (m, 2H), 2.05 – 1.89 (m, 2H), 1.79 – 1.65 (m, 2H); 13C NMR (126 MHz, DMSO) δ 162.76, 154.32, 150.10, 147.41, 138.78, 136.07, 131.27, 130.64, 124.43, 121.78, 115.78, 114.97, 113.81, 111.39, 30.33, 28.84, 22.22; HRMS (ESI) m/z calcd for C20H19N4 [M + H]+ 315.1610, found 315.1612.
4.1.2.17 5-(8,9,10,11-Tetrahydro-3H-pyrazolo[4,3-a]phenanthridin-7-yl)pyridin-2-amine (17)
Yellow solid (205 mg, 65%). 1H NMR (500 MHz, MeOD-d4) δ 8.51 (s, 1H), 8.16 (d, J = 2.1 Hz, 1H), 8.14 (dd, J = 9.0, 2.2 Hz, 1H), 7.88 (d, J = 9.1 Hz, 1H), 7.83 (d, J = 9.2 Hz, 1H), 7.09 (d, J = 9.0 Hz, 1H), 3.26 (t, J = 6.6 Hz, 2H), 2.87 (t, J = 6.1 Hz, 2H), 2.06 (ddt, J = 9.2, 6.5, 3.2 Hz, 2H), 1.87 – 1.84 (m, 2H); 13C NMR (126 MHz, MeOD) δ 155.32, 150.42, 145.84, 143.67, 143.48, 141.93, 138.13, 130.19, 126.90, 123.86, 122.70, 119.27, 115.60, 111.95, 29.80, 28.11, 21.86, 21.86; HRMS (ESI) m/z calcd for C19H18N5 [M + H]+ 316.1562, found 316.1559.
4.1.2.18 5-(8,9,10,11-Tetrahydro-3H-pyrazolo[4,3-a]phenanthridin-7-yl)pyrimidin-2-amine (18)
Yellow solid (152 mg, 48%).; 1H NMR (500 MHz, DMSO-d6) δ 8.61 – 8.47 (m, 3H), 7.84 (s, 2H), 6.85 (s, 2H), 3.35 – 3.30 (m, 2H), 2.94 – 2.85 (m, 2H), 2.04 – 1.95 (m, 2H), 1.83 – 1.73 (m, 2H); 13C NMR (126 MHz, DMSO) δ 163.24, 158.64, 152.40, 143.92, 142.53, 138.65, 136.38, 129.80, 129.66, 123.40, 121.74, 116.45, 114.46, 29.74, 28.82, 22.64, 22.59; HRMS (ESI) m/z calcd for C18H17N6 [M + H]+ 317.1515, found 317.1515.
4.1.2.19 4-(6,7,8,9-Tetrahydro-3H-pyrazolo[4,3-c]phenanthridin-5-yl)phenol (19)
Pale yellow solid (161 mg, 51%).1H NMR (500 MHz, MeOD-d4) δ 8.10 (s, 1H), 7.73 (d, J = 9.0 Hz, 1H), 7.59 (d, J = 9.0 Hz, 1H), 7.38 (d, J = 8.5 Hz, 2H), 6.92 (d, J = 8.6 Hz, 2H), 3.18 (t, J = 6.5 Hz, 2H), 2.76 (t, J = 6.2 Hz, 2H), 2.03 – 1.91 (m, 2H), 1.80 – 1.70 (m, 2H).; 13C NMR (126 MHz, MeOD) δ 159.00, 157.94, 143.72, 137.36, 134.24, 133.58, 131.34, 130.05, 130.02, 129.40, 125.19, 121.58, 119.04, 116.10, 114.76, 28.52, 26.20, 22.31, 21.98.; HRMS (ESI) m/z calcd for C20H18N3O [M + H]+ 316.1450, found 316.1459.
4.1.2.20 4-(8,9,10,11-Tetrahydro-1H-pyrazolo[3,4-a]phenanthridin-7-yl)phenol (20)
Off-white solid (182 mg, 58%). 1H NMR (500 MHz, DMSO-d6) δ 8.31 (s, 1H), 7.87 (d, J = 8.9 Hz, 1H), 7.51 (d, J = 8.9 Hz, 1H), 7.39 (d, J = 8.5 Hz, 2H), 6.85 (d, J = 8.5 Hz, 2H), 3.50 (t, J = 6.6 Hz, 2H), 2.79 (t, J = 6.1 Hz, 2H), 1.97 – 1.92 (m, 2H), 1.77 – 1.70 (m, 2H); 13C NMR (126 MHz, DMSO) δ 158.40, 157.95, 146.70, 142.55, 131.73, 130.87, 128.50, 124.50, 122.19, 119.87, 116.93, 115.17, 29.97, 29.12, 22.56; HRMS (ESI) m/z calcd for C20H18N3O [M + H]+ 316.1450, found 316.1443.
4.1.2.21 2,3-Difluoro-4-(8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridin-7-yl)phenol (21)
Off-white solid (177 mg, 50%). 1H NMR (500 MHz, DMSO-d6) δ 10.56 (s, 1H), 8.56 (s, 1H), 7.92 – 7.76 (m, 2H), 7.05 (td, J = 8.2, 2.0 Hz, 1H), 6.90 (td, J = 8.3, 1.6 Hz, 1H), 3.33–3.32 (m, 2H), 2.62 (t, J = 6.1 Hz, 2H), 1.96 (dt, J = 11.1, 5.9 Hz, 2H), 1.75 – 1.73 (m, 2H); 13C NMR (126 MHz, DMSO) δ 151.48, 149.77(d, J = 243.18 Hz), 147.03, 143.67, 142.40, 141.06 d, J = 240.66 Hz), 138.74, 136.40, 130.47, 129.50, 125.40, 122.18, 120.81, 116.39, 114.56, 113.32, 29.50, 27.48, 22.58, 22.19; HRMS (ESI) m/z calcd for C20H16F2N3O [M + H]+ 352.1261, found 352.1256.
4.2 Biological Evaluation
4.2.1 Materials
Haspin kinase was purchased from SignalChem (Richmond, BC, Canada). ADP-Glo™ kinase assay system and CellTiter-Blue® cell viability assay were purchased from Promega (Madison, WI, USA).
4.2.2 Cells and cell culture
HCT116, HeLa and A375 cells were used in this study. Routinely, HCT116 was cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS). HeLa and A375 cells were cultured in DMEM medium supplemented with 10% FBS. Cultures were incubated at 37 °C with 5% CO2.
4.2.3 Haspin kinase activity
The effect of compounds on haspin kinase activity was assayed using the ADP-Glo™ kinase assay system (Promega, Madison WI). The kinase reaction was performed in 384 well plates in a 5 μL volume containing reaction buffer (25 mM MOPS (pH 7.2) 12.5mM β-glycerol-phosphate, 25 mM MgCl2, 5 mM EGTA, 2 mM EDTA, 0.25 mM DTT and 0.1 mg/ml BSA), 10 μM ATP, 0.1 mg/mL casein protein with 100 nM compound and 5 ng/μL haspin kinase. The reaction was incubated for 3 h at room temperature (23 – 25 °C) after which the ADP-Glo™ kinase assay system was used to determine haspin activity following the manufacturer’s protocol.
4.2.4 Antiproliferative activity
To determine the effect of compounds on the proliferation of cancer cells, HCT116, HeLa and A375 cells were seeded into 96 well plates at 4000 cells/well for HCT116 and 2000 cells/well for HeLa and A375 cells in 190 μL of medium. After 18 h to 24 h, cultures were treated with 10 μL of compounds diluted in medium to yield a final concentration of 10 μM for the screening. For the dose response curves, compounds were serially diluted in DMSO before being further diluted into medium. The cultures were ended after 72 h by the addition of 10 μL CellTiter-Blue® cell viability assay (Promega, Madison WI). The plates were incubated for a further 4 hours before the viability assayed by measuring the fluorescence with excitation at 560 nm and emission at 590 nm on a BioTek Cytation 5 Cell Imaging multi-mode reader. Data were normalized to the DMSO control. Dose-response curves were generated by fitting data to a non-linear regression analysis using GraphPad Prism 5.0 Software (La Jolla CA).
4.2.5 Molecular docking analysis
Docking of HSD972 to haspin (PDB 3DLZ) [22] was performed using the Schrodinger software packages using default settings. Briefly, HSD972 was optimized for docking using the default settings of the LigPrep module. Haspin (PDB 3DLZ) was imported into Maestro 11.3.016 and the Protein preparation wizard was used to optimize the PDB file. The Receptor Grid Generation module was used to generate a grid around the active site, which was used in the Ligand docking module with the optimized ligands for docking. HSD972 and ADP were both viewed in the active site of Haspin at the same time to determine the binding position of HSD972 relative to ADP.
4.2.6 Cell cycle analysis
Cell cycle analysis was performed using propidium iodide as a nuclear stain. HCT116 cells (2 × 106) were seeded and incubated with HSD972 for overnight. Harvested cells were fixed with 70% ethanol following overnight incubation at −20 C°. In the next step, the cells were treated with propidium iodide and RNase and analyzed by flow cytometry (BD Bioscience).
4.2.7 Apoptosis
Caspase 3/7 assay was carried out using Caspase 3/7 fluorescence assay kit (Cayman Chemicals). Briefly, 5 × 105 HCT116 cells were seeded in 12 well plates. The cells were treated with HSD972 and were collected after 24, 48 and 72 hours. Fluorescence intensity was measured by BioTek Cytation 5 plate reader.
Supplementary Material
Highlights.
One flask synthesis of haspin kinase inhibitors via Doebner/Povarov reaction.
Most active inhibitor, HSD972, inhibits haspin with IC50 of 14 nM.
Novel pyrazolo[4,3-f]quinoline-containing haspin inhibitors could inhibit the proliferation of HCT116, Hela and A375 cell lines.
HSD972 induces a G2/M cell cycle arrest.
HSD972 causes an increase in caspase 3/7 activities in HCT 116 cells.
Acknowledgments
We thank Purdue University and Elks foundation for financial support. NMR and MS data were acquired by the NMR and MS facilities supported by NIH P30 CA023168.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
HOS is a co-founder of KinaRx LLC, a start-up company interested in developing oncology therapeutics.
References
- 1.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11(5):384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 2.Lund AH, van Lohuizen M. Epigenetics and cancer. Genes & Development. 2004;18(19):2315–2335. doi: 10.1101/gad.1232504. [DOI] [PubMed] [Google Scholar]
- 3.Rossetto D, Avvakumov N, Côté J. Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics. 2012;7(10):1098–108. doi: 10.4161/epi.21975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Z, Patel DJ. Small molecule epigenetic inhibitors targeted to histone lysine methyltransferases and demethylases. Q Rev Biophys. 2013;46(4):349–73. doi: 10.1017/S0033583513000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nebbioso A, Carafa V, Benedetti R, Altucci L. Trials with ‘epigenetic’ drugs: An update. Mol Oncol. 2012;6(6):657–682. doi: 10.1016/j.molonc.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hieu DT, Anh DT, Tuan NM, Hai PT, Huong LT, Kim J, Kang JS, Vu TK, Dung PTP, Han SB, Nam NH, Hoa ND. Design, synthesis and evaluation of novel N-hydroxybenzamides/N-hydroxypropenamides incorporating quinazolin-4(3H)-ones as histone deacetylase inhibitors and antitumor agents. Bioorg Chem. 2017;76:258–267. doi: 10.1016/j.bioorg.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 7.Han C, Li Z, Hou J, Wang Z, Xu D, Xue G, Kong L. Bioactivity evaluation of natural product α-mangostin as a novel xanthone-based lysine-specific demethylase 1 inhibitor to against tumor metastasis. Bioorg Chem. 2017;76:415–419. doi: 10.1016/j.bioorg.2017.12.004. [DOI] [PubMed] [Google Scholar]
- 8.Halsall JA, Turner BM. Histone deacetylase inhibitors for cancer therapy: An evolutionarily ancient resistance response may explain their limited success. Bioessays. 2016;38(11):1102–1110. doi: 10.1002/bies.201600070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morera L, Lübbert M, Jung M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin Epigenetics. 2016;8:57. doi: 10.1186/s13148-016-0223-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Dai J, Higgins JM. Haspin: a mitotic histone kinase required for metaphase chromosome alignment. Cell Cycle. 2005;4(5):665–8. doi: 10.4161/cc.4.5.1683. [DOI] [PubMed] [Google Scholar]
- 12.Dai J, Sultan S, Taylor SS, Higgins JM. The kinase haspin is required for mitotic histone H3 Thr 3 phosphorylation and normal metaphase chromosome alignment. Genes Dev. 2005;19(4):472–88. doi: 10.1101/gad.1267105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huertas D, Soler M, Moreto J, Villanueva A, Martinez A, Vidal A, Charlton M, Moffat D, Patel S, McDermott J, Owen J, Brotherton D, Krige D, Cuthill S, Esteller M. Antitumor activity of a small-molecule inhibitor of the histone kinase Haspin. Oncogene. 2012;31(11):1408–18. doi: 10.1038/onc.2011.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Antoni A, Maffini S, Knapp S, Musacchio A, Santaguida S. A small-molecule inhibitor of Haspin alters the kinetochore functions of Aurora B. J Cell Biol. 2012;199(2):269–84. doi: 10.1083/jcb.201205119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang F, Ulyanova NP, Daum JR, Patnaik D, Kateneva AV, Gorbsky GJ, Higgins JMG. Haspin inhibitors reveal centromeric functions of Aurora B in chromosome segregation. J Cell Biol. 2012;199(2):251–268. doi: 10.1083/jcb.201205106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patnaik D, Xian Jun, Glicksman MA, Cuny GD, Stein RL, Higgins JM. Identification of small molecule inhibitors of the mitotic kinase haspin by high-throughput screening using a homogeneous time-resolved fluorescence resonance energy transfer assay. J Biomol Screen. 2008;13(10):1025–34. doi: 10.1177/1087057108326081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han L, Wang P, Sun Y, Liu S, Dai J. Anti-melanoma activities of haspin inhibitor CHR-6494 deployed as a single agent or in a synergistic combination with MEK inhibitor. J Cancer. 2017;8(15):2933–2943. doi: 10.7150/jca.20319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Denmark SE, Venkatraman S. On the mechanism of the Skraup-Doebner-Von Miller quinoline synthesis. J Org Chem. 2006;71(4):1668–76. doi: 10.1021/jo052410h. [DOI] [PubMed] [Google Scholar]
- 19.Liu Q, Wang C, Li Q, Hou Y, Wu Y, Liu L, Chang W, Li J. Povarov reaction of cycloiminium formed in situ via hydroamination cycloisomerization of homopropargylic amines with electron-rich olefins. J Org Chem. 2017;82(2):950–958. doi: 10.1021/acs.joc.6b02496. [DOI] [PubMed] [Google Scholar]
- 20.Di Pietro O, Viayna E, Vicente-García E, Bartolini M, Ramón R, Juárez-Jiménez J, Clos MV, Pérez B, Andrisano V, Luque FJ, Lavilla R, Muñoz-Torrero D. 1,2,3,4-Tetrahydrobenzo[h][1,6]naphthyridines as a new family of potent peripheral-to-midgorge-site inhibitors of acetylcholinesterase: synthesis, pharmacological evaluation and mechanistic studies. Eur J Med Chem. 2014;73:141–52. doi: 10.1016/j.ejmech.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 21.Damon DB, Dugger RW, Magnus-Aryitey G, Ruggeri RB, Wester RT, Tu M, Abramov Y. Synthesis of the CETP inhibitor torcetrapib: The resolution route and origin of stereoselectivity in the iminium ion cyclization. Org Process Res Dev. 2006;10(6):464–471. [Google Scholar]
- 22.Eswaran J, Patnaik D, Filippakopoulos P, Wang F, Stein RL, Murray JW, Higgins JM, Knapp S. Structure and functional characterization of the atypical human kinase haspin. Proc Natl Acad Sci USA. 2009;106(48):20198–203. doi: 10.1073/pnas.0901989106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.