

Abstract
Viral quasispecies (QS) have long been considered to affect the efficiency of hepatitis C virus (HCV) antiviral therapy, but a correlation between QS diversity and treatment outcomes has not been established conclusively. We previously measured HCV QS diversity by genome-wide quantification of high-resolution mutation load in HCV genotype 1a patients achieving a sustained virological response (1a/SVR) or a null response (1a/null). The current study extended this work into HCV 1a patients experiencing relapse (1a/relapse, n = 19) and genotype 2b patients with SVR (2b/SVR, n = 10). The mean mutation load per patient in 2b/SVR and 1a/relapse was similar, respectively, to 1a/SVR (517.6 ± 174.3 vs 524 ± 278.8 mutations, P = 0.95) and 1a/null (829.2 ± 282.8 vs 805.6 ± 270.7 mutations, P = 0.78). Notably, a deleterious mutation load, as indicated by the percentage of non-synonymous mutations, was highest in 2b/SVR (33.2 ± 8.5 %) as compared with 1a/SVR (23.6 ± 7.8 %, P = 0.002), 1a/null (18.2 ± 5.1 %, P = 1.9 × 10− 7) or 1a/relapse (17.8 ± 5.3 %, P = 1.8 × 10− 6). In the 1a/relapse group, continuous virus evolution was observed with excessive accumulation of a deleterious load (17.8 ± 5.3 % vs 35.4 ± 12.9 %, P = 3.5 × 10− 6), supporting the functionality of Muller's ratchet in a treatment-induced population bottleneck. Taken together, the magnitude of HCV mutation load, particularly the deleterious mutation load, provides an evolutionary explanation for the emergence of multiple response patterns as well as an overall high SVR rate in HCV genotype 2 patients. Augmentation of Muller's ratchet represents a potential strategy to reduce or even eliminate viral relapse in HCV antiviral therapy.
Introduction
As with most RNA viruses, hepatitis C virus (HCV) presents a typical nature of viral quasispecies (QS), circulating as a heterogeneous population in infected individuals (Domingo et al., 2012). This is believed to be an essential factor modulating disease pathogenesis, mediating resistance to antiviral therapy and contributing to the difficulty in vaccine development. The intra-patient population heterogeneity of HCV, i.e. HCV QS, has thus been studied extensively for potential relevance to numerous clinical traits. However, it is difficult to reach a consensus in almost all HCV-associated clinical settings. For instance, slow disease progression or favourable response patterns in antiviral therapy appears to be correlated with either high (Farci et al., 2006; Li et al., 2010, 2011; Lyra et al., 2002; Qin et al., 2005; Sullivan et al., 2007) or low (Morishima et al., 2006; Wang et al., 2007) QS diversity or is independent of the HCV QS diversity (López-Labrador et al., 1999; Sandres et al., 2000). These discrepancies are hard to explain without the consideration of methodological shortcomings. One such shortcoming is the low resolution of mutation frequency of no more than 20 % in conventional methods to decipher viral QS diversity, such as gel-based shift assays or cloning/Sanger sequencing (Larder et al., 1993; Van Laethem et al., 1999). Under the approach of cloning/Sanger sequencing, we have demonstrated that among-patient HCV QS diversity presents an exponential distribution pattern that may account for statistical ambiguity in comparative analysis (Fan et al., 2009). By combining long reverse transcription (RT)-PCR and pyrosequencing, we recently measured HCV QS diversity by direct counting of HCV genome-wide mutations (Wang et al., 2014). At 1 % resolution of mutation frequency, the genome-wide HCV mutation load showed a normal distribution among HCV patients and was significantly different between two response patterns associated with IFN-based antiviral therapy: null and a sustained virological response (SVR) (Wang et al., 2014). Using this new index for HCV QS diversity, the current study extended our work to patients who were infected with HCV genotype 2 or relapsed from pegylated (peg)-IFN-α2a and ribavirin combination therapy. Our data provide a full scenario regarding the role of HCV QS in antiviral therapy, as well as interpretation in terms of evolutionary biology.
Results
HCV mutation load among the response patterns in antiviral therapy
The 454 sequencing of 48 samples (Table 1) produced a total of 1 405 351 reads with a mean read length at 437 bp. After the quality control, 1 147 434 reads (81.6 %) gave a mean base coverage of 1276.4 ± 570, which was deep enough for a saturated quantification of the HCV mutation load (Wang et al., 2014) (Fig. S1, available in the online Supplementary Material). Using our two-step protocol (Wang et al., 2014), 27 556 mutations were identified in the nearly full-length HCV coding region in the 48 samples. The mean mutation load per patient in the group of 1a/relapse_B (sampled at baseline) was 829.2 ± 282.8 mutations, which resembled the group 1a/null (805.6 ± 270.7) rather than the group 1a/SVR (524 ± 278.8 mutations) from our previous study (Wang et al., 2014). However, the mutation load at the time of viral relapse was significantly decreased in comparison with the baseline (829.2 ± 282.8 vs 348.7 ± 185 mutations, respectively; P = 3.8 × 10− 7) (Fig. 1a). Under the SVR pattern, the mutation load in HCV genotype 2b was similar to HCV 1a (517.6 ± 174.3 vs 524 ± 278.8 mutations, P = 0.95) (Fig. 1a). However, the percentage non-synonymous mutation load was very different between these two groups [33.2 ± 8.5 (2b) vs 23.6 ± 7.8 % (1a); P = 0.002] (Fig. 1d). Overall, the SVR of either HCV 2b or 1a had a much higher percentage non-synonymous mutation load than that from patients with null (18.2 ± 5.1 %) or relapse pattern (17.8 ± 5.3 %) (Fig. 1d). The highest percentage non-synonymous mutation load was found at the time of viral relapse in HCV 1a patients (35.4 ± 12.9 %) (Fig. 1d).
Table 1. Overview of demographic, virological, disease status and genetic information of the 29 patients enrolled in the study.
BMI, Body mass index; IS, Ishak scores; M, male; na, not available. HCV RNA titres are expressed as log values. The IL28B genotype was based on the reference single nucleotide polymorphism site rs129798. –, No HCV RNA detection.
| Patients | Response | Age | Sex | Race | Weight | BMI | Genotype | HCV RNA | IS | IL28 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| W00 | W60 | ||||||||||
| 1 | Relapse | 42 | M | White | 79.9 | 27.98 | 1a | 7.298 853 | 6.514 548 | 4 | CC |
| 2 | Relapse | 48 | M | White | 75.4 | 26.4 | 1a | 6.429 752 | 5.904 174 | 2 | CT |
| 3 | Relapse | 54 | M | White | 87.7 | 27.07 | 1a | 7.056 905 | 6.598 791 | 4 | CC |
| 4 | Relapse | 54 | M | White | 106.7 | 32.57 | 1a | 7.064 458 | 6.740 363 | 3 | CC |
| 5 | Relapse | 50 | M | White | 86.9 | 30.43 | 1a | 5.959 041 | 5.650 308 | 5 | CT |
| 6 | Relapse | 54 | M | White | 92.1 | 40.93 | 1a | 6.801 404 | 6.385 606 | 3 | CC |
| 7 | Relapse | 47 | M | White | 83.4 | 26.92 | 1a | 7.521 138 | 5.576 341 | 5 | CT |
| 8 | Relapse | 42 | M | White | 90.3 | 26.67 | 1a | 7.409 933 | 6.374 748 | 5 | na |
| 9 | Relapse | 48 | M | White | 84.9 | 27.41 | 1a | 6.816 241 | 6.840 733 | 4 | CC |
| 10 | Relapse | 43 | M | White | 76.2 | 25.17 | 1a | 6.484 3 | 5.944 976 | 3 | CT |
| 11 | Relapse | 47 | M | White | 71.3 | 23.02 | 1a | 6.988 113 | 6.766 413 | 5 | CC |
| 12 | Relapse | 44 | M | White | 85.2 | 28.47 | 1a | 6.766 413 | 5.619 093 | 4 | CC |
| 13 | Relapse | 52 | M | White | 110.2 | 28.69 | 1a | 6.986 324 | 6.158 362 | 3 | CC |
| 14 | Relapse | 47 | M | White | 69.4 | 22.92 | 1a | 7.008 6 | 6.997 823 | 3 | na |
| 15 | Relapse | 42 | M | White | 99.3 | 33.57 | 1a | 5.996 074 | 5.808 211 | 3 | CT |
| 16 | Relapse | 47 | M | White | 106.4 | 33.58 | 1a | 6.964 731 | 6.49276 | 3 | na |
| 17 | Relapse | 45 | M | White | 110.1 | 33.98 | 1a | 6.674 861 | 5.574 031 | 5 | CT |
| 18 | Relapse | 51 | M | White | 68.7 | 20.97 | 1a | 6.352 183 | 6.143 015 | 2 | CT |
| 19 | Relapse | 49 | M | White | 78.9 | 24.35 | 1a | 6.439 333 | 5.701 568 | 3 | CC |
| 20 | SVR | 40 | M | White | 86.7 | 28.64 | 2b | 6.959 995 | – | 3 | CT |
| 21 | SVR | 52 | M | White | 104.4 | 36.55 | 2b | 6.906 874 | – | 4 | CT |
| 22 | SVR | 56 | M | White | 89.9 | 26.55 | 2b | 7.037 426 | – | 3 | CC |
| 23 | SVR | 60 | M | White | 104 | 31.75 | 2b | 6.753 583 | – | 3 | CC |
| 24 | SVR | 51 | M | White | 118.5 | 36.98 | 2b | 6.181 844 | – | 4 | CT |
| 25 | SVR | 46 | M | White | 91.7 | 29.27 | 2b | 7.204 12 | – | 3 | CC |
| 26 | SVR | 49 | M | White | 100.8 | 32.91 | 2b | 7.139 879 | – | 3 | CC |
| 27 | SVR | 63 | F | White | 50.4 | 20.71 | 2b | 5.880 814 | – | 3 | CT |
| 28 | SVR | 64 | F | White | 52.7 | 20.33 | 2b | 6.558 709 | – | 2 | CC |
| 29 | SVR | 49 | M | White | 81.7 | 21.71 | 2b | 6.212 188 | – | 3 | CT |
Fig. 1.

Box and whisker plots showing response-based comparative analysis of HCV mutation load, including total mutation load (a), synonymous mutation load (b), non-synonymous mutation load (c) and the percentage of non-synonymous mutation load (d). Each group of data was represented by box (Second quartile, median and third quartile mutation loads) and whiskers (minimum and maximum mutation loads).
Of all four groups of patients (1a/SVR, 1a/null, 1a/relapse_B and 2b/SVR), 76 patients had IL28B genotypes available from the parent study (Lee et al., 2004). The HCV mutation load was lower in patients with the IL28B genotype CC (n = 29) than in those with the genotypes CT and TT (n = 47) (Fig. 2). However, the former had a percentage of non-synonymous mutation load significantly higher in comparison with the patients with IL-28B genotypes CT and TT (24.3 ± 10 vs 20 ± 6 %, P = 0.02) (Fig. 2).
Fig. 2.
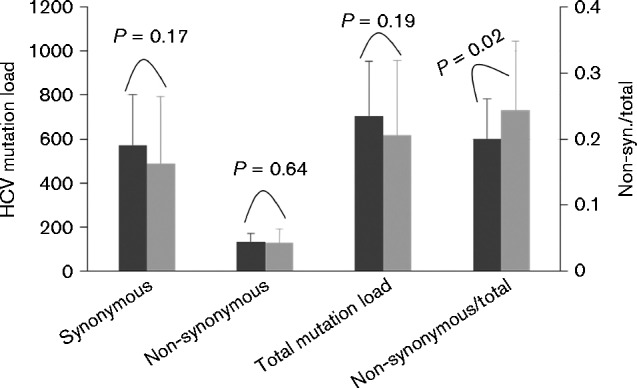
Comparison of the HCV mutation load between patients with IB28B genotype CC (n = 29, dark grey bars) and genotype CT or TT (n = 47, light grey bars). There was no statistical difference with regard to synonymous, non-synonymous or total HCV mutation load (left y-axis). However, the ratio of non-synonymous load to the total HCV mutation load was significantly higher in the genotype CC group than in the CT or TT group (P = 0.02, right y-axis).
Genome-wide structural comparison of HCV mutation loads
By including 36 665 mutations identified from 1a/SVR and 1a/null patients (Wang et al., 2014), we redefined the distribution pattern of a total of 64 221 mutations with regard to their mutation frequencies in the HCV QS population. Again, a power-law distribution pattern was confirmed (Kolmogorov–Smirnov = 0.093, α = 1.85, P = 0.846) (Fig. S2). On the curve, the low boundary of the number of mutations (χmin) was at 445, corresponding to a mutation frequency of 14.5 % (Fig. S2). Based on this cut-off value of mutation frequency, genome-wide structural analysis was conducted in three comparisons. First, while the 1a/null and 1a/relapse_B had similar mutational loads (805.6 ± 270.7 vs 829.2 ± 282.8 mutations, respectively; P = 0.78) (Fig. 1), sliding-window analysis revealed that major differences were located in the high-frequency synonymous load in which six domains achieved statistical significance (Fig. 3a). Secondly, under the SVR pattern, HCV 2b showed a much higher synonymous load, particularly in the low-frequency portion (Fig. 3b). Interestingly, the difference was found mainly in HCV non-structural genes except for NS5a (Fig. 3b). Finally, when comparing 1a/relapse_B with 1a/relapse_R (sampled at the time of viral relapse), the synonymous mutation load accounted for the major difference (Fig. 3c). At the time of viral relapse, the low-frequency synonymous load was shrunk dramatically throughout the entire HCV coding region (Fig. 3c). In the non-synonymous mutational load, a peak in the HCV hypervariable region 1 (HVR1), an 81-bp domain located at the 5′ end of the HCV E2 gene, was lost after viral relapse (Fig. 3c).
Fig. 3.


Comparative sliding-window analyses of the HCV mutation load in 1a/null (red) vs 1a/relapse_R (blue) (a), 1a/SVR (blue) vs 2b/SVR (red) (b) or 1a/relapse_R (red) vs 1a/relapse_B (blue) (c). In each analysis, HCV mutation load was stratified into four parts based on mutation nature [synonymous (right panels)/non-synonymous (left panels)] or the mutation frequency [high frequency (upper panels)/low frequency (lower panels)]. In each part, the HCV mutation loads, calculated as normalized Shannon entropy (left axes), were compared for statistical significance, expressed as P values (right axes) and represented by bars.
Continuous evolution of HCV in patients relapsing after antiviral therapy
In 19 patients who relapsed from peg-IFN/ribavirin therapy, phylogenetic analysis using nearly full-length HCV coding sequences confirmed the re-emergence of viral strains with continuous evolution (Fig. 4a). Comparing the consensus sequences derived at the baseline and at viral relapse, there were a total of 2527 substitutions with a mean of 133 substitutions per patient, ranging from 30 to 374. Of these, 14.6 % resulted in amino acid changes, with the hot spot located in the HCV HVR1 (Fig. S3). However, there was no apparent difference with regard to the numbers of HVR1 variants reconstructed at baseline and at the time of viral relapse (5.89 ± 2.33 vs 4.68 ± 2.45, P = 0.12). On the neighbour-joining tree with all HVR1 variants (n = 201), the HVR1 displayed either temporal or spatial topologies for individual patients (Fig. 4b).
Fig. 4.

Neighbour-joining trees constructed with either HCV consensus sequences (a) or HCV HVR1 QS variants (b) under the maximum composition likelihood model and gamma distribution of rate variation among sites (γ = 0.5). Filled circles and triangles represent the HCV consensus sequences derived at the baseline and viral relapse, respectively, while HVR1 QS variants are indicated by branch colours: black (baseline) and blue (viral relapse).
Lack of a correlation between treatment outcomes and HCV genomic domains
Using nearly full-length genomic sequences, all 19 HCV strains in patients relapsed from antiviral therapy belonged to HCV 1a clade 1 in the phylogenetic analysis (data not shown). These sequences, together with 19 and 21 HCV 1a clade 1 strains from 1a/null and 1a/SVR patients, respectively, in our previous study (Wang et al., 2014), were analysed using phylogenetic approaches to explore the potential roles of any HCV genomic domains in the treatment outcomes. The results showed that no HCV domain or nearly full-length coding region had both an association index (AI) and parsimony score (PS) of less than 0.01, suggesting a lack of statistical correlation (Fig. 5).
Fig. 5.
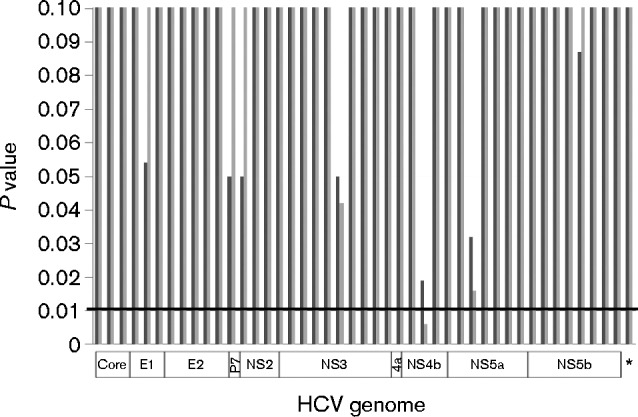
P values of the AI (dark grey bars) and PS (light grey bars) from HCV genome-wide BaTS analysis in a sliding-window manner. BaTS analysis was also performed with nearly full-length HCV coding sequences (8817 bp) as donated by the asterisk. None of HCV domains had both AL and PS values of less than 0.01, indicating the lack of statistical significance in terms of the treatment outcome-dependent phylogenetic clustering.
Discussion
Through the combination of long RT-PCR and next-generation sequencing, we have introduced high-resolution HCV genome-wide mutational load as a new index to quantify viral QS population diversity (Wang et al., 2014). The current study is an extension of our previous work by including 2b/SVR and 1a/relapse patients in the setting of HCV antiviral therapy. All four groups of patients (1a/SVR, 2b/SVR, 1a/null and 1a/relapse) were chosen from the Hepatitis C Antiviral Long-term Treatment against Cirrhosis (HALT-C) trial (Lee et al., 2004) with compatible demographic factors and disease status (Table 1). Both the experimental method and the study design allowed the role of HCV genomic heterogeneity in the antiviral therapy to be investigated in an unprecedented manner.
At the level of viral consensus sequences, the treatment outcomes (SVR, null and relapse) of IFN-based antiviral therapy did not correlate with the heterogeneity of any HCV domains (Fig. 5). Previous studies have suggested the existence of this kind of HCV domain, such as an IFN-sensitivity-determining region (Enomoto et al., 1996). As the phylogenetic approach did not require any reference sequences, methodological differences might explain the discrepancy between our findings and previous data. Indeed, so far, there is no explicit HCV target documented in IFN-mediated antiviral activity. In in vitro experiments, there are also no consistent mutational pathways found in the HCV genome in response to IFN treatment (Perales et al., 2013). Taken together, the genetic heterogeneity among HCV 1a strains has no apparent role in affecting the therapeutic efficiency of IFN.
Major findings come from the high-resolution quantification of HCV QS diversity. While the HCV mutation loads in the 2b/SVR and 1a/relapse_B groups were analogous, respectively, to those of the 1a/SVR and 1a/null groups, the percentage of non-synonymous mutation load presented an order of 2b/SVR>1a/SVR>1a/relapse_B or 1a/null (Fig. 1d). A high mutation load facilitates the evolution of viral populations in response to external pressure, such as antiviral therapy. Given its slightly deleterious nature (Eyre-Walker et al., 2002; Kimura, 1983), however, excessive non-synonymous mutations are detrimental to such an adaptive evolution (Peck, 1994). For the first time, our data thus provide an evolutionary interpretation as to why patients with HCV genotype 2 have overall high SVR rates in comparison with patients with HCV genotype 1a in IFN-based HCV antiviral therapy (Feld & Hoofnagle, 2005). As an index of deleterious mutation load, the magnitude of the percentage of non-synonymous mutation load also explains differential response patterns, at least in HCV 1a patients under antiviral therapy. It is worth noting that the percentage of non-synonymous mutation load correlated with IL28B genotype (Fig. 2), a documented host factor associated with treatment outcomes in IFN-based HCV antiviral therapy (Balagopal et al., 2010). IL28B-mediated inhibition of HCV replication dynamics contributes at least partially to a high deleterious mutation load in the HCV QS population (Zhang et al., 2011a). In addition, in each group of patients, presumptive deleterious mutation loads were not detected using consensus HCV sequences (Table 2), suggesting essential differences in viral QS population and consensus sequences in reflecting evolutionary forces.
Table 2. Selection pressures on the external and internal branches of the phylogenetic trees constructed with nearly full-length HCV coding sequences.
In comparison with the internal branches, similar or even smaller dN/dS (ratio of number of non-synonymous substitutions per non-synonymous site to the number of synonymous substitutions per synonymous site) values on the external branches indicated the lack of presumptive deleterious HCV mutation loads carried by patients with differential response patterns in HCV antiviral therapy.
| Response pattern | Genotype | Number of patients | Number of codons | dN/dS (ω) | ||
|---|---|---|---|---|---|---|
| Overall | Internal | External | ||||
| SVR | 2b | 10 | 2939 | 0.09885 | 0.13296 | 0.09387 |
| SVR | 1a | 30 | 2939 | 0.11774 | 0.11043 | 0.11904 |
| Null | 1a | 26 | 2939 | 0.1091 | 0.13944 | 0.10538 |
| Relapse_B | 1a | 19 | 2939 | 0.13607 | 0.1467 | 0.13487 |
| Relapse_R | 1a | 19 | 2939 | 0.12157 | 0.15704 | 0.11711 |
The survival of HCV depends solely on its continuous replication, as its genome does not become integrated into the host (Lindenbach & Rice, 2005). In patients relapsed from antiviral therapy, HCV might enter a population bottleneck in which the virus maintains a low-dynamic replication that is under the detection limit of current methods. Supporting this assertion, the low-frequency synonymous mutation load, closely relevant to viral replicative dynamics (Lemey et al., 2007), shrank significantly at the time of viral relapse (Fig. 2c). During the bottleneck, however, HCV experienced continuous evolution, as evidenced by the phylogenetic trees based on the consensus sequences (Fig. 4a) and HVR1 QS variants (Fig. 4b). More importantly, such an evolution was accompanied by a remarkable increase in the percentage of non-synonymous mutation load (Fig. 1d). Pieced together, these data support the functionality of Muller's ratchet in a treatment-induced viral population bottleneck (Chao, 1990). When a population bottleneck is long enough, Muller's ratchet will eventually drive virus extinction through excessive accumulation of a deleterious mutation load. Indeed, the extension of treatment duration in either IFN-based or IFN-free HCV antiviral therapy does enhance the SVR rates (Pearlman et al., 2007; Wyles et al., 2015). Ribavirin might play a similar role by accelerating the accumulation of a deleterious mutation load (Ortega-Prieto et al., 2013). The high predictive value of a rapid virological response in SVR could also be explained as a result of early onset of the viral population bottleneck (Poordad et al., 2008).
Viral relapse remains an issue, even in the era of directly acting antivirals (DAAs) in HCV antiviral therapy (Pawlotsky, 2015). Our findings thus have important implications. If Muller's ratchet also functions in the setting of DAA therapy, viral relapse could be eliminated by the modulation of treatment duration. Thus, a personalized treatment course of DAA therapy could benefit from measurement of pre-treatment HCV mutation load, monitoring the time of onset of the viral population bottleneck and novel methods capable of detecting low-dynamic HCV replication.
Methods
Patient samples
The current study consisted of two groups of patients registered in the HALT-C trial (Lee et al., 2004). All patients received 48-week full-dose peg-IFN-α2a/ribavirin combination therapy (Lee et al., 2004). One group comprised 10 patients who were infected with HCV genotype 2b and achieved SVR to the treatment (2b/SVR). The other group had 19 patients infected with HCV genotype 1a who relapsed after treatment cessation. Samples were collected prior to the treatment (1a/relapse_B) and at the time of viral relapse (1a/relapse_R). Demographic and other factors for these patients were well characterized (Table 1). The HCV genotype was determined by a line probe assay (Innogenetics) in the parental study. Upon receiving samples, the genotype was further confirmed by phylogenetic analysis of the HCV NS5b region through direct amplicon sequencing. HCV RNA titres in all patients were measured using a Roche Amplicor HCV Monitor (version 2.0). Of the 29 patients, 26 had IL28B genotype data available from the parental study (Table 1).
For collection of serum samples, written informed consent was obtained from each patient and was approved by the local Institutional Review Board in the parental studies (Lee et al., 2004). The research protocol for the use of these patient samples was reviewed and approved by the Saint Louis University Institutional Review Board (IRB protocol: SLU15565).
Quantification of the genome-wide HCV mutation load
Experimental methods and bioinformatics pipelines for the quantification of the HCV mutation load have been detailed in our previous study (Wang et al., 2014). Briefly, after total RNA extraction from patient serum, long RT-PCR was applied to amplify the nearly full-length HCV genome, a 9022 bp amplicon for HCV genotype 1a, using primers as described previously (Fan & Di Bisceglie, 2010). For patients infected with HCV genotype 2b, the same protocol was used to amplify a 9085 bp amplicon with the genotype 2b-specific primers QR2_2b_16 (5′-GCCCACGGTGAACCAC-3′; positions 9310–9325) and WF33_2b (5′-ACGCAGAAAGCGTCTAGCCAT-3′; positions 65–85) for RT and the first round of PCR, and WF5_2b (5′-CATAGTGGTCTGCGGAACCGGT-3′; positions 138–159) and WR55_2b (5′-ATGGCCGCCCTCCCTCCCTG-3′; positions 9203–9222) for the second round of PCR. Nucleotide numbering is according to HCV JCV-6 strain (GenBank accession no. AB047645). RT-PCR amplicons of the expected size were gel purified using a QIAEX II Gel Extraction kit (Qiagen) and subjected to library construction with a Rapid Library Preparation kit (Roche Applied Science), followed by 454 sequencing on a GS/FLX Titanium platform. After the quality control of sequencing reads, the genome-wide HCV mutation load was determined by a two-step protocol as described by Wang et al. (2014).
Structural analysis of the genome-wide HCV mutation load
By including data from previous studies, we first generated a histogram of mutational frequencies, followed by the determination of its distribution patterns (power law, log-normal, exponential and Poisson) using Clauset's method (Clauset et al., 2009). On the selective pattern, the low-bound point, a lower cut-off (χmin) for the scaling region, was calculated through the goodness-of-fit test based on the Kolmogorov–Smirnov statistic and likelihood ratios (Clauset et al., 2009). The value of χmin then served as an artificial point to assign mutations into the low- or high-frequency category. Based on a mutation's frequency (low or high) and nature (synonymous or non-synonymous), sliding-window analyses (windows size, 300 bp; overlap, 100 bp) were conducted over the HCV coding region from the core protein to NS5b. When necessary, statistical significance at each window was determined between the response patterns of IFN-based antiviral therapy.
Phylogenetic analysis
Multiple data analyses were performed using phylogenetic approaches. First, phylogenetic analysis was used to pursue the genetic origin of HCV QS variants that relapsed after the treatment cessation. Under the model of maximum composite likelihood, a neighbour-joining tree was constructed using nearly full-length HCV coding sequences assembled from the relapsers in the mega program (version 6) (Tamura et al., 2011). Secondly, for each group of patients (Table 2), the potentially deleterious mutation load at the nearly full-length HCV genome level was estimated by a phylogenetic approach (Pybus et al., 2007). In doing so, the best-fit nucleotide substitution model for each group of HCV sequences was determined through JModelTest (Darriba et al., 2012). Selected models were used for the simulation of maximum-likelihood (ML) trees in paup* (version 4.0b) (Swofford, 2002). The ML trees then served as the template to compute the mean numbers of non-synonymous (dN) and synonymous (dS) nucleotide substitutions per site (dN/dS ratio) under the ‘one-ratio’ model (all branches have the same dN/dS) from the codeml program in the PAML package (version 4) (Yang, 2007). Alternatively, the selection pressures on external and internal branches of each ML tree were quantified using the ‘two-ratio’ model, which allows a different dN/dS for internal and external branches. Finally, treatment outcomes (SVR, null and relapse) were examined to look for possible correlation with HCV domains, as described previously (Wang et al., 2014). Briefly, the nearly full-length HCV 1a coding region was scanned through a sliding window (size, 300 bp; overlap, 99 bp). Bayesian Markov chain Monte Carlo (MCMC) phylogenetic trees were simulated in the beast package under selective models from JModelTest as well as additional parameter settings, including a relaxed molecular clock, a Bayesian skyline coalescent prior and a total run of 10 million generations to reach relevant parameter convergence as estimated by Tracer (Drummond & Rambaut, 2007). The inferred MCMC trees, after discarding the first 10 % trees as burn-in, were used as the input to estimate the strength of HCV 1a strain clustering in terms of response pattern with 1000 replications in BaTS (Parker et al., 2008). Based on empirical and simulated data, P values of both the AI and PS of less than 0.01 were considered as having statistical significance (Parker et al., 2008; Zhang et al., 2011b).
Statistical analysis
Except for those statistical analyses stated, comparisons were done with a two-tailed, unpaired Student's t-test, χ2 test or Fisher's exact test. Where applicable, data are expressed as means ± sd. P < 0.05 was considered statistically significant.
Acknowledgements
This work was supported by the US National Institutes of Health (NIH) (grant DK80711 to X. F.).
Supplementary Data
Supplementary Data
Footnotes
Raw sequence data in fastq format from all 48 patient samples are archived in the NCBI Sequence Read Archive (SRA) under SRA accession no. SRP059615.
Three supplementary figures are available with the online Supplementary Material.
References
- Balagopal A., Thomas D. L., Thio C. L. (2010). IL28B and the control of hepatitis C virus infection Gastroenterology 1391865–1876 10.1053/j.gastro.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao L. (1990). Fitness of RNA virus decreased by Muller's ratchet Nature 348454–455 10.1038/348454a0. [DOI] [PubMed] [Google Scholar]
- Clauset A., Shalizi C. R., Newman M. E. J. (2009). Power-law distributions in empirical data SIAM Rev 51661–703 10.1137/070710111. [DOI] [Google Scholar]
- Darriba D., Taboada G. L., Doallo R., Posada D. (2012). jModelTest 2: more models, new heuristics and parallel computing Nat Methods 9772. 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo E., Sheldon J., Perales C. (2012). Viral quasispecies evolution Microbiol Mol Biol Rev 76159–216 10.1128/MMBR.05023-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J., Rambaut A. (2007). beast: Bayesian evolutionary analysis by sampling trees BMC Evol Biol 7214. 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto N., Sakuma I., Asahina Y., Kurosaki M., Murakami T., Yamamoto C., Ogura Y., Izumi N., Marumo F., Sato C. (1996). Mutations in the nonstructural protein 5A gene and response to interferon in patients with chronic hepatitis C virus 1b infection N Engl J Med 33477–82 10.1056/NEJM199601113340203. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A., Keightley P. D., Smith N. G., Gaffney D. (2002). Quantifying the slightly deleterious mutation model of molecular evolution Mol Biol Evol 192142–2149 10.1093/oxfordjournals.molbev.a004039. [DOI] [PubMed] [Google Scholar]
- Fan X., Di Bisceglie A. M. (2010). RT-PCR amplification and cloning of large viral sequences Methods Mol Biol 630139–149 10.1007/978-1-60761-629-0_10. [DOI] [PubMed] [Google Scholar]
- Fan X., Mao Q., Zhou D., Lu Y., Xing J., Xu Y., Ray S. C., Di Bisceglie A. M. (2009). High diversity of hepatitis C viral quasispecies is associated with early virological response in patients undergoing antiviral therapy Hepatology 501765–1772 10.1002/hep.23290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farci P., Quinti I., Farci S., Alter H. J., Strazzera R., Palomba E., Coiana A., Cao D., Casadei A. M., other authors (2006). Evolution of hepatitis C viral quasispecies and hepatic injury in perinatally infected children followed prospectively Proc Natl Acad Sci U S A 1038475–8480 10.1073/pnas.0602546103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feld J. J., Hoofnagle J. H. (2005). Mechanism of action of interferon and ribavirin in treatment of hepatitis C Nature 436967–972 10.1038/nature04082. [DOI] [PubMed] [Google Scholar]
- Kimura M. (1983). Neutral Theory of Molecular Evolution. 10.1017/CBO9780511623486Cambridge: Cambridge University Press. [DOI] [Google Scholar]
- Larder B. A., Kohli A., Kellam P., Kemp S. D., Kronick M., Henfrey R. D. (1993). Quantitative detection of HIV-1 drug resistance mutations by automated DNA sequencing Nature 365671–673 10.1038/365671a0. [DOI] [PubMed] [Google Scholar]
- Lee W. M., Dienstag J. L., Lindsay K. L., Lok A. S., Bonkovsky H. L., Shiffman M. L., Everson G. T., Di Bisceglie A. M., Morgan T. R., other authors (2004). Evolution of the HALT-C trial: pegylated interferon as maintenance therapy for chronic hepatitis C in previous interferon nonresponders Control Clin Trials 25472–492 10.1016/j.cct.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Lemey P., Kosakovsky Pond S. L., Drummond A. J., Pybus O. G., Shapiro B., Barroso H., Taveira N., Rambaut A. (2007). Synonymous substitution rates predict HIV disease progression as a result of underlying replication dynamics PLOS Comput Biol 3e29. 10.1371/journal.pcbi.0030029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Sullivan D. G., Feuerborn N., McArdle S., Bekele K., Pal S., Yeh M., Carithers R. L., Perkins J. D., Gretch D. R. (2010). Genetic diversity of hepatitis C virus predicts recurrent disease after liver transplantation Virology 402248–255 10.1016/j.virol.2010.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Hughes A. L., Bano N., McArdle S., Livingston S., Deubner H., McMahon B. J., Townshend-Bulson L., McMahan R., other authors (2011). Genetic diversity of near genome-wide hepatitis C virus sequences during chronic infection: evidence for protein structural conservation over time PLoS One 6e19562. 10.1371/journal.pone.0019562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbach B. D., Rice C. M. (2005). Unravelling hepatitis C virus replication from genome to function Nature 436933–938 10.1038/nature04077. [DOI] [PubMed] [Google Scholar]
- López-Labrador F. X., Ampurdanès S., Giménez-Barcons M., Guilera M., Costa J., Jiménez de Anta M. T., Sánchez-Tapias J. M., Rodés J., Sáiz J. C. (1999). Relationship of the genomic complexity of hepatitis C virus with liver disease severity and response to interferon in patients with chronic HCV genotype 1b infection [correction of interferon] Hepatology 29897–903 10.1002/hep.510290306. [DOI] [PubMed] [Google Scholar]
- Lyra A. C., Fan X., Lang D. M., Yusim K., Ramrakhiani S., Brunt E. M., Korber B., Perelson A. S., Di Bisceglie A. M. (2002). Evolution of hepatitis C viral quasispecies after liver transplantation Gastroenterology 1231485–1493 10.1053/gast.2002.36546. [DOI] [PubMed] [Google Scholar]
- Morishima C., Polyak S. J., Ray R., Doherty M. C., Di Bisceglie A. M., Malet P. F., Bonkovsky H. L., Sullivan D. G., Gretch D. R., other authors (2006). Hepatitis C virus-specific immune responses and quasi-species variability at baseline are associated with nonresponse to antiviral therapy during advanced hepatitis C J Infect Dis 193931–940 10.1086/500952. [DOI] [PubMed] [Google Scholar]
- Ortega-Prieto A. M., Sheldon J., Grande-Pérez A., Tejero H., Gregori J., Quer J., Esteban J. I., Domingo E., Perales C. (2013). Extinction of hepatitis C virus by ribavirin in hepatoma cells involves lethal mutagenesis PLoS One 8e71039. 10.1371/journal.pone.0071039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J., Rambaut A., Pybus O. G. (2008). Correlating viral phenotypes with phylogeny: accounting for phylogenetic uncertainty Infect Genet Evol 8239–246 10.1016/j.meegid.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Pawlotsky J. M. (2015). Hepatitis C treatment: the data flood goes on—an update from the Liver Meeting 2014 Gastroenterology 148468–479 10.1053/j.gastro.2015.01.002. [DOI] [PubMed] [Google Scholar]
- Pearlman B. L., Ehleben C., Saifee S. (2007). Treatment extension to 72 weeks of peginterferon and ribavirin in hepatitis c genotype 1-infected slow responders Hepatology 461688–1694 10.1002/hep.21919. [DOI] [PubMed] [Google Scholar]
- Peck J. R. (1994). A ruby in the rubbish: beneficial mutations, deleterious mutations and the evolution of sex Genetics 137597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perales C., Beach N. M., Gallego I., Soria M. E., Quer J., Esteban J. I., Rice C., Domingo E., Sheldon J. (2013). Response of hepatitis C virus to long-term passage in the presence of alpha interferon: multiple mutations and a common phenotype J Virol 877593–7607 10.1128/JVI.02824-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poordad F., Reddy K. R., Martin P. (2008). Rapid virologic response: a new milestone in the management of chronic hepatitis C Clin Infect Dis 4678–84 10.1086/523585. [DOI] [PubMed] [Google Scholar]
- Pybus O. G., Rambaut A., Belshaw R., Freckleton R. P., Drummond A. J., Holmes E. C. (2007). Phylogenetic evidence for deleterious mutation load in RNA viruses and its contribution to viral evolution Mol Biol Evol 24845–852 10.1093/molbev/msm001. [DOI] [PubMed] [Google Scholar]
- Qin H., Shire N. J., Keenan E. D., Rouster S. D., Eyster M. E., Goedert J. J., Koziel M. J., Sherman K. E., Multicenter Hemophilia Cohort Study Group (2005). HCV quasispecies evolution: association with progression to end-stage liver disease in hemophiliacs infected with HCV or HCV/HIV Blood 105533–541 10.1182/blood-2004-04-1452. [DOI] [PubMed] [Google Scholar]
- Sandres K., Dubois M., Pasquier C., Payen J. L., Alric L., Duffaut M., Vinel J. P., Pascal J. P., Puel J., Izopet J. (2000). Genetic heterogeneity of hypervariable region 1 of the hepatitis C virus (HCV) genome and sensitivity of HCV to alpha interferon therapy J Virol 74661–668 10.1128/JVI.74.2.661-668.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan D. G., Bruden D., Deubner H., McArdle S., Chung M., Christensen C., Hennessy T., Homan C., Williams J., other authors (2007). Hepatitis C virus dynamics during natural infection are associated with long-term histological outcome of chronic hepatitis C disease J Infect Dis 196239–248 10.1086/518895. [DOI] [PubMed] [Google Scholar]
- Swofford D. L. (2002). paup*. Phylogenetic analysis using parsimony (*and other methods), version 4 Sunderland, MA: Sinauer Associates. [Google Scholar]
- Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. (2011). mega5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods Mol Biol Evol 282731–2739 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laethem K., Van Vaerenbergh K., Schmit J. C., Sprecher S., Hermans P., De Vroey V., Schuurman R., Harrer T., Witvrouw M., other authors (1999). Phenotypic assays and sequencing are less sensitive than point mutation assays for detection of resistance in mixed HIV-1 genotypic populations J Acquir Immune Defic Syndr 22107–118 10.1097/00126334-199910010-00001. [DOI] [PubMed] [Google Scholar]
- Wang X. H., Netski D. M., Astemborski J., Mehta S. H., Torbenson M. S., Thomas D. L., Ray S. C. (2007). Progression of fibrosis during chronic hepatitis C is associated with rapid virus evolution J Virol 816513–6522 10.1128/JVI.02276-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Zhang X., Xu Y., Weinstock G. M., Di Bisceglie A. M., Fan X. (2014). High-resolution quantification of hepatitis C virus genome-wide mutation load and its correlation with the outcome of peginterferon-alpha2a and ribavirin combination therapy PLoS One 9e100131. 10.1371/journal.pone.0100131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyles D. L., Ruane P. J., Sulkowski M. S., Dieterich D., Luetkemeyer A., Morgan T. R., Sherman K. E., Dretler R., Fishbein D., other authors (2015). Daclatasvir plus sofosbuvir for HCV in patients coinfected with HIV-1 N Engl J Med 373714–725 10.1056/NEJMoa1503153. [DOI] [PubMed] [Google Scholar]
- Yang Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood Mol Biol Evol 241586–1591 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Zhang L., Jilg N., Shao R. X., Lin W., Fusco D. N., Zhao H., Goto K., Peng L. F., Chen W. C., Chung R. T. (2011a). IL28B inhibits hepatitis C virus replication through the JAK-STAT pathway J Hepatol 55289–298 10.1016/j.jhep.2010.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Ryu S. H., Xu Y., Elbaz T., Zekri A. R., Abdelaziz A. O., Abdel-Hamid M., Thiers V., Elena S. F., other authors (2011b). The Core/E1 domain of hepatitis C virus genotype 4a in Egypt does not contain viral mutations or strains specific for hepatocellular carcinoma J Clin Virol 52333–338 10.1016/j.jcv.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data