

Abstract
Oncolytic virotherapy is a unique cancer therapeutic that encompasses tumour cell lysis through both virus replication and programmed cell death (PCD) pathways. Nonetheless, clinical efficacy is relatively modest, likely related to the immunosuppressive tumour milieu. Our studies use the herpes simplex virus type 2 (HSV-2)-based oncolytic virus ΔPK that has documented anti-tumour activity associated with virus replication, PCD and cancer stem cell lysis. They are designed to examine whether ΔPK-mediated oncolysis includes the ability to reverse the immunosuppressive tumour microenvironment by altering the balance of cytokines directly secreted by the melanoma cells and to define its mechanism. Here, we show that melanoma cells secreted the immunosuppressive cytokine IL-10, and that secretion was inhibited by ΔPK through virus replication and c-Jun N-terminal kinase/c-Jun activation. ΔPK-induced IL-10 inhibition upregulated surface expression of MHC class I chain-related protein A, the ligand for the activating NKG2D receptor expressed on NK- and cytotoxic T-cells. Concomitantly, ΔPK also upregulated the secretion of inflammatory cytokines TNF-α, granulocyte macrophage colony-stimulating factor and IL-1β through autophagy-mediated activation of Toll-like receptor 2 pathways and pyroptosis, and it inhibited the expression of the negative immune checkpoint regulator cytotoxic T-lymphocyte antigen 4. Pharmacologic inhibition of these processes significantly reduces the oncolytic activity of ΔPK.
Introduction
Oncolytic viruses (OVs) originally developed to lyse tumour cells through selective replication can also induce distinct programmed cell death (PCD) pathways. However, clinical efficacy remains modest, likely related to the immunosuppressive tumour milieu (Aurelian, 2013). OVs developed from herpes simplex virus (oHSVs) are primarily based on HSV-1 and are typically deleted in the neurovirulence gene ICP34.5 and/or the large subunit of the ribonucleotide reductase (R1). Some are also deleted in ICP47, which functions in virus immune evasion, but the role of this deletion in oncolytic potential is still unclear (Chiocca & Rabkin, 2014). T-Vec, an oHSV armed with granulocyte macrophage colony-stimulating factor (GM-CSF), has recently passed phase III clinical trials in stage III and IV melanoma (Hersey & Gallagher, 2014).
HSV-2 differs from HSV-1 in that many of its replicative aspects depend on the protein kinase function of R1 (also known as ICP10PK), which is poorly conserved and non-functional in HSV-1. ICP10PK activates Ras signalling pathways and is required for virus growth in slowly replicating normal cells that have low levels of Ras activity (Smith et al., 1998, 2000). We have shown that an HSV-2 mutant deleted in ICP10PK (ΔPK) that was tolerated well in vaccine phase I/II clinical trials (Aurelian, 2004; Casanova et al., 2002) has strong oncolytic activity in melanoma cultures and xenografts, where it causes a profound and long-lasting reduction in tumour burden. In addition to virus replication, oncolytic activity is associated with calpain and caspase-3/7 activation, autophagy induction, cancer stem cell lysis, and intratumour influx of CD11b+ antigen-presenting cells (Colunga et al., 2010, 2014). However, the ability of ΔPK to inhibit the immunosuppressive tumour microenvironment by altering the balance of cytokines directly secreted by the melanoma cells (Kaufman et al., 2014) and to induce surface expression of immunostimulatory molecules (Mouawad et al., 2010) is still unknown. Our studies were designed to address this question and identify the molecular mechanisms involved in these processes.
Results
ΔPK increases transcription of death-associated and inflammatory genes
ΔPK replicates well in melanoma cells (Colunga et al., 2010, 2014), as confirmed by plaque assay (Fig. 1a) and immunoblotting with VP5 antibody (Fig. 1b). To examine whether ΔPK alters expression of cytokines and death-associated functions, cells were mock-infected with PBS or infected with ΔPK (m.o.i. 0.5 p.f.u. per cell), and RNA isolated at 24 h post-infection (p.i.) was analysed with inflammatory and cell death targeted oligoarrays. The results are shown in Fig. 1(c) for A2058 cells and in Fig. S1(a) (available in the online Supplementary Material) for LM cells that were established from a heterogeneous primary melanoma passaged only six to eight times before this study. Similar patterns of gene expression ( ± 12 %) were obtained in two separate experiments for each cell culture examined (Tables S1 and S2). ΔPK caused a marked upregulation of several genes, including inflammatory caspase-1, -4, and -5, the cytokines IL-1α, IL-1β, IL-6, IL-8, IL-12, TNF-α, lymphotoxin-α and GM-CSF, and mitogen-activated protein kinases (MAPKs) involved in both PCD and inflammatory processes. Amongst these are MAP3K7IP1 (also known as TAB1), which is involved in IL-1-induced signalling pathways (Wolf et al., 2011); MAP4K4, the upstream activator of c-Jun N-terminal kinase (JNK) (Machida et al., 2004) and its downstream target c-Jun; MAP2K6, the upstream activator of the pro-apoptotic p38MAPK (Han et al., 1996); and MAP3K14 (also known as NIK), which stimulates NFκB signalling common to TNF-α and IL-1 receptors (Vallabhapurapu et al., 2008). Whilst limited, direct comparison of ΔPK with HSV-2 indicated notable differences, including the failure of HSV-2 to upregulate TNF-α, IL-1β, GM-CSF, c-Jun, MAP3K14, Toll-like receptor (TLR)-2 and MyD88, and poor IL-10 inhibition relative to ΔPK (Fig. S2, Table S3).
Fig. 1.

ΔPK upregulates inflammatory and apoptotic genes and alters cytokine secretion. (a) Cells were infected with ΔPK (m.o.i. 0.5) in serum-free medium and virus titres were determined by plaque assay. (b) Protein extracts from mock- or ΔPK-infected (m.o.i. 0.5; 24 h) were immunoblotted with antibodies to VP5 and β-actin. Data are presented as mean ± sd densitometry units. (c) Total RNA from mock- or ΔPK-infected (m.o.i. 0.5; 24 h) A2058 cells was assayed with targeted microarrays. (d) Conditioned media from A375 cells mock- or ΔPK-infected (m.o.i. 0.5; 24 h) were assayed for cytokine levels by ELISA. Data are presented as mean ± sd. ***P < 0.001 versus mock, by one-way ANOVA.
ΔPK alters the balance of melanoma-secreted cytokines from immunosuppressive to inflammatory
The immunosuppressive tumour microenvironment contributes to the relatively poor clinical efficacy of OV treatment (de Aquino et al., 2015), but the contribution of direct melanoma cell secretion of immunosuppressive cytokines is still poorly understood. Our studies focused on the immunosuppressive cytokine IL-10 (Chen et al., 2014) and multiple inflammatory cytokines (TNF-α, IL-1β, GM-CSF). A2058 and A375 melanoma cells were mock-infected with PBS or infected with ΔPK (m.o.i. 1), and the conditioned media (24 h p.i.) were assayed for cytokine expression by ELISA. The data summarized in Fig. 1(d) for A375 cells and Fig. S1(b) for A2058 cells indicate that, despite their heterogeneity, the mock-infected cultures secreted similar levels of IL-10 (32.5 ± 0.84 and 25.9 ± 0.62 ng ml− 1 for A375 and A2058 cells, respectively), but not TNF-α, IL-1β or GM-CSF. By contrast, when infected with ΔPK the balance of the secreted cytokines was reversed, with a simultaneous decrease in the levels of IL-10, and a significant increase in the secretion of TNF-α, IL-1β and GM-CSF. Interestingly, the inhibition of the immunosuppressive cytokine IL-10 was similar in both cell lines (14.9 ± 1.7 and 8.3 ± 0.4 ng ml− 1 for A375 and A2058, respectively), potentially reflecting the role of virus replication in its inhibition. By contrast, the levels of induced inflammatory cytokines were ∼10-fold lower in A2058 than in A375 cells, suggesting that cell-type-specific factors modulated ΔPK-induced cytokine release.
Inhibition of IL-10 secretion is through JNK/c-Jun activation
Having seen that ΔPK induces the transcription of MAP4K4, JNK and c-Jun (Fig. 1c), we wanted to verify that this reflected its ability to activate the JNK/c-Jun pathway implicated in the regulation of inflammatory responses (Oltmanns et al., 2003). A375 cells were mock-infected with PBS or infected with ΔPK (m.o.i. 1), and protein extracts obtained at 1, 4 and 24 h p.i. were immunoblotted with antibody that recognized the phosphorylated JNK isoforms (pJNK1 and pJNK2/3). The blot was stripped and reprobed with β-actin antibody as gel loading control. Two bands corresponding to pJNK1 (46 kDa) and pJNK2/3 (54 kDa) were seen. Their levels were significantly higher in the ΔPK- than mock-infected cultures (Fig. 2a) and reduced by treatment with the JNK-specific inhibitor SP600125 (100 μM) (Fig. 2b).
Fig. 2.

ΔPK activates the JNK/c-Jun pathway. (a) A375 cells were mock- or ΔPK-infected (m.o.i. 0.5), and protein extracts were immunoblotted with antibodies to pJNK and β-actin. (b) Parallel cultures were infected as above and treated with SP600125 (SP) (100 μM) after virus adsorption (1 h, 4 °C), and protein extracts were similarly immunoblotted. (c) Extracts from A2058 cells mock- or ΔPK-infected (m.o.i. 0.5; 24 h) subsequently treated with or without SP600125, as in (b), were separated into nuclear (Nuc) and cytoplasmic (Cyto) fractions, and immunoblotted with antibodies to p-c-Jun and β-actin. Data are presented as mean ± sd densitometry units. ***P < 0.001 versus mock, by one-way ANOVA.
To confirm that ΔPK also activated c-Jun, protein extracts from duplicate A375 and A2058 cultures mock-infected or infected with ΔPK (m.o.i. 1; 24 h) in the absence or presence of SP600125 were separated into nuclear and cytoplasmic fractions, and immunoblotted with antibodies against phosphorylated c-Jun (p-c-Jun) and β-actin. As shown in Fig. 2(c) for A2058 cells, the levels of p-c-Jun were significantly increased in the ΔPK- as compared with the mock-infected cultures, primarily in the nuclear fraction, and upregulation was inhibited by SP600125. This was consistent with the established nuclear translocation of activated c-Jun (Angel & Karin, 1991) and indicated that ΔPK activates the JNK/c-Jun pathway. Significantly, SP600125 restored the levels of secreted IL-10 to those seen in the mock-infected controls, but had no effect on ΔPK-induced secretion of GM-CSF, TNF-α and IL-1β (Fig. 3a, b). The data indicated that ΔPK-induced activation of the JNK/c-Jun pathway inhibited IL-10 expression/secretion, but it did not affect the production of the inflammatory cytokines. ΔPK inhibited IL-10 secretion through virus replication and JNK/c-Jun activation, as evidenced by virus growth inhibition with SP600125 (Fig. 4d).
Fig. 3.

ΔPK-activated JNK/c-Jun inhibits IL-10 secretion and induces MICA expression. Conditioned media from (a) A2058 and (b) A375 cells mock- or ΔPK-infected (m.o.i. 1; 24 h), in the presence or absence of SP600125 (100 μM), added after adsorption (as described in Methods), were assayed for cytokine levels by ELISA. Data are presented as mean ± sd. (c) Protein extracts from A375 cells mock- or ΔPK-infected (m.o.i. 1; 24 h) in the absence or presence of recombinant human IL-10 (14 ng ml− 1) or SP600125 (100 μM), added after adsorption were immunoblotted with antibodies to MICA and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Data are presented as mean ± densitometry units. (d) A2058 cells mock-, ΔPK- or HSV-2-infected (m.o.i. 1; 24 h) were stained with MICA antibody and analysed by FACS. ***P < 0.001, by one-way ANOVA.
Fig. 4.

ΔPK induces inflammatory cytokine expression through autophagy-related TLR-2 activation. (a) Protein extracts from A375 cells mock- or ΔPK-infected (m.o.i. 1), followed by treatment with or without 3-MA (5 mM), were immunoblotted with antibody to TLR-2 (top) or MyD88 (bottom). (b) Extracts from A375 cells mock- or ΔPK-infected as in (a) were immunoblotted with antibody to NFκB p50. Data are presented as mean ± densitometry units. ***P < 0.001 and *P < 0.05, by one way ANOVA. (c) Conditioned media from A2058 cells mock- or ΔPK-infected (m.o.i. 1; 24 h) were assayed for cytokine levels by ELISA. Data are presented as mean ± sd. (d) A2058 cells were infected with ΔPK (m.o.i. 1; 24 h), followed by 3-MA, SP600125 or vehicle control treatment at 0 h p.i., and virus titres were determined by plaque assay. Protein extracts were immunoblotted with antibodies to VP5 or β-actin. Data are presented as mean ± sd. ***P < 0.001 by one-way ANOVA.
IL-10 inhibition induces expression of the MHC class I chain-related protein A (MICA)
MICA is a ligand for the activating receptor NKG2D expressed on NK-, γδ T-, cytotoxic αβ CD8+T- and NKT-cells. Its expression on tumour cells engages NKG2D, resulting in the cytotoxic killing of the target cells (Bauer et al., 1999). IL-10 facilitates melanoma cell escape from immune surveillance by decreasing MICA expression, which in turn reduces NKG2D-mediated cytotoxicity (Serrano et al., 2011). Having seen that ΔPK induced JNK-dependent inhibition of IL-10 expression (Fig. 2), we wanted to know whether this resulted in increased MICA expression. A2058 and A375 cells were mock-infected or infected with ΔPK (m.o.i. 1; 24 h) in the presence or absence of the JNK inhibitor SP600125 (100 μM) or recombinant IL-10 (14 ng ml− 1) and protein extracts were immunoblotted with anti-MICA followed by β-actin antibodies. The results are summarized in Fig. 3(c) for A375 cells. MICA was minimally expressed in mock-infected melanoma cells and its expression was significantly increased by ΔPK infection (Figs. 3c and S3). Both SP600125 and exogenously added IL-10 restored the MICA levels to those seen in the mock-infected cells (Figs. 3c and S3), indicating that ΔPK increased MICA expression through JNK-mediated IL-10 inhibition. Similar results were obtained in A2058 cells (Fig. S3). FACS analysis indicated that ΔPK-induced MICA was located on the cell surface and MICA was also induced by HSV-2, albeit at lower levels (Fig. 3d).
Inflammatory cytokines are upregulated through autophagy-dependent activation of TLR-2 signalling
TLRs are pattern recognition receptors that promote T-cell-mediated adaptive immunity (Akira et al., 2001; Bergsbaken et al., 2009; Inoue & Tani, 2014). TLR-2 induces inflammatory cytokine production by microglia, astrocytes, neutrophils and monocytes following HSV-1 intracranial infection (Aravalli et al., 2005; Villalba et al., 2012; Wang et al., 2012), but its induction in melanoma cells, in the context of oncolysis, and as related to direct cytokine secretion, is unknown. As autophagy was previously linked to cytokine secretion (Crişan et al., 2011; Harris, 2011), A2058 and A375 cells were mock-infected with PBS or infected with ΔPK (m.o.i. 1; 4 h) in the absence or presence of the autophagy inhibitor 3-methyladenine (3-MA) (5 mM) (Seglen & Gordon, 1982), and protein extracts were immunoblotted with antibodies to TLR-2, the TLR adaptor protein MyD88 or the mature NFκB subunit (p50) downstream of TLR-2 (Akira et al., 2001).
A 95–110 kDa doublet consistent with the mature TLR-2 (Kim et al., 2013) was seen in the ΔPK- but not mock-infected cells and its induction was inhibited by 3-MA (Fig. 4a). MyD88 was also seen in the ΔPK- but not mock-infected cultures, as shown in Fig. 4(a) for A375 cells, and this was accompanied by increased expression of NFκB p50, the latter also inhibited by 3-MA (Fig. 4b). Double-immunofluorescent staining of the mock- and ΔPK-infected cells with antibodies to TNF-α and NFκB p50 confirmed that ΔPK-induced TNF-α upregulation was associated with NFκB activation, as also evidenced by the co-localization of TNF-α with NFκB in ΔPK- but not mock-infected cells (Fig. S4). The levels of TNF-α, GM-CSF and IL-1β in the culture supernatants were also significantly higher in the ΔPK- than mock-infected cultures and this increase was blocked by ΔPK infection in the presence of 3-MA (Fig. 4c). Inflammatory cytokine secretion was unrelated to virus replication because virus growth was virtually identical in A2058 cells infected with ΔPK in the absence or presence of 3-MA (Fig. 4d). The data indicated that ΔPK-induced secretion of inflammatory cytokines by the melanoma cells was through autophagy-dependent TLR-2 activation. However, 3-MA only partially blocked the ability of ΔPK to upregulate NFκB (Fig. 4b), suggesting that inflammatory cytokine upregulation also included autophagy-dependent TLR-2 pathways other than MyD88/NFκB. 3-MA had no effect on ΔPK-induced IL-10 inhibition and similar results were obtained in A375 cells (data not shown).
ΔPK-induced pyroptosis contributes to IL-1β secretion
Pyroptosis is a caspase-1-dependent form of inflammatory cell death, which is activated after an initial NFκB -dependent priming step (Fernandes-Alnemri et al., 2007; Sutterwala et al., 2014). Having seen that the levels of secreted IL-1β were increased in ΔPK-infected cells, we wanted to know whether this involved pyroptosis-related caspase-1 activation. A375 and A2058 cells were mock- or ΔPK-infected (m.o.i. 1; 24 h), and immunoblotted with antibody to activated caspase-1. The results summarized in Fig. 5(a) indicate that caspase-1 was activated in the ΔPK- but not mock-infected cells, consistent with the oligoarray and ELISA findings (Figs 1c and 3a, b). Caspase-1 activation was accompanied by IL-1β production as evidenced by the loss of pro-IL-1β and its restored expression in cells infected with ΔPK in the presence of the caspase-1-specific inhibitor z-YVAD-fmk (Fig. 5b). Collectively, the data indicated that ΔPK-induced IL-1β secretion involved pyroptosome-dependent caspase-1 activation and pro-IL-1β cleavage.
Fig. 5.

ΔPK-induced pyroptosis activates caspase-1 resulting in mature IL-β production. (a) Extracts from A2058 cells mock- or ΔPK-infected (m.o.i. 1) were immunoblotted with caspase-1 (Casp-1) (p20) antibody. (b) Extracts from A2058 cultures mock- or ΔPK-infected (m.o.i. 1; 24 h) in the presence or absence of z-YVAD-fmk (YVAD) (100 μM) were immunoblotted with antibody to pro-IL-1β. Data are presented as mean ± sd densitometry units. ***P < 0.001 versus mock, by one-way ANOVA.
Altered cytokine balance contributes to ΔPK-induced melanoma cell death
To examine the contribution of the altered cytokine balance to ΔPK-induced melanoma cell death, A2058 and A375 cells were mock- or ΔPK-infected in the presence or absence of SP600125 (100 μM), which regulates JNK-dependent IL-10 expression, 3-MA (5 mM), which regulates autophagy-dependent TLR-2 expression, or z-YVAD-fmk (100 μM), which regulates pyroptosis-dependent IL-1β expression. Cell death was examined by Trypan blue exclusion at 48 h p.i. ΔPK-induced cell death was significantly inhibited by the addition of SP600125 (P < 0.001), 3-MA (P < 0.001) or z-YVAD-fmk (P = 0.002) in both A2058 (Fig. 6a) and A375 (Fig. 6b) cells, confirming the contribution of the pathways. Collectively, the data indicated that ΔPK-induced tumour cell death and modulation of the tumour microenvironment involved multiple regulatory pathways.
Fig. 6.

Cytokine modulation contributes to ΔPK-induced melanoma cell death. (a) A2058 and (b) A375 cultures were mock- or ΔPK-infected (m.o.i. 1) followed by treatment with or without SP600125 (100 μM, left), 3-MA (5 mM, middle) or z-YVAD-fmk (100 μM, right) and examined for cell death by Trypan blue exclusion at 48 h p.i. Dead cells (blue staining) were counted in four quadrants and the percentage of dead cells was calculated. Data are presented as mean ± sd. ***P < 0.001 and **P < 0.01, by one-way ANOVA.
MICA and TNF-α expression are upregulated in vivo
To examine whether tumour growth inhibition was associated with ΔPK-induced microenvironment alteration, A375 cells were implanted into BALB/c nude mice by subcutaneous injection into both flanks and when the tumours became palpable (day 7; ∼200 mm3), the animals were given intratumour injections (100 μl) of partially purified ΔPK (106 p.f.u.) or culture medium control, and tumour volume was monitored over time. Confirming our previous findings that ΔPK inhibited tumour growth (Colunga et al., 2010), mock-infected animals evidenced time-dependent tumour growth reaching maximal levels on day 27. ΔPK caused a significant (P < 0.001) decrease in tumour growth that was still seen on day 41 (Fig. 7b). Representative xenografts collected from the ΔPK-treated animals on day 28 were examined for MICA and TNF-α expression indicative of altered tumour microenvironment through inhibition of the immunosuppressive cytokine IL-10 and upregulation of inflammatory cytokines, respectively. Both MICA and TNF-α were seen in the ΔPK- but not mock-treated xenografts (Fig. 7b), and this was associated with the inhibition of tumour growth (Fig. 7a). The data were consistent with those obtained in cultured cells and are schematically represented in Fig. 8.
Fig. 7.

ΔPK inhibits tumour growth, associated with MICA and TNF-α upregulation. (a) A375 xenografts were given four intratumour injections of ΔPK (n = 6; 106 p.f.u.) or growth medium (n = 3) at weekly intervals (days 7–28). Tumour growth ratio (mean ± sem) is as described in Methods. Statistically significant differences between mock and ΔPK treatment were first seen at day 19 (P < 0.001, by two-way ANOVA). (b) Extracts from 28 day representative mock- or ΔPK-treated xenografts (n = 3 each) were immunoblotted with antibodies to MICA and β-actin (left panel). Each lane represents a tumour from a different animal. Data are presented as mean ± densitometry units. Xenograft duplicates were stained with TNF-α antibody by immunohistochemistry (right panel). Staining cells were counted in three randomly selected fields (50 mm2) and the mean ± number of positive cells per field was calculated. ***P < 0.001 versus mock, by one-way ANOVA.
Fig. 8.
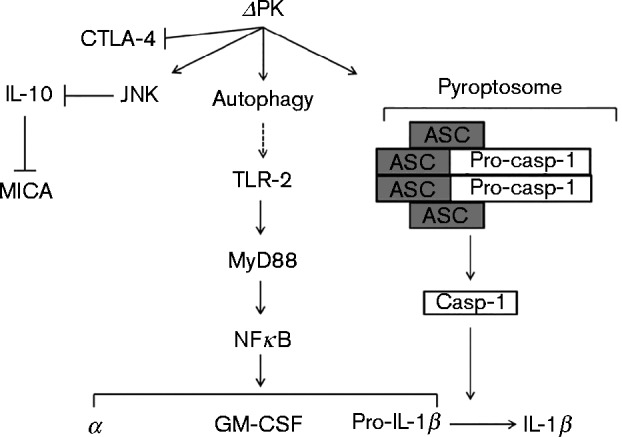
Schematic representation of the ΔPK activity in melanoma cells, which includes JNK/c-Jun-dependent inhibition of IL-10 resulting in MICA upregulation, induction of inflammatory cytokines through autophagy-mediated TLR-2 activation and pyroptosis, and CTLA-4 inhibition.
Discussion
The salient feature of the presented data is the finding that ΔPK alters the balance of cytokines directly secreted by the melanoma cells from an immunosuppressive to an inflammatory pattern, thereby increasing the expression of immunostimulatory molecules and cytotoxicity-associated functions. The following comments seem pertinent with respect to these findings.
Tumour immune evasion resulting from a highly immunosuppressive microenvironment is at least partially responsible for the overall limited clinical efficacy of virotherapy. In addition to infiltrating negative regulatory cells (Devaud et al., 2013), the tumour microenvironment contains immunosuppressive factors directly secreted by the tumour cells, including the cytokine IL-10 (Whiteside, 2002), which recruits immunomodulatory regulatory T-cells (Devaud et al., 2013). To overcome this problem, OVs were armed with inflammatory cytokines or delivered together with anti-tumour factors. Adenovirus- and HSV-based OVs armed with TNF-α were shown to have enhanced anti-tumour activity (Han et al., 2007; Hirvinen et al., 2015), as were HSV or vaccinia-based OVs armed with GM-CSF (Kaufman et al., 2014; Parviainen et al., 2015) and an oncolytic Newcastle disease delivered in combination with systemic cytotoxic T-lymphocyte antigen 4 (CTLA-4) blockade (Zamarin et al., 2014). However, individual cytokines deliver immunostimulatory signals in a relatively non-specific manner and they may even contribute to the establishment of immune tolerance, as shown for exogenously delivered GM-CSF (Mortha et al., 2014). Therefore, OVs that induce diverse anti-tumour effects including altered tumour cell secretion of multiple cytokines are particularly desirable.
Following on from previous findings that the strong oncolytic activity of ΔPK is associated with stimulation of multiple death-inducing pathways and virus replication (Colunga et al., 2014, 2010), our studies were designed to examine the potential contribution of an altered pattern of melanoma-secreted cytokines. We focused on the immunosuppressive cytokine IL-10 that inhibits antigen presentation (Buelens et al., 1997), and expression of MHC class II and co-stimulatory molecules (de Waal Malefyt et al., 1991), and promotes myeloid-derived suppressor cells and regulatory T-cell recruitment to the tumour microenvironment (Marvel & Gabrilovich, 2015). We also studied the inflammatory cytokines TNF-α and GM-CSF, which were individually associated with improved efficacy of virotherapy (Hersey & Gallagher, 2014; Hirvinen et al., 2015), and IL-1β, which induces robust and durable primary and secondary CD4+T-cell responses (Ben-Sasson et al., 2009). Despite their known heterogeneity, the studied melanoma cells constitutively secreted IL-10, but not TNF-α, IL-1β and GM-CSF, and this pattern was reversed by ΔPK infection. The ability of ΔPK to inhibit IL-10 secretion is through JNK/c-Jun pathway activation, as evidenced by: (i) increased levels of phosphorylated (activated) JNK and c-Jun, (ii) p-c-Jun nuclear translocation, and (iii) restored IL-10 expression through treatment with the JNK-specific inhibitor SP600125. This is in direct contrast to findings in monocytes/macrophages (Chanteux et al., 2007; Dobreva et al., 2009; Norkina et al., 2007), and it underscores the virus- and cell-type specificity of this response, and the importance of developing a better understanding of the role of OV therapy in inducing cytokine secretion in situ. Significantly, SP600125 also inhibited virus growth, indicating that IL-10 inhibition requires virus replication. However, similar IL-10 inhibition was not seen in HSV-2-infected melanoma cells, at least as determined by transcriptional array analysis (Fig. S2), and this is consistent with our earlier findings that the numbers of IL-10-producing T-cells are significantly lower in ΔPK- than HSV-2-infected immunocompetent animals (Gyotoku et al., 2002).
ΔPK infection induced the cell surface expression of MICA, the ligand for the NKG2D receptor expressed by NK- and cytotoxic T-cells (Bauer et al., 1999; Moretta et al., 2001), as confirmed by FACS analysis. Increase was blocked by both SP600125 and recombinant IL-10, indicating that ΔPK stimulates MICA expression through JNK/c-Jun-mediated IL-10 inhibition. MICA was also expressed in HSV-2-infected melanoma cells, but its levels were visibly lower than those seen for ΔPK, although virus replication was 100-fold higher (Colunga et al., 2010). The mechanism of HSV-2-induced MICA upregulation is still unknown. However, it is likely independent of JNK/c-Jun, because HSV-2 does not upregulate c-Jun (Fig. S2, Table S3) or activate the JNK/c-Jun pathway (Perkins et al., 2003). MICA expression was also upregulated in ΔPK-infected melanoma xenografts, potentially contributing to tumour growth inhibition (Fig. 7b) through NKG2D-dependent cytotoxicity by NK-, γδ T-, cytotoxic αβ CD8+T- or NKT-cells (Armeanu et al., 2005; Bauer et al., 1999; Skov et al., 2005). However, final conclusions must await the results of ongoing studies.
The TLR family plays a crucial role in antigen presentation, tumour clearance and pyroptosis, and promotes T-cell-mediated adaptive immunity (Bergsbaken et al., 2009; Inoue & Tani, 2014). In macrophages, TLR-2 promotes autophagy (Chuang et al., 2013), and intracranial HSV-1 infection stimulates TLR-2-dependent expression of the inflammatory cytokines IL-1β, IL-6 and TNF-α by microglia, astrocytes, neutrophils and monocytes (Aravalli et al., 2005; Villalba et al., 2012; Wang et al., 2012). However, in both macrophages and human foreskin fibroblasts, HSV-1 actually inhibits the secretion of mature IL-1β, apparently related to the trapping of caspase-1 by actin clusters (Johnson et al., 2013). HSV-2 decreases the expression of TLR-2 in human vaginal epithelial cells (Yao & Rosenthal, 2011) and it does not increase the number of T-cells that produce the inflammatory cytokine IFN-γ in immunocompetent animals (Gyotoku et al., 2002). Our transcriptional analysis indicates that, unlike ΔPK, HSV-2 does not upregulate TLR-2, MyD88, TNF-α, GM-CSF and IL-1β in melanoma cells (Fig. S2, Table S3). The exact contribution of the TLRs to tumour cell cytokine secretion and their role in virotherapy are still poorly understood. However, whilst co-stimulation with TLR ligands was recently shown to enhance the efficacy of OV therapy, it also sensitized the treated animals to cytokine shock-like response (Rommelfanger et al., 2013), underscoring the therapeutic advantage of an OV platform that directly stimulates TLR responses by the tumour cells.
We found that ΔPK induces the TLR-2/MyD88 pathway that culminates in the expression and nuclear translocation of the NFκB p50 active subunit, and the secretion of the inflammatory cytokines TNF-α, GM-CSF and IL-1β in the infected melanoma cells. Significantly, the ability of ΔPK to upregulate TLR-2 expression and cytokine secretion was autophagy-dependent and inhibited by treatment with the autophagy inhibitor 3-MA. This is consistent with previous findings that (i) autophagy contributes to innate immunity stimulation and cytokine secretion (Crişan et al., 2011; Harris, 2011), and (ii) ΔPK induces autophagy in melanoma cells (Colunga et al., 2010, 2014). However, whilst TLR-2 expression was strongly inhibited by 3-MA, the levels of activated NFκB were only partially decreased, suggesting that alternative autophagy-dependent and/or NFκB -independent pathways contribute to inflammatory cytokine production/secretion in ΔPK-infected melanoma cells. Indeed, MyD88/NFκB is an established pathway for TNF-α and IL-1β upregulation (Kissner et al., 2011; Lawrence, 2009), but TLR-2-initiated inflammatory amplification involving the signal adaptors TRADD/MAL/TRAF6 was also identified recently (Chang et al., 2014). We do not exclude the possibility that 3-MA has an off-target autophagy-unrelated effect that involves caspase-dependent cell death (Hou et al., 2012) and/or class I and II rather than class III phosphatidylinositide 3-kinase (Wu et al., 2010). Notwithstanding, the ability of 3-MA to inhibit ΔPK oncolytic activity confirms that autophagy-dependent TLR-2 pathways and inflammatory cytokine secretion contribute to its ability to cause tumour cell death, and it is unrelated to ΔPK replication, which was unaltered by 3-MA treatment.
Consistent with current findings that TLR activation induces pro-IL-1β expression (Hornung & Latz, 2010), which, in turn is cleaved and secreted by pyroptosome-activated caspase-1 (Fernandes-Alnemri et al., 2007), and pyroptosis contributes to tumour cell death (Bridle et al., 2010; Guo et al., 2014; Thorne, 2011), we found that ΔPK induced pyroptosome formation in the melanoma cells (through adapter protein ASC and pro-caspase-1 oligomerization) resulting in caspase-1 activation and pro-IL-1β cleavage. Cleavage was inhibited by the caspase-1-specific inhibitor z-YVAD-fmk that also inhibited the ability of ΔPK to induce melanoma cell death, confirming the contribution of pyroptosis to ΔPK-induced cell death. Interestingly, ΔPK also inhibited the expression of CTLA-4 (Fig. S5), which is constitutively expressed in several solid tumour-derived cells, including melanoma (Contardi et al., 2005; Shah et al., 2008), and behaves as a negative regulator of T-cell function (Teft et al., 2006). However, the mechanism of inhibition and its contribution to virotherapy are still under investigation.
In conclusion, our findings extend our original observations of ΔPK-mediated anti-tumour activity. They show that ΔPK switches the balance of cytokines that are directly secreted by the melanoma cells from immunosuppressive to inflammatory, thereby priming for the expression of functions, such as MICA, that are known to prime anti-tumour immune recognition and promote cytotoxicity (Fig. 8). Importantly, although the mutational landscape determines therapeutic sensitivity (Rizvi et al., 2015), ΔPK inhibits the immunosuppressive microenvironment in heterogeneous melanoma cultures that are molecularly distinct (Smith et al., 2012), and in immunocompetent mice and guinea pigs (Gyotoku et al., 2002; Wachsman et al., 2001). ΔPK is a particularly promising OV, because it was also well tolerated in phase I/II clinical studies (Aurelian, 2004; Casanova et al., 2002). However, the exact contribution of the modulated functions identified in these studies to cytotoxic anti-tumour immune responses is still unclear and final conclusions must await the results of ongoing studies.
Methods
Cells and viruses
A2058 and A375 cells were from the American Type Culture Collection, and grown in Dulbecco's modified Eagle's medium with 4.5 g glucose l− 1, 1500 mg sodium bicarbonate ml− 1, 4 mM glutamine and 10 % FBS (Gemini Bioproducts). ΔPK and the HSV-2 strain (G) from which it was established were described previously (Smith et al. 1998). ΔPK is deleted in the sequences that encode the independent protein kinase function in R1 (known as ICP10PK), which is required for virus growth. ΔPK expresses the kinase-negative R1 protein (p95) under the direction of the authentic R1 promoter. Following virus adsorption (1 h; 4 °C), the virus was removed, and the cells were overlaid with serum-free DMEM with or without the pharmacological inhibitors (0 h p.i.) and incubated at 37 °C for the indicated times. Virus titres were determined by plaque assay on Vero cells and the results are expressed as mean p.f.u. ml− 1 or mean p.f.u. per cell.
Antibodies, pharmacological inhibitors and reagents
Antibodies to caspase-1, IL-1β, pJNK, p-c-Jun and TLR-2 were from Cell Signaling Technology. Antibodies to β-actin, ASC, NFκB, MyD88, MICA, CTLA-4, TNF-α and glyceraldehyde 3-phosphate dehydrogenase were from Santa Cruz Biotechnology. Alexa Fluor 488- and 594-conjugated secondary antibodies were from Invitrogen. HRP-conjugated anti-rabbit and anti-mouse antibodies and mammalian-derived recombinant human IL-10 were from Cell Signalling Technologies, the caspase-1 inhibitor z-YVAD-fmk from Calbiochem, and the JNK inhibitor SP600125 and the autophagy inhibitor 3-MA from Sigma-Aldrich.
Microarray analysis
Total RNA was isolated and purified from mock- or ΔPK (m.o.i. 0.5 p.f.u. per cell; 24 h)-infected cells using a RNeasy kit (Qiagen). Biotin-labelled cRNA target was prepared with a TrueLabelling-AMP 2.0 kit (SABiosciences), and hybridized onto inflammatory and cell death targeted oligoarrays (Oligo GEArrays; SABiosciences) according to the manufacturer's instructions. Data were analysed with GEArray Expression Analysis Suite 2.0 software (SABiosciences) and were expressed as heatmaps derived from relative gene expression analysis.
ELISA
Conditioned media were assayed with ELISA kits (eBioscience) according to the manufacturer's instructions.
Cell death assay
Cell death was determined using Trypan blue staining. The percentage of dead (blue) cells was calculated relative to the total cell numbers in four independent fields using a haemocytometer.
Cytoplasmic/nuclear separation and immunoblotting
Separation of cytoplasmic/nuclear fractions and immunoblotting were done as described previously (Bollino et al., 2015; Wales et al., 2008).
Immunofluorescence and immunohistochemistry
Cells grown on glass coverslips were fixed with 4 % paraformaldehyde (30 min; room temperature) and permeabilized (2 min; 4 °C) with 0.1 % Triton X-100 in 0.1 % sodium citrate buffer. The coverslips were incubated overnight at 4 °C with primary antibody diluted in 5 % BSA and 5 % normal goat serum, washed in PBS/0.1 % Tween 20, and exposed to fluorochrome-labelled secondary antibodies (1 h; 37 °C). Slides were mounted in Vectashield with DAPI (Vector) and visualized with an Olympus BX50 fluorescent microscope. Stained cells (>250) were counted in four randomly selected fields. For FACS analysis, cells were resuspended (106 cells per tube) in FACS buffer (PBS with 0.5 % BSA and 2 mM EDTA), stained with MICA antibody (30 min; 4 °C) and FITC-conjugated secondary antibody (30 min; 4 °C; in the dark), and fixed with 1 % paraformaldehyde. Data were collected on an LSR II cytometer (BD Biosciences) and analysed with FCS Express Version 3 (De novo Software). Tumour sections (20 μm thick) were post-fixed (30 min) in 4 % paraformaldehyde, treated (10 min) with 0.3 % H2O2 to remove endogenous peroxidases, permeabilized and blocked (1 h) in blocking solution (10 % goat serum, 1 % BSA and 0.3 % Triton X-100 in PBS). Primary antibody (overnight; 4 °C) was followed by HRP-conjugated secondary antibody diluted in 5 % goat serum and 5 % BSA (1 h). The reaction was developed with ImmPACT DAB substrate (Vector), and sections were counterstained with Mayer's haematoxylin (Sigma-Aldrich) and visualized under bright-field conditions with an Olympus BX50 microscope. Stained cells were counted in representative 50 μm2 fields and the percentage of positive cells was calculated relative to the total cells per field.
In vivo studies
The Animal Care and Use Committee of the University of Maryland School of Medicine approved these studies. Male BALB/c nu/nu mice (6–8 weeks old) were from Charles River Laboratories. Xenografts were established by subcutaneous injection of A375 cells (107 in 100 μl) into both the left- and right-hind flanks. When the tumours became palpable (∼200 mm3; day 7), animals were randomly assigned to treatment groups consisting of partially purified ΔPK (100 μl; 106 p.f.u.) or virus-free culture medium in 100 μl. Four intratumour injections were given at weekly intervals. Every other day, minimum and maximum perpendicular tumour axes were measured, and tumour volume was calculated as described previously (Colunga et al., 2010). Data are expressed as tumour growth ratio, calculated by dividing each tumour volume measured over time by the initial tumour volume on day 7. Animals were maintained under pathogen-free conditions and were euthanized when their tumours reached 1.5 cm in any one direction. Tumours were collected after euthanasia, and processed for immunoblotting and immunohistochemistry.
Statistical analysis
ANOVA used SigmaPlot 11.0 for Windows (Systat Software). One-way ANOVA was followed by pairwise comparison with the Holm–Sidak test.
Acknowledgements
These studies were supported by a grant from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (AR053512) to L. A. A. C. was supported by the National Cancer Institute (CA154087).
Supplementary Data
Supplementary Data
Footnotes
Supplementary material is available with the online Supplementary Material.
References
- Akira S., Takeda K., Kaisho T. (2001). Toll-like receptors: critical proteins linking innate and acquired immunity Nat Immunol 2675–680 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- Angel P., Karin M. (1991). The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation Biochim Biophys Acta 1072129–157. [DOI] [PubMed] [Google Scholar]
- Aravalli R. N., Hu S., Rowen T. N., Palmquist J. M., Lokensgard J. R. (2005). Cutting edge: TLR2-mediated proinflammatory cytokine and chemokine production by microglial cells in response to herpes simplex virus J Immunol 1754189–4193 10.4049/jimmunol.175.7.4189. [DOI] [PubMed] [Google Scholar]
- Armeanu S., Bitzer M., Lauer U. M., Venturelli S., Pathil A., Krusch M., Kaiser S., Jobst J., Smirnow I., other authors (2005). Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate Cancer Res 656321–6329 10.1158/0008-5472.CAN-04-4252. [DOI] [PubMed] [Google Scholar]
- Aurelian L. (2004). Herpes simplex virus type 2 vaccines: new ground for optimism? Clin Diagn Lab Immunol 11437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurelian L. (2013). Oncolytic virotherapy: the questions and the promise Oncolytic Virother 201319–29 10.2147/OV.S39609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer S., Groh V., Wu J., Steinle A., Phillips J. H., Lanier L. L., Spies T. (1999). Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA Science 285727–729 10.1126/science.285.5428.727. [DOI] [PubMed] [Google Scholar]
- Ben-Sasson S. Z., Hu-Li J., Quiel J., Cauchetaux S., Ratner M., Shapira I., Dinarello C. A., Paul W. E. (2009). IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation Proc Natl Acad Sci U S A 1067119–7124 10.1073/pnas.0902745106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsbaken T., Fink S. L., Cookson B. T. (2009). Pyroptosis: host cell death and inflammation Nat Rev Microbiol 799–109 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollino D., Balan I., Aurelian L. (2015). Valproic acid induces neuronal cell death through a novel calpain-dependent necroptosis pathway J Neurochem 133174–186 10.1111/jnc.13029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridle B. W., Stephenson K. B., Boudreau J. E., Koshy S., Kazdhan N., Pullenayegum E., Brunelliere J., Bramson J. L., Lichty B. D., Wan Y. (2010). Potentiating cancer immunotherapy using an oncolytic virus Mol Ther 181430–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buelens C., Verhasselt V., De Groote D., Thielemans K., Goldman M., Willems F. (1997). Interleukin-10 prevents the generation of dendritic cells from human peripheral blood mononuclear cells cultured with interleukin-4 and granulocyte/macrophage-colony-stimulating factor Eur J Immunol 27756–762 10.1002/eji.1830270326. [DOI] [PubMed] [Google Scholar]
- Casanova G., Cancela R., Alonzo L., Benuto R., Magana Mdel. C., Hurley D. R., Fishbein E., Lara C., Gonzalez T., other authors (2002). A double-blind study of the efficacy and safety of the ICP10deltaPK vaccine against recurrent genital HSV-2 infections Cutis 70235–239. [PubMed] [Google Scholar]
- Chang Y. L., Chen T. H., Wu Y. H., Chen G. A., Weng T. H., Tseng P. H., Hsieh S. L., Fu S. L., Lin C. H., other authors (2014). A novel TLR2-triggered signalling crosstalk synergistically intensifies TNF-mediated IL-6 induction J Cell Mol Med 181344–1357 10.1111/jcmm.12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanteux H., Guisset A. C., Pilette C., Sibille Y. (2007). LPS induces IL-10 production by human alveolar macrophages via MAPKinases- and Sp1-dependent mechanisms Respir Res 871. 10.1186/1465-9921-8-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Zheng L., He W., Qiu M., Gao L., Liu J., Huang A. (2014). Cotransfection with IL-10 and TGF-β1 into immature dendritic cells enhances immune tolerance in a rat liver transplantation model Am J Physiol Gastrointest Liver Physiol 306G575–G581 10.1152/ajpgi.00283.2013. [DOI] [PubMed] [Google Scholar]
- Chiocca E. A., Rabkin S. D. (2014). Oncolytic viruses and their application to cancer immunotherapy Cancer Immunol Res 2295–300 10.1158/2326-6066.CIR-14-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang S. Y., Yang C. H., Chou C. C., Chiang Y. P., Chuang T. H., Hsu L. C. (2013). TLR-induced PAI-2 expression suppresses IL-1β processing via increasing autophagy and NLRP3 degradation Proc Natl Acad Sci U S A 11016079–16084 10.1073/pnas.1306556110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colunga A. G., Laing J. M., Aurelian L. (2010). The HSV-2 mutant DeltaPK induces melanoma oncolysis through nonredundant death programs and associated with autophagy and pyroptosis proteins Gene Ther 17315–327 10.1038/gt.2009.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colunga A., Bollino D., Schech A., Aurelian L. (2014). Calpain-dependent clearance of the autophagy protein p62/SQSTM1 is a contributor to ΔPK oncolytic activity in melanoma Gene Ther 21371–378 10.1038/gt.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contardi E., Palmisano G. L., Tazzari P. L., Martelli A. M., Fala F., Fabbi M., Kato T., Lucarelli E., Donati D., other authors (2005). CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction Int J Cancer 117538–550. [DOI] [PubMed] [Google Scholar]
- Crişan T. O., Plantinga T. S., van de Veerdonk F. L., Farcaş M. F., Stoffels M., Kullberg B. J., van der Meer J. W., Joosten L. A., Netea M. G. (2011). Inflammasome-independent modulation of cytokine response by autophagy in human cells PLoS One 6e18666. 10.1371/journal.pone.0018666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Aquino M. T., Malhotra A., Mishra M. K., Shanker A. (2015). Challenges and future perspectives of T cell immunotherapy in cancer Immunol Lett 166117–133 10.1016/j.imlet.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Waal Malefyt R., Haanen J., Spits H., Roncarolo M. G., te Velde A., Figdor C., Johnson K., Kastelein R., Yssel H., de Vries J. E. (1991). Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression J Exp Med 174915–924 10.1084/jem.174.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaud C., John L. B., Westwood J. A., Darcy P. K., Kershaw M. H. (2013). Immune modulation of the tumor microenvironment for enhancing cancer immunotherapy OncoImmunology 2e25961. 10.4161/onci.25961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobreva Z. G., Miteva L. D., Stanilova S. A. (2009). The inhibition of JNK and p38 MAPKs downregulates IL-10 and differentially affects c-Jun gene expression in human monocytes Immunopharmacol Immunotoxicol 31195–201 10.1080/08923970802626276. [DOI] [PubMed] [Google Scholar]
- Fernandes-Alnemri T., Wu J., Yu J. W., Datta P., Miller B., Jankowski W., Rosenberg S., Zhang J., Alnemri E. S. (2007). The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation Cell Death Differ 141590–1604 10.1038/sj.cdd.4402194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z. S., Liu Z., Bartlett D. L. (2014). Oncolytic immunotherapy: dying the right way is a key to eliciting potent antitumor immunity Front Oncol 474. 10.3389/fonc.2014.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyotoku T., Ono F., Aurelian L. (2002). Development of HSV-specific CD4+ Th1 responses and CD8+ cytotoxic T lymphocytes with antiviral activity by vaccination with the HSV-2 mutant ICP10DeltaPK Vaccine 202796–2807 10.1016/S0264-410X(02)00199-8. [DOI] [PubMed] [Google Scholar]
- Han J., Lee J. D., Jiang Y., Li Z., Feng L., Ulevitch R. J. (1996). Characterization of the structure and function of a novel MAP kinase kinase (MKK6) J Biol Chem 2712886–2891 10.1074/jbc.271.6.2886. [DOI] [PubMed] [Google Scholar]
- Han Z. Q., Assenberg M., Liu B. L., Wang Y. B., Simpson G., Thomas S., Coffin R. S. (2007). Development of a second-generation oncolytic Herpes simplex virus expressing TNFalpha for cancer therapy J Gene Med 999–106 10.1002/jgm.999. [DOI] [PubMed] [Google Scholar]
- Harris J. (2011). Autophagy and cytokines Cytokine 56140–144 10.1016/j.cyto.2011.08.022. [DOI] [PubMed] [Google Scholar]
- Hersey P., Gallagher S. (2014). Intralesional immunotherapy for melanoma J Surg Oncol 109320–326 10.1002/jso.23494. [DOI] [PubMed] [Google Scholar]
- Hirvinen M., Rajecki M., Kapanen M., Parviainen S., Rouvinen-Lagerstrom N., Diaconu I., Nokisalmi P., Tenhunen M., Hemminki A., Cerullo V. (2015). 10.1089/hum.2014.069Immunological effects of a tumor necrosis factor-alpha armed oncolytic adenovirus Hum Gene Ther. [DOI] [PubMed] [Google Scholar]
- Hornung V., Latz E. (2010). Critical functions of priming and lysosomal damage for NLRP3 activation Eur J Immunol 40620–623 10.1002/eji.200940185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou H., Zhang Y., Huang Y., Yi Q., Lv L., Zhang T., Chen D., Hao Q., Shi Q. (2012). Inhibitors of phosphatidylinositol 3′-kinases promote mitotic cell death in HeLa cells PLoS One 7e35665. 10.1371/journal.pone.0035665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H., Tani K. (2014). Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments Cell Death Differ 2139–49 10.1038/cdd.2013.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K. E., Chikoti L., Chandran B. (2013). Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes J Virol 875005–5018 10.1128/JVI.00082-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman H. L., Ruby C. E., Hughes T., Slingluff C. L., Jr (2014). Current status of granulocyte-macrophage colony-stimulating factor in the immunotherapy of melanoma J Immunother Cancer 211. 10.1186/2051-1426-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C., Ho D. H., Suk J. E., You S., Michael S., Kang J., Joong Lee S., Masliah E., Hwang D., other authors (2013). Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia Nat Commun 41562. 10.1038/ncomms2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissner T. L., Ruthel G., Cisney E. D., Ulrich R. G., Fernandez S., Saikh K. U. (2011). MyD88-dependent pro-inflammatory cytokine response contributes to lethal toxicity of staphylococcal enterotoxin B in mice Innate Immun 17451–462 10.1177/1753425910374092. [DOI] [PubMed] [Google Scholar]
- Lawrence T. (2009). The nuclear factor NF-kappaB pathway in inflammation Cold Spring Harb Perspect Biol 1a001651. 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida N., Umikawa M., Takei K., Sakima N., Myagmar B. E., Taira K., Uezato H., Ogawa Y., Kariya K. (2004). Mitogen-activated protein kinase kinase kinase kinase 4 as a putative effector of Rap2 to activate the c-Jun N-terminal kinase J Biol Chem 27915711–15714 10.1074/jbc.C300542200. [DOI] [PubMed] [Google Scholar]
- Marvel D., Gabrilovich D. I. (2015). Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected J Clin Invest 1253356–3364 10.1172/JCI80005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretta A., Bottino C., Vitale M., Pende D., Cantoni C., Mingari M. C., Biassoni R., Moretta L. (2001). Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis Annu Rev Immunol 19197–223 10.1146/annurev.immunol.19.1.197. [DOI] [PubMed] [Google Scholar]
- Mortha A., Chudnovskiy A., Hashimoto D., Bogunovic M., Spencer S. P., Belkaid Y., Merad M. (2014). Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis Science 3431249288. 10.1126/science.1249288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouawad R., Sebert M., Michels J., Bloch J., Spano J. P., Khayat D. (2010). Treatment for metastatic malignant melanoma: old drugs and new strategies Crit Rev Oncol Hematol 7427–39 10.1016/j.critrevonc.2009.08.005. [DOI] [PubMed] [Google Scholar]
- Norkina O., Dolganiuc A., Shapiro T., Kodys K., Mandrekar P., Szabo G. (2007). Acute alcohol activates STAT3, AP-1, and Sp-1 transcription factors via the family of Src kinases to promote IL-10 production in human monocytes J Leukoc Biol 82752–762 10.1189/jlb.0207099. [DOI] [PubMed] [Google Scholar]
- Oltmanns U., Issa R., Sukkar M. B., John M., Chung K. F. (2003). Role of c-jun N-terminal kinase in the induced release of GM-CSF, RANTES and IL-8 from human airway smooth muscle cells Br J Pharmacol 1391228–1234 10.1038/sj.bjp.0705345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parviainen S., Ahonen M., Diaconu I., Kipar A., Siurala M., Vaha-Koskela M., Kanerva A., Cerullo V., Hemminki A. (2015). GMCSF-armed vaccinia virus induces an antitumor immune response Int J Cancer 1361065–1072. [DOI] [PubMed] [Google Scholar]
- Perkins D., Pereira E. F., Aurelian L. (2003). The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) functions as a dominant regulator of apoptosis in hippocampal neurons involving activation of the ERK survival pathway and upregulation of the antiapoptotic protein Bag-1 J Virol 771292–1305 10.1128/JVI.77.2.1292-1305.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi N. A., Hellmann M. D., Snyder A., Kvistborg P., Makarov V., Havel J. J., Lee W., Yuan J., Wong P., other authors (2015). Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer Science 348124–128 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommelfanger D. M., Compte M., Diaz R. M., Ilett E., Alvarez-Vallina L., Thompson J. M., Kottke T. J., Melcher A., Vile R. G. (2013). The efficacy versus toxicity profile of combination virotherapy and TLR immunotherapy highlights the danger of administering TLR agonists to oncolytic virus-treated mice Mol Ther 21348–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seglen P. O., Gordon P. B. (1982). 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes Proc Natl Acad Sci U S A 791889–1892 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano A. E., Menares-Castillo E., Garrido-Tapia M., Ribeiro C. H., Hernández C. J., Mendoza-Naranjo A., Gatica-Andrades M., Valenzuela-Diaz R., Zúñiga R., other authors (2011). Interleukin 10 decreases MICA expression on melanoma cell surface Immunol Cell Biol 89447–457 10.1038/icb.2010.100. [DOI] [PubMed] [Google Scholar]
- Shah K. V., Chien A. J., Yee C., Moon R. T. (2008). CTLA-4 is a direct target of Wnt/beta-catenin signaling and is expressed in human melanoma tumors J Invest Dermatol 1282870–2879 10.1038/jid.2008.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skov S., Pedersen M. T., Andresen L., Straten P. T., Woetmann A., Odum N. (2005). Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B Cancer Res 6511136–11145 10.1158/0008-5472.CAN-05-0599. [DOI] [PubMed] [Google Scholar]
- Smith C. C., Peng T., Kulka M., Aurelian L. (1998). PK domain of the large subunit of herpes simplex virus type 2 ribonucleotide reductase (ICP10) is required for immediate-early gene expression and virus growth J Virol 729131–9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. C., Nelson J., Aurelian L., Gober M., Goswami B. B. (2000). Ras-GAP binding and phosphorylation by herpes simplex virus type 2 RR1 PK (ICP10) and activation of the Ras/MEK/MAPK mitogenic pathway are required for timely onset of virus growth J Virol 7410417–10429 10.1128/JVI.74.22.10417-10429.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. C., Lee K. S., Li B., Laing J. M., Hersl J., Shvartsbeyn M., Aurelian L. (2012). Restored expression of the atypical heat shock protein H11/HspB8 inhibits the growth of genetically diverse melanoma tumors through activation of novel TAK1-dependent death pathways Cell Death Dis 3e371. 10.1038/cddis.2012.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutterwala F. S., Haasken S., Cassel S. L. (2014). Mechanism of NLRP3 inflammasome activation Ann N Y Acad Sci 131982–95 10.1111/nyas.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teft W. A., Kirchhof M. G., Madrenas J. (2006). A molecular perspective of CTLA-4 function Annu Rev Immunol 2465–97 10.1146/annurev.immunol.24.021605.090535. [DOI] [PubMed] [Google Scholar]
- Thorne S. H. (2011). Immunotherapeutic potential of oncolytic vaccinia virus Immunol Res 50286–293 10.1007/s12026-011-8211-4. [DOI] [PubMed] [Google Scholar]
- Vallabhapurapu S., Matsuzawa A., Zhang W., Tseng P. H., Keats J. J., Wang H., Vignali D. A., Bergsagel P. L., Karin M. (2008). Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling Nat Immunol 91364–1370 10.1038/ni.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba M., Hott M., Martin C., Aguila B., Valdivia S., Quezada C., Zambrano A., Concha M. I., Otth C. (2012). Herpes simplex virus type 1 induces simultaneous activation of Toll-like receptors 2 and 4 and expression of the endogenous ligand serum amyloid A in astrocytes Med Microbiol Immunol (Berl) 201371–379 10.1007/s00430-012-0247-0. [DOI] [PubMed] [Google Scholar]
- Wachsman M., Kulka M., Smith C. C., Aurelian L. (2001). A growth and latency compromised herpes simplex virus type 2 mutant (ICP10DeltaPK) has prophylactic and therapeutic protective activity in guinea pigs Vaccine 191879–1890 10.1016/S0264-410X(00)00446-1. [DOI] [PubMed] [Google Scholar]
- Wales S. Q., Laing J. M., Chen L., Aurelian L. (2008). ICP10PK inhibits calpain-dependent release of apoptosis-inducing factor and programmed cell death in response to the toxin MPP+ Gene Ther 151397–1409 10.1038/gt.2008.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. P., Bowen G. N., Zhou S., Cerny A., Zacharia A., Knipe D. M., Finberg R. W., Kurt-Jones E. A. (2012). Role of specific innate immune responses in herpes simplex virus infection of the central nervous system J Virol 862273–2281 10.1128/JVI.06010-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteside T. L. (2002). Tumor-induced death of immune cells: its mechanisms and consequences Semin Cancer Biol 1243–50 10.1006/scbi.2001.0402. [DOI] [PubMed] [Google Scholar]
- Wolf A., Beuerlein K., Eckart C., Weiser H., Dickkopf B., Müller H., Sakurai H., Kracht M. (2011). Identification and functional characterization of novel phosphorylation sites in TAK1-binding protein (TAB) 1 PLoS One 6e29256. 10.1371/journal.pone.0029256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. T., Tan H. L., Shui G., Bauvy C., Huang Q., Wenk M. R., Ong C. N., Codogno P., Shen H. M. (2010). Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase J Biol Chem 28510850–10861 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X. D., Rosenthal K. L. (2011). Herpes simplex virus type 2 virion host shutoff protein suppresses innate dsRNA antiviral pathways in human vaginal epithelial cells J Gen Virol 921981–1993 10.1099/vir.0.030296-0. [DOI] [PubMed] [Google Scholar]
- Zamarin D., Holmgaard R. B., Subudhi S. K., Park J. S., Mansour M., Palese P., Merghoub T., Wolchok J. D., Allison J. P. (2014). Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy Sci Transl Med 6226–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data