

Abstract
Vaccinia virus (VACV) genes are characterized as either essential or non-essential for growth in culture. It seems intuitively obvious that if a gene can be deleted without imparting a growth defect in vitro it does not have a function related to basic replication or spread. However, this interpretation relies on the untested assumption that there is no redundancy across the genes that have roles in growth in cell culture. First, we provide a comprehensive summary of the literature that describes the essential genes of VACV. Next, we looked for interactions between large blocks of non-essential genes located at the ends of the genome by investigating sets of VACVs with large deletions at the genomic termini. Viruses with deletions at either end of the genome behaved as expected, exhibiting only mild or host-range defects. In contrast, combining deletions at both ends of the genome for the VACV Western Reserve (WR) strain caused a devastating growth defect on all cell lines tested. Unexpectedly, we found that the well-studied VACV growth factor homologue encoded by C11R has a role in growth in vitro that is exposed when 42 genes are absent from the left end of the VACV WR genome. These results demonstrate that some non-essential genes contribute to basic viral growth, but redundancy means these functions are not revealed by single-gene-deletion mutants.
Introduction
Vaccinia virus (VACV) is the most extensively studied member of the poxvirus family, which also counts amongst its number the causative agents of smallpox and human monkeypox. VACV strains have dsDNA genomes of ∼200 kb. The broad in vitro host range of VACV demands a high level of independence from host cell functions and so the VACV genome encodes most of the proteins required for the production of infectious virions. The set of genes required for propagation in mammalian cells under ideal conditions in vitro are described in the literature as the essential genes of poxviruses. As a large DNA virus, VACV also encodes genes that are dispensable in cell culture. Many of these non-essential genes have been shown to act as virulence factors in animal models of infection.
Essential genes as defined by conservation and large deletions
Phylogenetic analyses have shown that the conserved genes of poxviruses cluster in the central region of the genome (Upton et al., 2003; Gubser et al., 2004; Lefkowitz et al., 2006). Conversely, the variable termini contain many virulence factors, as well as gene fragments, most of which are assumed to be non-functional (Fig. S1, available in the online Supplementary Material). As many of the non-essential genes are found within the variable termini of the genome, it has been possible to obtain viable VACV mutants that contain large deletions (Moss et al., 1981; Panicali et al., 1981; Paez & Esteban, 1985; Kotwal & Moss, 1988; Perkus et al., 1989, 1991; Blake et al., 1995; Symons et al., 1995). The ability to isolate these viruses provides much of the evidence for the oft-repeated statement that genes at the termini of the VACV genome are non-essential for replication in cell culture, but rigorous studies of these viruses in multiple cell lines is lacking.
The VACV strain with the largest targeted deletions was produced by Perkus et al. (1991). Two blocks, containing 38 ORFs (C23L–F4L at the left) and 17 ORFs (B13R–B29R at the right), were deleted from the termini of the genome of VACV Copenhagen to create vP811. A second virus, named vP759, has only the 17 genes at the right end deleted. Additionally, both vP811 and vP759 are deleted for J2R (thymidine kinase), A26L (A-type inclusion locus) and A56R (haemagglutinin) (Fig. 1a). Importantly, the human host-range gene C7L was used as a selectable marker for deletion of the right terminal genes from vP811 and so remains intact in this virus. The original report suggested that these viruses are capable of forming plaques on the human cell line MRC-5 (Perkus et al., 1991). Later reports showed that vP811 can form infectious virions on HeLa (Fagan-Garcia & Barry, 2011) and establish a persistent infection of Jurkat cells (Melana & Pogo, 2005). Furthermore, in single-step growth curves, vP811 replicates as well as Copenhagen in BGMK and A549 cells, but forms smaller plaques than Copenhagen on BS-C-1, A549 and BGMK cells (Burles et al., 2014; Sumner et al., 2014). Poor replication in a multiple-step growth curve in BGMK cells, a small plaque phenotype and an observation of fewer actin tails all suggested that whilst viable in culture, vP811 has a defect in virus spread (Burles et al., 2014).
Fig. 1.

Position of large deletions in recombinant viruses. (a) HindIII map of the VACV Copenhagen genome, showing the position of the deletions at the termini of the genome in either vP811 or vP759. Short grey lines represent the thymidine kinase gene, A-type inclusion locus region and haemagglutinin gene that are deleted from both strains. The gpt selectable marker is present at the left end of vP811 and the right end of vP759. The human host-range gene C7L is present at the right terminus of vP811. (b) HindIII map of the VACV WR genome, showing the position of the deletions at the termini of the genome in six of the recombinant viruses produced. The EGFP/bsd fusion gene and VACWR021 (C7L) are present at the left end of all recombinant viruses carrying ΔLeft, and the mCherry gene was inserted at the right end to select for deletions ΔRight, ΔITRr or ΔUniqueR. (c) WR and Copenhagen orthologues relevant to (a, b). The genes delimiting the large deletions are the same across VACV strains, but some of the genes within these blocks differ due to strain differences.
Essential genes as defined by single-gene-deletion mutants
By far the majority of work contributing to our knowledge of VACV genes with functions related to growth in vitro has used viruses with single genes deleted. We made a comprehensive survey of this literature and assigned each of the 223 ORFs of the Western Reserve (WR) strain and 207 ORFs of the Copenhagen strain as essential or non-essential for replication in cell culture (Fig. S1a). Commonly, genes are defined as non-essential if a viable deletion mutant can be isolated. However, by this definition a single gene deletion that causes a 1000-fold reduction in infectious virus still results in the gene being considered non-essential. As replication defects may be additive and we were interested in VACV mutants missing multiple genes, we have used a more strict definition, where a gene is considered essential (i.e. has a role in cell culture) if its deletion results in a decrease in virus titre>10-fold in either a single- or multiple-step growth curve. All genes involved in wrapped virion production, actin tail formation and extracellular virion release have also been defined as essential. As plaque size can be reduced without a measurable decrease in virus production (Dobson et al., 2014; Dobson & Tscharke, 2014), other genes that influence plaque size, but not replication, have been defined as non-essential. By this definition, 93 genes are required for VACV replication in cell culture, whilst 108 and 94 ORFs from WR and Copenhagen, respectively, are non-essential (Fig. S1a). Tables S1–S5 detail the classification of genes as above on a gene-by-gene basis supported by selected references (Table S1, essential genes; Table S2, non-essential genes; Table S3, genes defined as non-essential on the basis of viruses with large deletions; Table S4, genes with no published deletion; Table S5, host-range genes).
As expected, the essential genes cluster at the centre of the genome, like the conserved genes of poxviruses (Upton et al., 2003; Lefkowitz et al., 2006) (Fig. S1b). In general, the correlation between conserved genes and genes that cannot be deleted from the VACV genome is good. However, five conserved genes (G6R, I5L, F16L, D10R and A14.5L) have been defined as non-essential because single-gene-deletion mutants were created successfully (Betakova et al., 2000; Parrish & Moss, 2006; Senkevich et al., 2008; Sood et al., 2008; Senkevich et al., 2011; Liu et al., 2014). Four of these genes are known to affect virulence after infection of mice, suggesting that these genes were conserved because they carry out a crucial role in vivo (Betakova et al., 2000; Senkevich et al., 2008; Sood et al., 2008; Liu et al., 2014).
The VACV literature as summarized above does not cover the entire genome. There are 18 ORFs across the VACV WR and Copenhagen genomes for which no deletion mutant has been reported. Furthermore, there are an additional 51 ORFs that are considered non-essential due to being deleted in large blocks of sequence, but where the resulting viruses are not well characterized. Of these 69 ORFs, 33 appear to be gene fragments (http://www.poxvirus.org/), but this still leaves 36 poorly characterized genes. Perhaps more significantly, implicit in the traditional gene-by-gene approach, is the assumption that each gene contributes a unique function. This premise underpins the common interpretation that if a gene can be deleted its function is unrelated to basic replication and spread in culture. However, it is known that there can be redundancy between host-range genes, which are those required for growth in particular cell lines (Meng et al., 2009). Other functions such as inhibition of apoptosis are redundant to some degree as well, with a hierarchy of importance across viral genes (Veyer et al., 2014). This suggests that similar redundancy might exist for functions more generally required for growth in culture, but surprisingly this possibility has never been tested explicitly. To do this here, we examined a set of VACVs with large deletions at the genomic termini. Large deletions at the right or the left end of the genome alone gave rise to host-range defects. However, combining these deletions caused a severe general replication defect, suggesting redundancy exists between these blocks of genes. We then provide a specific example of a gene previously defined as non-essential for growth in culture (C11R) that behaves like an essential gene in the context of a large deletion at the other end of the genome.
Results
Deletion of 55 ORFs from the genomic termini of VACV Copenhagen reduces virus replication on multiple cell lines
We began experiments using vP811 and vP759, the genomes of which are described in Fig. 1. Consistent with previous reports (Burles et al., 2014; Sumner et al., 2014), vP811 produced only very small foci on monolayers of BS-C-1 cells, but the plaques formed by vP759 were also significantly smaller than VACV Copenhagen (Fig. 2). The replication capacity of vP811 and vP759 was then compared with the parental VACV strain Copenhagen in multiple-step growth curves (m.o.i. 0.01) on six commonly used mammalian cell lines (Fig. 3). We collected cell-associated and supernatant virus to ensure that we were not missing any infectious progeny. On every cell line tested, the titre of each virus present at 72 h post-infection (p.i.) exceeded the input titre. This was consistent with the conclusion that vP811 remains viable in vitro, despite the deletion of 58 genes. However, vP811 exhibited a substantial replication defect on each of the six mammalian cell lines tested, producing less virus than either Copenhagen or vP759 from the same cell type. The extent of the replication defect of vP811 varied depending on cell type. The most severe defect was seen on HeLa cells in which vP811 produced 200-fold less virus than vP759 or Copenhagen. However, on Vero cells, vP811 replicated almost as well as vP759 and Copenhagen. Overall, however, the defect in replication for vP811 was >10-fold on most common cell lines, so the genes deleted in this virus cannot be classified as non-essential by our strict definition. On all cell lines except MRC-5, the virus titre recovered after vP759 infection was not different to infection with Copenhagen.
Fig. 2.
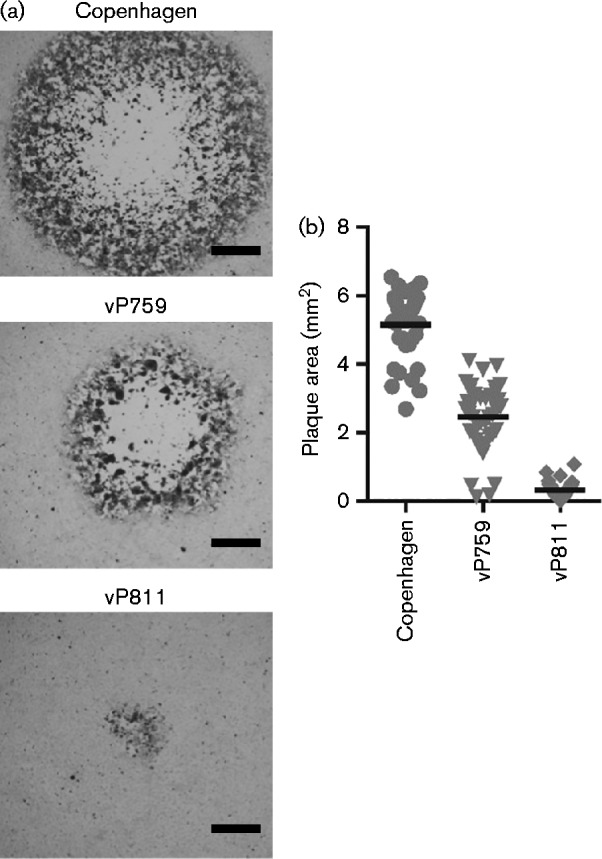
Size and morphology of plaques formed by VACV strains Copenhagen, vP759 and vP811 on BS-C-1 cells. (a) Representative immunostained plaques formed on BS-C-1 over 72 h by Copenhagen, vP759 and vP811. Bar, 0.5 mm. (b) Area of immunostained plaques produced on BS-C-1 cells (n = 40). The horizontal line represents mean plaque area. Statistical significance was determined using one-way ANOVA (P < 0.0001, n = 40) with Tukey's multiple comparison test (all pairs are significantly different, P < 0.001).
Fig. 3.

vP811 has a severe replication defect on multiple cell lines. (a) Replication rates of vP759 (squares) and vP811 (triangles) were compared with Copenhagen (circles) in multiple-step growth curves (m.o.i. 0.01) in the cell lines shown. The 0 h sample was collected immediately after removing the initial inoculum and addition of fresh media. The yield of cell-associated virus (black) and virus present in the supernatant (grey) was determined separately. Data are mean ± sem from three independent wells. Error bars are plotted, but in most cases are obscured by the data points. (b) Data from (a) presented as the total (sum of cell-associated and supernatant virus, except MRC-5 and 293A) fold increase of virus over the inoculum at 48 h p.i. Cop, Copenhagen.
Deletion of multiple genes from the left or right terminus of the VACV WR genome
Three possibilities might explain the more severe replication defect of vP811 than vP759: (i) a gene (genes) at the left end of the genome is (are) important for replication in vitro; (ii) genes within the left and right termini provide complementary functions; and (iii) an unintended mutation to an essential gene at the centre of the genome was acquired during production of vP811. We sought to distinguish between these possibilities by producing a new set of large deletion viruses. VACV WR was chosen as the parent, because it has been the most commonly used strain in studies of VACV gene function (Fig. 1b). The first two viruses produced were WRΔLeft and WRΔRight that had deletions at the right and left ends of the genome, respectively. The deletion boundaries for these viruses were based on those of vP811 (Perkus et al., 1991), but the number and exact set of genes deleted were not identical due to the different arrangement of genes at the termini of Copenhagen and WR (Fig. 1). WRΔLeft had 42 ORFs between VACWR001 (C23L) and VACWR043 (F4L) deleted and replaced with EGFP/bsd (blasticidin) (Wong et al., 2011) and VACWR021 (C7L); WRΔRight had 25 ORFs between VACWR195 (B13R) and VACWR218 (B29R) replaced with mCherry. We maintained 3701 bp of non-coding DNA at the termini, including the essential concatemer resolution sequence, terminal hairpin and four blocks containing direct repeat elements (Merchlinsky & Moss, 1989; Merchlinsky, 1990; Garcia & Moss, 2001). We used the two fluorescent marker genes (EGFP and mCherry) to counter potential difficulties associated with isolating recombinant viruses with a growth defects using drug selection alone (Da Fonseca & Moss, 2003; Parrish & Moss, 2006; Senkevich et al., 2009). The inclusion of the host-range gene VACWR021 (C7L) in all viruses with deletions at the left end of the genome was to maintain consistency with vP811 and to avoid a human cell host-range defect.
Next, WRΔLeft and WRΔRight were used as parents to make two identical, but independently derived viruses with large deletions at both genomic termini; these were called WRΔLR and WRΔRL, respectively. These viruses were exceptionally difficult to isolate and grow, and their identification on monolayers was only possible with the aid of fluorescence from EGFP and mCherry. Multiple attempts to amplify these on a variety of cell lines yielded titres of the order of 106 p.f.u. ml− 1. This is orders of magnitude lower than for the parent viruses with single deletions and vP811, the equivalent deletion virus in strain Copenhagen.
Large deletions at both ends of the VACV genome cause a tiny plaque phenotype
The area of plaques formed by WRΔLR, WRΔRL, WRΔLeft, WRΔRight and WR on BS-C-1 cells were measured from micrographs of immunostained plaques (Fig. 4). The plaques formed by all deletion viruses were significantly smaller than those of WR (P < 0.05). Deletion of the 42 ORFs from the left end of the genome had a greater effect on plaque size than deletion of 25 ORFs from the right terminus (P < 0.05). However, the combination of both deletions had a far more severe impact than would be predicted by adding the phenotypes of the single end deletions. This suggested that one or more genes at the left and right genomic termini have complementary functions in plaque formation, such that loss of those from one end alone has a modest effect, but the combined loss is devastating.
Fig. 4.

Plaque morphology of WRΔLR and WRΔRL on BS-C-1 cells. (a) Representative immunostained plaques formed on BS-C-1 monolayers after 72 h by VACV recombinants; (b) higher magnification images of plaques from WRΔLR and WRΔRL. Bar, 0.5 mm. (c) Area of plaques produced by the viruses (n = 30); the horizontal line indicates the mean plaque area. All pairs are significantly different except WRΔLR and WRΔRL (ANOVA with Tukey's post-test P < 0.05).
Large deletions at both ends of the VACV genome cause a severe replication defect
The small amounts of WRΔLR and WRΔRL able to be grown limited replication analyses with these viruses to a single time point under multiple-step (low m.o.i.) conditions. All four deletion viruses had a replication defect of greater than an order of magnitude on at least two cell lines (Fig. 5). The viruses with a deletion at only one end of the genome had cell-specific or host-range defects. In spite of plaque size differences, the virus titres obtained at 48 h p.i. on BS-C-1 cells after infection with WRΔLeft or WRΔRight were comparable with the parental virus WR. However, replication defects were observed for WRΔLeft on BHK-21, WRΔRight on Vero and both strains on MRC-5. The host-range defect of WRΔLeft for growth on BHK-21 was likely due to the deletion of K3L, which is required for replication on these cells (Langland & Jacobs, 2002), but no host-range gene required for replication on Vero cells has been described in the B13R–B29R region. In contrast to the single end deletions, WRΔLR and WRΔRL had extremely severe growth defects on all four cell lines tested, with increases over the inoculum generally restricted to ≤ 10-fold. To place this poor replication in context, vP811 was able to generate increases over inoculum of 100- to 1000-fold on these cell lines. Together, the data revealed that there are functions required for the replication of VACV in culture encoded by redundant sets of genes found at opposing ends of the genome.
Fig. 5.

Replication is severely decreased in WR recombinants with large deletions at the left and right end. Growth of viruses on the indicated cell lines was measured at 48 h p.i. after low m.o.i. (0.01) infection. The titre of (a) cell-associated virus and (b) virus released into the supernatant was determined by plaque assay on BS-C-1 cells with immunostaining. Data are mean ± sem from three independent wells. (c) The sum of results in (a, b) presented as the total fold increase of virus at 48 h p.i. over the inoculum.
More than one gene at the right terminal contributes to plaque size
To further explore the interplay of genes found at the VACV genomic termini, we set out to identify which regions at the right terminus are important for replication on BS-C-1 cells when 42 ORFs are missing from the left end of the genome. The ends of the VACV genome are inverted terminal repeats (ITRs) that contains some ORFs, so it is possible that one of these diploid genes is essential, but the function is protected by having copies at both ends. Alternatively, a gene (or genes) in the unique region immediately to the left of the right ITR could encode a function complementary to a non-homologous gene at the left end of the genome. Finally, a combination of genes from both the ITR and unique regions may be involved. To distinguish between these possibilities, we produced two recombinant viruses using WRΔLeft as the parent: (i) WRΔLΔITRr has the genes corresponding to the right ITR of WR (VACWR207–VACWR218) removed in addition to the left end deletion and (2) WRΔLΔUniqueR has the left end deletion with the right ITR intact, but is missing genes VACWR195–VACWR206.
The size and morphology of plaques formed on BS-C-1 cell monolayers by the two viruses, WRΔLΔUniqueR and WRΔLΔITRr were similar (Fig. 6). Both viruses formed plaques slightly larger than WRΔLR, but significantly smaller than the parent virus WRΔLeft. Therefore, it appears that at least one gene within the right ITR, and a gene (or genes) within the Right Unique region (VACWR195–VACWR206) are required to support replication and/or spread of WRΔLeft on BS-C-1 cells.
Fig. 6.
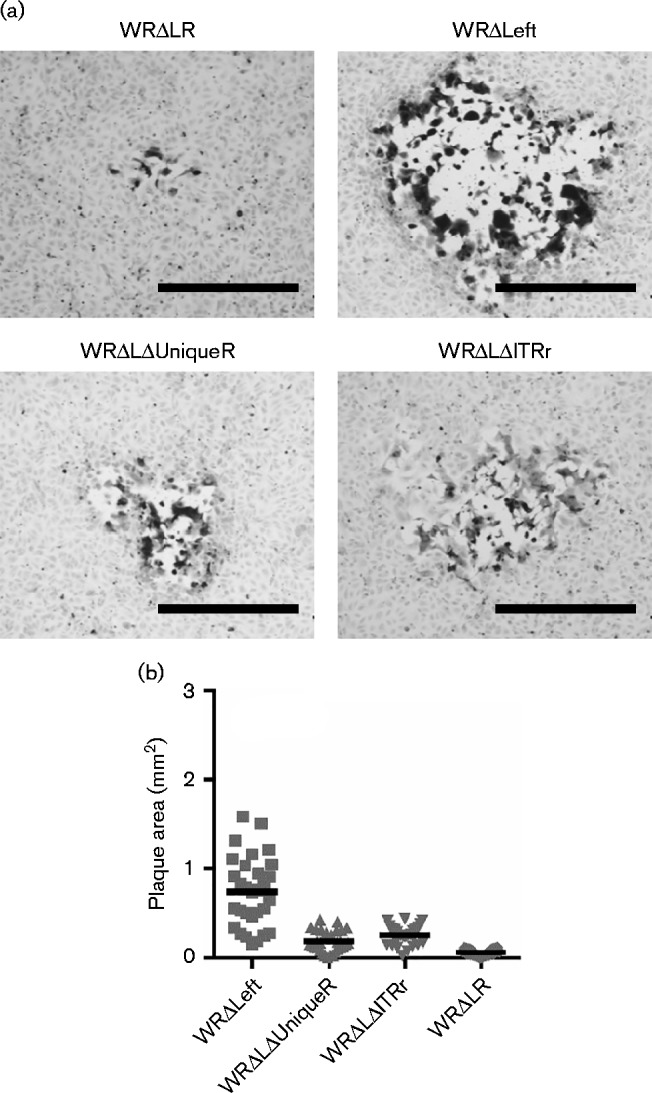
Plaque morphology of WRΔLΔUniqueR and WRΔLΔITRr on BS-C-1 cells. Immunostained plaques formed on BS-C-1 cell monolayers over 72 h. (a) Representative plaques. Bar, 0.5 mm. (b) Areas of plaques (n = 30); horizontal lines represent the mean. Areas for all other viruses significantly smaller than WRΔLeft. using one-way ANOVA with Tukey's multiple comparison test (P < 0.001).
Marker rescue identifies C11R as a gene of interest in the right ITR
The ITRs of VACV WR contain 12 annotated ORFs (annotated as VACWR001–VACWR012 at the left and VACWR207–VACWR218 at the right), but only two of these are not truncated versions of larger genes found in cowpox virus. The two intact genes, i.e. VACWR209 and VACWR210, are equally good candidates as genes with a redundant role that support replication. Deletion of these genes from both ITRs of VACV WR suggested both genes have modest roles in VACV plaque formation on BS-C-1, but only minor impact on replication (Buller et al., 1988b; Fahy et al., 2008). VACWR209 is known as C10L based on Copenhagen (as used here) and C16L in WR, and has a several defined roles in vivo and in vitro (Fahy et al., 2008; Mazzon et al., 2013, 2015; Peters et al., 2013). VACWR210, referred to here as C11R, encodes the well-characterized VACV epidermal growth factor (VGF). VGF is secreted from infected cells and binds to the epidermal growth factor receptor (EGFR, ErbB-1), resulting in cell proliferation that may support virus replication or inhibit apoptosis (Brown et al., 1985; Stroobant et al., 1985; Twardzik et al., 1985; Buller et al., 1988a; Tzahar et al., 1998; Andrade et al., 2004; Postigo et al., 2009).
Three recombinant viruses were made, with either C11R, C10L or both restored to WRΔLΔITRr along with their native promoters (named WRΔLI/C11R, WRΔLI/C10L or WRΔLI/C11RC10L). These viruses allowed us to study the effect of each of these candidate genes in a virus that lacked 42 ORFs from the left end and all other ORFs in the right ITR. All viruses in this set containing C11R replicated to high titres in BS-C-1 cells after infection at low m.o.i., whilst the two viruses lacking C11R, i.e. WRΔLΔITRr and WRΔLI/C10L, had a replication defect compared with WRΔLeft (Fig. 7a, b). Likewise, the plaques formed by the recombinant viruses containing a copy of C11R (WRΔLI/C11R and WRΔLI/C11RC10L) were the same size as WRΔLeft plaques. In contrast, restoring C10L alone to WRΔLΔITRr did not affect plaque size and restoration of C10L along with C11R did not increase plaque size more than addition of C11R alone (Fig. 7c).
Fig. 7.
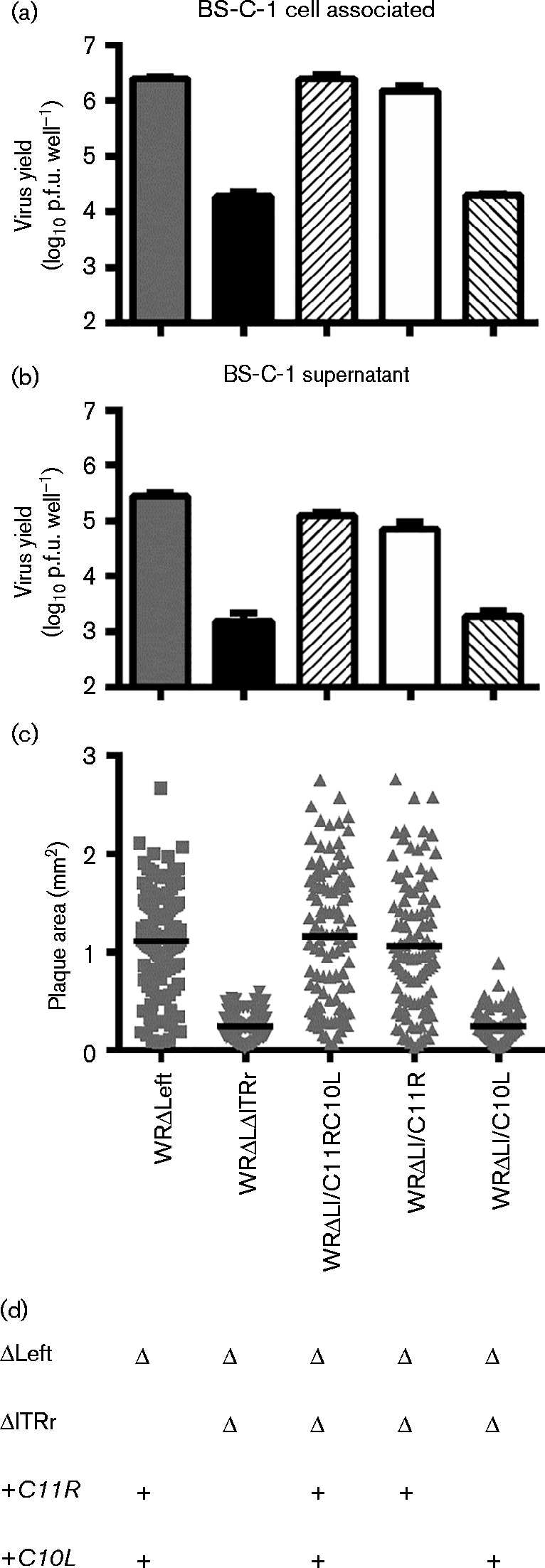
Restoration of C11R rescues the replication and plaque formation defect of WRΔLΔITRr. Virus production and plaque formation by WRΔLI/C11RC10L, WRΔLI/C11R and WRΔLI/C10L was compared with the parental strain, WRΔLΔITRr and WRΔLeft. The amount of (a) cell associated virus and (b) virus released into the supernatant after 48 h following a low m.o.i. (0.01) infection. Data are mean ± sem of three independent wells. (c) Area of immunostained plaques formed on BS-C-1 cells over 72 h. Horizontal lines indicate mean plaque area. Data are representative of two independent experiments. WRΔLeft and the other C11R-containing viruses were significantly different from the two viruses lacking C11R (P < 0.0001). (d) Summary of the genotypes of the recombinant viruses.
As a further, independent test of the role of C11R in supporting replication and plaque size when genes from the left end of the genome were missing, we restored C11R alone to WRΔLR (to create WRΔLR/C11R). As expected, based on our observations of WRΔLΔUniqueR, restoring C11R to WRΔLR resulted in enhanced replication on BS-C-1 cells after infection at low m.o.i. (P < 0.0001, t-test). However, the titre achieved by 48 h p.i. remained lower than WRΔLeft and WRΔLI/C11R (Fig. 8a, b). Insertion of C11R into WRΔLR did not increase plaque size dramatically and whilst the mean plaque area was larger than for the parent, this was not statistically significant due to large variation, despite measuring 100 plaques for each virus (Fig. 8c).
Fig. 8.
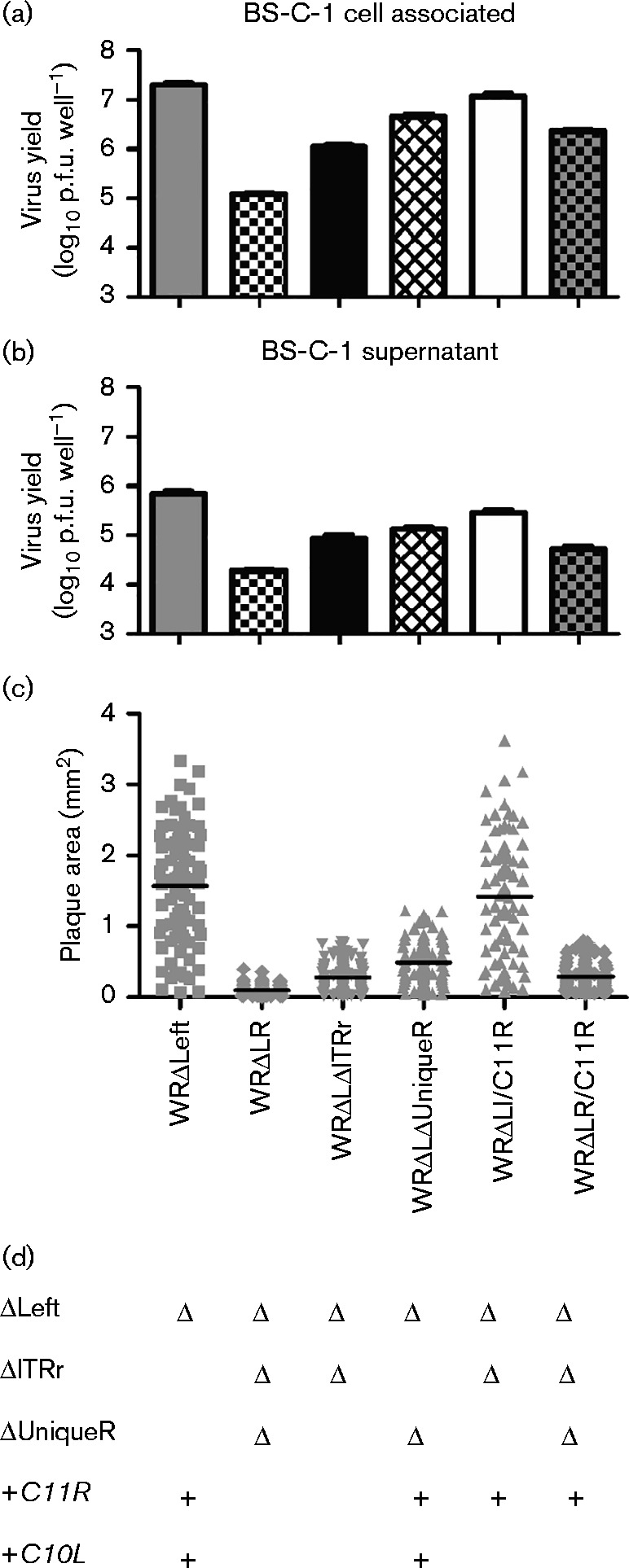
Restoration of C11R partially rescues the replication and plaque formation defect of WRΔLR. Virus production and plaque formation by WRΔLR/C11R was compared with the parental strain, WRΔLR, and three viruses containing C11R (WRΔLeft, WRΔLΔUniqueR and WRΔLI/C11R) and WRΔLΔITRr, which lacks C11R. The amount of (a) cell-associated virus and (b) virus released into the supernatant after 48 h following a low multiplicity m.o.i. (0.01) infection. Data are mean ± sem of three independent wells. The experiment was repeated using the second lineage of WRΔLR/C11R. (c) Area of immunostained plaques formed on BS-C-1 cells over 72 h. Horizontal lines indicate mean plaque area. WRΔLeft significantly different from all viruses except WRΔLI/C11R (p < 0.0001); WRΔLR significantly different from all viruses except WRΔLΔITRr and WRΔLR/C11R (p < 0.0001). Data are representative of two independent experiments. (d) Summary of the genotypes of the recombinant viruses.
Thus, (i) these data identify C11R as a gene with a role in growth in vitro, but this role is redundant due to complementation by another gene at the opposite end of the genome; (ii) this property is shared by one or more additional genes from near the right ITR; and (iii) all of these functions are required for normal VACV growth and plaque morphology.
Discussion
We have shown that the deletion of multiple genes from both ends of the genome of VACV WR causes a severe general replication defect, whilst either deletion alone impacts host range more than growth in general. This finding demonstrates that genes at the termini of VACV and most likely other poxviruses can encode functions required for growth in culture, but are non-essential due to complementation provided by genes from the opposite end of the genome. Such redundancy or complementation clearly requires consideration when interpreting the results obtained with single-gene-deletion viruses. The comparison between vP811 and WRΔLR/WRΔRL further highlights this complexity. We speculate that large terminal deletions also expose a hidden impact of genetic differences in otherwise highly conserved genes that lie in the central portion of the genomes. Considerations such as these are likely to be important in understanding the attenuation of strains such as modified vaccinia Ankara (MVA), which is genetically complex and cannot be explained by the major deletions (Wyatt et al., 1998; Meisinger-Henschel et al., 2010; Dimier et al., 2011; Dobson & Tscharke, 2014).
The boundaries, gene content and size of the ITRs differ widely amongst VACV strains. Furthermore, recombinant viruses with large deletions involving the genomic termini tend to be unstable, so we were concerned this might affect our viruses. For all recombinant VACV WR strains described here, the ITR has been reduced from 10 195 bp (GenBank accession number NC_006998) to only 3701 bp of non-coding sequence. Several of the viruses (e.g. WRΔLR and WRΔRL) were designed with EGFP/bsd and mCherry at opposite ends of the genome, but a few plaques that retained the tiny phenotype could be found where one of these fluorescent markers was apparently lost. Careful PCR and DNA sequence analysis found that stocks of these viruses were never pure and contained viruses with terminal rearrangements as minor contaminants. Some viruses had markers at the expected termini, but others had EGFP/bsd at the right end or mCherry at the left end of the genome, suggesting some viruses contain just one of the markers, but at both ends of the genome. These rearrangements were likely due to inter- or intra-genomic recombination events that can occur during poxvirus replication. The possibility that this instability might have contributed to the replication defect is a caveat for our conclusions, but it seems unlikely that this is the main cause of our findings in that (i) most of the virus had the expected termini, and (ii) defects and rescues occurred in a predictable manner in our deletions of smaller regions at the right (including the ITR and Right Unique regions) and addition of C11R, respectively. The presence of these unwanted recombinants may have contributed to the variability in plaque size, but again this will have been limited by their low level in our stocks.
C11R encodes a well-characterized protein and provides an excellent example of the role of genetic redundancy. Loss of the two copies C11R from WR has little effect on replication in BS-C-1 cells (Postigo et al., 2009); however, in contrast, we found substantial differences in replication on the same cells when C11R was absent from WRs that also had large deletions at the left end of the genome. This suggests that at least one unrelated VACV gene from the left end is able to provide a function that is equivalent to, or can substitute for, that of C11R. Despite only having a limited effect on virus growth in vitro, at least one copy of C11R is found in almost all sequenced strains of VACV, many of which have experienced extensive passage in cell culture. Qin et al. (2013) proposed that for C11R to be seen in most VACV strains, despite its position in a region with a high rate of gene loss, the presence of C11R must be advantageous in both cell culture and artificial passage in vivo. This proposition is supported by our data. In terms of gene interactions with C11R, the simplest hypothesis is that there is another VACV gene that acts in the EGFR signalling pathway. However, there are many possibilities. For example, the induction of a proliferative state by C11 may compensate for the loss of other viral proteins that scavenge or produce components required for virus replication. This could explain the greater effect C11R has on replication when many other genes are missing from the termini of VACV WR. In support of this possibility, deletion of C11R and J2R (encoding thymidine kinase) produces a stronger replication defect than the loss of either gene alone (McCart et al., 2001). At the same time, deletion of C11R from MVA has been shown to slightly decrease activation of the antiviral NFκB response (Martin et al., 2012). Together, these findings highlight the many complex interactions possible amongst VACV genes.
Overall, the analysis of the VACV WR recombinants produced here shows redundancy at the termini of the VACV genome that can confound the assignment of viral genes as essential or non-essential. This complexity cannot be uncovered through the use of viruses with loss of only a single functional gene. We speculate that similar complexity and genetic redundancy is probably a common feature of other poxviruses and perhaps other viruses with large DNA genomes. Whilst we have not demonstrated that the complementation provided by genes at the left and right ends of the genome is due to provision of the same functions, this is the simplest explanation of the data. Assuming this might be the case, we suggest that in addition to the concept of essential genes, an appreciation of the essential functions required for viral replication is important. An essential function may be fulfilled by the presence of a single essential gene, one member of a collection of non-essential genes or even a host-encoded factor in selected cell lines.
Methods
Cells and virus
BHK-21 cells were maintained in minimum essential medium (Gibco/Invitrogen) with l-glutamine and 10 % FBS. All other cell lines, including BS-C-1, 293A, Vero, MRC-5 and HeLa, were maintained in Dulbecco's modified Eagle's medium (DMEM;Gibco/Invitrogen) with l-glutamine and 10 % FBS (D10). VACV stocks were grown in BHK-21 or BS-C-1 cells in DMEM with l-glutamine and 2 % FBS (D2), and purified by centrifugation through a 36 % sucrose cushion. Virus titres were determined by plaque assay on BS-C-1 cells. Plaques were identified by staining with crystal violet or immunostaining with an anti-MVA antibody followed by an anti-rabbit secondary antibody conjugated to HRP (Staib et al., 2004). Viruses were kindly provided by Michelle Barry (vP811 and vP759; University of Alberta, Canada), Geoffrey Smith (Copenhagen; University of Cambridge, UK) and Bernard Moss (WR; National Institutes of Health, USA).
Plaque formation
Confluent cell monolayers grown in D10 in 9.5 cm2 wells were infected with 50–100 p.f.u. virus in 1 ml D2 media. After 2 h at 37 °C, the inoculum was replaced with a semisolid overlay of D2+0.4 % carboxymethyl cellulose (D2-CMC). Cells were fixed and immunostained as described above. Plaque area was measured using photographs of immunostained foci and Image J version 1.46j (http://imagej.nih.gov/ij/).
Multiple-step growth analysis
Confluent cell monolayers in 9.5 cm2 wells were infected with 1 × 104 p.f.u. virus in 1 ml D2 media (m.o.i. 0.01) for 1 h at 37 °C. Unabsorbed virus was removed, the cell monolayer washed once and 2 ml fresh media was added. The 0 h samples were harvested immediately after addition of fresh media. Further wells were harvested at 24, 48 or 72 h p.i. Cells were harvested into existing media, and centrifuged at 931 g for 10 min to separate cell-associated and supernatant virus. Cell-associated and supernatant virus were titrated separately as described above.
Generation of recombinant VACV
All recombinant viruses were generated by homologous recombination between the VACV genome of the appropriate parent virus and linearized plasmid DNA in infected cells. All homology arms in plasmids were generated by PCR and were inserted into a vector using In-Fusion cloning (Clontech). Primers for these PCRs included 5′ extensions of 15 nt identical to sequences flanking a unique restriction site in the vector. Some plasmids were made using three- or four-way In-Fusion reactions and in these cases relevant primers included 15 nt of complementary sequence to allow joining in the In-Fusion reactions. The sequence of primers used to amplify inserts for In-Fusion cloning are listed in Table S6. The cloning process to produce the plasmids used to generate each recombinant virus is described in Table S7. The 293A cells were infected with the appropriate parent virus (m.o.i. 0.05) followed by transfection 1 h later with 1 μg linearized plasmid DNA using Lipofectamine 2000 (Invitrogen). DNA/Lipofectamine mix was replaced after 2 h at 37 °C with 2 ml per well D2 media. Cells were harvested 2 days later and virus was released by three cycles of freeze/thawing. Large blocks of genes at the left end of the genome were replaced by EGFP/bsd (e.g. WRΔLeft) and at the right (e.g. WRΔRight) by mCherry, both under the control of the poxvirus strong synthetic promoter. Plaque purification was carried out on BS-C-1 cells in phenol red-free D2-CMC by screening for the expression of the expected fluorescent proteins. For marker rescue, the fluorescent marker gene at the right end of the genome was replaced by VACV DNA encoding selected genes under the control of the native promoters and plaques expressing only a single fluorescent reporter were picked. For all viruses, PCR screening was used to confirm the correct modification had occurred and identify plaque picks free of parental virus. DNA sequencing of the surrounding region and insertions was carried out for all recombinants produced. Two lineages of each recombinant virus were isolated from independent infection/transfection experiments to control for mutations elsewhere in the genome. WRΔLR and WRΔRL acted as independent lineages, but were produced from different parent viruses, WRΔLeft and WRΔRight, respectively.
Sequence analysis
Sanger sequencing was carried out on PCR products or plasmid DNA at the ACRF Biomolecular Resource Facility (Canberra, Australia).
Statistical analysis
Statistical comparisons were performed using Prism version 5.01 (GraphPad).
Acknowledgements
The authors would like to thank Bernard Moss, Michelle Barry and Geoffrey Smith for generously providing the VACV strains analysed here. This work was funded in part by National Institutes of Health grant R01AI067401 and an Australian Research Council Future Fellowship (FT110100310) to D.C.T.
Supplementary Data
Supplementary Data
Footnotes
One supplementary figure and seven supplementary tables are available with the online Supplementary Material.
References
- Andrade A.A., Silva P.N., Pereira A.C., De Sousa L.P., Ferreira P.C., Gazzinelli R.T., Kroon E.G., Ropert C., Bonjardim C.A. (2004). The vaccinia virus-stimulated mitogen-activated protein kinase (MAPK) pathway is required for virus multiplication Biochem J 381437–446 10.1042/BJ20031375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betakova T., Wolffe E.J., Moss B. (2000). The vaccinia virus A14.5L gene encodes a hydrophobic 53-amino-acid virion membrane protein that enhances virulence in mice and is conserved among vertebrate poxviruses J Virol 744085–4092 10.1128/JVI.74.9.4085-4092.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake N.W., Kettle S., Law K.M., Gould K., Bastin J., Townsend A.R.M., Smith G.L. (1995). Vaccinia virus serpins B13R and B22R do not inhibit antigen presentation to class I-restricted cytotoxic T lymphocytes J Gen Virol 762393–2398 10.1099/0022-1317-76-9-2393. [DOI] [PubMed] [Google Scholar]
- Brown J.P., Twardzik D.R., Marquardt H., Todaro G.J. (1985). Vaccinia virus encodes a polypeptide homologous to epidermal growth factor and transforming growth factor Nature 313491–492 10.1038/313491a0. [DOI] [PubMed] [Google Scholar]
- Buller R.M.L., Chakrabarti S., Moss B., Fredricksont T. (1988a). Cell proliferative response to vaccinia virus is mediated by VGF Virology 164182–192 10.1016/0042-6822(88)90635-6. [DOI] [PubMed] [Google Scholar]
- Buller R.M.L., Chakrabarti S., Cooper J.A., Twardzik D.R., B. Moss. (1988b). Deletion of the vaccinia virus growth factor gene reduces virus virulence J Virol 62866–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burles K., Irwin C.R., Burton R.-L., Schriewer J., Evans D.H., Buller R.M., Barry M. (2014). Initial characterization of vaccinia virus B4 suggests a role in virus spread Virology 456–457108–120 10.1016/j.virol.2014.03.019. [DOI] [PubMed] [Google Scholar]
- Da Fonseca F., Moss B. (2003). Poxvirus DNA topoisomerase knockout mutant exhibits decreased infectivity associated with reduced early transcription Proc Natl Acad Sci U S A 10011291–11296 10.1073/pnas.1534874100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimier J., Ferrier-Rembert A., Pradeau-Aubreton K., Hebben M., Spehner D., Favier A.-L., Gratier D., Garin D., Crance J.-M., Drillien R. (2011). Deletion of major nonessential genomic regions in the vaccinia virus Lister strain enhances attenuation without altering vaccine efficacy in mice J Virol 855016–5026 10.1128/JVI.02359-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson B.M., Tscharke D.C. (2014). Truncation of gene F5L partially masks rescue of vaccinia virus strain MVA growth on mammalian cells by restricting plaque size J Gen Virol 95466–471 10.1099/vir.0.058495-0. [DOI] [PubMed] [Google Scholar]
- Dobson B.M., Procter D.J., Hollett N.A., Flesch I.E.A., Newsome T.P., Tscharke D.C. (2014). Vaccinia virus F5 is required for normal plaque morphology in multiple cell lines but not replication in culture or virulence in mice Virology 456-457145–156 10.1016/j.virol.2014.03.020. [DOI] [PubMed] [Google Scholar]
- Fagan-Garcia K., Barry M. (2011). A vaccinia virus deletion mutant reveals the presence of additional inhibitors of NF-kappaB J Virol 85883–894 10.1128/JVI.01267-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahy A.S., Clark R.H., Glyde E.F., Smith G.L. (2008). Vaccinia virus protein C16 acts intracellularly to modulate the host response and promote virulence J Gen Virol 892377–2387 10.1099/vir.0.2008/004895-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia A.D., Moss B. (2001). Repression of vaccinia virus Holliday junction resolvase inhibits processing of viral DNA into unit-length genomes J Virol 756460–6471 10.1128/JVI.75.14.6460-6471.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubser C., Hué S., Kellam P., Smith G.L. (2004). Poxvirus genomes: a phylogenetic analysis J Gen Virol 85105–117 10.1099/vir.0.19565-0. [DOI] [PubMed] [Google Scholar]
- Kotwal G.J., Moss B. (1988). Analysis of a large cluster of nonessential genes deleted from a vaccinia virus terminal transposition mutant Virology 167524–537. [PubMed] [Google Scholar]
- Langland J.O., Jacobs B.L. (2002). The role of the PKR-inhibitory genes, E3L and K3L, in determining vaccinia virus host range Virology 299133–141 10.1006/viro.2002.1479. [DOI] [PubMed] [Google Scholar]
- Lefkowitz E.J., Wang C., Upton C. (2006). Poxviruses: past, present and future Virus Res 117105–118 10.1016/j.virusres.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Liu S.W., Wyatt L.S., Orandle M.S., Minai M., Moss B. (2014). The D10 decapping enzyme of vaccinia virus contributes to decay of cellular and viral mRNAs and to virulence in mice J Virol 88202–211 10.1128/JVI.02426-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S., Harris D.T., Shisler J. (2012). The C11R gene, which encodes the vaccinia virus growth factor, is partially responsible for MVA-induced NF-κB and ERK2 activation J Virol 869629–9639 10.1128/JVI.06279-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzon M., Peters N.E., Loenarz C., Krysztofinska E.M., Ember S.W.J., Ferguson B.J., Smith G.L. (2013). A mechanism for induction of a hypoxic response by vaccinia virus Proc Natl Acad Sci U S A 11012444–12449 10.1073/pnas.1302140110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzon M., Castro C., Roberts L.D., Griffin J.L., Smith G.L. (2015). A role for vaccinia virus protein C16 in reprogramming cellular energy metabolism J Gen Virol 96395–407 10.1099/vir.0.069591-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCart J.A., Ward J.M., Lee J., Hu Y., Alexander H.R., Libutti S.K., Moss B., Bartlett D.L. (2001). Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes Cancer Res 618751–8757. [PubMed] [Google Scholar]
- Meisinger-Henschel C., Späth M., Lukassen S., Wolferstätter M., Kachelriess H., Baur K., Dirmeier U., Wagner M., Chaplin P., other authors (2010). Introduction of the six major genomic deletions of modified vaccinia virus Ankara (MVA) into the parental vaccinia virus is not sufficient to reproduce an MVA-like phenotype in cell culture and in mice J Virol 849907–9919 10.1128/JVI.00756-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melana S.M., Pogo B.G. (2005). Molecular characterization of Jurkat cells persistently infected with vaccinia virus mutant vp811 Intervirology 4889–96 10.1159/000081734. [DOI] [PubMed] [Google Scholar]
- Meng X., Jiang C., Arsenio J., Dick K., Cao J., Xiang Y. (2009). Vaccinia virus K1L and C7L inhibit antiviral activities induced by type I interferons J Virol 8310627–10636 10.1128/JVI.01260-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchlinsky M. (1990). Mutational analysis of the resolution sequence of vaccinia virus DNA: essential sequence consists of two separate AT-rich regions highly conserved among poxviruses J Virol 645029–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchlinsky M., Moss B. (1989). Nucleotide sequence required for resolution of the concatemer junction of vaccinia virus DNA J Virol 634354–4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B., Winters E., Cooper J.A. (1981). Deletion of a 9,000-base-pair segment of the vaccinia virus genome that encodes nonessential polypeptides J Virol 40387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez E., Esteban M. (1985). Interferon prevents the generation of spontaneous deletions at the left terminus of vaccinia virus DNA J Virol 5675–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panicali D., Davis S.W., Mercer S.R., Paoletti E. (1981). Two major DNA variants present in serially propagated stocks of the WR strain of vaccinia virus J Virol 371000–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish S., Moss B. (2006). Characterization of a vaccinia virus mutant with a deletion of the D10R gene encoding a putative negative regulator of gene expression J Virol 80553–561 10.1128/JVI.80.2.553-561.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkus M.E., Limbach K., Paoletti E. (1989). Cloning and expression of foreign genes in vaccinia virus, using a host range selection system J Virol 633829–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkus M.E., Goebel S.J., Davis S.W., Johnson G.P., Norton E.K., Paoletti E. (1991). Deletion of 55 open reading frames from the termini of vaccinia virus Virology 180406–410 10.1016/0042-6822(91)90047-F. [DOI] [PubMed] [Google Scholar]
- Peters N.E., Ferguson B.J., Mazzon M., Fahy A.S., Krysztofinska E., Arribas-Bosacoma R., Pearl L.H., Ren H., Smith G.L. (2013). A mechanism for the inhibition of DNA-PK-mediated DNA sensing by a virus PLoS Pathog 9e1003649. 10.1371/journal.ppat.1003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postigo A., Martin M.C., Dodding M.P., Way M. (2009). Vaccinia-induced epidermal growth factor receptor-MEK signalling and the anti-apoptotic protein F1L synergize to suppress cell death during infection Cell Microbiol 111208–1218 10.1111/j.1462-5822.2009.01327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L., Liang M., Evans D.H. (2013). Genomic analysis of vaccinia virus strain TianTan provides new insights into the evolution and evolutionary relationships between Orthopoxviruses Virology 44259–66 10.1016/j.virol.2013.03.025. [DOI] [PubMed] [Google Scholar]
- Senkevich T.G., Wyatt L.S., Weisberg A.S., Koonin E.V., Moss B. (2008). A conserved poxvirus NlpC/P60 superfamily protein contributes to vaccinia virus virulence in mice but not to replication in cell culture Virology 374506–514 10.1016/j.virol.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkevich T.G., Koonin E.V., Moss B. (2009). Predicted poxvirus FEN1-like nuclease required for homologous recombination, double-strand break repair and full-size genome formation Proc Natl Acad Sci U S A 10617921–17926 10.1073/pnas.0909529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkevich T.G., Koonin E.V., Moss B. (2011). Vaccinia virus F16 protein, a predicted catalytically inactive member of the prokaryotic serine recombinase superfamily, is targeted to nucleoli Virology 417334–342 10.1016/j.virol.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood C.L., Ward J.M., Moss B. (2008). Vaccinia virus encodes I5, a small hydrophobic virion membrane protein that enhances replication and virulence in mice J Virol 8210071–10078 10.1128/JVI.01355-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staib C., Drexler I., Sutter G. (2004). Construction and isolation of recombinant MVA Methods Mol Biol 26977–99 10.1385/1-59259-789-0:077. [DOI] [PubMed] [Google Scholar]
- Stroobant P., Rice A.P., Gullick W.J., Cheng D.J., Kerr I.M., Waterfield M.D. (1985). Purification and characterization of vaccinia virus growth factor Cell 42383–393 10.1016/S0092-8674(85)80133-1. [DOI] [PubMed] [Google Scholar]
- Sumner R.P., Maluquer de Motes C., Veyer D.L., Smith G.L. (2014). Vaccinia virus inhibits NF-κB-dependent gene expression downstream of p65 translocation J Virol 883092–3102 10.1128/JVI.02627-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons J.A., Alcamí A., Smith G.L. (1995). Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species specificity Cell 81551–560 10.1016/0092-8674(95)90076-4. [DOI] [PubMed] [Google Scholar]
- Twardzik D.R., Brown J.P., Ranchalis J.E., Todaro G.J., Moss B. (1985). Vaccinia virus-infected cells release a novel polypeptide functionally related to transforming and epidermal growth factors Proc Natl Acad Sci U S A 825300–5304 10.1073/pnas.82.16.5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzahar E., Moyer J.D., Waterman H., Barbacci E.G., Bao J., Levkowitz G., Shelly M., Strano S., Pinkas-Kramarski R., other authors (1998). Pathogenic poxviruses reveal viral strategies to exploit the ErbB signaling network EMBO J 175948–5963 10.1093/emboj/17.20.5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton C., Slack S., Hunter A.L., Ehlers A., Roper R.L. (2003). Poxvirus orthologous clusters: toward defining the minimum essential poxvirus genome J Virol 777590–7600 10.1128/JVI.77.13.7590-7600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veyer D.L., Maluquer de Motes C., Sumner R.P., Ludwig L., Johnson B.F., Smith G.L. (2014). Analysis of the anti-apoptotic activity of four vaccinia virus proteins demonstrates that B13 is the most potent inhibitor in isolation and during viral infection J Gen Virol 952757–2768 10.1099/vir.0.068833-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong Y.C., Lin L.C.W., Melo-Silva C.R., Smith S.A., Tscharke D.C. (2011). Engineering recombinant poxviruses using a compact GFP-blasticidin resistance fusion gene for selection J Virol Methods 171295–298 10.1016/j.jviromet.2010.11.003. [DOI] [PubMed] [Google Scholar]
- Wyatt L.S., Carroll M.W., Czerny C.P., Merchlinsky M., Sisler J.R., Moss B. (1998). Marker rescue of the host range restriction defects of modified vaccinia virus Ankara Virology 251334–342 10.1006/viro.1998.9397. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data