

Summary
Interferons (IFNs) are produced in response to virus infection, and induce an anti-viral state in virtually all cell types. In addition to upregulating the transcription of genes that inhibit virus replication, type I (or −α/β) IFNs also act to orchestrate the adaptive immune response to virus infection. Recently a new family of anti-viral cytokines, the type III (or −λ) IFNs, has been identified which activate the same anti-viral pathways via a distinct receptor. Although the identical transcription factor, IFN Stimulated Gene Factor 3 (ISGF3), is activated by either IFN-α/β or IFN-λ signaling, differences in the induction and action of these two cytokine families are beginning to be appreciated. In this article we review this emerging body of literature on the differing roles these cytokines play in host defense of the mucosal surface. Although many viruses enter the body through the respiratory and gastrointestinal tracts, we have focused the discussion on influenza A virus (IAV), respiratory syncytial virus (RSV) and rotavirus (RV), three ubiquitous human pathogens that target the epithelial lining and are associated with a major disease burden.
Keywords: interferon, influenza A virus, Respiratory Syncytial Virus, Rotavirus, IFN induction, Stat signaling
Introduction
Interferons were discovered and named for their anti-viral effect in the 1950s (1). The first IFN to be cloned was IFN-β (2, 3) followed by cloning of several IFN-αs (4-6) that laid the foundation of the family of Type I IFNs. They are produced in response to virus infection, and induce an anti-viral state in virtually all cell types. More recently it has become evident that, in addition to upregulating the transcription of genes that inhibit virus replication, these cytokines also act to orchestrate the adaptive immune response to virus infection. Most recently, a new family of anti-viral cytokines, the type III (or −λ) IFNs, have been identified which activate the same anti-viral pathways via a distinct receptor (7). Although the identical transcription factors, primarily the IFN Stimulated Gene Factor 3 (ISGF3) are activated by either IFN-α/β or IFN-λ signaling, differences in the induction and action of these two cytokine families are beginning to be appreciated. In this article we review this emerging body of literature and the differing roles these cytokines play in host defense of the mucosal surface.
Although many viruses enter the body through the respiratory and gastrointestinal tracts, we will focus on influenza A virus (IAV), respiratory syncytial virus (RSV) and rotavirus (RV), three ubiquitous human pathogens that target the epithelial lining and are associated with a major disease burden. RSV is a member of the Paramyxoviridae family. Paramyxoviruses make up a family of enveloped, single-stranded RNA viruses with a negative polarity genome, and are the cause of the majority of viral pneumonias in the pediatric population (8). In the developed world, respiratory syncytial virus (RSV) accounts for up to 60% of these, followed by parainfluenza viruses (PIV) 1, 2, 3, and 4 at ∼20%, the newly discovered human metapneumovirus (HMPV) at ∼10%, and influenza virus accounting for approximately 8%. RSV alone, the major cause of bronchiolitis and pneumonia in infants, accounts for an estimated 86,000 pediatric hospitalizations in this country annually (9, 10).
Rotaviruses are members of the Reoviridae family which is made up of nonenveloped viruses with segmented, dsRNA genomes. The RV particles consist of a triple-layered capsid surrounding the 11 dsRNA segments which make up the genome. Human rotaviruses are transmitted by the oral fecal route, and are estimated to cause up to 50% of severe diarrheal disease worldwide (11). As with RSV, severe disease is seen primarily in children, with more than 500,000 deaths annually in children below the age of 5 (11).
Here we discuss the biochemistry of the IFNs and their receptors, the signaling pathways activated by IFNs, mechanisms by which each virus induces IFN production, the effects of IFNs on the infected epithelial surface, and the ways in which these cytokines influence the development of adaptive immunity.
Ligands
Type I
The most thoroughly studied, and probably the most biologically important type I IFNs are IFN-β and IFN-α, often collectively referred to as IFN-α/β. In humans there is one IFN-β and 13 functional IFN-αs, defined by structural similarity. IFN −ω,κ,ε sequences are all more distantly related to each other and to IFN-β and IFN-α than IFN-β and the 13 IFN-αs are to one another. IFN-ω is abundantly produced by leukocytes, and has physical and biological activities similar to IFN-αs. IFN-κ is unusual in that its expression is restricted to keratinocytes (12) and, unique among type I interferons, it is reported to be cell associated, exerting its effects in the immediate vicinity of the cells producing it (13). IFN-ε plays a unique role in antiviral protection of the female reproductive tract (14).
Type III
The IFN-λs, (IFN-λ1, −λ 2 and −λ3; also known as IL29, IL28A and IL28B, respectively) constitute the type III IFN family. Structurally more similar to the cytokines of the IL10 family than to the type I IFNs (15), they interact with a distinct receptor complex (see below). There is no crossreactivity between type I and type III IFNs and their corresponding receptors. Somewhat analogous totype I IFN-αs relative to IFN-β, IFN-λ2 and IFN-λ3 are structurally more closely related to one another than either is to IFN-λ1. Again, although all IFN-λs interact with the same receptor complex, subtly different biological activities have been reported.
Recently the existence of a fourth type III IFN, designated λ4, has been deduced, largely from genetic data (16). Adjacent to the gene for IFN-λ3, the IFN-λ4 gene is similarly organized, with four introns. It is apparently expressed in only a fraction of the human population, due to a widespread genetic polymorphism introducing a 5′- proximal frameshift. Surprisingly, the presence of the functional version of the gene is correlated with a poor response to IFN-α treatment for chronic hepatitis C virus (HCV) infection.
Type I and type III Interferon signaling
IFN receptors, as members of the class II family of cytokine receptors, consist of two chains of transmembrane glycoproteins, one of which (generically denoted subclass R1) has a relatively long intracytoplasmic domain (ICD) the other of which (R2) has a relatively short ICD (17). (The R1 and R2 type subunits of the type I IFN receptor are termed, for historical reasons, IFNAR2 and IFNAR1, respectively). The subunit with the longer ICD serves as a docking site for the tyrosine kinase, Jak1. In the case of both type I and type III interferons, the monomeric ligand binds the extracellular domain of the higher affinity R1, which then recruits the lower affinity R2 to form the ternary complex (17). The formation of this complex initiates the signaling cascades. An abbreviated summary of these pathways associated with type I IFN signaling is depicted in Figure 1.
Figure 1. Type I IFN signaling pathways.

All known IFN signaling pathways are initiated by Jak kinase tyrosine phosphorylation of substrate proteins. In the canonical Jak-STAT pathway (pink), Jak kinases phosphorylate STAT1 and STAT2 promoting formation of ISGF3. Phosphorylation of STAT1, 3, 4 and 5 also generates pSTAT homo- and heterodimers (SIFs). Activation of the MAPK-p38 pathway (yellow) is initiated by phosphorylation of vav or another GEF. Activation of PI3K pathways (blue), including mTOR is initiated by phosphorylation of IRS1/2.
Summary of Jak-STAT pathway in type I IFN response
Docking sites for Jak1 and Tyk2 have been identified on IFNAR2 and IFNAR1, respectively. In both cases, the docking sites are in the ICD, immediately proximal to the transmembrane domain (18, 19). Binding of the ligand changes the association of the two receptor subunits, and the approximation of the two associated kinases, which activates their auto- and trans- tyrosine phosphorylation.
Multiple sites within the cytoplasmic domains of the IFNAR receptor complex have been identified as Jak phosphorylation targets and potential docking sites for STAT1 and/or STAT2.
Although the intracellular domain of human IFNAR2 is sufficient for the recruitment and activation of STAT1, STAT2 and STAT3 (20, 21) with Y337 and Y512 playing critical and independent roles (22), Y466 on IFNAR1 was also reported to play a role in STAT2 activation (23, 24) was identified. In addition, the deletion of amino acids 525 to 544 of the IFNAR1 intracellular domain created receptor which produced an enhanced response (25). In mouse cells, a tyrosine residue near the C-terminus of IFNAR2 (Y511) appears to be the critical site (26). Jak kinase mediated phosphorylation of STAT1 and STAT2 on tyrosines Y701 and Y690, respectively, promotes their dimerization via reciprocal phosphotyrosine – SH2 interactions.
The dimeric STAT complex is joined by a third protein, IRF9, to form the trimeric complex, ISGF3. ISGF3 is then translocated into the nucleus, where it recognizes a consensus DNA motif, termed IFN stimulated response element (ISRE) and enhances the transcription of IFN-stimulated genes (ISG). The molecular mechanisms by which ISGF3 modulates transcriptional activity have begun to be elucidated. Activated STAT complexes interact with histone acetyltransferases CBP/p300 (27) and GCN5 (28), and at least one deacetylase (28, 29), (30), and with chromatin remodeling factor BRG1 (31).
The formation of the heterotrimeric ISGF3 represents an exception to the rule of Jak-STAT signal transduction. More typically, the activation of class II cytokine receptors results in the formation of phosphorylated STAT dimers. These complexes, collectively designated as SIFs (Sis-Inducing Factors) are translocated to the nucleus and exert transcriptional control upon binding to specific DNA sequences. The activated IFNAR complex is also capable of generating these complexes. While the IFNAR complex preferentially binds STAT1 and STAT2, and the association of IRF9 seems to be a property unique to STAT2, other members of the STAT family are also capable of docking and activation, forming homo- and heterodimeric complexes (32). Not surprisingly, pSTAT1 homodimers – SIF-C, the same signal transducer produced in response to type II IFN (IFN-γ) – is among these complexes and presumably accounts for much of the overlap between the sets of genes induced by both types of IFN. The extent to which these different activated STAT complexes are formed and compete with or modulate the activity of ISGF3 is a function of the availability of the STAT monomers which, in turn, reflects the cell type as well as its milieu, including the combined actions of other cytokines (33)
In addition to the action of the Jak kinases, as outlined above, the formation of ISGF3 involves the activity of multiple other enzymes. The function of ISGF3 as a transcriptional activator depends on serine phosphorylation of STAT1 at residue S727. The identity of the kinase responsible for S727 phosphorylation remains controversial (34)(Pilz et al. 2003).
Another serine phosphorylation on STAT1, at position S708, has been identified. This phosphorylation, mediated by IKK-ε, appears to render STAT1 less likely to homodimerize, and more likely to heterodimerize with STAT2 (Ng, Friedman 2011). Further, though ISGF3 formation is not dependent on S708 phosphorylation, it is also required for induction of a subset ISGs and full antiviral activity (35, 36).
The acetylation of multiple sites on STAT1 and STAT2 has recently been reported (37-39), but the role this modification in the positive or negative transcriptional regulation of ISGF3's activity is not clear (40). Tang et al. (41) identified IFNAR2 residue K399 as a key target for the acetyltransferase, CBP, and proposed that this modification was important in the recruitment of IRF9. Caution is warranted, however, by the fact that that residue is not phylogenetically conserved.
Nonphosphorylated STATs
The duration of the canonical ISGF3 signal, at least in experimental systems, is surprisingly short. Within an hour of IFN treatment, levels of phosphorylated STAT1/2 and ISGF3 activity, as measured in EMSA assays, markedly decrease. Yet the transcriptional enhancement of at least some target genes is maintained for days. The durable enhancement of transcription of a subset of IFN inducible genes – including that for STAT1 itself - has been ascribed to the action of unphosphorylated STAT1 (42, 43).
Additional signaling pathways triggered by type I IFN
PI3K
PI3K links the IFN signal to at least two additional kinase cascades. Activated by association with Tyk2 phosphorylated IRS1/2, protein/lipid kinase class Ia PI3K phosphorylates protein kinase C-delta (PKC-δ), which has a number of targets and has been implicated in S727 phosphorylation of STAT1 (34, 44). PI3K also activates the downstream kinase Akt, which in turn activates the pleotropic mTOR pathway, which has far-reaching consequences for gene expression, cell metabolism, and cell survival.
MAPK p38
In hematopoietic cells the GTP exchange factor (GEF), vav, upon phosphorylation by activated Tyk2 binds to and activates rac1, which in turn activates MAPK p38 (45). While vav expression is restricted to hematopoietic cells, it is likely that functionally related GEFs perform a similar function in other cells.
CRKL
CrkL (CT10 regulator of kinase L) is an adaptor protein that integrates signals important in tumorigenesis of breast cancer. Grumbach et al. (46) showed that it docks and is phosphorylated at IFNAR much like a STAT, and in fact forms a heterodimer with phosphorylated STAT5 (A or B) which, like dimeric pSTATs, is transported to the nucleus, and activates the transcription of a set of genes distinct from those activated by ISGF3.
IFN induced modulation of translation
Historically, research into the mechanisms of IFN induction of an antiviral state has focused on transcriptional control. Recent studies, however, have demonstrated a direct – i.e. not dependent on transcriptional induction - effect on translational control in cells responding to IFN. The mTOR pathway is activated by IFN-activated PI3K. Downstream of activated mTOR, phosphorylation of ribosomal S6 kinase and 4E-BP1 modify the translational environment in favor of cap-dependent translation. 4E-BP1 is also targeted by the MAPK effector, RSK1, by IFN independently of mTOR (47). The translation of some ISGs has been shown to depend on IFN-induced modifications of translational initiation factors (48)
Interferon Stimulated Genes
The genes induced by IFN stimulation have profound consequences on cell metabolism, cell survival and proliferation and modulation of adaptive immune responses in addition to direct antiviral effects. Dozens of microarray studies with human and mouse cells have demonstrated that IFN stimulation results in the up-regulation of hundreds of genes. The results of these studies have been curated and made available on an internet searchable database, Interferome 2.0 (49).
Direct antiviral effects of only a small fraction of them have been studied in any detail, recently reviewed by Schoggins and colleagues (50). These include mechanisms that result in interference with translation (e.g. PKR), degradation of RNA (e.g. OAS) (Sadler and Williams 2008) interference with viral assembly and release (e.g. viperin, (51, 52)). Many of the antiviral effects, however, are subtle, and it seems that effective viral control generally reflects “death by a thousand cuts” rather than one or two fatal blows. This may be an inevitable consequence of the degree to which viruses rely on host machinery for their replication.
In addition to gene products with direct antiviral effects, many gene products up-regulated by IFN have indirect effects. Upregulation of genes involved in MHC-I antigen presentation, for instance, renders cells more readily recognized by immune lymphocytes (53). One of the most prominently upregulated ISGs is ISG15, a 17kd protein that modifies the function of > 100 host proteins by ubiquitin-like conjugation (54), termed “ISGylation”. Many genes involved in the IFN pathway – including those for IFN-α, STAT1, STAT2 and IRF9 – are up-regulated in response to IFN stimulation, amplifying the IFN signal by positive feedback. At the same time, the stage is set for subsequent reversal of the IFN-stimulated state by induction of multiple negative feedback mechanisms.
Negative regulation
After the massive reorganization of cellular metabolism and machinery triggered by IFN, long term survival of the cell requires a well-tuned control mechanism to ensure the proper magnitude and duration of the reorganization and a subsequent return to the pre-induction pattern of gene expression. This is accomplished by the reversal of the activating posttranslational modifications on the components of signal transduction, and by reversal of their elevated concentrations by targeted proteolysis. Effectors of these processes are induced as ISGs, constituting a negative feedback loop. The most prominent of the negative regulatory factors is SOCS1. This protein binds to the IFNAR1-Tyk2 complex, and directly inhibits kinase activity (55). Another IFN induced protein, ubp43, binds to IFNAR2 and prevents the association with Jak1 (56).
TC45 is a protein tyrosine phosphatase that interacts with ISGF3 in the nucleus, reversing the Y701 phosphorylation of STAT1 (57). PIAS1 inactivates STAT1 by promoting SUMOylation (58)
Type III IFN signaling
The IFNAR genes are ubiquitously expressed, so that all nucleated cells are responsive to IFNα/β (59). The specificity of the type III IFN receptor resides in its R1 chain, which has been identified as IFNLR1. The R2 component is IL10R2, which is shared with the receptors for the IL10 family of cytokines (60). IFNLR1 is expressed in a limited range of cells, primarily epithelial, and only these cells are responsive to IFN-λ (61-65).
The molecular details of the activation of IFNAR are known in considerably more detail than those of the relatively recently discovered IFNLR. Nevertheless, STAT recruitment and activation through the intracellular domain of IFLR1 has been delineated. Two tyrosine-based motifs, surrounding Y343 and Y517, of IFNLR1 can independently mediate STAT2 activation by IFN-λs. Interestingly, these motifs within the IFNLR1 demonstrate some resemblance to sequences within IFNAR chains implicated in STAT recruitment, revealing the molecular mechanism underlying the ability of both types of IFNs to induce similar patterns of STAT activation (Dumoutier et al., 2004). The Y343-based motif of IFNLR1 (YIEPPS) shows some similarity with that surrounding Y466 of IFNAR1 (YVFFPS), whereas the C-terminal amino acid sequence of IFNLR1 containing Y517 (YMARstop) is very similar to the C-terminal amino acid sequence of IFNAR2 containing Tyr512 (YIMRstop). Activation of STAT4, and to some extent STAT1 and STAT3 activation, was independent of IFNLR1 tyrosine residues (66).
In addition to recruiting the Jaks and STATs central to the canonical ISGF3 pathway, IFNAR has been reported to interact directly with at least a dozen other proteins that are thought to modulate the ISGF3 pathway and/or mediate independent pathways (see Table 1).
Table 1. Proteins reported to interact directly with IFNAR1/2 ICD.
| Protein | Function | IFNAR subunit | PMID |
|---|---|---|---|
| Jak1 | Jak tyrosine kinase | 2 | (19) |
| Tyk2 | Jak tyrosine kinase | 1 | (153) |
| Erk2 | MAP kinase | 2 | (154)7569900 |
| STAT1 | signal transducer/transcription activator | 2 | (155) |
| STAT2 | signal transducer/transcription activator | 1 | (23) (156) (157) |
| STAT2 | signal transducer/transcription activator | 2 | (158) |
| STAT3 | signal transducer/transcription activator | 1 | (159) |
| STAT4 | signal transducer/transcription activator | ? | (160) |
| STAT5 | signal transducer/transcription activator | ? | (46) |
| STAT6 | signal transducer/transcription activator | 1 | (161) |
| IRF9 | transcriptional activator | 2 | (162) |
| CBP | Transcriptional co-activator, acetyltransferase | 2 | (162) |
| CRKL | adaptor protein | 1 | (46) |
| PI3K | protein/lipid kinase | 1 | (163) |
| PKCβ | protein kinase | 2 | (164) |
| PRMT1 | protein arginine methyl transferase | 1 | (165) |
| PRKD2 | protein kinase | 1 | (166) |
| PTP1D | protein tyrosine phosphatase | 1 | (167) |
| RACK1 | scaffold protein | 1, 2 | (155) |
| HOS | ubiquitinating enzyme | 1 | (168) |
| USP43 | ISG15 isopeptidase, kinase inhibitor | 2 | (56) |
The strength of the maximal signal induced by exposing cultured cells to saturating levels of cytokine, as measured either by degree of antiviral protection or by degree of STAT phosphorylation, has generally been more modest with IFN-λ than with IFN-α/β (60, 67, 68). Qualitatively, however, the downstream effects of ligand binding in both cases – STAT1 and STAT2 phosphorylation, formation of functional ISGF3, RSK1 activation - are thus far indistinguishable. Because the molecular details, so far as they are known, of IFNLR activation are parallel to those of IFNAR activation, a similar set of interacting molecules is expected to be involved with the IFNLR complex.
Thus, in light of the similarity of the known and inferred biochemical events associated with the type I and type III interferon receptors, it is surprising that the amino acid sequences of their cytoplasmic tails bear no identifiable homology. The phylogenetic conservation of the two systems strongly suggests that the functions they serve are not entirely overlapping.
IFN induction during mucosal virus infection
The innate immune system detects pathogens by a class of molecules known as pattern recognition receptors or PRRs. PRRs recognize pathogen associated molecular patterns (PAMPs) within microbial components. Toll-like receptors (TLRs) comprise a family of PRRs that detect a broad range of pathogens including bacteria, fungi, parasites and viruses, as well as specific host molecules known as DAMPs (danger associated molecular patterns) that serve as indicators of the damage inflicted to the host by pathogens. TLRs were found based upon their homology to the Drosophila toll, a gene which is essential for dorsoventral patterning during embryogenesis (69) (70). There are 13 mammalian members and 12 murine members of the TLR family. Discovery of these proteins, as well as a number of intracytoplasmic PRRs, has provided us with significant insight into the mechanisms by which cells detect virus infection. The details of this recognition and response have been extensively reviewed (71-75) (76). Our goal in these pages is first, to summarize what is known about the role of TLRs and the cytosolic PRRs, or retinoic acid-induced gene-like receptors (RLRs), in sensing mucosal virus infection, and secondly, how signaling through these receptors contributes to IFN induction. To sharpen our focus we will concentrate on what is known about TLR and RLR interactions with IAV, RSV and RV which target the epithelium lining the lung (IAV, RSV) or intestine. The major players in IFN induction by these viruses are pictured in Figure 2.
Figure 2. IFN-α/β induction.
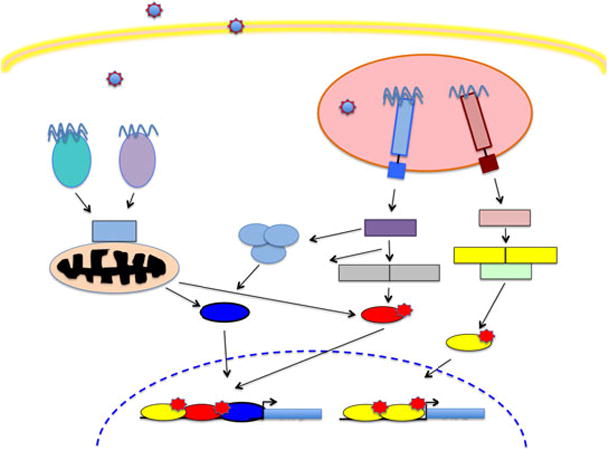
Although additional PRRs beyond those shown here participate in the IFN induction pathway, only the components known to recognize the RNA viruses discussed in this review are pictured here. Viral nucleic acids are recognized by both TLRs and RLRs. TLR7 is located in the endosomal compartment of DCs, and encounters ssRNA following virus infection or uptake of viral components by phagocytosis. Signaling by TLR 7 requires the adaptor molecule MyD88 which forms a complex with IRAK1, IRAK4 and TRAF6. TLR3 is expressed in the endosomal compartment of many cell types including DCs, macrophages and epithelial cells, and is activated by binding its ligand, dsRNA. TRIF is the adaptor protein required for TLR3 signaling, mediating activation of NF-κB by the canonical IKK kinases α, β and γ, and phosphorylation of IRF3 by the noncanonical IKK kinases TBK1 and IKKε. Alternatively, these IKK kinases can be activated by binding of ssRNA or dsRNA to the intracytoplasmic sensors RIG-I and MDA5.. RNA binding by either RIG-I or MDA-5 results in recruitment of the adaptor mitochondrial antiviral-signaling protein (MAVS) which dimerizes, and recruits additional adaptor proteins that activate the transcription factors NF-κB, IRF3 and IRF7. These activated transcription factors then translocate to the nucleus where they bind to promoter elements upstream of IFN-α and IFN-β gene leading to their transcriptional upregulation.
TLRs and RLRs in virus sensing
The primary TLRs involved in RNA virus sensing are TLR3 and TLR7 which recognize dsRNA and ssRNA respectively. The cytosolic RNA-helicase domain containing proteins RIG-I and melanoma differentiation-associated gene 5 (MDA5) also recognize these ligands, and also activate signaling cascades resulting in type I and type III IFN induction. The relative contribution of these pathways to IFN induction in the course of any given virus infection remains controversial. The TLRs are type I transmembrane proteins consisting of an extracellular domain with multiple leucine-rich repeats (LRRs), a transmembrane domain, and a cytoplasmic portion containing the conserved Toll/Interleukin-1 receptor (TIR) domain. It is through this TIR domain that TLRs initiate their intracellular signaling pathways. Double-stranded RNA, a virus component or an intermediate formed as part of virus replication, is the specific ligand recognized by TLR3 (77), and ligand binding by the extracellular domain results in TLR dimerization, and recruitment of a TIR-domain-containing adaptor protein. Four different adaptor proteins have been found to associate with the TLRs and these are: myeloid differentiation factor 88 (MyD88), MyD88 adaptor-like protein (MAL), TIR-domain-containing adaptor-inducing interferon-β (TRIF) and TRIF-related adaptor molecule (TRAM) (78). TRIF is the adaptor protein which interacts with the TIR domain of TLR3, and this association initiates a signaling cascade that leads to the production of IFNs, inflammatory cytokines and chemokines. TLR3 is the only member of this family that binds directly to TRIF and does not signal through MyD88. TRIF binding leads to the activation of the transcription factor IRF3, required for type I IFN induction, as well as NFκB and AP-1 which also regulate transcription of other inflammatory cytokines such as TNF-α and IL-6 (79, 80). TRIF acts by recruiting a number of other proteins including TNF receptor associated factor 3 (TRAF3) (81) and TRAF6 (82, 83). TRAF3 interacts with TRAF family member-associated NFκB activator (TANK), which forms a complex with the kinases TANK-binding kinase 1 (TBK1) and IKKε. TBK1 and IKKε phosphorylate IRF3 (84, 85), which then dimerizes and moves into the nucleus where it binds enhancer elements within the IFN-β promoter. TLR7 stimulation by ssRNA binding results in the recruitment of MyD88, which forms a signaling complex with IL-1 receptor associated kinases 1 and 4 (IRAK1 and IRAK4), and TRAF6. TRAF6 activation leads to NFκB activation via TGF-β activated kinase 1 (TAK1), and IRF7 is bound and phosphorylated by IRAK1 (86). IRF7 is an IFN-stimulated gene in most cell types, but is constitutively expressed at high levels in plasmacytoid dendritic cells (pDCs) (87, 88). Thus, only in the pDCs will TLR7 stimulation result in the rapid production of high levels of type I IFN, a function which distinguishes pDCs from other immune cell types. When present, IRF7 can also be phosphorylated by TBK1 and IKKε (84, 85) Activated IRF7, as a homodimer or partnering with IRF3, stimulates transcription of multiple IFN-α genes as well as IFN-β and IFN-λ genes (87, 89, 90) (91, 92).
The RIG-I like receptors, cytoplasmic sensors of viral RNAs, were first identified in 2004 (93). RIG-I recognizes 5′-triphosphate containing RNAs (94, 95), a distinguishing feature of negative strand of RNA virus genomes, including those of IAV and RSV, as well as viral transcripts. RIG-I also binds short dsRNA molecules, while MDA5 recognizes longer dsRNA molecules. While RIG-I is essential for IAV (96) or RSV (97) recognition by infected epithelial cells, both RIG-I and MDA5 mediate IFN induction in RV infected cells (98, 99). RNA binding by either RIG-I or MDA-5 results in recruitment of the adaptor mitochondrial antiviral-signaling protein (MAVS) which is inserted into the outer mitochondrial membrane. Following interaction with RIG-I or MDA-5, MAVS dimerizes and recruits additional adaptor proteins that activate the transcription factors NF-κB, IRF3 and IRF7 (74).
Many of the studies of TLR function have focused on immune phagocytic cells (100), and their role in IFN induction in infected epithelial cells is less certain. The literature regarding TLR expression in non-immune cells is somewhat confusing as RNA and protein detected in tissue homogenates may be derived from any number of cell types, but immunohistochemistry studies using human tissues have now shown that TLR3 is expressed by both lung and gut epithelium. Unlike phagocytic cells, where TLR3 is found only in endosomes TLR3 is present at the basolateral surface of ileal and colonic mucosa (101), and at the apical and basolateral surfaces of tracheal mucosa (102). TLR7 was found at low levels on the apical surface of human tracheal epithelium, but its expression in enterocytes has not been reported.
Although neither IAV nor RSV possesses a dsRNA genome, many viral RNAs are recognized by TLR3 via small stretches of dsRNA in the ss genomic segments, and TLR7 activation by IAV genomic RNA has been demonstrated (103). IAV infects target cells via the endosomal pathway, and acidification within that compartment is a necessary part of its replication process. Viral RNA exposed during that process may encounter TLR3 in the endosome, but this interaction does not appear to play a major role in type I IFN induction by this infection. When mice deficient in TLR7 were infected with IAV, they showed a marked decrease in serum IFN-α levels consistent with the inability of TLR7-/- pDCs to synthesize IFN-α in response to IAV. MyD88 was essential for IFN induction in pDCs, and there was no such impairment in pDCs isolated from TLR3-/- animals (104). This is consistent with data from our group showing a sharp decrease of type I IFN in the lungs of pDC depleted, IAV-infected animals (105). Although IFN-λ levels were not measured in that study, pDCs derived from bone marrow produced an order of magnitude more IFN-λ than IFN-α following IAV exposure in vitro (106), suggesting that they are also the major source of this cytokine in vivo.
Conversely, these studies also show that other cell types are also a significant, if not the primary, source of IFNs following IAV infection in vivo, on the order of 30 – 50% l (104, 105). Given that the respiratory epithelium expresses TLR3, is there a role for this molecule in IFN production by this cell type in response to IAV, or is that mediated primarily by the RLRs? Using a human bronchial epithelial cell line, BEAS-2B, Le Goffic et al. were able to demonstrate that, in response to IAV infection, TLR3 stimulation contributed predominantly to the production of proinflammatory cytokines, such as IL-6 and IL-8, and less so to the production of IFN-β (107). Like TLR3, the cytosolic sensor RIG-I triggered both NF-kB and IRF3 dependent responses, but with a relatively greater impact on the production of the IFN-β (107). This was also true for conventional DCs, with opposite results observed for pDCs (108). Thus, there is redundancy in the ability of a cell to recognize and respond to IAV infection, but the events triggered by TLR3 and RIG-I are not the same. This complexity is underscored by the observation that TLR3-/- mice infected with IAV had a survival advantage when compared with wild type (WT) animals, despite a higher viral load and a marked decrease in the rate of viral clearance (109).
Unlike IAV, studies with TLR3-/- mice have not shown a requirement for TLR3 in RSV clearance, and virus levels were unchanged in the absence of this molecule (110). This may not be surprising in light of differing mechanisms of RSV and IAV for cell entry. Paramyxoviruses, including RSV, are able to fuse with the plasma membrane delivering the contents of the viral envelope directly into the cytoplasm. Intranasal RSV infection of MAVS-/- mice, which lack a functional RLR pathway, produced no detectable IFN-α, IFN-β, or IL-6 suggesting that, as for IAV, the RLR pathway is both necessary and sufficient for induction of innate immune responses by RSV (111). Thus RSV does not trigger either TLR3 or TLR7, consistent with the observation that pDC depletion in RSV-infected animals did not change IFN levels (105). IFN production by pDCs in response to IAV does not require active infection; UV-inactivated IAV is sufficient, but not so for RSV (105). IFN induction by RSV in human pDCs requires actively replicating virus but not endosomal acidification (112). Thus, unlike IAV, RSV does not induce type I IFN via TLR3 or TLR7. Although TLR4 activation by RSV has been reported (113, 114), the impact of this interaction on IFN induction in vivo remains unclear.
An important theme running through this discussion is the identity of cell types producing IFNs in the infected host. One approach to this problem has been the generation of an IFN-α6 reporter mouse and characterization of GFP expressing cells as infection progressed (115). As expected, the pattern of IFN induction observed differed with different viral pathogens, but IAV, RSV and RV were not investigated in this study. In describing IFN induction by RV, Sen et al. (116) have fractionated cells from the small intestine of infected animals, and measured type I IFN induction in epithelial, stromal, and hematopoietic cell types by single cell qRT-PCR. This analysis demonstrated that the1000×-fold induction of IFN-β transcript seen in total intestinal RNA was largely derived from the CD45+ cell population rather than the infected enterocytes. RV infection of TLR3-/- mice produced wild type levels of serum IFN-β, while none was detected in infected MAVS-/- mice, and enterocyte IFN production was MAVS rather than TLR3 dependent (99). A similar result was seen with RV infection of MAVS-/- MEFs. This is an important distinction, as the dsRNA RV genome would be expected to activate TLR3. Although the precise mechanism by which RV enters the cell has not been determined, the consensus seems to be that virus directly penetrates the enterocyte plasma membrane (117), and so may not encounter TLR3. While TLR3 may not have a role in IFN induction by RV, recognition of RV components by TLR7 or TLR9, the TLRs expressed by human pDCs, has not been ruled out. Type I IFN induction by exposure of human pDCs to live or inactivated RV provides evidence that TLRs, or an as yet unknown receptor, is also important part of the innate immune response to this pathogen. Additional studies are needed to clarify the sources of IFN in the RV-infected host, as well as the specific pathways at work in each cell type (118).
In conclusion, while the same cast of characters is involved in IFN induction by each virus, the precise pathway differs in each infection, both in epithelial and immune cell compartments (Table 2). While the pathways triggered in each cell type are becoming increasingly clear, the relative contribution of each cell to total serum and tissue IFN levels in each infection in vivo requires further study.
Table 2.
Triggers of IFN Synthesis by Virus
| Virus | Infected Cell | pDC |
|---|---|---|
| IAV | RIG-I | TLR7 |
| RSV | RIG-I | RIG-I |
| RV | RIG-I + MDA5 | TLRX? |
Differential induction of type i and type iii IFNs
As the reader will note, our discussion so far has focused on type I rather than type III IFN induction pathways. The earliest studies of IFN-λ induction suggested that these cytokines were induced by the same stimuli and the same activated transcription factors that regulate type I IFN expression (91, 92). IFN-β gene regulation has been extensively characterized (119) and requires promoter binding of NF-κB, activated IRFs, and the ATF-2/c-Jun AP-1 complex. IRF3 is the predominant IRF recruited at early times post infection, IRF7 can function in its place but requires IFN-signaling for its expression. Activated IRF7 then drives expression of the IFN-α genes (120). Like the IFN-α genes, IFN-λ2 and −λ3 expression largely depends upon on IRF7, while early IFN-λ1 synthesis is regulated by IRF3 and NF-κB binding. However, the IFN-β and −λ1 promoters, and the IFN-α and IFN-λ2,3 promoters are not identical (See Figure 3), and examples of differential regulation are emerging. Multiple aspects of this differential induction remain to be explored. One major difference is the ability of IFN-λ1 promoter to be activated by NF-κB alone in the absence of activated IRFs (121). Although transcriptional regulation of human and mouse IFN-λ2 and -3 genes has not been thoroughly investigated, their promoters contain predicted NF-κB binding sites and therefore maybe also activated by NF-κB alone. This may explain the finding that high levels of IFN-λ are present in IFNAR-/- IAV-infected mice (106), a result not predicted if the synthesis of high levels of IFN-λ2, -3 requires IFN-β or IFN-λ1 mediated upregulation of IRF7, as the model shown in Figure 4 would predict. One possibility is that the bulk of IFN-λ is produced by pDCs with high constitutive levels of IRF7, but this would not explain preferential IFN-λ synthesis by IAV infected respiratory epithelial cells derived from IFNAR-/- mice (102) or the compelling observation that only IFN-λ is produced by IAV-infected human type II pneumocytes (122). In addition, the available data do not explain how live, or inactivated, IAV treatment of pDCs stimulated 10-fold more type III than type I IFN production (106). Nonetheless, despite our current lack of insight into precisely when and how IFN −α, -β or -λ synthesis is induced or inhibited, the existence of two independent antiviral defense mechanisms adds an additional layer of host protection.
Figure 3. Promoter/enhancer recognition sequences associated with type I and type III IFN genes.
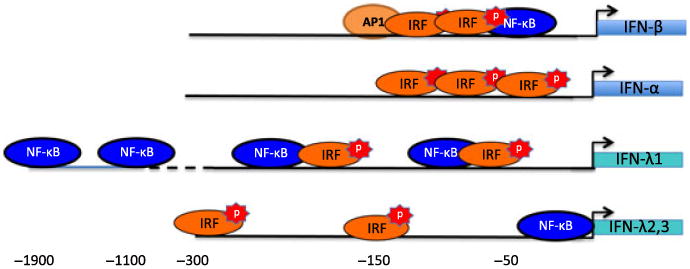
The promoters directing IFN-β, -α, -λ1, and λ2,3 have binding sites for IRFs and NF-κB, but are not as similar as was originally thought. IFN-β gene activation requires binding of IRF3 and/or IRF7 as well as NF-κB and AP1, while the analogous IFN-λ1 promoter can be activated by either stimulus. Upegulation of IFN-α or –λ2,3 gene transcription is largely IRF7 mediated.
Figure 4. Two-step induction of type I and type III IFNs.
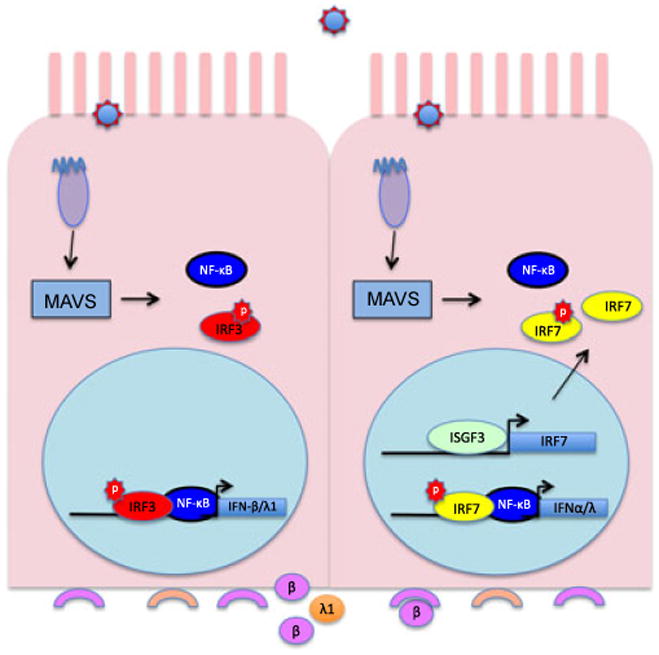
, Upregulation of IFN mRNA synthesis requires the availability of activated NF-κB and IRFs. When an epithelial cell or fibroblast is infected by virus, RLRs activate NF-κB and IRF3, which then move into the nucleus and upregulate IFN-β and IFN-λ1 transcription. These ligands are then secreted from the basolateral surface of the cell. IFNAR and IFNLR on adjacent cells bind secreted IFNs, leading to synthesis of ISGs including IRF7. The availability of IRF7 allows amplification of the IFN response when infection of the IFN-exposed cell triggers its activation, translocation, dimerization, and IFN promoter binding. Although IFN-α synthesis by fibroblasts (which lack the IFNLR) requires prior IFN-β synthesis (or IFN-α/β treatment), this is not true for IFN-λ responsive epithelial cells. In the absence of IRF3 activation and IFN-β production, alternative pathways allow IFN-λ induction in the absence of IRF3 activation.
Type I and type III IFN function in mucosal virus infection
Antiviral effects
As we have discussed, both IFN families are produced in response to virus infection, and both activate ISGF3, the transcription factor responsible for ISRE binding and induction of a specific set of ISGs. The first important difference noted between the two IFN systems is the expression pattern of their IFN receptors. Whereas type I IFN receptors are expressed in most cell types, IFNLR1 demonstrates a more restricted pattern of expression, limiting responses to type III IFNs primarily to epithelial cells of the respiratory, gastro-intestinal and reproductive tracts (61-63, 65, 123). This limitation may explain the observation that the IFN-λ antiviral system alone cannot provide full protection against systemic virus infections; for systemic infections in vivo, a functional type I IFN response is required. This became evident, in retrospect, when systemic infection was observed in IFNAR-/- mice infected with the mouse-adapted, WSN strain of IAV (124). While H1N1 infections are limited to the lung by their requirement for a lung tryptase for maturation, a NA mutation in the WSN strain allows HA cleavage outside of the lung, and IFN-α/β signaling is required to limit infection. In contrast, both type I and type III IFN systems are capable of providing effective antiviral protection in vivo against infections targeting epithelial cells expressing both types of IFN receptors. This was inferred from the lack of increased sensitivity of IFNAR-/- mice to IAV (125), RSV (126) or RV (127), then demonstrated using IFNLR-/- animals. From these data, we and others (62-65, 125, 126, 128) concluded initially that any differential function of these two cytokine families in terms of epithelial antiviral protection was likely due to the availability of ligand. However, more recently published studies suggest that the ligand-centric view is oversimplified.
It now appears that antiviral protection of intestinal epithelial cells against RV mainly relies on the action of the type III IFN antiviral system. Unlike IAV infection of the lung, where either type I or type III IFNs are sufficient for wild type levels of protection, Pott et al. (129) demonstrated that mice lacking a functional IFN-λ receptor complex were less able to control of oral rotavirus infection. Consistent with this observation, only systemic administration of IFN-λ, but not IFN-α, was able to suppress RV replication in the intestinal epithelium. Thus, the type III IFN system has a unique function in antiviral protection of intestinal epithelium that is independent of, and not overlapping with, the type I IFN antiviral system. This is a fascinating development, particularly in light of data showing that IECs, in vitro and ex vivo, responded equally well to both types of IFNs (129). Thus, expression of the IFNAR or IFNLR per se does not fully explain the functional specificity of type I and type III IFNs in the antiviral response of epithelial tissues to virus infection. The basis for this specificity has yet to be established.
Immunomodulatory Effects
It is now well established that, in addition to the antiviral properties of type I IFNs, these cytokines play a major role in orchestrating the adaptive immune response to virus infection. This occurs indirectly via upregulation of cytokine and chemokine synthesis, but also through direct action on NK cells, DCs and lymphocytes. Type I IFNs enhance the ability of DCs to present antigen (130-134), and have been shown to be important for cross priming of T cells and specific antibody production (132, 135). These effects in immune cells are mediated by activation of STAT1, but also STATs 3, 4 and 5, with different outcomes dependent upon the relative levels of each STAT protein (136-138). The pattern of STAT activation has been shown to be determined by the cell type, the cytokine milieu, and for T cells, activation through the T cell receptor (33, 138). While the implications of these nuanced responses to type I IFNs are just now emerging, it will also be important to understand how the presence of type III IFNs impacts these events.
Efforts have been made to determine whether immune cells express the IFNLR and, if so, the manner of their response to IFN-λ. When levels of IFNLR mRNA were measured in monocytes, T, B and NK cells isolated from PBMCs, the highest expression was found in B cells (139) (140, 141), and this correlated with weak STAT1 and STAT3 phosphorylation in response to IFN-λ1 or IFN-λ2, but no detectable upregulation of MHC class I expression. Plasmacytoid DCs (pDCs) express high levels of IFNLR (141) and their response to both type I and type III IFNs was demonstrated by IFN-λ -induced upregulation of CD80 and ICOS-L (142). Data for monocytes is not entirely consistent. While Witte et al. found low basal levels of the IFNLR message (139), that was further down-regulated in immature GM-CSF/IL-4-treated monocyte-derived DCs (MDDCs) or mature MDDCs that were further treated with TNF-α/IL-1β (143). Mennechet and Uze (144) reported contradictory result, with strong upregulation of IFNLR mRNA under similar differentiation conditions.
Flow cytometric analyses have also given conflicting results. High levels of IFNLR expression, and STAT1 phosphorylation in response to IFN-λ treatment, are consistently reported for pDCs (145), but data on IFN-λ responsiveness of monocytes and mDCs is conflicting (Yin et al., 2012} (146) (147) (143) (144). Overall, the current data suggest that the level of IFNLR expression does not always correlate with the levels of cell surface IFNLR expression, which in turn does not always indicate responsiveness of the cells to IFN-λ. Because the second chain of the IFN-λ receptor complex, IL-10R2, is ubiquitously expressed, the lack of responsiveness of cells with detectable levels of cell surface IFN-λR1 expression could potentially be explained by expression of a signal-incompetent splice variant of IFNLR (68). This splice variant of IFN-λR1 lacks the membrane proximal region of the intracellular domain that is expected to interact with Jak1 and therefore is expected to be non-functional. Although this possibility remains to be experimentally demonstrated, expression of both functional and non-functional splice variants have been detected in pDCs (141). Thus while many uncertainties remain to be resolved, it appears that pDCs, B cells, and perhaps other DC subsets, are sensitive to IFN-λs, whereas monocytes, T and NK cells are not. Therefore, despite reports of direct effects of IFN-λ on T cells (148, 149), it seems likely that the observed IFN-λ modulation of T cell function (144, 146, 150) (151) (152) is likely to be indirect, occurring through mechanisms that remain to be elucidated. Reported effects are varied, including both promotion (144), and inhibition. (152), of T regulatory cell development, as well as suppression of Th2 and Th17 responses (150). Thus, while there is compelling evidence for the notion that this cytokine family has effects beyond innate antiviral protection, the mechanisms employed have yet to be discovered.
Concluding Remarks
It has long been appreciated that innate IFNs induced by virus infection of epithelial cells are important for induction of the antiviral state, and for the shaping of adaptive immune responses. The coexistence of two parallel IFN pathways, with the newly recognized type III receptor limited primarily to the gastrointestinal and respiratory tracts, is a remarkable example of convergent evolution that underscores the need for careful modulation of the inflammatory response at these barrier surfaces. Multiple similarities between the type I and type III IFN pathways have recently been described, with the newest studies of IFN-λ biology now focused on understanding the differences between these systems.
Acknowledgments
This work was supported by grants from the NIH to SVK and JED; AI104669 and AI82994.
Footnotes
The authors declare no competing financial interests.
References
- 1.Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proceedings of the Royal Society of London Series B, Containing papers of a Biological character. 1957;147:258–267. doi: 10.1098/rspb.1957.0048. [DOI] [PubMed] [Google Scholar]
- 2.Weissenbach J, et al. Two interferon mRNAs in human fibroblasts: in vitro translation and Escherichia coli cloning studies. Proceedings of the National Academy of Sciences of the United States of America. 1980;77:7152–7156. doi: 10.1073/pnas.77.12.7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taniguchi T, Fujii-Kuriyama Y, Muramatsu M. Molecular cloning of human interferon cDNA. Proceedings of the National Academy of Sciences of the United States of America. 1980;77:4003–4006. doi: 10.1073/pnas.77.7.4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weck PK, Apperson S, May L, Stebbing N. Comparison of the antiviral activities of various cloned human interferon-alpha subtypes in mammalian cell cultures. The Journal of general virology. 1981;57:233–237. doi: 10.1099/0022-1317-57-1-233. [DOI] [PubMed] [Google Scholar]
- 5.Streuli M, Hall A, Boll W, Stewart WE, 2nd, Nagata S, Weissmann C. Target cell specificity of two species of human interferon-alpha produced in Escherichia coli and of hybrid molecules derived from them. Proceedings of the National Academy of Sciences of the United States of America. 1981;78:2848–2852. doi: 10.1073/pnas.78.5.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wetzel R, et al. Properties of a human alpha-interferon purified from E. coli extracts. Journal of interferon research. 1981;1:381–390. doi: 10.1089/jir.1981.1.381. [DOI] [PubMed] [Google Scholar]
- 7.Kotenko SV. IFN-lambdas. Curr Opin Immunol. 2011;23:583–590. doi: 10.1016/j.coi.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sinaniotis CA. Viral pneumoniae in children: incidence and aetiology. Paediatric Resp Rev. 2004;5:S197–200. doi: 10.1016/s1526-0542(04)90037-1. [DOI] [PubMed] [Google Scholar]
- 9.Hall CB. Respiratory syncytial virus and parainfluenza virus. N Engl J Med. 2001;344:1917–1928. doi: 10.1056/NEJM200106213442507. [DOI] [PubMed] [Google Scholar]
- 10.Paramore LC, Ciuryla V, Ciesla G, Liu L. Economic impact of respiratory syncytial virus-related illness in the US: an analysis of national databases. Pharmacoeconomics. 2004;22:275–284. doi: 10.2165/00019053-200422050-00001. [DOI] [PubMed] [Google Scholar]
- 11.Greenberg HB, Estes MK. Rotaviruses: from pathogenesis to vaccination. Gastroenterology. 2009;136:1939–1951. doi: 10.1053/j.gastro.2009.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LaFleur DW, et al. Interferon-kappa, a novel type I interferon expressed in human keratinocytes. J Biol Chem. 2001;276:39765–39771. doi: 10.1074/jbc.M102502200. [DOI] [PubMed] [Google Scholar]
- 13.Buontempo PJ, Jubin RG, Buontempo CA, Wagner NE, Reyes GR, Baroudy BM. Antiviral activity of transiently expressed IFN-kappa is cell-associated. J Interferon Cytokine Res. 2006;26:40–52. doi: 10.1089/jir.2006.26.40. [DOI] [PubMed] [Google Scholar]
- 14.Fung KY, et al. Interferon-epsilon protects the female reproductive tract from viral and bacterial infection. Science. 2013;339:1088–1092. doi: 10.1126/science.1233321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gad HH, Dellgren C, Hamming OJ, Vends S, Paludan SR, Hartmann R. Interferon-lambda is functionally an interferon but structurally related to the interleukin-10 family. J Biol Chem. 2009;284:20869–20875. doi: 10.1074/jbc.M109.002923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prokunina-Olsson L, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet. 2013;45:164–171. doi: 10.1038/ng.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langer JA, Cutrone EC, Kotenko S. The Class II cytokine receptor (CRF2) family: overview and patterns of receptor-ligand interactions. Cytokine & growth factor reviews. 2004;15:33–48. doi: 10.1016/j.cytogfr.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 18.Yan H, Piazza F, Krishnan K, Pine R, Krolewski JJ. Definition of the interferon-alpha receptor-binding domain on the TYK2 kinase. J Biol Chem. 1998;273:4046–4051. doi: 10.1074/jbc.273.7.4046. [DOI] [PubMed] [Google Scholar]
- 19.Usacheva A, et al. Contribution of the Box 1 and Box 2 motifs of cytokine receptors to Jak1 association and activation. J Biol Chem. 2002;277:48220–48226. doi: 10.1074/jbc.M205757200. [DOI] [PubMed] [Google Scholar]
- 20.Kotenko SV, Izotova LS, Mirochnitchenko OV, Lee C, Pestka S. The intracellular domain of interferon-alpha receptor 2c (IFN-alphaR2c) chain is responsible for Stat activation. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:5007–5012. doi: 10.1073/pnas.96.9.5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nadeau OW, et al. The proximal tyrosines of the cytoplasmic domain of the beta chain of the type I interferon receptor are essential for signal transducer and activator of transcription (Stat) 2 activation. Evidence that two Stat2 sites are required to reach a threshold of interferon alpha-induced Stat2 tyrosine phosphorylation that allows normal formation of interferon-stimulated gene factor 3. J Biol Chem. 1999;274:4045–4052. doi: 10.1074/jbc.274.7.4045. [DOI] [PubMed] [Google Scholar]
- 22.Wagner TC, et al. Interferon signaling is dependent on specific tyrosines located within the intracellular domain of IFNAR2c. Expression of IFNAR2c tyrosine mutants in U5A cells. J Biol Chem. 2002;277:1493–1499. doi: 10.1074/jbc.M108928200. [DOI] [PubMed] [Google Scholar]
- 23.Yan H, et al. Phosphorylated interferon-alpha receptor 1 subunit (IFNaR1) acts as a docking site for the latent form of the 113 kDa STAT2 protein. EMBO J. 1996;15:1064–1074. [PMC free article] [PubMed] [Google Scholar]
- 24.Leung S, Qureshi SA, Kerr IM, Darnell JE, Jr, Stark GR. Role of STAT2 in the alpha interferon signaling pathway. Mol Cell Biol. 1995;15:1312–1317. doi: 10.1128/mcb.15.3.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gibbs VC, Takahashi M, Aguet M, Chuntharapai A. A negative regulatory region in the intracellular domain of the human interferon-alpha receptor. J Biol Chem. 1996;271:28710–28716. doi: 10.1074/jbc.271.45.28710. [DOI] [PubMed] [Google Scholar]
- 26.Zhao W, et al. A conserved IFN-alpha receptor tyrosine motif directs the biological response to type I IFNs. J Immunol. 2008;180:5483–5489. doi: 10.4049/jimmunol.180.8.5483. [DOI] [PubMed] [Google Scholar]
- 27.Bhattacharya S, et al. Cooperation of Stat2 and p300/CBP in signalling induced by interferon-alpha. Nature. 1996;383:344–347. doi: 10.1038/383344a0. [DOI] [PubMed] [Google Scholar]
- 28.Paulson M, Press C, Smith E, Tanese N, Levy DE. IFN-Stimulated transcription through a TBP-free acetyltransferase complex escapes viral shutoff. Nature cell biology. 2002;4:140–147. doi: 10.1038/ncb747. [DOI] [PubMed] [Google Scholar]
- 29.Nusinzon I, Horvath CM. Interferon-stimulated transcription and innate antiviral immunity require deacetylase activity and histone deacetylase 1. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:14742–14747. doi: 10.1073/pnas.2433987100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang HM, et al. Induction of interferon-stimulated gene expression and antiviral responses require protein deacetylase activity. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9578–9583. doi: 10.1073/pnas.0400567101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang M, Qian F, Hu Y, Ang C, Li Z, Wen Z. Chromatin-remodelling factor BRG1 selectively activates a subset of interferon-alpha-inducible genes. Nature cell biology. 2002;4:774–781. doi: 10.1038/ncb855. [DOI] [PubMed] [Google Scholar]
- 32.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 33.van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25:361–372. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 34.Uddin S, et al. Protein kinase C-delta (PKC-delta) is activated by type I interferons and mediates phosphorylation of Stat1 on serine 727. J Biol Chem. 2002;277:14408–14416. doi: 10.1074/jbc.M109671200. [DOI] [PubMed] [Google Scholar]
- 35.Perwitasari O, Cho H, Diamond MS, Gale M., Jr Inhibitor of kappaB kinase epsilon (IKK(epsilon)), STAT1, and IFIT2 proteins define novel innate immune effector pathway against West Nile virus infection. J Biol Chem. 2011;286:44412–44423. doi: 10.1074/jbc.M111.285205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tenoever BR, Ng SL, Chua MA, McWhirter SM, Garcia-Sastre A, Maniatis T. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science. 2007;315:1274–1278. doi: 10.1126/science.1136567. [DOI] [PubMed] [Google Scholar]
- 37.Wieczorek M, Ginter T, Brand P, Heinzel T, Kramer OH. Acetylation modulates the STAT signaling code. Cytokine & growth factor reviews. 2012;23:293–305. doi: 10.1016/j.cytogfr.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 38.Kramer OH, et al. Acetylation of Stat1 modulates NF-kappaB activity. Genes Dev. 2006;20:473–485. doi: 10.1101/gad.364306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kramer OH, Heinzel T. Phosphorylation-acetylation switch in the regulation of STAT1 signaling. Molecular and cellular endocrinology. 2010;315:40–48. doi: 10.1016/j.mce.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 40.Antunes F, Marg A, Vinkemeier U. STAT1 signaling is not regulated by a phosphorylation-acetylation switch. Mol Cell Biol. 2011;31:3029–3037. doi: 10.1128/MCB.05300-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang X, et al. Acetylation-dependent signal transduction for type I interferon receptor. Cell. 2007;131:93–105. doi: 10.1016/j.cell.2007.07.034. [DOI] [PubMed] [Google Scholar]
- 42.Cheon H, Stark GR. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:9373–9378. doi: 10.1073/pnas.0903487106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheon H, Yang J, Stark GR. The functions of signal transducers and activators of transcriptions 1 and 3 as cytokine-inducible proteins. J Interferon Cytokine Res. 2011;31:33–40. doi: 10.1089/jir.2010.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pilz A, Ramsauer K, Heidari H, Leitges M, Kovarik P, Decker T. Phosphorylation of the Stat1 transactivating domain is required for the response to type I interferons. EMBO reports. 2003;4:368–373. doi: 10.1038/sj.embor.embor802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uddin S, et al. The Rac1/p38 mitogen-activated protein kinase pathway is required for interferon alpha-dependent transcriptional activation but not serine phosphorylation of Stat proteins. J Biol Chem. 2000;275:27634–27640. doi: 10.1074/jbc.M003170200. [DOI] [PubMed] [Google Scholar]
- 46.Grumbach IM, et al. Engagement of the CrkL adaptor in interferon alpha signalling in BCR-ABL-expressing cells. British journal of haematology. 2001;112:327–336. doi: 10.1046/j.1365-2141.2001.02556.x. [DOI] [PubMed] [Google Scholar]
- 47.Kroczynska B, et al. Regulatory effects of ribosomal S6 kinase 1 (RSK1) in IFNlambda signaling. J Biol Chem. 2011;286:1147–1156. doi: 10.1074/jbc.M110.183566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaur S, et al. Dual regulatory roles of phosphatidylinositol 3-kinase in IFN signaling. J Immunol. 2008;181:7316–7323. doi: 10.4049/jimmunol.181.10.7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rusinova I, et al. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 2013;41:D1040–1046. doi: 10.1093/nar/gks1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schoggins JW, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jouvenet N, et al. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. Journal of virology. 2009;83:1837–1844. doi: 10.1128/JVI.02211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fitzgerald KA. The interferon inducible gene: Viperin. J Interferon Cytokine Res. 2011;31:131–135. doi: 10.1089/jir.2010.0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jamaluddin M, et al. IFN-beta mediates coordinate expression of antigen-processing genes in RSV-infected pulmonary epithelial cells. American journal of physiology. 2001;280:L248–257. doi: 10.1152/ajplung.2001.280.2.L248. [DOI] [PubMed] [Google Scholar]
- 54.Zhang D, Zhang DE. Interferon-stimulated gene 15 and the protein ISGylation system. J Interferon Cytokine Res. 2011;31:119–130. doi: 10.1089/jir.2010.0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Piganis RA, et al. Suppressor of cytokine signaling (SOCS) 1 inhibits type I interferon (IFN) signaling via the interferon alpha receptor (IFNAR1)-associated tyrosine kinase Tyk2. J Biol Chem. 2011;286:33811–33818. doi: 10.1074/jbc.M111.270207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Malakhova OA, et al. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006;25:2358–2367. doi: 10.1038/sj.emboj.7601149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.ten Hoeve J, et al. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol. 2002;22:5662–5668. doi: 10.1128/MCB.22.16.5662-5668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Song L, Bhattacharya S, Yunus AA, Lima CD, Schindler C. Stat1 and SUMO modification. Blood. 2006;108:3237–3244. doi: 10.1182/blood-2006-04-020271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Navarro S, Colamonici OR, Llombart-Bosch A. Immunohistochemical detection of the type I interferon receptor in human fetal, adult, and neoplastic tissues. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 1996;9:150–156. [PubMed] [Google Scholar]
- 60.Kotenko SV, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nature immunology. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 61.Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS pathogens. 2008;4:e1000017. doi: 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mordstein M, et al. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. Journal of virology. 2010;84:5670–5677. doi: 10.1128/JVI.00272-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pulverer JE, et al. Temporal and spatial resolution of type I and III interferon responses in vivo. Journal of virology. 2010;84:8626–8638. doi: 10.1128/JVI.00303-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ank N, et al. An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J Immunol. 2008;180:2474–2485. doi: 10.4049/jimmunol.180.4.2474. [DOI] [PubMed] [Google Scholar]
- 65.Mordstein M, et al. Interferon-lambda contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS pathogens. 2008;4:e1000151. doi: 10.1371/journal.ppat.1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dumoutier L, Tounsi A, Michiels T, Sommereyns C, Kotenko SV, Renauld JC. Role of the interleukin (IL)-28 receptor tyrosine residues for antiviral and antiproliferative activity of IL-29/interferon-lambda 1: similarities with type I interferon signaling. J Biol Chem. 2004;279:32269–32274. doi: 10.1074/jbc.M404789200. [DOI] [PubMed] [Google Scholar]
- 67.Meager A, Visvalingam K, Dilger P, Bryan D, Wadhwa M. Biological activity of interleukins-28 and -29: comparison with type I interferons. Cytokine. 2005;31:109–118. doi: 10.1016/j.cyto.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 68.Sheppard P, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nature immunology. 2003;4:63–68. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- 69.Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–983. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 70.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 71.Arpaia N, Barton GM. Toll-like receptors: key players in antiviral immunity. Current opinion in virology. 2011;1:447–454. doi: 10.1016/j.coviro.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annual review of immunology. 2011;29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- 73.Barber GN. Cytoplasmic DNA innate immune pathways. Immunological Reviews. 2011;243:99–108. doi: 10.1111/j.1600-065X.2011.01051.x. [DOI] [PubMed] [Google Scholar]
- 74.Rathinam VA, Fitzgerald KA. Cytosolic surveillance and antiviral immunity. Current opinion in virology. 2011;1:455–462. doi: 10.1016/j.coviro.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rathinam VA, Fitzgerald KA. Innate immune sensing of DNA viruses. Virology. 2011;411:153–162. doi: 10.1016/j.virol.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Randall RE, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. The Journal of general virology. 2008;89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- 77.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 78.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 79.Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nature immunology. 2003;4:161–167. doi: 10.1038/ni886. [DOI] [PubMed] [Google Scholar]
- 80.Yamamoto M, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 81.Hacker G, Suttner K, Harada H, Kirschnek S. TLR-dependent Bim phosphorylation in macrophages is mediated by ERK and is connected to proteasomal degradation of the protein. Int Immunol. 2006;18:1749–1757. doi: 10.1093/intimm/dxl109. [DOI] [PubMed] [Google Scholar]
- 82.Sato S, et al. Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappa B and IFN-regulatory factor-3, in the Toll-like receptor signaling. J Immunol. 2003;171:4304–4310. doi: 10.4049/jimmunol.171.8.4304. [DOI] [PubMed] [Google Scholar]
- 83.Jiang Z, Mak TW, Sen G, Li X. Toll-like receptor 3-mediated activation of NF-kappaB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-beta. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:3533–3538. doi: 10.1073/pnas.0308496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fitzgerald KA, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nature immunology. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 85.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 86.Uematsu S, et al. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. J Exp Med. 2005;201:915–923. doi: 10.1084/jem.20042372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Marie I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998;17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Izaguirre A, et al. Comparative analysis of IRF and IFN-alpha expression in human plasmacytoid and monocyte-derived dendritic cells. Journal of leukocyte biology. 2003;74:1125–1138. doi: 10.1189/jlb.0603255. [DOI] [PubMed] [Google Scholar]
- 89.Sato M, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 90.Honda K, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 91.Onoguchi K, et al. Viral infections activate types I and III interferon genes through a common mechanism. J Biol Chem. 2007;282:7576–7581. doi: 10.1074/jbc.M608618200. [DOI] [PubMed] [Google Scholar]
- 92.Osterlund P, Pietila T, Veckman V, Kotenko S, Julkunen I. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-lambda) genes. J Immunol. 2007;179:3434–3442. doi: 10.4049/jimmunol.179.6.3434. [DOI] [PubMed] [Google Scholar]
- 93.Yoneyama M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nature immunology. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 94.Pichlmair A, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 95.Hornung V, et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 96.Garcia-Sastre A. Induction and evasion of type I interferon responses by influenza viruses. Virus Res. 2011;162:12–18. doi: 10.1016/j.virusres.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bao X, Liu T, Spetch L, Kolli D, Garofalo RP, Casola A. Airway epithelial cell response to human metapneumovirus infection. Virology. 2007;368:91–101. doi: 10.1016/j.virol.2007.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sen A, Pruijssers AJ, Dermody TS, Garcia-Sastre A, Greenberg HB. The early interferon response to rotavirus is regulated by PKR and depends on MAVS/IPS-1, RIG-I, MDA-5, and IRF3. Journal of virology. 2011;85:3717–3732. doi: 10.1128/JVI.02634-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Broquet AH, Hirata Y, McAllister CS, Kagnoff MF. RIG-I/MDA5/MAVS are required to signal a protective IFN response in rotavirus-infected intestinal epithelium. J Immunol. 2011;186:1618–1626. doi: 10.4049/jimmunol.1002862. [DOI] [PubMed] [Google Scholar]
- 100.Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. 2010;32:305–315. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 101.Abreu M. TLR signalling in the intestinal epithelium: how bactrerial recognition shapes intestinal function. Nat Rev Immunol. 2010:131–144. doi: 10.1038/nri2707. [DOI] [PubMed] [Google Scholar]
- 102.Ioannidis I, Ye F, McNally B, Willette M, Flano E. Toll-like receptor expression and induction of type I and type III interferons in primary airway epithelial cells. Journal of virology. 2013;87:3261–3270. doi: 10.1128/JVI.01956-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 104.Lund JM, et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jewell NA, et al. Differential type I interferon induction by respiratory syncytial virus and influenza a virus in vivo. Journal of virology. 2007;81:9790–9800. doi: 10.1128/JVI.00530-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jewell NA, et al. Lambda interferon is the predominant interferon induced by influenza A virus infection in vivo. Journal of virology. 2010;84:11515–11522. doi: 10.1128/JVI.01703-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Le Goffic R, et al. Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J Immunol. 2007;178:3368–3372. doi: 10.4049/jimmunol.178.6.3368. [DOI] [PubMed] [Google Scholar]
- 108.Kato H, et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 109.Le Goffic R, et al. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS pathogens. 2006;2:e53. doi: 10.1371/journal.ppat.0020053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rudd BD, et al. Deletion of TLR3 alters the pulmonary immune environment and mucus production during respiratory syncytial virus infection. J Immunol. 2006;176:1937–1942. doi: 10.4049/jimmunol.176.3.1937. [DOI] [PubMed] [Google Scholar]
- 111.Bhoj V, et al. MAVS and MyD88 are essential for innate immunity but not cytotoxic T lymphocyte response against respiratory syncytial virus. PNAS. 2008;105:14046–14051. doi: 10.1073/pnas.0804717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hornung V, et al. Replication-dependent potent IFN-alpha induction in human plasmacytoid dendritic cells by a single-stranded RNA virus. J Immunol. 2004;173:5935–5943. doi: 10.4049/jimmunol.173.10.5935. [DOI] [PubMed] [Google Scholar]
- 113.Kurt-Jones EA, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nature immunology. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 114.Rallabhandi P, et al. Analysis of TLR4 polymorphic variants: new insights into TLR4/MD-2/CD14 stoichiometry, structure, and signaling. J Immunol. 2006;177:322–332. doi: 10.4049/jimmunol.177.1.322. [DOI] [PubMed] [Google Scholar]
- 115.Kumagai Y, et al. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity. 2007;27:240–252. doi: 10.1016/j.immuni.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 116.Sen A, et al. Innate immune response to homologous rotavirus infection in the small intestinal villous epithelium at single-cell resolution. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:20667–20672. doi: 10.1073/pnas.1212188109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Estes MK, Kapikian AZ. Rotaviruses. In: Knipe DM, Howley PM, editors. Fields Virology. 5th. Philadelphia, PA: Lippincott, Williams & Wilkins; 2007. pp. 1917–1974. [Google Scholar]
- 118.Deal EM, Jaimes MC, Crawford SE, Estes MK, Greenberg HB. Rotavirus structural proteins and dsRNA are required for the human primary plasmacytoid dendritic cell IFNalpha response. PLoS pathogens. 2010;6:e1000931. doi: 10.1371/journal.ppat.1000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Maniatis T, et al. Structure and function of the interferon-beta enhanceosome. Cold Spring Harb Symp Quant Biol. 1998;63:609–620. doi: 10.1101/sqb.1998.63.609. [DOI] [PubMed] [Google Scholar]
- 120.Levy DE, Marie IJ. How viruses elicit interferon production: Triggering the innate immune response to viral infection. In: Chaugeux JP, Palese P, editors. Modulation of host gene expression and of innate immunity by viruses. Dordrecht: Kluwer Academic Publishers; 2005. [Google Scholar]
- 121.Thomson SJ, et al. The role of transposable elements in the regulation of IFN-lambda1 gene expression. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:11564–11569. doi: 10.1073/pnas.0904477106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang J, et al. Differentiated human alveolar type II cells secrete antiviral IL-29 (IFN-lambda1) in response to influenza A infection. J Immunol. 2009;182:1296–1304. doi: 10.4049/jimmunol.182.3.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lasfar A, et al. Characterization of the mouse IFN-lambda ligand-receptor system: IFN-lambdas exhibit antitumor activity against B16 melanoma. Cancer research. 2006;66:4468–4477. doi: 10.1158/0008-5472.CAN-05-3653. [DOI] [PubMed] [Google Scholar]
- 124.Garcia-Sastre A, et al. The role of interferon in influenza virus tissue tropism. Journal of virology. 1998;72:8550–8558. doi: 10.1128/jvi.72.11.8550-8558.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Price G, Gaszewska-Mastarlarz A, Moskophidis D. The role of alpha/beta and gamma interferons in development of immunity to influenza A virus in mice. Journal of virology. 2000;74:3996–4003. doi: 10.1128/jvi.74.9.3996-4003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Johnson TR, et al. Role for innate IFNs in determining respiratory syncytial virus immunopathology. J Immunol. 2005;174:7234–7241. doi: 10.4049/jimmunol.174.11.7234. [DOI] [PubMed] [Google Scholar]
- 127.Feng N, et al. Role of interferon in homologous and heterologous rotavirus infection in the intestines and extraintestinal organs of suckling mice. Journal of virology. 2008;82:7578–7590. doi: 10.1128/JVI.00391-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. Journal of virology. 2006;80:4501–4509. doi: 10.1128/JVI.80.9.4501-4509.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Pott J, et al. IFN-lambda determines the intestinal epithelial antiviral host defense. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7944–7949. doi: 10.1073/pnas.1100552108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ito T, Amakawa R, Inaba M, Ikehara S, Inaba K, Fukuhara S. Differential regulation of human blood dendritic cell subsets by IFNs. J Immunol. 2001;166:2961–2969. doi: 10.4049/jimmunol.166.5.2961. [DOI] [PubMed] [Google Scholar]
- 131.Montoya M, et al. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood. 2002;99:3263–3271. doi: 10.1182/blood.v99.9.3263. [DOI] [PubMed] [Google Scholar]
- 132.Le Bon A, Schiavoni G, D'Agostino G, Gresser I, Belardelli F, Tough DF. Type i interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity. 2001;14:461–470. doi: 10.1016/s1074-7613(01)00126-1. [DOI] [PubMed] [Google Scholar]
- 133.Padovan E, Spagnoli GC, Ferrantini M, Heberer M. IFN-alpha2a induces IP-10/CXCL10 and MIG/CXCL9 production in monocyte-derived dendritic cells and enhances their capacity to attract and stimulate CD8+ effector T cells. Journal of leukocyte biology. 2002;71:669–676. [PubMed] [Google Scholar]
- 134.Mattei F, Schiavoni G, Belardelli F, Tough DF. IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J Immunol. 2001;167:1179–1187. doi: 10.4049/jimmunol.167.3.1179. [DOI] [PubMed] [Google Scholar]
- 135.Le Bon A, et al. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nature immunology. 2003;4:1009–1015. doi: 10.1038/ni978. [DOI] [PubMed] [Google Scholar]
- 136.van Boxel-Dezaire AH, Zula JA, Xu Y, Ransohoff RM, Jacobberger JW, Stark GR. Major differences in the responses of primary human leukocyte subsets to IFN-beta. J Immunol. 2010;185:5888–5899. doi: 10.4049/jimmunol.0902314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gil MP, Salomon R, Louten J, Biron CA. Modulation of STAT1 protein levels: a mechanism shaping CD8 T-cell responses in vivo. Blood. 2006;107:987–993. doi: 10.1182/blood-2005-07-2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gil MP, et al. Regulating type 1 IFN effects in CD8 T cells during viral infections: changing STAT4 and STAT1 expression for function. Blood. 2012;120:3718–3728. doi: 10.1182/blood-2012-05-428672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Witte K, et al. Despite IFN-lambda receptor expression, blood immune cells, but not keratinocytes or melanocytes, have an impaired response to type III interferons: implications for therapeutic applications of these cytokines. Genes Immun. 2009;10:702–714. doi: 10.1038/gene.2009.72. [DOI] [PubMed] [Google Scholar]
- 140.Doyle SE, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology (Baltimore, Md) 2006;44:896–906. doi: 10.1002/hep.21312. [DOI] [PubMed] [Google Scholar]
- 141.Zhang S, Kodys K, Li K, Szabo G. Human Type 2 Myeloid Dendritic Cells Produce Interferon-lambda and Amplify Interferon-alpha in Response to Hepatitis C Virus Infection. Gastroenterology. 2013;144:414–425 e417. doi: 10.1053/j.gastro.2012.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Megjugorac NJ, Gallagher GE, Gallagher G. Modulation of human plasmacytoid DC function by IFN-lambda1 (IL-29) Journal of leukocyte biology. 2009;86:1359–1363. doi: 10.1189/jlb.0509347. [DOI] [PubMed] [Google Scholar]
- 143.Wolk K, et al. Maturing dendritic cells are an important source of IL-29 and IL-20 that may cooperatively increase the innate immunity of keratinocytes. Journal of leukocyte biology. 2008;83:1181–1193. doi: 10.1189/jlb.0807525. [DOI] [PubMed] [Google Scholar]
- 144.Mennechet FJ, Uze G. Interferon-lambda-treated dendritic cells specifically induce proliferation of FOXP3-expressing suppressor T cells. Blood. 2006;107:4417–4423. doi: 10.1182/blood-2005-10-4129. [DOI] [PubMed] [Google Scholar]
- 145.Yin Z, et al. Type III IFNs are produced by and stimulate human plasmacytoid dendritic cells. J Immunol. 2012;189:2735–2745. doi: 10.4049/jimmunol.1102038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Dolganiuc A, et al. Type III interferons, IL-28 and IL-29, are increased in chronic HCV infection and induce myeloid dendritic cell-mediated FoxP3+ regulatory T cells. PLoS One. 2012;7:e44915. doi: 10.1371/journal.pone.0044915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Liu BS, Janssen HL, Boonstra A. IL-29 and IFNalpha differ in their ability to modulate IL-12 production by TLR-activated human macrophages and exhibit differential regulation of the IFNgamma receptor expression. Blood. 2011;117:2385–2395. doi: 10.1182/blood-2010-07-298976. [DOI] [PubMed] [Google Scholar]
- 148.Dai J, Megjugorac NJ, Gallagher GE, Yu RY, Gallagher G. IFN-lambda1 (IL-29) inhibits GATA3 expression and suppresses Th2 responses in human naive and memory T cells. Blood. 2009;113:5829–5838. doi: 10.1182/blood-2008-09-179507. [DOI] [PubMed] [Google Scholar]
- 149.He S, et al. Interferon-lambda mediates oral tolerance and inhibits antigen-specific, T-helper 2 cell-mediated inflammation. Gastroenterology. 2011;141:249–258. doi: 10.1053/j.gastro.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 150.Koltsida O, et al. IL-28A (IFN-lambda2) modulates lung DC function to promote Th1 immune skewing and suppress allergic airway disease. EMBO molecular medicine. 2011;3:348–361. doi: 10.1002/emmm.201100142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Morrow MP, et al. Unique Th1/Th2 phenotypes induced during priming and memory phases by use of interleukin-12 (IL-12) or IL-28B vaccine adjuvants in rhesus macaques. Clin Vaccine Immunol. 2010;17:1493–1499. doi: 10.1128/CVI.00181-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Morrow MP, et al. IL-28B/IFN-lambda 3 drives granzyme B loading and significantly increases CTL killing activity in macaques. Molecular therapy : the journal of the American Society of Gene Therapy. 2010;18:1714–1723. doi: 10.1038/mt.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Richter MF, Dumenil G, Uze G, Fellous M, Pellegrini S. Specific contribution of Tyk2 JH regions to the binding and the expression of the interferon alpha/beta receptor component IFNAR1. J Biol Chem. 1998;273:24723–24729. doi: 10.1074/jbc.273.38.24723. [DOI] [PubMed] [Google Scholar]
- 154.David M, Petricoin E, 3rd, Benjamin C, Pine R, Weber MJ, Larner AC. Requirement for MAP kinase (ERK2) activity in interferon alpha- and interferon beta-stimulated gene expression through STAT proteins. Science. 1995;269:1721–1723. doi: 10.1126/science.7569900. [DOI] [PubMed] [Google Scholar]
- 155.Usacheva A, et al. The WD motif-containing protein receptor for activated protein kinase C (RACK1) is required for recruitment and activation of signal transducer and activator of transcription 1 through the type I interferon receptor. J Biol Chem. 2001;276:22948–22953. doi: 10.1074/jbc.M100087200. [DOI] [PubMed] [Google Scholar]
- 156.Nguyen VP, et al. Stat2 binding to the interferon-alpha receptor 2 subunit is not required for interferon-alpha signaling. J Biol Chem. 2002;277:9713–9721. doi: 10.1074/jbc.M111161200. [DOI] [PubMed] [Google Scholar]
- 157.Uddin S, Chamdin A, Platanias LC. Interaction of the transcriptional activator Stat-2 with the type I interferon receptor. J Biol Chem. 1995;270:24627–24630. doi: 10.1074/jbc.270.42.24627. [DOI] [PubMed] [Google Scholar]
- 158.Bluyssen HA, et al. Combinatorial association and abundance of components of interferon-stimulated gene factor 3 dictate the selectivity of interferon responses. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:5645–5649. doi: 10.1073/pnas.92.12.5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yang CH, et al. Direct association of STAT3 with the IFNAR-1 chain of the human type I interferon receptor. J Biol Chem. 1996;271:8057–8061. doi: 10.1074/jbc.271.14.8057. [DOI] [PubMed] [Google Scholar]
- 160.Matikainen S, Sareneva T, Ronni T, Lehtonen A, Koskinen PJ, Julkunen I. Interferon-alpha activates multiple STAT proteins and upregulates proliferation-associated IL-2Ralpha, c-myc, and pim-1 genes in human T cells. Blood. 1999;93:1980–1991. [PubMed] [Google Scholar]
- 161.Gupta S, Jiang M, Pernis AB. IFN-alpha activates Stat6 and leads to the formation of Stat2:Stat6 complexes in B cells. J Immunol. 1999;163:3834–3841. [PubMed] [Google Scholar]
- 162.Huang J, et al. Inhibition of type I and type III interferons by a secreted glycoprotein from Yaba-like disease virus. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:9822–9827. doi: 10.1073/pnas.0610352104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rani MR, et al. Catalytically active TYK2 is essential for interferon-beta-mediated phosphorylation of STAT3 and interferon-alpha receptor-1 (IFNAR-1) but not for activation of phosphoinositol 3-kinase. J Biol Chem. 1999;274:32507–32511. doi: 10.1074/jbc.274.45.32507. [DOI] [PubMed] [Google Scholar]
- 164.Du Z, Shen Y, Yang W, Mecklenbrauker I, Neel BG, Ivashkiv LB. Inhibition of IFN-alpha signaling by a PKC- and protein tyrosine phosphatase SHP-2-dependent pathway. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:10267–10272. doi: 10.1073/pnas.0408854102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Abramovich C, Yakobson B, Chebath J, Revel M. A protein-arginine methyltransferase binds to the intracytoplasmic domain of the IFNAR1 chain in the type I interferon receptor. EMBO J. 1997;16:260–266. doi: 10.1093/emboj/16.2.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zheng H, Qian J, Varghese B, Baker DP, Fuchs S. Ligand-stimulated downregulation of the alpha interferon receptor: role of protein kinase D2. Mol Cell Biol. 2011;31:710–720. doi: 10.1128/MCB.01154-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.David M, Zhou G, Pine R, Dixon JE, Larner AC. The SH2 domain-containing tyrosine phosphatase PTP1D is required for interferon alpha/beta-induced gene expression. J Biol Chem. 1996;271:15862–15865. doi: 10.1074/jbc.271.27.15862. [DOI] [PubMed] [Google Scholar]
- 168.Kumar KG, Tang W, Ravindranath AK, Clark WA, Croze E, Fuchs SY. SCF(HOS) ubiquitin ligase mediates the ligand-induced down-regulation of the interferon-alpha receptor. EMBO J. 2003;22:5480–5490. doi: 10.1093/emboj/cdg524. [DOI] [PMC free article] [PubMed] [Google Scholar]